
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIlluminating the mechanistic impacts of an Fe-quaterpyridine functionalized crystalline poly(triazine imide) semiconductor for photocatalytic CO2 reduction†
Scott
McGuigan
 a,
Stephen J.
Tereniak
b,
Avery
Smith
c,
Subhendu
Jana
a,
Carrie L.
Donley
b,
Leonard
Collins
d,
Nandan
Ghorai
e,
Yixuan
Xu
e,
Emmanuel Adu
Fosu
c,
Simon
Suhr
f,
Hannah R. M.
Margavio
g,
Hyuenwoo
Yang
g,
Gregory N.
Parsons
g,
Patrick L.
Holland
f,
Elena
Jakubikova
c,
Tianquan
Lian
e and
Paul A.
Maggard
*a
a,
Stephen J.
Tereniak
b,
Avery
Smith
c,
Subhendu
Jana
a,
Carrie L.
Donley
b,
Leonard
Collins
d,
Nandan
Ghorai
e,
Yixuan
Xu
e,
Emmanuel Adu
Fosu
c,
Simon
Suhr
f,
Hannah R. M.
Margavio
g,
Hyuenwoo
Yang
g,
Gregory N.
Parsons
g,
Patrick L.
Holland
f,
Elena
Jakubikova
c,
Tianquan
Lian
e and
Paul A.
Maggard
*a
aDepartment of Chemistry and Biochemistry, Baylor University, Waco, TX 76798, USA. E-mail: Paul_Maggard@baylor.edu
bDepartment of Chemistry, University of Chapel Hill, Chapel Hill, NC 27599, USA
cDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695, USA
dMolecular Education, Technology and Research Innovation Center, North Carolina State University, Raleigh, NC 27695, USA
eDepartment of Chemistry, Emory University, Atalanta, GA 30322, USA
fDepartment of Chemistry, Yale University, New Haven, CT 06520, USA
gDepartment of Chemical Engineering, North Carolina State University, Raleigh, NC 27695, USA
First published on 17th June 2025
Abstract
The strategy of incorporating earth-abundant catalytic centers into light-absorbing architectures is desirable from the viewpoint of low cost, low toxicity, and versatility for activating small molecules to produce solar-based fuels. Herein, we show that an Fe-quaterpyridine molecular catalyst can be anchored to a light-absorbing, crystalline, carbon nitride (PTI), to yield a molecular-catalyst/material hybrid, Fe-qpy-PTI, capable of facilitating CO2 reduction to CO selectively (up to ∼97–98%) in aqueous solution under low-intensity light irradiation. This hybrid material leverages the ability of the Fe-qpy catalyst to bind CO2 upon a one-electron reduction, as achieved by transfer of excited electrons from the carbon-nitride semiconductor. At a low incident power density of only 50 mW cm−2, the catalytic activity of the hybrid material was measured across a range of catalyst loadings from 0.1–3.8 wt%, yielding CO rates of up to 596 μmol g−1 h−1 for a 3.8 wt% loading during a 3 h experiment. Over the course of 8 h, the hybrid material attained a CO evolution rate of 608 μmol g−1 h−1 and 305 turnovers for a TOF of ∼38 h−1 and an apparent quantum yield of 2.6%. Higher light intensities provided an initial increase in activity but negatively impacted photocatalytic rates with time, with an AQY of 0.6% at 150 mW cm−2 and 0.4% at 250 mW cm−2. Transient absorption spectroscopy results showed electron survival probabilities consistent with the trends in observed product rates. Computational modeling was also used to evaluate and understand the mechanistic pathway of the high product selectivity for CO versus H2. These results thus help unveil key factors for leveraging the mechanistic understanding of molecular catalysts for CO2 reduction for pairing with light absorbing semiconductors and establishing optimal conditions to attain maximal rates in aqueous solution.
I. Introduction
There is a drive in the world of solar fuels catalysis to leverage mechanistic understanding of molecular catalysts for their synergistic functionalization of robust, light absorbing, semiconductors.1–3 Surfaces such as silicon, TiO2, and graphitic carbon nitride (g-CNx) have been explored as semiconducting scaffolds to facilitate light absorption and subsequent charge transfer to surface bound catalysts for activating small molecules such as CO2 and water.4–19 Iron represents an attractive metal center for molecular catalysis, owing to its ubiquitous presence in enzymes and minerals.20–22 It is also earth abundant, readily accessible, and possesses synthetic versatility. Based on its coordination environment, it has demonstrated the capability to catalyze oxidative or reductive reactions. These include water oxidation, C–H bond oxidation, the hydrogen evolution reaction, oxygen reduction to hydrogen peroxide, nitrogen (N2 or NO3−) reduction to ammonia, and most pertinent to this study, CO2 reduction.23–38 Other first-row transition metals recently investigated for electrocatalytic CO2 reduction include Mn, Co, Ni, Cr, and Cu, with each showing various levels of reactivity and product rates.39 While Fe has been found to be less favorable in some regards owing to its tight substrate binding, this study provides an alternative perspective for better harnessing its potential reactivity and product selectivity, such as has been reported more commonly for Co-based and Ni-based molecular catalysts.40 Herein, we use an Fe-quaterpyridine molecular complex immobilized on a crystalline carbon nitride material, i.e., poly(triazine imide) lithium chloride (PTI-LiCl), to investigate selective CO2 reduction driven by light irradiation. The PTI-LiCl material possesses a highly-ordered and crystalline structure for efficient diffusion of charge carriers as well as a functional polymeric framework that can interact with the ligand functional groups of molecular catalysts at its surfaces. Because of these desirable properties, PTI-LiCl has been the subject of much recently reported photocatalytic research demonstrating notable improvements in efficiency and performance compared to other amorphous carbon nitrides.41–43 For example, a recent study involving PTI crystallites as the light-absorbing semiconductor, together with a Co(bpy)32+ molecular catalyst, exhibited CO evolution in mixed media (67% acetonitrile, 33% water, and 33% triethanolamine) with 12.9% apparent quantum yield (AQY).44The choice of catalyst and semiconductor pairing in this work follows from our previous research which demonstrated among the highest observed rates and selectivity currently reported for photocatalytic CO2 reduction in aqueous media using a Co-quaterpyridine molecular catalyst attached to the surfaces of PTI-LiCl.45 Robert et al. have published recent synthetic advancements of this quaterpyridine ligand that facilitates a covalent attachment to g-CNx for high turnover numbers (>2500) in an acetonitrile-based solution.36–38,46 The Fe-centered analogue of this quaterpyridine species has also been shown to electrocatalytically reduce CO2 to CO at high rates with an applied bias. A proposed mechanism of the CO2 to CO reduction reaction on the Fe2+ site has been reported by Head-Gordon and colleagues based on computational results.35 The proposed mechanism consists of two ligand-based reductions stemming from low lying π* orbitals of the quaterpyridine ligand, thus allowing for delocalization of electron density from CO2 into the unoccupied Fe-based orbitals. These interactions permit CO2 binding to the metal center upon an initial 1e− reduction of the molecular complex. Prior to this report, the mechanistic impact of the molecular catalyst has not been well investigated within the context of a light absorbing hybrid framework.
The attachment of the Fe-qpy complex to the surfaces of PTI-LiCl crystallites for photocatalytic CO2 reduction is investigated and described herein. This study reveals the impact of its alternate mechanistic pathway on attaining maximal AQY using increased surface loadings and low light irradiation, as represented in Scheme 1 for the general proposed mechanism of anchoring the Fe-qpy complex to the surface of PTI-LiCl. This hybrid material was investigated for its activity and selectivity in aqueous solution, for which the competitive hydrogen evolution reaction (HER) becomes a more predominant factor and has yet to be evaluated. This study also probes the impact of different molecular-level mechanisms that can result in disparate activity trends dependent on the catalyst loading and the light intensity. These results contrast with our previous research using the analogous Co-quaterpyridine catalyst, wherein a loss of activity occurs at higher surface loadings due to the requirement of generating doubly-reduced catalyst sites for subsequent CO2 coordination and reduction to CO.45 Further, optimal reaction parameters are elucidated for attaining maximal reduction rates and catalyst stability.
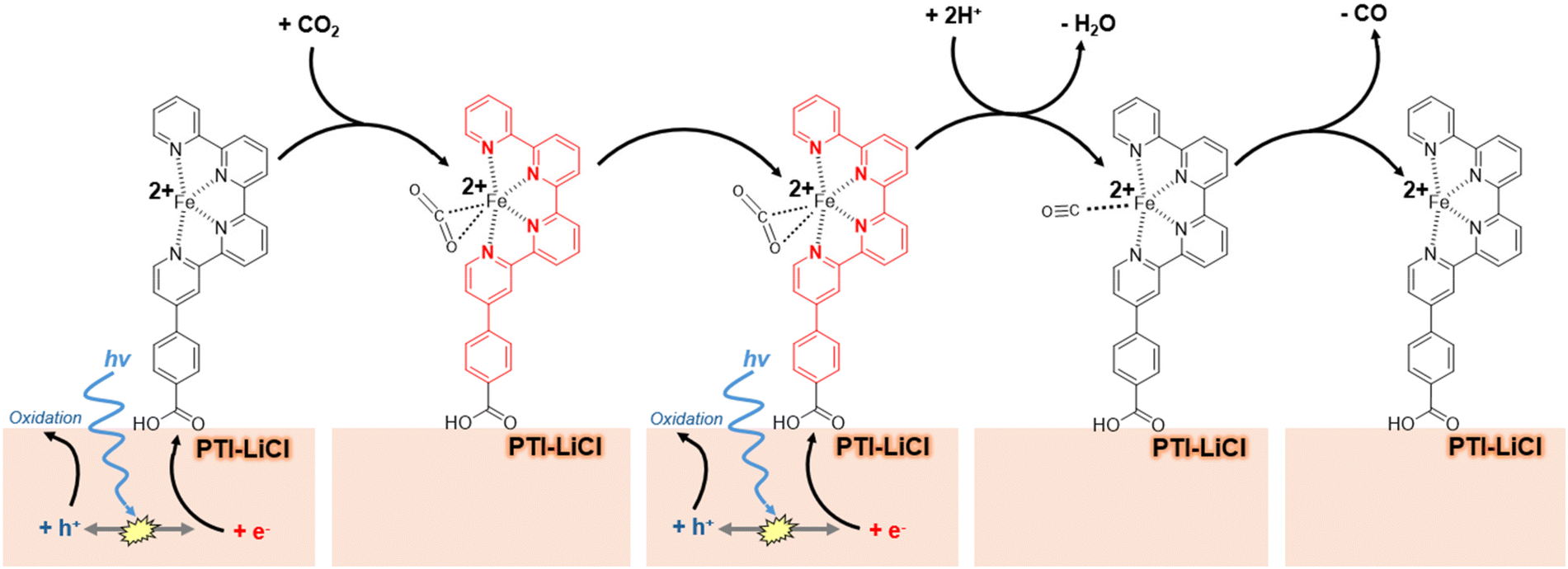 | ||
| Scheme 1 Reduction mechanism for CO2 mediated by an Fe-qpy complex, as envisaged on the light absorbing PTI scaffold. The red-colored ligand indicates a reduced molecular complex. | ||
II. Experimental section
A. Materials
Materials for the synthesis of PTI-LiCl included melamine from Sigma-Aldrich 99%, lithium chloride reagent grade from Fisher Science Education, potassium chloride from Fluka Chemical (>99.5%) or Fisher Chemical (Assay 99.7%), and dimethyl sulfoxide from Millipore Sigma. Anhydrous FeCl2 (99.5% trace metal basis) from BTC Chemicals was used for coordination into the quaterpyridine framework, and ligand materials were sourced as mentioned in ESI.† For photocatalysis measurements, all solutions used ultrapure water, potassium bicarbonate from MP Biomedicals (ACS grade) or Fisher Chemical (99–101.5%), and sodium sulfite (98+%) from Acros Chemical. Carbon dioxide grade 4.0, nitrogen grade 5.0, and argon grade 5.0 from Arc3 gases. NMR analysis utilized 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) from Sigma Aldrich and deuterium oxide (99.9%) from Millipore Sigma or Cambridge Isotope Laboratories, Inc.B. Synthesis and characterization
Crystalline PTI-LiCl was synthesized by mixing melamine with a eutectic flux of KCl (45 at%) and LiCl (55 at%) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mole ratio of carbon nitride precursor to salt flux in ambient conditions with a mortar and pestle. Large batches were made using 1 g of melamine as starting material. The mixture was then transferred into a 140 °C oven overnight to remove any absorbed moisture. The mixture was broken up to break up any large chunks before transferring to a fused silica reaction vessel with ample head space to account for ammonia evolution. This vessel was then sealed under vacuum before reacting in a box furnace. It was heated to 470 °C (7–8 °C per minute) and held at this temperature for 36 h. It was then slowly cooled (2 °C h−1) below the flux melting point at 350 °C and then allowed to radiatively cool to room temperature. The material was then removed from the vessel and washed thoroughly with DI water and lastly with acetone to allow for room temperature drying. Lastly, the product was then thoroughly mixed with mortar and pestle. Powder X-ray Diffraction (PXRD) was conducted on a PANalaytical Empyrean – Linear Detector and non-ambient environment instrument using a Cu Kα source, current of 40 mA, voltage of 45 kV, operating with Bragg–Brentano reflection optics and a PIXcelD detector. FTIR was conducted on a Cary 630 Agilent Spectrometer in the range of 4000 to 400 cm−1. UV-vis Diffuse Reflectance (DR) spectra were collected on a Shimadzu UV-3600 UV-vis-NIR spectrophotometer with an integration sphere range of 200–1000 nm and pressed BaSO4 disks as the reflectance backgrounds. Scanning Electron Microscopy (SEM) was conducted using a Field Emission Scanning Electron Microscope – Hitachi SU8700 with an Everhart-Thomley secondary electron detector, an upper secondary electron detector, and an in-lens backscattered electron detector. XPS data was collected on a Kratos Supra + system with a monochromatic Kα X-ray source operated at 150 W. A charge neutralizer was used to prevent charging when necessary, and all spectra were corrected to the C 1s peak at 284.6 eV. Survey and high-resolution scans were acquired at pass energies of 80 and 20 eV, respectively, and the analyzed spot size was 300 × 700 μm. BET surface area calculations were conducted using a Micrometrics 3Flex instrument on dried PTI-LiCl. N2 adsorption–desorption isotherms were analyzed at 77 K and using relative pressures from 0.05 to 0.3 to analyze specific surface area.
:5 mole ratio of carbon nitride precursor to salt flux in ambient conditions with a mortar and pestle. Large batches were made using 1 g of melamine as starting material. The mixture was then transferred into a 140 °C oven overnight to remove any absorbed moisture. The mixture was broken up to break up any large chunks before transferring to a fused silica reaction vessel with ample head space to account for ammonia evolution. This vessel was then sealed under vacuum before reacting in a box furnace. It was heated to 470 °C (7–8 °C per minute) and held at this temperature for 36 h. It was then slowly cooled (2 °C h−1) below the flux melting point at 350 °C and then allowed to radiatively cool to room temperature. The material was then removed from the vessel and washed thoroughly with DI water and lastly with acetone to allow for room temperature drying. Lastly, the product was then thoroughly mixed with mortar and pestle. Powder X-ray Diffraction (PXRD) was conducted on a PANalaytical Empyrean – Linear Detector and non-ambient environment instrument using a Cu Kα source, current of 40 mA, voltage of 45 kV, operating with Bragg–Brentano reflection optics and a PIXcelD detector. FTIR was conducted on a Cary 630 Agilent Spectrometer in the range of 4000 to 400 cm−1. UV-vis Diffuse Reflectance (DR) spectra were collected on a Shimadzu UV-3600 UV-vis-NIR spectrophotometer with an integration sphere range of 200–1000 nm and pressed BaSO4 disks as the reflectance backgrounds. Scanning Electron Microscopy (SEM) was conducted using a Field Emission Scanning Electron Microscope – Hitachi SU8700 with an Everhart-Thomley secondary electron detector, an upper secondary electron detector, and an in-lens backscattered electron detector. XPS data was collected on a Kratos Supra + system with a monochromatic Kα X-ray source operated at 150 W. A charge neutralizer was used to prevent charging when necessary, and all spectra were corrected to the C 1s peak at 284.6 eV. Survey and high-resolution scans were acquired at pass energies of 80 and 20 eV, respectively, and the analyzed spot size was 300 × 700 μm. BET surface area calculations were conducted using a Micrometrics 3Flex instrument on dried PTI-LiCl. N2 adsorption–desorption isotherms were analyzed at 77 K and using relative pressures from 0.05 to 0.3 to analyze specific surface area.
The quaterpyridine ligand with an attached benzoic acid (qpy) functional group was synthesized according to a previous synthetic method and is described in detail in the ESI.† Coordination of the Fe(II) cation was accomplished by dissolving anhydrous FeCl2 (60.4 mg) in a N2 purged methanol (10 mL) solvent before adding the qpy ligand (61.8 mg). The mix was stirred for 24 h at room temperature under N2 atmosphere before separating the Fe-qpy with a 0.45 μm filter (vacuum filtration) under argon flow. Additional methanol was used to rinse away uncoordinated FeCl2, and the final product was further dried under vacuum for 6 h to completely remove excess solvent before being transferred to an argon-filled glovebox for storage. “Zero-field” Mössbauer measurements were performed using a SEE Co. MS4 Mössbauer spectrometer integrated with Janis SVT-400T He/N2 cryostat for measurements at 80 K with a small 0.07 T applied magnetic field. Isomer shifts were determined relative to alpha-iron at 298 K. Mössbauer spectra were fit using the program MossA using Lorentzian doublets. For Mössbauer spectroscopy, samples were packed in Delrin sample cups and loaded into the spectrometer at 77 K. FTIR of the dried complex was accomplished with a Cary 630 Agilent Spectrometer in the range of 4000 to 400 cm−1. XPS was conducted using a Kratos Supra + system with a monochromatic Kα X-ray source operated at 150 W. A charge neutralizer was used to prevent charging when necessary, and spectra were corrected to the C 1s peak at 284.6 eV. Survey and high-resolution scans were acquired at pass energies of 80 and 20 eV, respectively, and the analyzed spot size was 300 × 700 μm. UV-Vis-NIR spectra was additionally taken of the molecular complex dissolved in a N2 purged DMSO solution on an Agilent Cary Series UV-Vis-NIR spectrophotometer with glass cuvettes having a 1 cm path length.
Lastly, the hybrid Fe-qpy-PTI material was prepared by dissolving the Fe-qpy catalyst into an N2 purged DMSO solvent to yield a starting solution with a 3.66 mM concentration. The solution was then mixed with stoichiometric amounts of PTI-LiCl depending on the desired surface loading. These particle suspensions were then stirred at room temperature in an N2-purged solution for 4 h before being transferred to a Schlenk line for vacuum drying. Once dried the materials were quickly ground in mortar/pestle and then sealed under Ar for longer term storage. The samples were characterized further with XPS analysis as previously described to confirm that the molecular catalyst was detected on the surface of the material. Graphs obtained of the Fe 2p signals utilize a background subtraction and smoothing line for the lower concentration hybrid samples. Powder XRD was used to assess the crystal structure before and after the procedure for molecular catalyst attachment to the surface (Fig. 3d).
C. Characterization of catalyst loading
The Fe-qpy catalyst was first dissolved in N2-purged DMSO and diluted to a 1.46 mM concentration. It was then mixed with a stoichiometric amount of PTI-LiCl powder before allowing it to settle over a couple of days. A small aliquot of the solution was then removed via syringe and centrifuged to remove all residual carbon nitride material from the bulk solution. Lastly, the final solution was then extracted for LC-MS analysis. An Orbitrap Exactice Plus mass spectrometer (Thermo Scientific, Bremen, DE) connected to an UltiMate 3000 liquid chromatography system (Thermo Scientific, Germering, DE) was used to measure complex concentrations. Each sample was diluted by a factor of 1:100 into methanol before injecting 20 μL onto a Hypersil Gold guard cartridge (1 mm × 2.1 mm C18). Compounds were eluted isocritically with 90% methanol and 10% water containing 0.1% formic acid at a flow rate of 100 μL min−1. Mass spectrometer parameters were set as follows: 3.5 kV positive ion mode spray voltage, ion transfer tube temperature of 320 °C, auxiliary gas flow of 8.0, sheath gas flow of 25.0, S-lens RF level of 55, 100% normalized AGC Target, mass resolution of 140 K at m/z 200, and scan range of m/z 515 to 565. The [M]+ ion at m/z 556.0165 was extracted from each chromatogram using a 5 ppm mass window and integrated.
D. Suspended particle photocatalysis
The solution was prepared by mixing 20 mL of 0.5 M KHCO3 with 0.2 M Na2SO3 and purged with CO2 gas for 20 minutes before being added to the reaction vessel with the powder sample of mass 10 mg, unless otherwise specified. The solution and vessel were then purged for a further 10 min before final venting of the headspace with CO2 and sealing off to ambient air with a water bubble. The reaction vessel was then placed in the reaction setup for suspended particle photocatalysis with either no light, 50, 150, or 250 mW cm−2 irradiation power density with a 390 nm LED lamp. Cooling fans are used to maintain a headspace temperature under 28 °C unless otherwise specified. A liquid aliquot was removed before the reactions took place for a comparison against the final liquid concentration. A two-point analysis of the headspace was conducted with initial sampling after 1 h to account for the equilibration of the reaction vessel and initial headspace purging, if needed, with the final gas sample being taken after the duration of the experiment. Gas products were evaluated using manual gas insertion with gas-tight syringe into an SRI 8610C gas chromatograph with Ar carrier gas. Scans were compared against calibration curves generated with standards to evaluate composition of headspace and photogenerated products. 1H NMR analyses of the liquid reaction solvent were evaluated using a Bruker Avance NEO 600 MHz NMR and a reaction mix consisting of 350 μL with reaction solution, D2O (100 μL), and a 0.00028 M 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS) solution (150 μL) as the standard.E. Transient absorption spectroscopy
Transient absorption measurements were performed using regenerative Ti: Sapphire amplifier femtoseconds laser system (Astrella, coherent). The output of the amplifier laser is 5 W, centered at 800 nm, pulse width 35 femtoseconds, and repetition rate 1 kHz. The amplified beam was split into two beams (90:10) by the beam splitter. 90% of the output was fed to an optical parametric amplifier (OPA) to generate a pump beam. The OPA (Light conversion LLC) can generate pump light from 260 to 1600 nm pump wavelength, we can set pump wavelength depending on the desired expectation. We have used 350 nm pump wavelength excitation for all transient experiments. The remaining 10% of the output was used to generate white light probe, the fundamental probe light went through the delay stage for the pump probe experiments. We used sapphire windows to generate white light visible probe. An electrical chopper has been employed to chop pump beams in half in frequency, which is 500 Hz, and natural density filter were used to control pump fluence. The transient pump–probe data was acquired by using a Helios Fire spectrometer (Ultrafast System) and Helios Fire software and analyzed with the Surface Explorer software. All samples were prepared inside a glovebox. First, we dispersed all the samples in 2 mL methanol in a centrifuge tube. Then, we drop casted samples on the sapphire substrate and used another sapphire substrate on the top of the sample. Finally, we inserted it into a Harrick IR cell. We sealed the IR cell with blue tack for further experiments. We have presented the sample preparation procedures schematically in Scheme S1.†
F. Computational techniques
All calculations were done using the Gaussian 16 software package Revision A.0347 with B3LYP functional48–50 including Grimme's D2 dispersion correction.51 Solvation effects were accounted for using the implicit PCM solvation model52 with water as the solvent. Geometry optimization and frequency calculations were done using the split valence triple-ξ (TZ) 6-311G* basis sets for C, N, O and H elements, and the Stuttgart-Dresden (SDD) effective core potential with the associated basis set was employed for Fe.53,54 An ultrafine grid was used for all calculations. Full optimization of all minima on the PES was done without symmetry constraints. The nature of all optimized structures was verified by frequency calculations, that is, all optimized structures had only real frequencies associated with all vibrations. Wavefunction stability tests were performed on all complexes, and only complexes with stable wavefunctions are reported. The electronic state of open shell complexes was further characterized using natural orbital (NO) analysis.55 The UV-Vis spectrum of PTI was modeled with time-dependent density functional theory (TD-DFT),56,57 utilizing the same model chemistry as the ground state calculations. Note that, a single sheet of PTI was used in the excited state calculations. The stick spectra were broadened using Lorentzian functions with a half-width-at-half maximum (HWHM) of 0.14 eV.III. Results and discussion
A. Synthesis and characterization of the Fe-qpy-PTI hybrid material
The components and assembly of the hybrid architecture (Fe-qpy-PTI) followed procedures similar to a prior report.45 First, crystalline PTI-LiCl was synthesized from melamine via a eutectic flux melt at high temperatures. The PTI framework in this material is stabilized by Li cations and Cl anions that coordinate within the pore network of the two-dimensional carbon nitride and between its layers.58,59Fig. 1 (panels a and b) show SEM and optical images of the hexagonal crystallites obtained and the color corresponding to the measured bandgap (3.18 eV). Additional characterization is consistent with its extended and local structural features, as given in Fig. S2 to S4 and Table S1 in the ESI.† The functionalized quaterpyridine (qpy) molecular catalyst was synthesized according to literature methods.19,45 Coordination of Fe to the quaterpyridine ligand was performed by mixing with FeCl2 and N2-purged methanol for 24 h. Mössbauer spectroscopy was used to assess the Fe oxidation state. A Mössbauer spectrum of the coordinated Fe center, shown in Fig. 1c, can be fitted with two doublets. The major doublet accounts for 76% and shows an isomer shift of 1.10 mm s−1 and a quadrupolar splitting of 2.04 mm s−1. These are typical values for a high-spin Fe(II) center. The minor doublet showed an isomer shift of 0.42 mm s−1 and a quadrupolar splitting of 1.15 mm s−1. These values indicate the presence of high-spin Fe(III) accounting for 24% of the total signal. The latter is likely present because of a small amount of oxidation that occurs during the filtration and drying process. Fourier-transform infrared spectroscopic (FTIR) analysis shows that the ligand structure was maintained throughout this coordination, with some shifts in the C–N stretching region (1650–900 cm−1) due to Fe coordination to the quaterpyridine ligand (Fig. 1d). X-ray photoelectron spectroscopy (XPS) analysis yielded elemental contributions consistent with the Fe-containing catalyst (Table S2†). The Fe 2p region of the XPS spectrum showed a mixture of Fe chemical states that were difficult to quantify owing to analytical complexities when differentiating between Fe(II) and Fe(III), but is consistent with the Mössbauer oxidation state measurements (Fig. 1e).60 | ||
| Fig. 1 Characterization results including (a) SEM image of PTI-LiCl crystallites, (b) optical image of PTI-LiCl powder, (c) Mössbauer spectroscopy of the Fe center after coordination to the qpy ligand, (d) FTIR spectra of the qpy ligand before (red dots) and after (black dots with fitted red line) Fe coordination, and (e) XPS data for the Fe-qpy molecular catalyst (Fe 2p region). | ||
The Fe-qpy molecular catalyst and PTI-LiCl were next combined in solution, as shown in Fig. 2a (inset) with its measured UV-Vis spectrum. To probe the interaction between the two, liquid chromatography mass spectrometry (LC-MS) of the dissolved molecular catalyst was measured before and after mixing with PTI-LiCl, Fig. 2c. A calibration curve, mass spectrometry signal, and chromatograph of the pure DMSO solvent (Fig. S7 and S8†) confirmed the dissolution of the Fe-qpy complex and provided a reference point to measure the remnant catalyst after mixing. This method shows that about 40.0 μmol of Fe-qpy attached per gram of carbon nitride. The Brunauer–Emmett–Teller (BET) method gave the isotherm shown in Fig. 2b, which yielded a specific surface area of 17.8 m2 g−1 and a calculated 1.4 Fe-qpy molecules anchored per nm2 of PTI-LiCl surface area. This loading density was consistent with attachment of the carboxylic acid group to the basic N sites throughout the carbon nitride. Unfavorable surface crowding is avoided only when the ligand is oriented perpendicular to the surface, Fig. S9.†
 | ||
| Fig. 2 (a) UV-vis spectra of Fe-qpy dissolved in DMSO with a picture of the solution (inset), (b) BET adsorption isotherm for specific surface area of the PTI-LiCl powder, and (c) LC-MS chromatographs of the solution of dissolved Fe-qpy before and after mixing with PTI-LiCl, and calculated values for the catalyst surface loading. | ||
A range of catalyst loadings on the PTI-LiCl powder was prepared to attain a range of catalyst surface loadings for investigation of its impact on photocatalytic rates and the product selectivity. Stoichiometric suspensions were prepared in the range of 0.1 to 3.8 wt% and mixed for 4 h before vacuum drying to remove the DMSO solvent, as illustrated in Fig. 3a. XPS of the dried material showed an Fe 2p signal characteristic of the starting molecular catalyst, which increases with higher loading amounts on PTI-LiCl. This is shown in Fig. 3b with the loading of 0.3 wt% representing the lowest concentration at which the Fe 2p signal was detectable above the background. The C 1s signal (284.5 eV) associated with the quaterpyridine backbone (C–C bonding) also increases with increased catalyst loading. This differs from the C 1s signal (287.6 eV) that correlates with the C–N groups of the underlying PTI-LiCl framework in Fig. 3c. Powder XRD results shown in Fig. 3d confirm that the diffraction pattern for PTI-LiCl remained predominantly unchanged, and thus that its bulk crystalline structure was maintained throughout the preparation of the hybrid material.
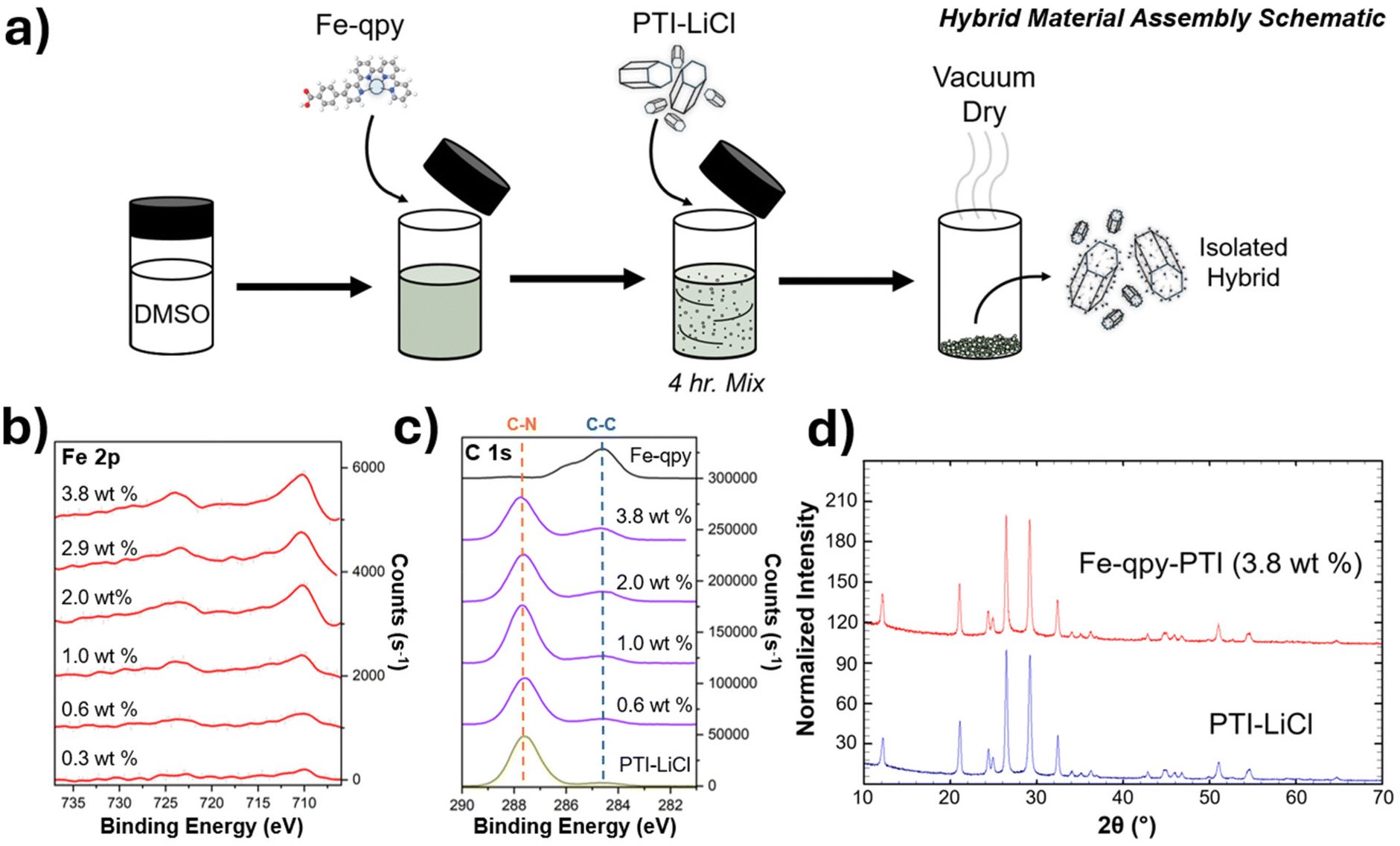 | ||
| Fig. 3 (a) Schematic of hybrid material assembly showing isolation of material for testing, (b) increasing Fe 2p XPS signal as observed in Fig. 1 with increased surface loading of the Fe-qpy molecular catalyst, (c) C 1s XPS spectra showing increased intensity related to the qpy backbone on the surface of the hybrid material, and (d) PXRD patterns of pure PTI-LiCl and after a high loading of molecular catalyst on the surface. | ||
B. Suspended particle photocatalysis
Photocatalytic activity over the range of surface catalyst loadings was measured in a fused-silica reaction vessel irradiated by a 390 nm LED lamp at a relatively low 50 mW cm−2 intensity, as further described in the ESI.† The photocatalytic rates are plotted in Fig. 4a, showing two characteristic regions associated with changes in the molecular catalyst surface loading. With increasing Fe-qpy surface loadings from 0.1 to 1.0 wt% there was a dramatic increase in CO2 reduction rates to 513 μmol CO g−1 h−1. This was accompanied by an increase in the product selectivity for CO and decreasing turnover frequencies (TOF), from 62 h−1 at 0.1 wt% loading of Fe-qpy to 30 h−1 at a 1.0 wt%. The second region, located between 1.0–3.8 wt% of Fe-qpy, exhibited a relative plateau of photcatalytic activity with only a slow gradual increase towards the highest loadings, between 500–600 μmol CO g−1 h−1. Further, throughout this higher loading region the hybrid material exhibited the highest selectivity for CO between 97 to 98% for all samples measured. The calculated apparent quantum yield (AQY) reached above 2.0% for Fe-qpy loadings beyond 1 wt%, with a maximum AQY of 2.4% at the highest loading of 3.8 wt%. These rates for CO2 reduction are among the state-of-art for carbon nitride hybrids for photocatalysis, particularly at the lower irradiation intensity that is more compatible with the diffuse solar spectrum. Specifically, these rates are about 30 to 40% of the highest currently reported, using only about 17 to 30% of the light intensity of prior studies.15,44,45,61 Control studies were conducted using a 0.9 wt% Fe-qpy loading of PTI-LiCl, as well as for the individual components of Fe-qpy-PTI respectively. The results are listed in Table 1. When the CO2 gas was replaced with Ar, i.e., only H2 evolution is observed. There was also no detectable CO evolution when the individual components are subjected to the CO2-saturated photocatalytic reaction conditions, with only a small amount of H2 and HCOO− found when using PTI-LiCl. Further, reduction products were not observed when testing Fe-qpy alone under irradiation.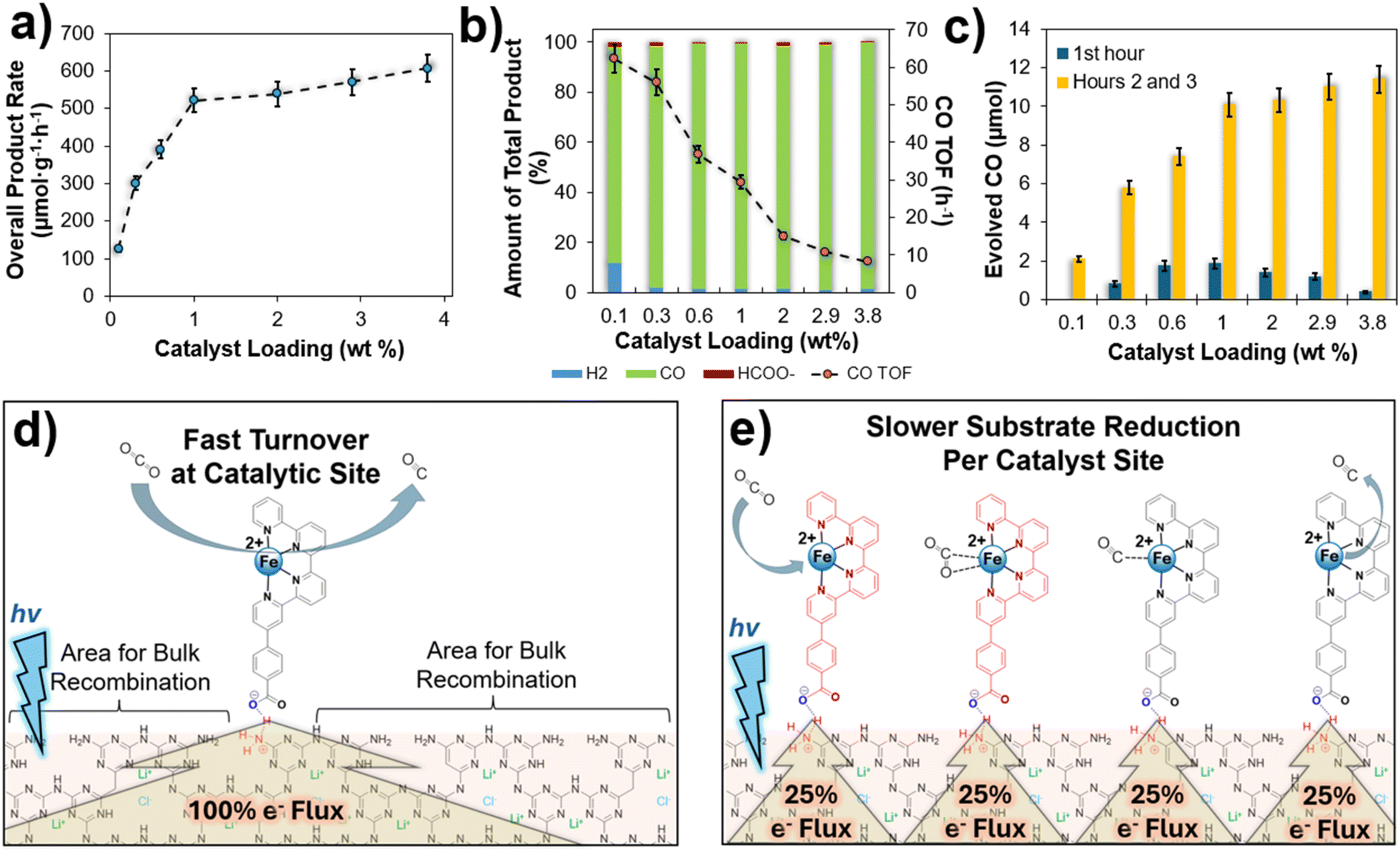 | ||
| Fig. 4 (a) Photocatalytic rates of the Fe-qpy-PTI hybrid material as a function of catalyst loading from 0.1 to 3.8 wt%, (b) product selectivity across the range of samples with associated TOF values for CO2 reduction to CO, (c) comparison of evolved CO after the first hour of catalysis and over the next two hours, (d) schematic of isolated Fe-qpy catalyst site in the case of low loading and higher turnover frequency from receiving a larger share of the photo-generated electrons, and (e) analogous schematic for a higher Fe-qpy loading with the stepwise reduction when receiving a smaller percentage. Red-colored quaterpyridine ligands indicate a reduced molecular complex, with each complex shown at a different step of the mechanism. | ||
Thus, the control experiments generally confirmed that the measured reduction products were obtained by the light-driven reduction of CO2 to CO, as mediated by the excited-state electron transfer from the semiconductor to the Fe-qpy molecular catalyst. When using the Fe-qpy-PTI hybrid, the incident photons are absorbed by the PTI-LiCl semiconductor with the excited electrons diffusing to the Fe-qpy molecular catalysts at the surfaces. Here, the carboxylic acid group of the ligand facilitates attachment to the surface as well as helps passivate the surface defects, as reported in prior studies.62,63 This has been probed by transient absorption spectroscopy below, described below. Electron transfer to the Fe-qpy catalyst facilitates subsequent CO2 coordination and reduction to CO and concurrent suppression of the hydrogen evolution reaction.
Fundamental mechanisms hypothesized for the reduction of CO2 by molecular catalysts can help by providing valuable insights to understand the effects of reaction conditions, such as light intensity and loading dependence, on photocatalytic rates and selectivity.35,45 At low catalyst loadings of Fe-qpy onto PTI (<1 wt%), the highest TOF values are observed. Under these conditions the more isolated catalyst sites can be driven by higher rates of electron transfer at the surface. These conditions lead to the reduction of CO2 at maximal turnover frequencies by the catalyst, but with potential losses in efficiency arising from surface-mediated recombination over the bare surface regions. When the surface loading of the catalyst is increased from 1.0 to 3.8 wt%, a small, gradual increase in photocatalytic CO2 reduction rates occurs with a decrease in the TOF. This occurs as the photogenerated charge carriers are increasingly diluted over a higher proportion of catalyst sites that cover a larger fraction of the surfaces.64–66 In contrast to these findings, the previously reported Co-qpy-PTI hybrid showed a significant decline in rates at the higher catalyst loadings.40 Mechanistic studies show that while the CO2 is predicted to coordinate to the Fe-qpy catalyst after reduction by a single electron, the Co-qpy catalyst requires reduction by two electrons prior to CO2 coordination. Thus, mechanistic pathways mediated by multi-electron steps can be particularly susceptible to losses from back electron transfer and recombination events under conditions where the electron transfer rates from the semiconductor are sub-optimal. Another impact of the mechanistic difference for the two-electron reduction of CO2 is revealed when comparing the amount of CO formed during the first hour, versus hours two and three, as shown in Fig. 4c. Over the first hour, the 1.0 wt% Fe-qpy sample exhibited the highest CO product rate. In the following two hours, the Fe-qpy-PTI hybrid materials with higher catalyst loadings attained significantly higher rates. This is likely the result of a longer induction period necessary to build up a larger amount of two-electron reduced catalysts sites to start achieving maximal catalyst turnover at the higher catalyst loadings. Fig. 4d and e schematically represent the low and high catalyst surface loadings respectively.
The photocatalysis rates for CO2 reduction reported here are competitive with current carbon nitride hybrid materials used for photocatalysis, particularly with respect to the lower light intensity used compared to most studies.15,44,45,61 Prior research reports have also optimized the design of the reactor setup to attain the highest AQYs, with less emphasis on maximizing rates and product selectivity. Future developments of a more tightly engineered and efficient reactor setup, while not the predominant focus of the current study, could likely lead to improvements in the calculated AQY metrics in this work.
C. Transient absorption spectroscopy
Transient absorption spectroscopy (TAS) measurements were performed on the Fe-qpy-PTI architectures with catalyst loadings of 0.0, 0.1, 1.0, 2.0 and 2.9 wt%. Thin films of these materials were prepared via a drop cast method into a transparent sapphire window (Scheme S1†). TAS measurements were conducted in the visible (500–760 nm) and near-infrared (950–1200 nm) regions following excitation with a 350 nm laser pump. The TA spectra revealed a broad bleach signal from 500 to 760 nm (Fig. S12†) in all samples. Based on literature precedents for PTI-LiCl, this is attributed to stimulated emission.67,68 The transient kinetics in 500–580 nm probing wavelength region, as shown in Fig. 5a, exhibited negligible dependence on catalyst loading, which is consistent with our previous report.45 However, TA spectra in the near infrared probing (950–1200 nm) wavelength, shown in Fig. S13,† contained extended featureless photo-induced absorption signals, which are attributed to photogenerated electrons in the carbon nitride framework.69,70 TAS decay kinetics at 950–1200 nm as a function of catalyst loading are presented in Fig. 5b. Unlike visible-light probing, the decay kinetics of electrons in this region became slower when Fe-qpy catalyst loading increased from 0.0 wt% to 0.1 wt% and were significantly extended at high catalyst loadings 1.0 wt% to 2.9 wt%. Decay kinetics at low catalyst loadings can be well fitted by a power law relationship, (ΔA(t)∝t−β) as in previous studies.71,72 However, the data deviated from the power law relationship at higher loadings (1.0 wt% to 2.9 wt% Fe-qpy). The fits are shown in Fig. 5b. | ||
| Fig. 5 TAS data of Fe-qpy-PTI films with different loadings of the Fe-qpy catalyst. (a) Decay kinetics measured using a detection range of 500–580 nm, (b) decay kinetics measured using a detection range of 950–1200 nm, (c) comparison of overall product rate and electron survival lifetime with catalyst loading, and (d) a schematic of electron excitation and transfer with and without Fe-qpy on PTI. | ||
The long-lived electrons are hypothesized to be responsible for the catalytic reactions, and the electron survival probabilities are defined as the relative amplitude of the TA signal at 100–1000 ps compared to the initial amplitude at 0.1–0.3 ps. In Fig. 5c, the electron survival probability and overall product rate are compared from suspended particle photocatalysis as a function of catalyst loading. Each exhibits similar trends, with a rapid increase with catalyst loading up to 1.0 wt% and showing smaller changes upon further increases in loading. The initial increase of electron lifetime can be attributed to surface state passivation by the adsorbed molecular complex, enabling photogenerated electrons to be transferred to the adsorbed catalysts that drive reduction events, circumventing recombination as represented in Fig. 5d.45 At further increased surface concentrations the catalyst sites can function as rapid electron acceptors and may limit electron lifetimes.45 The competition of these effects leads to the trend observed in the electron survival probability between 1.0–2.9 wt% Fe-qpy loadings, without a continued increase in lifetime. Interestingly, at these high loadings the electron survival probability is significantly prolonged and does not appreciably decrease. Here, the unique orbital interactions between the Fe(II) center and the ligand is hypothesized to prolong the survivability of these photogenerated charge carriers from the ligand centered reduction, maintaining the catalytic efficiency observed at 1.0–3.8 wt% Fe-qpy surface loadings.
D. Computational analysis
Density functional theory (DFT) calculations were performed to provide further insights into the catalytic mechanism, the thermodynamics of the CO2 reduction steps to CO with the Fe-qpy-PTI hybrid in water (using the implicit PCM73 solvation model), and a comparison with the hydrogen evolution pathway. The DFT calculations suggest that [Fe(qpy)]2+ is in the quintet ground electronic state, which is consistent with the experimental Mössbauer data. The spin density and natural orbital analysis showed all unpaired electrons localized on the Fe metal center with empty low-lying quaterpyridine π* orbitals, suggesting electron transfer from PTI* into these orbitals (Fig. 6a). Photocatalytic reduction of CO2 to CO is found to proceed via two main steps: (i) the photoexcitation step (Fig. S16†) followed by a photo-induced electron transfer to the catalyst and (ii) the main catalytic cycle. Of note, a molecular model of the PTI network was used for the light absorber (Fig. S14†). The photoexcitation step involves an initial excitation of the PTI molecular sheet, leading to the formation of the excited PTI* species, which transfers an electron to Fe-qpy to form the oxidized PTI+ intermediate, that is lastly regenerated (reduced) by a sacrificial reagent.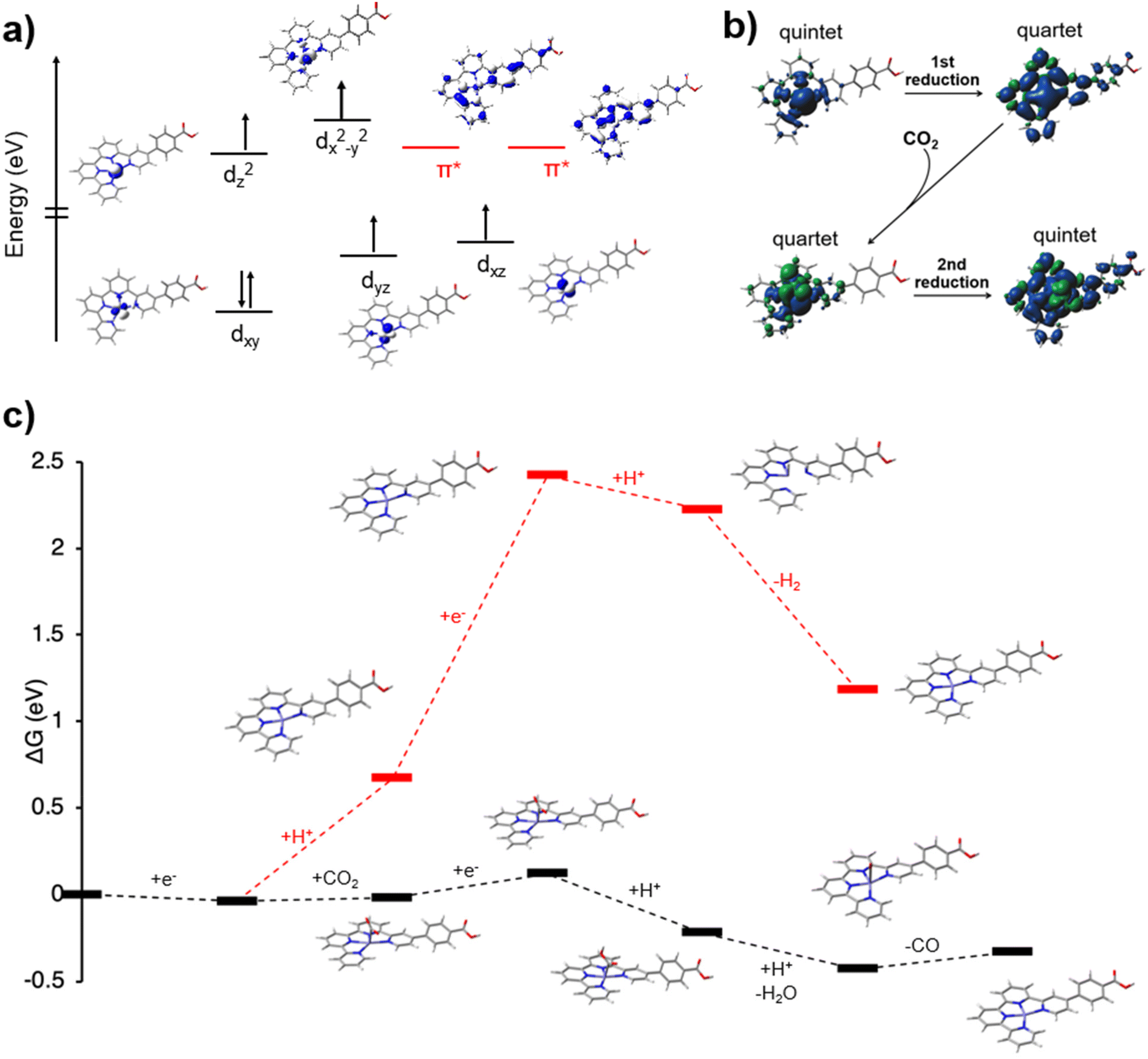 | ||
| Fig. 6 Computational CO2 reduction mechanism on Fe-qpy, including (a) ground state electronic structure of the Fe-qpy complex exhibiting a quintet d-electron orientation and empty low-lying qpy orbitals, (b) spin density in the different steps of the 2e− reduction process, and (c) a thermodynamic free energy profile for CO2RR (black) and HER (red) pathways under an applied excitation energy of −3.19 eV. | ||
One of the key questions in CO2 photocatalysis is the sequence of the catalyst reduction (electron transfer) and CO2 binding steps. The spin density during reduction and the mechanistic pathway obtained from these calculations is outlined in Fig. 6b and c, with the Fe-qpy complex modeled as receiving excited electrons from PTI for reduction. Note that the free energy profiles shown are with respect to the applied excitation energy (3.19 eV). In agreement with the previous literature precedent35 as well as our experimental findings, the active catalyst is found to be the [Fe(qpy)]+ quartet, wherein the reduction was quaterpyridine ligand-centered as shown in Fig. 6b with significant electron density contributions located on the quaterpyridine ligand. The reduction of [Fe(qpy)]2+ to [Fe(qpy)]+ was followed by CO2 binding (ΔG = +0.02 eV). The CO2 binding step was then followed by the second excited-state electron transfer from PTI*, where the electron was again transferred to the empty qpy ligand π* orbitals (Fig. 6b). Upon reaching the doubly-reduced [Fe(qpy)(CO2)] intermediate, subsequent steps proceeded via a protonation of the bound CO2 to form COOH, followed by concerted protonation and water dissociation steps (ΔG = −0.21 eV). A possible stepwise loss of hydroxyl followed by protonation to form H2O is endergonic (∼1.66 eV), which suggested water, rather than hydroxide, as the byproduct. Although the CO dissociation was uphill, the thermodynamic stability of previous steps mitigated this and resulted in an overall exergonic CO release (Fig. 6c).
Lastly, the mechanistic pathway was calculated for the competing hydrogen evolution reaction (HER) on the [Fe(qpy)]2+ complex. The computational results showed that while the initial reduction was comparable for the two pathways, the second electron reduction is significantly more favorable for the CO2RR route (0.14 eV) than for the HER route (1.75 eV). After this second reduction step for the HER pathway the remaining mechanistic pathway was overall exergonic, including H binding to the Fe metal center before H2 formation and release. However, this remained less favorable due to the higher energy input required for the second reduction. These results are consistent with the experimental data in which CO2 reduction to CO dominates the reactivity. Given the calculated energies for the HER pathway, these results do not eliminate the possibility of H2 formation, as observed in the control studies when CO2 was absent from the reaction. Though, the selective reduction of CO2 to CO is shown to be the energetically preferred pathway for the Fe-qpy molecular catalyst.
E. Impact of incident power density on catalytic activity
Photocatalytic rates were also investigated at higher irradiation intensities to assess the impact of higher rates of excited charge carriers reaching the surface Fe-qpy catalyst sites. The Fe-qpy-PTI materials with higher catalyst loadings should have the capacity to attain higher rates, in theory, as previously described for the Co-qpy-PTI hybrid material.45 The impact of increased catalyst loading could potentially be accentuated for the Fe-qpy complex, by leveraging its capability to bind the CO2 substrate upon its first electron reduction. Interestingly, initial experiments at a higher photon flux (150 mW cm−2) showed significantly lower activity than expected (Fig. S21†). This result suggested that higher incident light intensities may cause decomposition to the hybrid. To test this hypothesis, photocatalysis reactions were conducted on a hybrid material with 0.9 wt% Fe-qpy catalyst loading at three different light intensities with an extended reaction time of 8 h, shown in Fig. 7a. At 50 mW cm−2 irradiation intensity, the CO evolution rate was 608 μmol g−1 h−1 with 97% selectivity for 305 total turnovers. The resultant AQY was calculated to be 2.6%, slightly increased from the 3 h reaction experiments. Significantly decreased rates were observed at 150 mW cm−2 and at 250 mW cm−2, showing a lower selectivity for CO and lower AQY values of 0.6% and 0.4%, respectively. However, the rate of product evolution during the first hour of each reaction strongly correlated with the increased light intensity, as shown in Fig. 7b. Their rates of CO evolution drastically decreased over the duration of the experiment, to less than 50% of the rate observed at 50 mW cm−2. The trend in the photocatalytic rates versus time for the Fe-qpy-PTI hybrid at the three different light intensities is illustrated in Fig. 7c, with sustainable activity associated with the lowest light intensity. The sample temperature was monitored and remained similar with each experiment (±7 °C), and so any decomposition was unlikely to arise from sample heating. Removal of the cooling fan from the experimental setup gave only a slight decrease in efficiency, Fig. S22,† with other reaction conditions kept constant. This deactivation is also unlikely to be associated with the durability of the PTI-LiCl scaffold, as prior studies have demonstrated its exceptional durability for 32 h under similar reaction conditions. While the mechanism of catalyst deactivation is unclear, it can most likely be attributed to the Fe-qpy catalyst.45,74 Recent research has suggested that a potential contributor may be over-reduction to an unstable Fe0-CO complex, which decomposes, halting further catalysis.75 Nonetheless, while this behavior is considered unfavorable to access the highest possible turnover numbers capable for this molecular catalyst, the ability of this hybrid interaction to effectively utilize low incident power densities makes it an attractive option for solar applications. On average, normal solar irradiance at the surface of the earth is 1 Sun (AM 1.5 G or 100 mW cm−2), but with only ∼50.3% representing ultraviolet and visible light wavelenths.76,77 For solar-driven applications, light-harvesting systems will be needed with operational efficiency and stability at lower light intensities, such as in relatively cloudy environments or with increased levels of light scattering.78–80 | ||
| Fig. 7 (a) Photocatalytic rates and selectivity as a function of applied irradiation intensity over the course of an 8 h measurement, (b) initial gaseous product evolution (first hour) of the 8 h experiments showing differing selectivity than the remaining duration of the experiments, and (c) schematic of the light intensity dependent stability of the Fe-qpy-PTI hybrid over time. | ||
IV. Conclusions
The Fe-qpy-PTI hybrid material was found to be photocatalytically active for selective CO2 reduction to CO as suspended particles in aqueous solution under relatively low light intensities. The measured rates of CO evolution reached up to 596 μmol g−1 h−1 in 3 h, demonstrating high selectivity (>97%) at a low incident power density of 50 mW cm−2. An increase in CO evolution was observed as the loading of Fe-qpy on PTI-LiCl was increased from 0.1 to 1.0 wt%, with efficiency maintained and slight rate increases observed beyond this concentration. Time-resolved spectroscopy analysis indicated that at higher concentrations (1.0–2.9 wt%), the decay rates from excited electronic states were slower compared to bare PTI-LiCl or to the lower 0.1 wt% loading, consistent with the photocatalysis rates. Computational studies suggest a viable mechanistic pathway for CO2 reduction in aqueous reaction medium, highlighting the coordination of CO2 by the Fe-qpy complex and the energetically favorable CO2 reduction reaction over the hydrogen evolution reaction. In experiments with a higher photon flux, the hybrid material exhibited deactivation over time, leading to a reduced overall activity and selectivity, despite an initially faster rate. By utilizing lower-intensity irradiation, the hybrid material's lifetime was extended, achieving a CO evolution rate of 608 μmol g−1 h−1 over 8 hours. This research demonstrates the precedent of leveraging the fundamental understanding of mechanistic pathways in molecular catalysts for CO2 reduction to achieve a synergistic integration with light-absorbing semiconductors under optimal reaction reactions.Author contributions
Formal analysis, investigation, visualization, and writing – all authors: SM, ST, AS, SJ, CD, LC, NG, YX, EF, SS, HM, HY, GP, PH, EJ, TL and PM; project administration and supervision – GP, PH, EJ, TL and PM.Conflicts of interest
The authors have no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported as part of the Center for Hybrid Approaches in Solar Energy to Liquid Fuels (CHASE), an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0021173. Research efforts were performed in part at the Molecular Education, Technology and Research Innovation Center (METRIC) at NC State University and performed in part at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-2025064). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI). This work used the High-Performance Research Computing FASTER cluster at Texas A&M University through allocation CHE240107 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program,81 which is supported by U.S. National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296. Additional efforts were performed at the University of North Carolina's Chapel Hill Analytical and Nanofabrication Laboratory, CHANL, a member of the North Carolina Research Triangle Nanotechnology Network, RTNN, which is supported by the National Science Foundation, Grant ECCS-2025064, as part of the National Nanotechnology Coordinated Infrastructure, NNCI. The authors thank the University of North Carolina's Department of Chemistry NMR Core Laboratory for the use of their NMR spectrometers, particularly the instrument funded under the National Science Foundation Grant No. CHE-1828183. The authors thank the University of North Carolina's Department of Chemistry Mass Spectrometry Core Laboratory for the use of their mass spectrometer funded by the National Science Foundation under Grant No. CHE-1726291.References
- B. Zhang and L. Sun, Artificial Photosynthesis: Opportunities and Challenges of Molecular Catalysts, Chem. Soc. Rev., 2019, 48(7), 2216–2264 RSC.
- X. Liang, X. Cao, W. Sun and Y. Ding, Recent Progress in Visible Light Driven Water Oxidation Using Semiconductors Coupled with Molecular Catalysts, ChemCatChem, 2019, 11(24), 6190–6202 CrossRef CAS.
- T. Morikawa, S. Sato, K. Sekizawa, T. Arai and T. M. Suzuki, Molecular Catalysts Immobilized on Semiconductor Photosensitizers for Proton Reduction toward Visible-Light-Driven Overall Water Splitting, ChemSusChem, 2019, 12(9), 1807–1824 CrossRef CAS PubMed.
- H.-Y. Liu, C. C. Cody, J. A. Jayworth, R. H. Crabtree and G. W. Brudvig, Surface-Attached Molecular Catalysts on Visible-Light-Absorbing Semiconductors: Opportunities and Challenges for a Stable Hybrid Water-Splitting Photoanode, ACS Energy Lett., 2020, 5(10), 3195–3202 CrossRef CAS.
- X. Jia, E. Stewart-Jones, J. L. Alvarez-Hernandez, G. P. Bein, J. L. Dempsey, C. L. Donley, N. Hazari, M. N. Houck, M. Li, J. M. Mayer, H. S. Nedzbala and R. E. Powers, Photoelectrochemical CO2 Reduction to CO Enabled by a Molecular Catalyst Attached to High-Surface-Area Porous Silicon, J. Am. Chem. Soc., 2024, 146(12), 7998–8004 CrossRef CAS PubMed.
- G. P. Bein, M. A. Stewart, E. A. Assaf, S. J. Tereniak, R. N. Sampaio, A. J. M. Miller and J. L. Dempsey, Methyl Termination of P-Type Silicon Enables Selective Photoelectrochemical CO2 Reduction by a Molecular Ruthenium Catalyst, ACS Energy Lett., 2024, 9(4), 1777–1785 CrossRef CAS.
- M. Laurans, J. A. L. Wells and S. Ott, Immobilising Molecular Ru Complexes on a Protective Ultrathin Oxide Layer of P-Si Electrodes towards Photoelectrochemical CO2 Reduction, Dalton Trans., 2021, 50(30), 10482–10492 RSC.
- X. Jia, H. S. Nedzbala, S. R. Bottum, J. F. Cahoon, J. J. Concepcion, C. L. Donley, A. Gang, Q. Han, N. Hazari, M. C. Kessinger, M. R. Lockett, J. M. Mayer, B. Q. Mercado, G. J. Meyer, A. J. Pearce, C. L. Rooney, R. N. Sampaio, B. Shang and H. Wang, Synthesis and Surface Attachment of Molecular Re(I) Complexes Supported by Functionalized Bipyridyl Ligands, Inorg. Chem., 2023, 62(5), 2359–2375 CrossRef CAS PubMed.
- T. V. Pho, M. V. Sheridan, Z. A. Morseth, B. D. Sherman, T. J. Meyer, J. M. Papanikolas, K. S. Schanze and J. R. Reynolds, Efficient Light-Driven Oxidation of Alcohols Using an Organic Chromophore–Catalyst Assembly Anchored to TiO2, ACS Appl. Mater. Interfaces, 2016, 8(14), 9125–9133 CrossRef CAS.
- H. Shimakoshi and Y. Hisaeda, A Hybrid Catalyst for Light-Driven Green Molecular Transformations, ChemPlusChem, 2017, 82(1), 18–29 CrossRef CAS PubMed.
- C. Liu, T. Jin, M. E. Louis, S. A. Pantovich, S. L. Skraba-Joiner, T. Rajh and G. Li, Molecular Deposition of a Macrocyclic Cobalt Catalyst on TiO2 Nanoparticles, J. Mol. Catal. A: Chem., 2016, 423, 293–299 CrossRef CAS.
- T. E. Rosser, C. D. Windle and E. Reisner, Electrocatalytic and Solar-Driven CO2 Reduction to CO with a Molecular Manganese Catalyst Immobilized on Mesoporous TiO2, Angew. Chem., Int. Ed., 2016, 55(26), 7388–7392 CrossRef CAS.
- M. S. Choe, S. Choi, S.-Y. Kim, C. Back, D. Lee, H. S. Lee, C. H. Kim, H.-J. Son and S. O. Kang, A Hybrid Ru(II)/TiO2 Catalyst for Steadfast Photocatalytic CO2 to CO/Formate Conversion Following a Molecular Catalytic Route, Inorg. Chem., 2021, 60(14), 10235–10248 CrossRef CAS.
- C. D. Windle, E. Pastor, A. Reynal, A. C. Whitwood, Y. Vaynzof, J. R. Durrant, R. N. Perutz and E. Reisner, Improving the Photocatalytic Reduction of CO2 to CO through Immobilisation of a Molecular Re Catalyst on TiO2, Chem. – Eur. J., 2015, 21(9), 3746–3754 CrossRef CAS PubMed.
- B. Shang, F. Zhao, C. Choi, X. Jia, M. Pauly, Y. Wu, Z. Tao, Y. Zhong, N. Harmon, P. A. Maggard, T. Lian, N. Hazari and H. Wang, Monolayer Molecular Functionalization Enabled by Acid–Base Interaction for High-Performance Photochemical CO2 Reduction, ACS Energy Lett., 2022, 7(7), 2265–2272 CrossRef CAS.
- B. Das, M. Devi, S. Deb and S. S. Dhar, Boosting Photocatalytic Property of Graphitic Carbon Nitride with Metal Complex Fabrication for Efficient Degradation of Organic Pollutants, Chemosphere, 2023, 323, 138230 CrossRef CAS PubMed.
- S. Roy and E. Reisner, Visible-Light-Driven CO2 Reduction by Mesoporous Carbon Nitride Modified with Polymeric Cobalt Phthalocyanine, Angew. Chem., Int. Ed., 2019, 58(35), 12180–12184 CrossRef CAS.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Visible-Light-Driven CO2 Reduction with Carbon Nitride: Enhancing the Activity of Ruthenium Catalysts, Angew. Chem., Int. Ed., 2015, 54(8), 2406–2409 CrossRef CAS.
- B. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T.-C. Lau, J. Bonin and M. Robert, Efficient Visible-Light-Driven CO2 Reduction by a Cobalt Molecular Catalyst Covalently Linked to Mesoporous Carbon Nitride, J. Am. Chem. Soc., 2020, 142(13), 6188–6195 CrossRef CAS PubMed.
- D. Buongiorno and G. D. Straganz, Structure and Function of Atypically Coordinated Enzymatic Mononuclear Non-Heme-Fe(II) Centers, Coord. Chem. Rev., 2013, 257(2), 541–563 CrossRef CAS PubMed.
- F. W. Outten and E. C. Theil, Iron-Based Redox Switches in Biology, Antioxid. Redox Signal., 2008, 11(5), 1029–1046 CrossRef PubMed.
- J. Huang, A. Jones, T. D. Waite, Y. Chen, X. Huang, K. M. Rosso, A. Kappler, M. Mansor, P. G. Tratnyek and H. Zhang, Fe(II) Redox Chemistry in the Environment, Chem. Rev., 2021, 121(13), 8161–8233 CrossRef CAS PubMed.
- M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, A Pentanuclear Iron Catalyst Designed for Water Oxidation, Nature, 2016, 530(7591), 465–468 CrossRef CAS PubMed.
- J. Lloret-Fillol and M. Costas, Chapter One - Water Oxidation at Base Metal Molecular Catalysts, in Advances in Organometallic Chemistry, ed. P. J. Pérez, Academic Press, 2019, vol. 71, pp. 1–52 Search PubMed.
- A. C. Lindhorst, S. Haslinger and F. E. Kühn, Molecular Iron Complexes as Catalysts for Selective C–H Bond Oxygenation Reactions, Chem. Commun., 2015, 51(97), 17193–17212 RSC.
- W. T. Eckenhoff, Molecular Catalysts of Co, Ni, Fe, and Mo for Hydrogen Generation in Artificial Photosynthetic Systems, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- F. Gärtner, A. Boddien, E. Barsch, K. Fumino, S. Losse, H. Junge, D. Hollmann, A. Brückner, R. Ludwig and M. Beller, Photocatalytic Hydrogen Generation from Water with Iron Carbonyl Phosphine Complexes: Improved Water Reduction Catalysts and Mechanistic Insights, Chem. – Eur. J., 2011, 17(23), 6425–6436 CrossRef PubMed.
- M. J. Rose, H. B. Gray and J. R. Winkler, Hydrogen Generation Catalyzed by Fluorinated Diglyoxime–Iron Complexes at Low Overpotentials, J. Am. Chem. Soc., 2012, 134(20), 8310–8313 CrossRef CAS PubMed.
- S. Kaur-Ghumaan, L. Schwartz, R. Lomoth, M. Stein and S. Ott, Catalytic Hydrogen Evolution from Mononuclear Iron(II) Carbonyl Complexes as Minimal Functional Models of the [FeFe] Hydrogenase Active Site, Angew. Chem., Int. Ed., 2010, 49(43), 8033–8036 CrossRef CAS PubMed.
- C. Costentin, H. Dridi and J.-M. Savéant, Molecular Catalysis of O2 Reduction by Iron Porphyrins in Water: Heterogeneous versus Homogeneous Pathways, J. Am. Chem. Soc., 2015, 137(42), 13535–13544 CrossRef CAS PubMed.
- R. A. Ghiladi, R. M. Kretzer, I. Guzei, A. L. Rheingold, Y.-M. Neuhold, K. R. Hatwell, A. D. Zuberbühler and K. D. Karlin, (F8TPP)FeII/O2 Reactivity Studies {F8TPP = Tetrakis(2,6-Difluorophenyl)Porphyrinate(2−)}: Spectroscopic (UV−Visible and NMR) and Kinetic Study of Solvent-Dependent (Fe/O2 = 1:1 or 2:1) Reversible O2-Reduction and Ferryl Formation, Inorg. Chem., 2001, 40(23), 5754–5767 CrossRef CAS PubMed.
- A. Fürstner, Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion, ACS Cent. Sci., 2016, 2(11), 778–789 CrossRef PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Catalytic Conversion of Nitrogen to Ammonia by an Iron Model Complex, Nature, 2013, 501(7465), 84–87, DOI:10.1038/nature12435.
- A. Genoux, M. Pauly, C. L. Rooney, C. Choi, B. Shang, S. McGuigan, M. S. Fataftah, Y. Kayser, S. C. B. Suhr, S. DeBeer, H. Wang, P. A. Maggard and P. L. Holland, Well-Defined Iron Sites in Crystalline Carbon Nitride, J. Am. Chem. Soc., 2023, 145(38), 20739–20744 CrossRef CAS PubMed.
- M. Loipersberger, D. G. A. Cabral, D. B. K. Chu and M. Head-Gordon, Mechanistic Insights into Co and Fe Quaterpyridine-Based CO2 Reduction Catalysts: Metal–Ligand Orbital Interaction as the Key Driving Force for Distinct Pathways, J. Am. Chem. Soc., 2021, 143(2), 744–763 CrossRef CAS PubMed.
- C. Cometto, R. Kuriki, L. Chen, K. Maeda, T.-C. Lau, O. Ishitani and M. Robert, A Carbon Nitride/Fe Quaterpyridine Catalytic System for Photostimulated CO2-to-CO Conversion with Visible Light, J. Am. Chem. Soc., 2018, 140(24), 7437–7440 CrossRef CAS PubMed.
- Z. Guo, S. Cheng, C. Cometto, E. Anxolabéhère-Mallart, S.-M. Ng, C.-C. Ko, G. Liu, L. Chen, M. Robert and T.-C. Lau, Highly Efficient and Selective Photocatalytic CO2 Reduction by Iron and Cobalt Quaterpyridine Complexes, J. Am. Chem. Soc., 2016, 138(30), 9413–9416 CrossRef CAS PubMed.
- Y. Wei, L. Chen, H. Chen, L. Cai, G. Tan, Y. Qiu, Q. Xiang, G. Chen, T. Lau and M. Robert, Highly Efficient Photocatalytic Reduction of CO2 to CO by in Situ Formation of a Hybrid Catalytic System Based on Molecular Iron Quaterpyridine Covalently Linked to Carbon Nitride, Angew. Chem., Int. Ed., 2022, 61(11), e202116832 CrossRef CAS PubMed.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Identification of Champion Transition Metals Centers in Metal and Nitrogen-Codoped Carbon Catalysts for CO2 Reduction, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- C. Liu, T. R. Cundari and A. K. Wilson, CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis, J. Phys. Chem. C, 2012, 116(9), 5681–5688 CrossRef CAS.
- H. Zhuzhang, X. Liang, J. Li, S. Xue, Y. Lin, B. Sa, S. Wang, G. Zhang, Z. Yu and X. Wang, Precise Manipulation on the Structural Defects of Poly (Triazine Imide) Single Crystals for Efficient Photocatalytic Overall Water Splitting, Angew. Chem., 2025, e202421861 CAS.
- G. Zou, Q. Wang, G. Ye, Z. Pan, S. Wang, M. Anpo and G. Zhang, Copolymerization of Poly (Triazine Imide) Single Crystal Nanoplates with Enhanced Charge Transfer for Efficient Photocatalytic Overall Water Splitting, Adv. Funct. Mater., 2024, 2420899 CrossRef.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Rh/Cr2O3 and CoOx Cocatalysts for Efficient Photocatalytic Water Splitting by Poly (Triazine Imide) Crystals, Angew. Chem., 2023, 135(37), e202304694 CrossRef.
- F. Liu, J. Deng, B. Su, K.-S. Peng, K. Liu, X. Lin, S.-F. Hung, X. Chen, X. F. Lu, Y. Fang, G. Zhang and S. Wang, Poly(Triazine Imide) Crystals for Efficient CO2 Photoreduction: Surface Pyridine Nitrogen Dominates the Performance, ACS Catal., 2025, 15(2), 1018–1026 CrossRef CAS.
- S. McGuigan, S. J. Tereniak, C. L. Donley, A. Smith, S. Jeon, F. Zhao, R. N. Sampaio, M. Pauly, L. Keller, L. Collins, G. N. Parsons, T. Lian, E. A. Stach and P. A. Maggard, Discovery of a Hybrid System for Photocatalytic CO2 Reduction via Attachment of a Molecular Cobalt-Quaterpyridine Complex to a Crystalline Carbon Nitride, ACS Appl. Energy Mater., 2023, 6(20), 10542–10553 CrossRef CAS.
- C. Cometto, L. Chen, P.-K. Lo, Z. Guo, K.-C. Lau, E. Anxolabéhère-Mallart, C. Fave, T.-C. Lau and M. Robert, Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs Co Quaterpyridine Complexes upon Mechanistic Studies, ACS Catal., 2018, 8(4), 3411–3417 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- A. D. Becke, A New Mixing of Hartree–Fock and Local Density–functional Theories, J. Chem. Phys., 1993, 98(2), 1372–1377 CrossRef CAS.
- A. D. Becke, Density–functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- A. D. Becke, Density-Fnnctional Exchange-Energy Approximation with Correct Asymptotic Behavior, 1988, Vol. 38 Search PubMed.
- S. Grimme, Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General Formalism, J. Chem. Phys., 2010, 132(11), 114110 CrossRef PubMed.
- A. D. McLean and G. S. Chandler, Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Row Atoms, Z=11–18, J. Chem. Phys., 1980, 72(10), 5639–5648 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy–adjusted Ab Initio Pseudopotentials for the First Row Transition Elements, J. Chem. Phys., 1987, 86(2), 866–872 CrossRef CAS.
- P.-O. Löwdin, Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction, Phys. Rev., 1955, 97(6), 1474–1489 CrossRef.
- M. E. Casida, Time-Dependent Density Functional Response Theory for Molecules, in Recent Advances In Density Functional Methods: (Part I), World Scientific, 1995, pp. 155–192 Search PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold, J. Chem. Phys., 1998, 108(11), 4439–4449 CrossRef CAS.
- M. Pauly, J. Kröger, V. Duppel, C. Murphey, J. Cahoon, B. V. Lotsch and P. A. Maggard, Unveiling the Complex Configurational Landscape of the Intralayer Cavities in a Crystalline Carbon Nitride, Chem. Sci., 2022, 13(11), 3187–3193 RSC.
- E. Wirnhier, M. Döblinger, D. Gunzelmann, J. Senker, B. V. Lotsch and W. Schnick, Poly(Triazine Imide) with Intercalation of Lithium and Chloride Ions [(C3N3)2(NHx,Li1−x)3·LiCl]: A Crystalline 2D Carbon Nitride Network, Chem. – Eur. J., 2011, 17(11), 3213–3221 CrossRef CAS PubMed.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving Surface Chemical States in XPS Analysis of First Row Transition Metals, Oxides and Hydroxides: Sc, Ti, V, Cu and Zn, Appl. Surf. Sci., 2010, 257(3), 887–898 CrossRef CAS.
- S. Bhowmik, S. J. Phukan, N. K. Sah, M. Roy, S. Garai and P. K. Iyer, Review of Graphitic Carbon Nitride and Its Composite Catalysts for Selective Reduction of CO2, ACS Appl. Nano Mater., 2021, 4(12), 12845–12890 CrossRef CAS.
- K. Hara, Y. Dan-oh, C. Kasada, Y. Ohga, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Effect of Additives on the Photovoltaic Performance of Coumarin-Dye-Sensitized Nanocrystalline TiO2 Solar Cells, Langmuir, 2004, 20(10), 4205–4210 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, R. Humphry-Baker, J. E. Moser and M. Grätzel, Molecular–scale Interface Engineering of TiO2 Nanocrystals: Improve the Efficiency and Stability of Dye–sensitized Solar Cells, Adv. Mater., 2003, 15(24), 2101–2104 CrossRef CAS.
- J. Z. Zhang, Interfacial Charge Carrier Dynamics of Colloidal Semiconductor Nanoparticles, J. Phys. Chem. B, 2000, 7239–7253 CrossRef CAS . ACS Publications.
- G. Rothenberger, J. Moser, M. Graetzel, N. Serpone and D. K. Sharma, Charge Carrier Trapping and Recombination Dynamics in Small Semiconductor Particles, J. Am. Chem. Soc., 1985, 107(26), 8054–8059 CrossRef CAS.
- S. Morab, M. M. Sundaram and A. Pivrikas, Review on Charge Carrier Transport in Inorganic and Organic Semiconductors, Coatings, 2023, 13(9), 1657 CrossRef CAS.
- E. Gibson, A. Co and G. Meyer, Light-Driven and Electrochemical CO2 Reduction, ACS Appl. Energy Mater., 2024, 7(5), 1684–1686 CrossRef CAS.
- K. L. Corp and C. W. Schlenker, Ultrafast Spectroscopy Reveals Electron-Transfer Cascade That Improves Hydrogen Evolution with Carbon Nitride Photocatalysts, J. Am. Chem. Soc., 2017, 139(23), 7904–7912 CrossRef CAS PubMed.
- J. Xue, M. Fujitsuka and T. Majima, Near Bandgap Excitation Inhibits the Interfacial Electron Transfer of Semiconductor/Cocatalyst, ACS Appl. Mater. Interfaces, 2020, 12(5), 5920–5924 CrossRef CAS PubMed.
- W. Tang, J. Meng, T. Ding, H. Huang, C. Tan, Q. Zhong, H. Cao, Q. Li, X. Xu and J. Yang, Efficient Interfacial Charge Transfer Enables Nearly 100% Selectivity for Solar-Light-Driven CO2 Conversion, ACS Catal., 2024, 14(10), 7506–7513 CrossRef CAS.
- W. Yang, R. Godin, H. Kasap, B. Moss, Y. Dong, S. A. J. Hillman, L. Steier, E. Reisner and J. R. Durrant, Electron Accumulation Induces Efficiency Bottleneck for Hydrogen Production in Carbon Nitride Photocatalysts, J. Am. Chem. Soc., 2019, 141(28), 11219–11229 CrossRef CAS PubMed.
- R. Godin, Y. Wang, M. A. Zwijnenburg, J. Tang and J. R. Durrant, Time-Resolved Spectroscopic Investigation of Charge Trapping in Carbon Nitrides Photocatalysts for Hydrogen Generation, J. Am. Chem. Soc., 2017, 139(14), 5216–5224 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- L. Lin, H. Ou, Y. Zhang and X. Wang, Tri-s-Triazine-Based Crystalline Graphitic Carbon Nitrides for Highly Efficient Hydrogen Evolution Photocatalysis, ACS Catal., 2016, 6(6), 3921–3931 CrossRef CAS.
- G. Dai and J. Liu, Fe-Quaterpyridine Complex: A Comprehensive DFT Study on the Mechanism of CO2-to-CO Conversion, J. Mater. Sci., 2020, 55(29), 14301–14314 CrossRef CAS.
- W. Tan, S. Li, X. Liu, G. Liu and Z. Liu, Perfect Solar Light Absorption and Efficient Photo-Thermal Generation, IEEE Photonics Technol. Lett., 2023, 35(24), 1415–1418 CAS.
- V. Esen, Ş. Sağlam and B. Oral, Light Sources of Solar Simulators for Photovoltaic Devices: A Review, Renewable Sustainable Energy Rev., 2017, 77, 1240–1250 CrossRef CAS.
- S. K. Solanki, N. A. Krivova and J. D. Haigh, Solar Irradiance Variability and Climate, Annu. Rev. Astron. Astrophys., 2013, 51, 311–351 CrossRef CAS.
- D. S. Kumar, G. M. Yagli, M. Kashyap and D. Srinivasan, Solar Irradiance Resource and Forecasting: A Comprehensive Review, IET Renew. Power Gener., 2020, 14(10), 1641–1656 CrossRef.
- H. Horvath, Light Absorption in the Atmosphere, in Light Scattering Reviews 10: Light Scattering and Radiative Transfer, Springer, 2016, pp. 235–289 Search PubMed.
- T. J. Boerner, S. Deems, T. R. Furlani, S. L. Knuth and J. Towns, ACCESS: Advancing Innovation: NSF's Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support, in Practice and Experience in Advanced Research Computing (PEARC ‘23), ACM, New York, NY, USA. Portland, OR, USA, 2023, p. 4 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qi00859j |
| This journal is © the Partner Organisations 2025 |