
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Half-sandwich scandium methylidenes†
Gernot T. L.
Zug
,
Sylvia A.
Zeiner
,
Jonas
Reuter
,
Hartmut
Schubert
,
Cäcilia
Maichle-Mössmer
and
Reiner
Anwander
 *
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de
First published on 4th April 2025
Abstract
AlMe3 sticks, GaMe3 quits. A series of Lewis acid stabilized scandium methylidenes with commercially available cyclopentadienyl ligands CpR (CpR = C5Me5 (Cp*), C5Me4SiMe3 (Cp′)) were synthesized. The-salt metathesis reaction of new half-sandwich dichloride precursors CpRScCl2(μ-Cl)Li(thf)3 with LiAlMe4 and AlMe3 at ambient temperature yielded [CpRSc(AlMe4)Cl]2. [Cp′Sc(AlMe4)Cl]2 was further methylated at ambient temperature to yield Cp′Sc(AlMe4)Me. At 70 °C, the reaction of CpRScCl2(μ-Cl)Li(thf)3 with LiAlMe4 and AlMe3 led to the formation of Lewis acid stabilized Sc/Al2 methylidenes CpRSc(CH2)(AlMe3)2. The new mixed Sc/Al/Ga methylidene Cp′Sc(CH2)(AlMe3)(GaMe3) was obtained from the reaction of Cp′Sc(AlMe4)Me with GaMe3. When heated, complex Cp′Sc(CH2)(AlMe3)(GaMe3) converted into the Sc/Al methylidene [Cp′Sc(CH2)2AlMe]3via release of the comparatively weak Lewis acid GaMe3 and methane. The core of trimeric [Cp′Sc(CH2)2AlMe]3 can be described as a triscandacyclohexane {Sc(CH2)}3 stabilized by a trialacyclohexane {Al(CH2)}3via Pearson hard/hard matching. Complexes CpRSc(CH2)(AlMe3)2 and [Cp′Sc(CH2)2AlMe]3 differ in rigidity, thermal stability and reactivities toward ketones and Lewis bases. The isolated methylidenes were analyzed by 1H, 13C{1H}, 45Sc, and variable temperature 1H NMR spectroscopy, SC-XRD, IR spectroscopy, and elemental analysis. Complexes CpRSc(CH2)(AlMe3)2 feature pronounced Sc⋯HC α-agostic interactions. The reaction of CpRScCl2(μ-Cl)Li(thf)3 with LiAlMe4 and AlMe3 was investigated via in situ1H and 45Sc NMR spectroscopy.
Introduction
It is now 50 years since Schrock reported on the synthesis and structural characterization of tantalum methylidene Cp2Ta(CH2)Me as the first isolable molecular metal-methylidene complex.1 Soon after in 1978, Tebbe described the heterobimetallic titanium methylidene Cp2Ti(CH2)(Cl)AlMe2,2 which ever since advanced organic synthesis and, more specifically, excelled as Tebbe's reagent in functional group tolerant methylenation reactions.3 Early mechanistic and structural investigations have also contributed to an in-depth understanding of the role of the organoaluminum component.4 For example, while Schrock pointed out that Cp2Ta(CH2)Me does reversibly form an adduct with AlMe3,1 ideal performance of Tebbe's compound is achieved in the presence of a Lewis base like NEt3, resulting in the displacement of “stabilizing” Me2AlCl and the formation of transient [Cp2Ti![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2].5 Not surprisingly, research into Tebbe-like nucleophilic carbene chemistry also triggered feasibility studies on discrete methylidene complexes of metals adjacent to titanium. More recent studies include the structural characterization of the first monometallic, terminal methylidenes of zirconium(IV) and vanadium(V), (PNP)Zr(OAr)(CH2) and (PNP)V(NAr)(CH2) (PNP = N[2-P(iPr)2-4-methylphenyl]2, Ar = 2,6-iPr2C6H3) by the group of Mindiola.6,7 The latter two synthesis protocols do not involve any organoaluminum components. In contrast, the first discrete scandium methylidene complex (PNP)Sc(CH2)(AlMe3)2,8 also reported by the Mindiola group in 2008 draws not only on kinetic stabilization of the [ScCH2] moiety via a sterically demanding PNP pincer but also Lewis acid/base interactions with AlMe3 (Chart 1, I).8–10 We have previously shown that such multifaceted stabilization is also applicable for the larger rare-earth metals (Ln) when employing bulky hydrotris(3-R′-5-R-pyrazolyl)borato scorpionate ligands TpR′,R and either AlMe3 or GaMe3, e.g., (TptBu,Me)La(CH2)(EMe3)2 (E = Al, Ga) and (TpMe,Me)Sm(CH2)(AlMe3)2.9 For these larger metals, the ubiquitous cyclopentadienyl ancillary ligand only led to trimetallic clusters of the type (C5Me5)3Ln3(μ-CH2)X4 (X = Cl, Br).11,12 Compared to d-transition-metal methylidenes, the bonding in rare-earth-metal methylidenes is increasingly ionic and stabilization of the highly polarized Ln–C(methylidene) bond is achieved via cluster formation involving bridging CH22− moieties. The cluster size is greatly impacted by the steric shielding of the ancillary ligand. It is also noteworthy that except for the cuboidal cluster [(C5Me5)Sc(μ-CH2)]4 (Chart 1, II, Cheng, 2021)13 all previously reported methylidene complexes of the smallest rare-earth metal scandium feature non-cyclopentadienyl ancillary ligands (Chart 1, I and III–VI).13–18 The formation of tetrametallic IIvia thermal treatment (90 °C) of [(C5Me5)Sc(CH3)(μ-CH3)]2 clearly parallels the transformation of the Petasis reagent Cp2Ti(CH3)2, which generates transient [Cp2TiCH2] upon heating at 60–80 °C.19
CH2].5 Not surprisingly, research into Tebbe-like nucleophilic carbene chemistry also triggered feasibility studies on discrete methylidene complexes of metals adjacent to titanium. More recent studies include the structural characterization of the first monometallic, terminal methylidenes of zirconium(IV) and vanadium(V), (PNP)Zr(OAr)(CH2) and (PNP)V(NAr)(CH2) (PNP = N[2-P(iPr)2-4-methylphenyl]2, Ar = 2,6-iPr2C6H3) by the group of Mindiola.6,7 The latter two synthesis protocols do not involve any organoaluminum components. In contrast, the first discrete scandium methylidene complex (PNP)Sc(CH2)(AlMe3)2,8 also reported by the Mindiola group in 2008 draws not only on kinetic stabilization of the [ScCH2] moiety via a sterically demanding PNP pincer but also Lewis acid/base interactions with AlMe3 (Chart 1, I).8–10 We have previously shown that such multifaceted stabilization is also applicable for the larger rare-earth metals (Ln) when employing bulky hydrotris(3-R′-5-R-pyrazolyl)borato scorpionate ligands TpR′,R and either AlMe3 or GaMe3, e.g., (TptBu,Me)La(CH2)(EMe3)2 (E = Al, Ga) and (TpMe,Me)Sm(CH2)(AlMe3)2.9 For these larger metals, the ubiquitous cyclopentadienyl ancillary ligand only led to trimetallic clusters of the type (C5Me5)3Ln3(μ-CH2)X4 (X = Cl, Br).11,12 Compared to d-transition-metal methylidenes, the bonding in rare-earth-metal methylidenes is increasingly ionic and stabilization of the highly polarized Ln–C(methylidene) bond is achieved via cluster formation involving bridging CH22− moieties. The cluster size is greatly impacted by the steric shielding of the ancillary ligand. It is also noteworthy that except for the cuboidal cluster [(C5Me5)Sc(μ-CH2)]4 (Chart 1, II, Cheng, 2021)13 all previously reported methylidene complexes of the smallest rare-earth metal scandium feature non-cyclopentadienyl ancillary ligands (Chart 1, I and III–VI).13–18 The formation of tetrametallic IIvia thermal treatment (90 °C) of [(C5Me5)Sc(CH3)(μ-CH3)]2 clearly parallels the transformation of the Petasis reagent Cp2Ti(CH3)2, which generates transient [Cp2TiCH2] upon heating at 60–80 °C.19
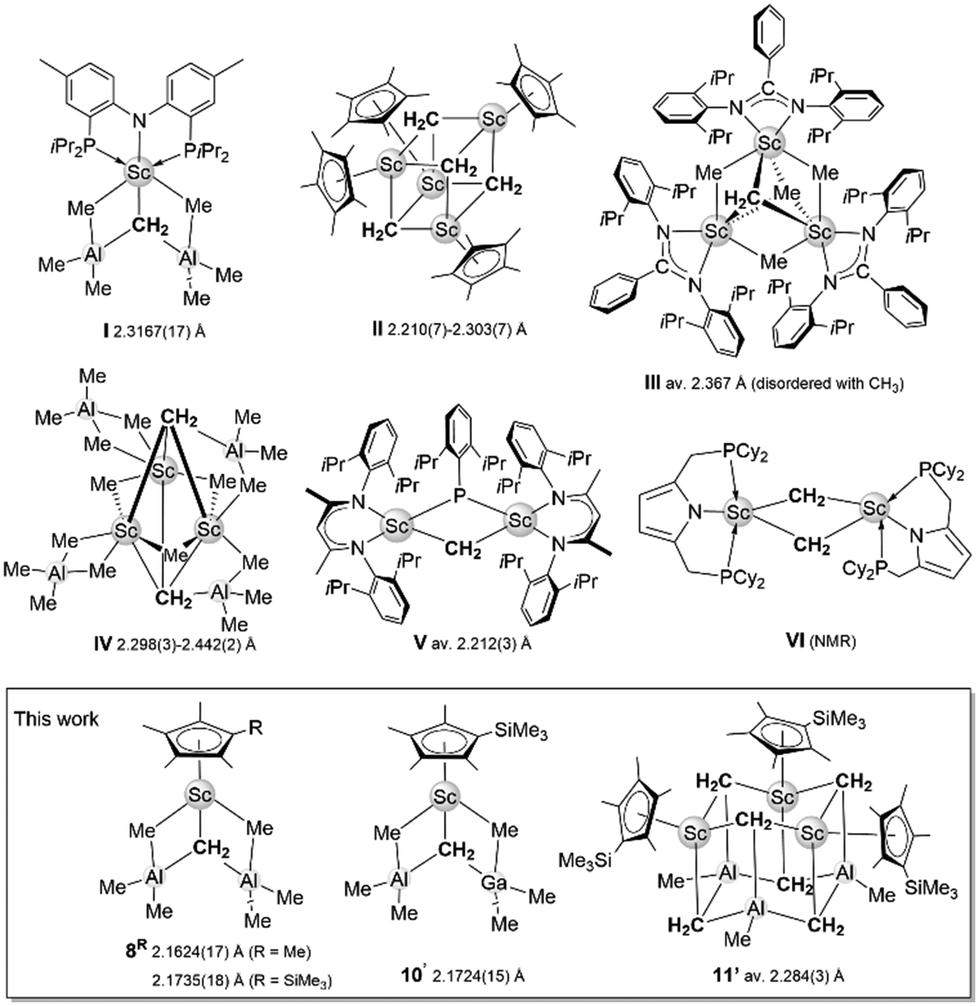 | ||
| Chart 1 Known scandium methylidene complexes, including Sc–C(methylidene) distances. | ||
Many of the isolated nucleophilic metal methylidenes were probed in the methylenation of ketones and compared to Tebbe's reagent.
Given the feasibility of the half-sandwich complex (C5Me5)Sc(AlMe4)2,18 although decomposing at temperatures >–20 °C, we wondered about the existence of discrete scandium methylidene complexes with the commercially available ligands Cp* (C5Me5) and Cp′ (C5Me4SiMe3). This might bring us a step closer to a scandium analog of the cyclopentadienyl-supported Tebbe reagent. Further, fathoming any group 13 effect (AlMe3versus GaMe3) might unveil distinct methylidene formations.9b,20
Here, we present discrete AlMe3-stabilized half-sandwich scandium methylidenes of the known motif I. Additionally, we present a novel mixed scandium aluminum gallium methylidene complex of motif I. This trimetallic complex decomposes under the release of GaMe3 and CH4 granting access to a hexametallic {Sc3Al3} cluster motif which is new in rare-earth-metal chemistry.
Results and discussion
Half-sandwich scandium chloride precursors
Initial investigations bore on the known metathesis reaction of ScCl3(thf)3 and Cp′Li (Cp′ = C5Me4SiMe3) to yield Cp′ScCl2(thf)221 as well on Okuda's work on half-sandwich methyl scandium complexes (vide infra).22 In our hands, the metathesis reactions of ScCl3(thf)3 and CpRLi (CpR = C5Me5, C5Me4SiMe3) yielded repeatedly crystals of ate complexes CpRScCl2(μ-Cl)Li(thf)3 (1R) (Scheme 1). While 1′ was obtained in 74% yield, the initial low yield of 1* could be increased from 31% to 55% by raising the reaction temperature to 40 °C and additional toluene extraction. Complexes 1′ and 1* are isostructural exhibiting a piano stool configuration, and were further analyzed by 1H, 13C{1H}, 7Li, and 45Sc NMR spectroscopy in C6D6, as well as elemental analysis. Compound 1* loses THF at prolonged exposure to vacuum. | ||
| Scheme 1 Synthesis of scandium chloride precursors CpRScCl2(μ-Cl)Li(thf)3 (1R) and their reactions with AlMe3 to yield [CpRScCl2]4 (2R). | ||
Targeting a potential Tebbe-like reactivity, the dichlorido precursors were treated with 5.2 (1′) or 5.5 (1*) equivalents of AlMe3. Not unexpectedly, addition of AlMe3 to 1R did not afford alkylation but displacement/precipitation of LiCl only,23 along with the formation of white [CpRScCl2]4 (2R). The reaction mixture was heated to 130 °C for one day, yielding a pale-yellow solution. Upon cooling to ambient temperature, complexes 2R crystallized and were analyzed by single-crystal X-ray diffraction (SC-XRD). Tetrameric 2R are isostructural to other half-sandwich rare-earth-metal dihalides including [CpRScI2]4.21b,24 Although LiCl is not included in the crystal structure, 2R showed a 7Li NMR signal even after crystalline isolation. Treatment of 2R with THF yielded 1R (Scheme 1).
Metathesis reactions of CpRScCl2(μ-Cl)Li(thf)3 with LiAlMe4
In 2008, the group of Okuda reported on metathesis reactions of Cp′ScCl2(thf)2 with two equivalents of LiAlMe4. This led to the successful isolation of [Cp′ScMe2]2.22 When AlMe3 was added in the metathesis reaction, an “orange oil” was obtained at ambient temperature, then depicted as [Cp′Sc(AlMe4)2]. The orange oil could not be identified unequivocally. The actual congener Cp*Sc(AlMe4)2 was isolated by our group in 2019, showing high sensitivity toward temperatures above −20 °C.18 This sparked our interest in re-investigating Okuda′s orange oil. Accordingly, complexes 1R were reacted with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 in toluene at ambient temperature overnight. This resulted in the isolation of mixed chlorido tetramethylaluminato complexes [CpRSc(AlMe4)Cl]2 (3R), which were analyzed by 1H, 13C{1H}, and 45Sc NMR spectroscopy in C6D6 and toluene-d8, SC-XRD, as well as elemental analysis. Complexes 3R crystallized as chlorido-bridged dimers.11b,25 The 45Sc NMR spectra displayed three signals (cf. ESI†). The 1H NMR spectra, however, did not indicate multiple species. Therefore, we assumed that 3R are subject to monomer-dimer equilibria or to the formation of even larger aggregates in solution which is only seen in the 45Sc NMR spectra. A variable temperature (VT) NMR study in toluene-d8 lent support to this hypothesis (cf. ESI†). The spectrum at 60 °C revealed only one broad peak in the methyl region. At −80 °C, four sharp peaks appeared, assignable to the monomer and the dimer. The VT 45Sc NMR spectra show that the peak at about 210 ppm increases with lower temperatures, whereas the peak at about 400 ppm gets bigger with higher temperatures. We were hoping that these mixed chlorido/tetramethylaluminato complexes 3R may directly lead to methylidene complexes, since the congeners of the larger rare-earth metals form trinuclear methylidenes when reacted with THF.11 In the case of the small scandium however, treatment of 3R with THF did not form such methylidenes. Instead, undefined decomposition occurred, and, on one occasion, the extremely sensitive aluminate cleavage product [Cp*ScMeCl(thf)]2 could be identified via a crystal snapshot.Prolonging the reaction of 1′ with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 in toluene for at least two days led to the isolation of [Cp′Sc(AlMe4)Me] (4′) (Scheme 2). Compound 4′ features the orange oil reported by Okuda (vide supra),22 also in our hands opposing successful crystallization. To prove its identity, 4′ was analyzed by 1H, 13C{1H}, and 45Sc NMR spectroscopy in C6D6. The 1H NMR signal of the Sc–CH3 protons of 4′ appeared as one broad peak, suggesting a high fluxionality, as known for tetramethylaluminato and methyl ligands. Moreover, this broad peak showed integrals between 15 and 22 which is less than the expected 24 for putative [Cp′Sc(AlMe4)2]. The higher integral numbers (compared to 15 in 4′) originate from residual AlMe3(thf) as the oil had to be evacuated and co-evaporated with toluene extensively in order to get rid of all AlMe3(thf). The 45Sc NMR resonance of 4′ was detected at 404.2 ppm, shifted considerably to lower field compared to Cp*Sc(AlMe4)2 (245.1 ppm).18 This downfield shift of 4′ suggests that it indeed is not putative [Cp′Sc(AlMe4)2]. To strengthen our hypothesis and without a crystal structure on hand, we envisaged derivatization reactions to further prove the identity of 4′ (Scheme 2). First, we reacted [Cp′ScMe2]2 with 1 equiv. AlMe3 which led to the sole quantitative formation of 4′. Compound 4′ reacted with TMEDA according to a donor-induced aluminate cleavage yielding single crystals of (tmeda)(AlMe3)2,26 verified by a unit cell check. The 1H and 45Sc NMR spectra of this reaction identified [Cp′ScMe2]2 as the coproduct (cf., ESI†).22 Reacting 4′ with the donor DMAP afforded the monomeric adduct Cp′ScMe2(dmap) (5′), which was analyzed by 1H, 13C{1H}, and 45Sc NMR spectroscopy, SC-XRD, and elemental analysis. The 1H NMR spectrum of 5′ in C6D6 revealed that coproduct (dmap)AlMe3 could not be removed entirely by crystallization. (dmap)AlMe3 itself was crystallized from the supernatant and identified by a unit cell check.27 To rule out that the orange oil itself is a methylidene complex, 4′ was treated with cyclohexanone. This led to the identification of dimeric cyclohexenolate complex [Cp′ScMe(OC6H9)]2 (6′) via a crystal snapshot (cf., ESI†). The small quantity of 6′ was also analyzed by 1H and 45Sc NMR spectroscopy in C6D6.
 | ||
| Scheme 2 Syntheses of Cp′Sc(AlMe4)Me (4′) using various precursors, and reactivities of Cp′Sc(AlMe4)Me (4′). | ||
In order to skip the isolation of 1R, we probed a one-pot reaction of ScCl3(thf)3, 1.1 equiv. Cp′H and 5 equiv. MeLi in THF. This led to the new half-sandwich scandium trimethyl ate complex [Li(thf)4][Cp′ScMe3] (7′), which was analyzed by 1H, 13C{1H}, 7Li, and 45Sc NMR spectroscopy in C6D6, SC-XRD, and elemental analysis. Complex 7′ is isostructural with [Li(tmeda)2][Cp*LuMe3], which was obtained from a one-pot reaction of LuCl3, NaCp*, and MeLi.28 When treated with 6 equiv. AlMe3, complex 7′ gave 4′ in essentially quantitative yield. Compound 4′ was also obtained from the reaction of half-sandwich dibenzyl complex Cp′Sc(CH2C6H4NMe2-o)2 with trimethylaluminum, further proving the orange oil 4′ to exhibit the connectivity [Cp′Sc(AlMe4)Me]. Compound 4′ could not be isolated in pure form. It contained at least 5% impurities such as 8′ (vide infra) or Me2Al(CH2C6H4NMe2-o).
Monomeric methylidenes CpRSc(CH2)(AlMe3)2
The thermal behavior of 4′ at temperatures >70 °C indicated release of methane but yielded a red oil containing an undefined mixture of products. Similarly, performing the metathesis reaction of 1R with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 at an increased temperature of 70 °C led to methane formation, yielding a red oil as well. However, now repeated re-crystallization of that red oil from an n-hexane solution gave methylidenes CpRSc(CH2)(AlMe3)2 (8R) as colorless crystals in moderate yields of 66% (R = Cp*) and 47% (R = Cp′) (Scheme 3, Fig. 1 and Fig. S74†). Complexes 8R were analyzed by 1H, 13C{1H}, and 45Sc NMR spectroscopy in C6D6 and toluene-d8, SC-XRD, DRIFTS, and elemental analysis. Compounds 8R feature Lewis acid stabilized methylidenes with the recurring structural motif [Ln(CH2)(EMe3)2]+ (Ln = rare-earth metal, E = Al, Ga) as detected for (PNP)Sc(CH2)(AlMe3)2 (I) and TptBu,MeLn(CH2)(EMe3)2 (Ln = La, Ce, Nd; E = Al, Ga; Ln = Sm; E = Ga).8–10 The 1H NMR spectrum of 8R at ambient temperature displayed the methylidene protons as two broad signals (Table 1: δ = 0.95, −0.01 ppm for 8′, δ = 0.84, −0.13 ppm for 8*). The slight upfield shift of one methylidene proton points to Sc⋯HC α-agostic interactions. The methyl groups appeared as one singlet (δ = −0.33 ppm for 8′, δ = −0.43 ppm for 8*), accounting for a rapid exchange of bridging and terminal methyl groups at ambient temperature. To further elucidate this fluxional behavior in solution, a variable temperature 1H NMR study was conducted between −80 and +80 °C (cf., ESI†). At low temperatures, the methylidene signals get sharper and geminal coupling of the two inequivalent protons is detected. The bridging and the two terminal methyl groups split into three signals at −80 °C. At 80 °C, the methylidene signals coalesce into one very broad peak. Crucially, the spectroscopic signature of the methylidene moiety is distinct from that reported for (PNP)Sc(CH2)(AlMe3)2 (I), which showed a singlet at 0.69 ppm at ambient temperature shifting to 0.75 ppm at −50 °C.8 | ||
| Scheme 3 Synthesis of AlMe3-stabilized methylidenes CpRSc(CH2)(AlMe3)2 (8R) via tandem metathesis/thermally induced methyl deprotonation. | ||
 | ||
| Fig. 1 Crystal structure of 8*. Atomic displacement ellipsoids were set at 50% probability. Hydrogen atoms except methylidene hydrogen atoms omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sc1–C11 2.1624(17), Al1–C11 2.0914(18), Al2–C11 2.0647(17), Sc1–H11A 2.17(2), Sc1–C11–H11A 77.2(13) (ESI†). Complex 8′ is isostructural (Fig. S74†). | ||
| Compound | CN | Sc–C [Å] | 13C{1H} [ppm] | 1H [ppm] | Ref. |
|---|---|---|---|---|---|
| L1 = PhC(NC6H4iPr2-2,6). L2 = N2C3HAriPr2-1,5-Me2-2,4. L3 = NC4H2(CH2PCy2)2-2,5. NNFc = 1,1′-fc(NSitBuMe2)2. Structures of I–VI are depicted in Chart 1. | |||||
| Cp*Sc(μ3-CH2)(AlMe3)2 (8*) | 6 | 2.1624(17) | 53.2 | 0.84(br)/−0.13(br) | This work |
| Cp′Sc(μ3-CH2)(AlMe3)2 (8′) | 6 | 2.1735(18) | 53.0 | 0.95(br)/−0.01(br) | This work |
| Cp′Sc(μ3-CH2)(AlMe3)(GaMe3) (10′) | 6 | 2.1724(15) | 53.1 | 0.94(br)/0.10(br) | This work |
| [Cp′Sc(μ3-CH2)(Al(μ3-CH2)Me)]3 (11′) | 6 | 2.245(3)–2.3338(18) | 81.2/35.0 | 0.73(d)/−0.13(s)/−1.37(d) | This work |
| (PNP)Sc(μ3-CH2)(AlMe3)2 (I) | 6 | 2.3167(17) | 28.8 | 0.69(s) | 8 |
| [Cp*Sc(μ3-CH2)]4 (II) | 6 | 2.210(7)–2.303(7) | — | 1.71(s) | 13 |
| L13Sc3(μ3-CH2)(CH3)4 (III) | 6 | 2.367 | 123.9 | 3.32(s) | 15 |
| Sc3(μ4-CH2)2(μ2-CH3)3(AlMe4)2(AlMe3)2 (IV) | 6 | 2.298(3)–2.442(2) | 78.9 | 2.06(s) | 18 |
| L22Sc2(μ2-CH2)(μ2-PC6H3iPr2-2,6) (V) | 5 | 2.193(3)/2.232(3) | — | 5.62(d, 3JPH) | 17 |
| [L3Sc(μ2-CH2)]2 (VI) | 5 | — | — | 1.51/1.48/1.13/1.09 | 16 |
| (NNFc)Sc(μ3-CH2)(AlMe3)2 | 5 | 2.6921(18) | −4.7 | −1.42(d) | 35 |
The 13C{1H} NMR spectra of 8R at ambient temperature show peaks at 53.2 (8*) and 53.0 ppm (8′) for the methylidene carbon atoms (Table 1), rather comparable to the respective shift of TptBu,MeLa(CH2)(AlMe3)2 (δ = 50.9 ppm)9a than of (PNP)Sc(CH2)(AlMe3)2 (I, 28.8 ppm). The signals of the methylidene carbon atoms are broadened due to 1J coupling to 7/2 and 5/2 nuclei of 45Sc and 27Al, respectively. The peaks were assigned to the methylidene carbon atom by 1H-13C HSQC NMR coupling with the methylidene protons. The Sc⋯HC α-agostic interaction is also seen in the SC-XRD analysis with short Sc–H(agostic) distances of 2.17(2) Å (8*) and 2.14(3) Å (8′). The Sc–C(methylidene)–H(agostic) angles are 77.2(13)° (8*) and 75.1(16)° (8′). The Sc–C(methylidene) distances are 2.1624(17) Å (8*) and 2.1735(18) Å (8′) (Table 1 and Fig. 1). To the best of our knowledge, these are the shortest distances of scandium to a methylidene carbon atom detected so far. In accordance with the NMR spectroscopic signatures, the Sc–C(methylidene) distance is markedly shorter than the 2.3167(17) Å detected for (PNP)Sc(CH2)(AlMe3)2 (I).8 The short Sc–C(methylidene) distance and the 1J coupling hint to comparatively strong Sc–methylidene interactions in 8R. The Al–C(methylidene) distances of 8* are 2.0647(17) Å and 2.0914(18) Å, and of 8′ are 2.0898(18) Å and 2.0879(18) Å. Compared to (PNP)Sc(CH2)(AlMe3)2 (I, 2.0387(18) Å and 2.0369(18) Å), the longer Al–C(methylidene) distances in 8R support the assumption of a stronger Sc–C(methylidene) interaction in 8R compared to (PNP)Sc(CH2)(AlMe3)2.
The complexes 8R and TptBu,MeLa(CH2)(AlMe3)2 show α-agostic interactions, fluxionality at ambient temperature, geminal coupling of the methylidene protons at −80 °C and a similar 13C{1H} NMR shift of the methylidene carbon atom (vide supra). Thus, one could draw the comparison of the fragments [CpRSc]2+ and [TptBu,MeLa]2+. The congener with the smaller yttrium, [TptBu,MeY(CH2)(AlMe3)2], is elusive since the reaction comes to a halt at TptBu,MeYMe(AlMe4).9a It seems that the steric bulk of the ancillary ligand (L) has to match the size of the rare-earth-metal cation (Ln) in order to obtain methylidene complexes from the intermediary formed [LLn(AlMe4)2]. Such size match is the case for the fragments [CpRSc]2+ and [TptBu,MeLa]2+. In 8R, the steric requirement is achieved with the commercially available ligands C5Me5 and C5Me4SiMe3.
To gain more insight into the formation mechanism of 8′, the reaction of 1′ with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 was monitored by 1H and 45Sc NMR spectroscopy without agitation of the reaction mixture (Fig. 2 and Scheme 4). At initial ambient temperature, the chlorido-bridged dimer 3′ is detected as the main species along with different unidentified species, maybe differently sized clusters of [Cp′ScCl2] (2′). Upon heating to 70 °C, full conversion into the mixed methyl/tetramethylaluminato complex 4′ evolved within 5 minutes. Over the course of two hours at 70 °C, methylidene complex 8′ emerged as the main product. Relating to the 45Sc NMR spectra, at the beginning, the peak pattern of 3′ appeared in the range between 200 and 420 ppm. Then, after 5 minutes at 70 °C, complex 4′ (δ = 404.2 ppm) emerged as the main intermediate (Scheme 4). During prolonged heating at 70 °C, 8′ became the main product already after 2 h (δ = 272.6 ppm). Consequently, 8′ could be obtained according to another reaction path by treating isolated 4′ in n-hexane with excess AlMe3 at 70 °C.
 | ||
| Fig. 2 Monitoring of the reaction of 1′ with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 in a J. Young-valved NMR tube without agitation via in situ1H (top) and 45Sc (bottom) NMR spectroscopy in C6D6, beginning with 3′ (blue), then intermediate 4′ (red), and final product 8′ (yellow). Unmarked spectra were measured (after cooling) at 26 °C. Spectra marked with an asterisk (*) were measured at 70 °C. | ||
 | ||
| Scheme 4 Mechanism of reaction of 1′ with LiAlMe4 and AlMe3 with intermediates 3′ and 4′ to yield the final product 8′. | ||
On one occasion, the red supernatant of precipitated 8′ yielded very few colorless crystals of another methylidene complex, [(Cp′ScCH2)3LiAlMeCl3]n (9′). The obtained small quantity of 9′ was analyzed by 1H and 45Sc NMR spectroscopy in C6D6, and SC-XRD. Compound 9′ consists of three [Cp′ScCH2] units which are linked to a one-dimensional coordination polymer via LiAlMeCl3 moieties (cf., ESI†). The 1H NMR spectrum of 9′ shows two very broad signals at 0.97 ppm and 1.36 ppm attributable to the methylidene protons. Compound 9′ revealed a very broad 45Sc NMR signal at 429.2 ppm being in stark contrast to the chemical shift of 8′ (δ = 272.6 ppm), and more in the range of cluster Sc3(μ4-CH2)2(μ2-CH3)3(AlMe4)2(AlMe3)2 (Chart 1, IV, δ = 388.3/331(2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) ppm). However, the amount of pure isolated 9′ was not sufficient for further analytics and various attempts failed to reproduce 9′. The Cp*-analog compound [Cp*Sc(AlMe4)Me] (4*) was not encountered. Prolonged reaction times of 1* with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 at ambient temperature only led to the isolation of [Cp*Sc(AlMe4)Cl]2 (3*), alongside small amounts of 8*. A reaction of Cp*Sc(CH2C6H4NMe2-o)2 with 3 equiv. AlMe3 at ambient temperature did not yield a distinct product nor 1H and 45Sc NMR spectra similar to 4′.
:1) ppm). However, the amount of pure isolated 9′ was not sufficient for further analytics and various attempts failed to reproduce 9′. The Cp*-analog compound [Cp*Sc(AlMe4)Me] (4*) was not encountered. Prolonged reaction times of 1* with 3 equiv. LiAlMe4 and 4 equiv. AlMe3 at ambient temperature only led to the isolation of [Cp*Sc(AlMe4)Cl]2 (3*), alongside small amounts of 8*. A reaction of Cp*Sc(CH2C6H4NMe2-o)2 with 3 equiv. AlMe3 at ambient temperature did not yield a distinct product nor 1H and 45Sc NMR spectra similar to 4′.
GaMe3 in place of AlMe3: formation of a trimeric Lewis acid stabilized scandium methylidene
GaMe3 is a weaker Lewis acid than AlMe3, according to Pearson's HSAB concept.29 This was key to the isolation of otherwise elusive compounds by our group. For example, GaMe3 gets easily displaced in imido complexes TptBu,MeLn(NAr)(GaMe3) (Ln = Nd, Sm; Ar = C6H3iPr2-2,6) via treatment with THF to yield the terminal imides.9b The AlMe3-containing congener did not show the same reactivity. In another case, GaMe3 was released under reduced pressure from TptBu,MeLn(Me)(GaMe4) to yield the terminal methyl complex TptBu,MeLnMe2.30 Thus, we envisaged organogallium instead of organoaluminum-stabilized scandium methylidenes. The initial reactions of Cp′Sc(CH2C6H4NMe2-o)2 with 3 or 4 equiv. GaMe3 at ambient temperature yielded a mixture of ill-defined products. Since the instability of elusive [Cp′Sc(GaMe4)2] was safe to assume or any subsequently evolved methylidene complex might also be unstable, we targeted a mixed aluminum/gallium compound.Given the formation of Sc/Al2 methylidene 8′ in a reaction of 4′ with AlMe3 at 70 °C, 4′ was treated with GaMe3 in a J. Young-valved NMR tube and heated to 70 °C (Scheme 5). After 6.5 h, the heterotrimetallic methylidene Cp′Sc(CH2)(AlMe3)(GaMe3) (10′) was evidenced as the main product through a 45Sc NMR chemical shift similar to 8′ (275.3 vs. 272.6 ppm). Compound 10′ was (re-)crystallized from a saturated n-hexane solution several times, and analyzed by 1H, 13C{1H}, and 45Sc NMR spectroscopy in toluene-d8, SC-XRD, and elemental analysis. Compound 10′ is isotypic to 8′ and features a very similar Sc–C(methylidene) distance (2.1724(15) vs. 2.1735(18) Å). The 1H NMR spectrum of 10′ shows a broad peak for each AlMe3 and GaMe3 moiety. While the shift of the AlMe3 moiety equals that of 8R, the GaMe3 signal appeared at lower field (−0.17 ppm). The 1H NMR spectroscopic monitoring also revealed that 10′ decomposes slowly at ambient temperature via methane evolution. This lack of stability may be due to GaMe3 being a softer Lewis acid compared to AlMe3,29 once more underpinning the limited coordination capability of GaMe3 in Pearson-type hard/hard metal–ligand pairs. Following this trend, AlMe3 instantly replaces GaMe3 when AlMe3 was added to 10′ to form 8′.
 | ||
| Scheme 5 Reactions of Cp′Sc(AlMe4)Me (4′) with AlMe3 or GaMe3 to form Sc/Al2 methylidene 8′ or mixed Sc/Al/Ga methylidene 10′ and its reaction to [Cp′Sc(CH2)2AlMe]3 (11′). | ||
The decomposition of 10′ is complete after 3 days at 80 °C. During this time, 10′ releases GaMe3 and another equivalent of CH4. Afterwards, crystals of the new Sc/Al methylidene [Cp′Sc(CH2)2AlMe]3 (11′) could be isolated, devoid of any gallium component (Scheme 5). Thus, the decomposition pathway of 10′ favors the formation of a second methylidene over the formation of a methylidyne.13,31 Compound 11′ was analyzed by 1H, 13C{1H}, and 45Sc NMR spectroscopy in C6D6, SC-XRD, DRIFTS, and elemental analysis. The molecular structure of 11′ can be described as a trimer of a half-sandwich scandium methylidene stabilized by a trimeric aluminum methyl methylidene (Fig. 3).
 | ||
| Fig. 3 Crystal structure of 11′. Atomic displacement ellipsoids were set at 50% probability. Hydrogen atoms except methylidene hydrogen atoms omitted for clarity. Selected interatomic distances [Å] and angles [°]: Sc1–C21 2.246(3), Sc1–C24 2.322(3), Al1–C21 2.054(3), Al1–C24 2.015(3), Sc1–H21A 2.29(3), Sc1–H24B 2.41(3), Sc1–C21–H21A 79.6(17) (ESI†). | ||
Overall, 11′ features a Sc3Al3(CH2)6 distorted hexagonal prismatic core, or in other words a triscandacyclohexane ring {Sc3(CH2)3} faced with a trialacyclohexane ring {Al3(CH2)3} through Pearson-type hard/hard matching. In terms of nuclearity, 11′ ranges between the homometallic parent methylidene complex [Cp′Sc(CH2)]4 (II)13 and dodecameric [MeAl(CH2)]12.32 The core structure of 11′ with two chair-like six-membered rings is a common motif for aluminum imides.33 For rare-earth-metal chemistry, it has been rarely observed. A crystal snapshot of homometallic Cp*6Y6(CH2)4Me4, which was obtained via thermolysis of [Cp*YMe2]3, comes close,34 but organo-heterobimetallic Ln/Al clusters exhibiting this structural motif have not been encountered to this date.
The 1H NMR spectrum of 11′ shows one signal set for the three Cp′ ligands and one signal set for the three methyl groups, accounting for a C3 symmetry in solution. The methylidene moieties show three signals. The methylidene protons of the {Sc3(CH2)3} ring appeared as a singlet at −0.13 ppm at ambient temperature. In contrast, the 1H NMR signal of the methylidene protons of the {Al3(CH2)3} ring is split at 0.73 and −1.37 ppm, mainly due to the distinct axial and equatorial ring positions. This splitting might also involve weak α-agostic interactions of one methylidene proton with scandium. In contrast to the rather fluxional methylidene moieties in 8R and 10′, the 1H NMR methylidene signals in 11′ do not coalesce into one signal even at 120 °C. Instead, the methylidene protons of the {Al3(CH2)3} ring show geminal coupling at ambient temperature and at low temperatures, arising from the rigid chair conformation.
Considering the 45Sc NMR chemical shifts, that of 11′ (δ = 396.7 ppm) is shifted downfield compared to 8′ (δ = 272.6 ppm), and hence similar to 9′ (δ = 429.2 ppm). Like observed for 9′, the 45Sc NMR signal of 11′ is much broader than that of 8′ (FWHM: 5566 Hz vs. 2715 Hz, cf., ESI†). The 13C{1H} NMR spectrum of 11′ at ambient temperature shows two broadened signals for the methylidene carbon atoms of each {M3(CH2)3} ring (M = Sc: δ = 81.2 ppm, Al: δ = 35.0 ppm). Similarly, compound Sc3(μ4-CH2)2(μ2-CH3)3(AlMe4)2(AlMe3)2 (Chart 1, IV) featuring two Sc3Al(μ4-CH2) coordination environments revealed a 13C{1H} (methylidene) resonance at 78.9 ppm.18 Apparently, the 13C{1H} NMR chemical shifts of the methylidene carbon atoms are strongly influenced by the number of adjacent rare-earth-metal and aluminum centers. For example, [Ln3(μ3-CH2)] moieties revealed the largest downfield shifts (e.g. (NSiMe3AriPr)3Y3(μ2-Me)3(μ3-Me)(μ3-CH2)(thf)3, δ = 100.2 ppm).12a Changing the ratio of coordinating metal centers from Sc2Al in 11′ (δ = 81.2 ppm) to ScAl2 in 8′ with a very short Sc–C(methylidene) distance (2.1735(18) Å) causes a significant shift to higher field (δ = 53.0 ppm). The signal of the methylidene carbon atom [ScAl2(μ3-CH2)] in 11′ (δ = 35.0 ppm) featuring a longer Sc–C(methylidene) distance (2.246(3) Å) is similar to the methylidene shifts in (PNP)Sc(CH2)(AlMe3)2 (I, δ = 28.77 ppm)8 and (TiPTAC)Y(Me3AlCH2AlMe3)(μ-MeAlMe3) (δ = 34.2 ppm).10c An even higher upfield chemical shift was observed by the group of Diaconescu for the moiety [ScAl2(μ3-CH2)] in (NNFc)Sc(CH2)(AlMe3)2 (δ = −4.7 ppm; NNFc = 1,1′-fc(NSitBuMe2)2), in accordance with a very long Sc–C(methylidene) distance of 2.6921(18) Å.35 The latter chemical shift is very close to the 13C{1H} NMR shift of the methylidene carbon atoms in [MeAl(CH2)]12 (δ = −5.1 ppm).32
The Sc–C(methylidene) distances in 11′ (2.245(3)–2.3338(15) Å) are elongated compared to the monomeric structures of 8R (2.1624(17)/2.1735(18) Å) and 10′ (2.1724(15) Å) (Fig. 3). Consequently, the Sc–H(agostic) distances in 11′ (2.24(4) Å/2.29(3) Å) are longer than those in 8R (2.17(2) Å/2.14(3) Å), but the Sc–C(methylidene)–H(agostic) angles (79.6(17)°/76(2)°) are similar to those in 8R (77.2(13)° for 8*, 75.1(16)° for 8′). The {Al3(CH2)3} ring shows a chair-like conformation with methylidene protons in distinct axial or equatorial positions, whereas the {Sc3(CH2)3} ring is distorted due to the steric pressure of the Cp′ ligands. Here, the methylidene protons don't assume the classical axial or equatorial positions. This may go hand-in-hand with a high mobility of the increasingly ionic methylidene moiety, resulting in only one signal for the two methylidene protons in the 1H NMR spectrum (vide supra). A strategy for reducing the ratio of stabilizing aluminum moieties per rare-earth-metal center to 1:1 was described for the Sc/Al silylalkylidene complex (PNP)Sc(μ2-CHSiMe3)(μ2-Me)[Al(Me)(CH2SiMe3)] by the group of Mindiola.36 There, the chemical shift of the methylidene carbon atom was noted as a broad signal at 137 ppm, corresponding to a very short Sc–CHSiMe3 distance of 2.0845(14) Å. The heterobimetallic complex Cp*Ir(μ2-CHSiMe3)(μ2-NtBu)Sc(Cp′)(py)x displayed similar spectroscopic and structural signatures (x = 0: 13C{1H}, 114.4 ppm; x = 1: Sc–C, 2.139(9) Å).37 For further comparison, scandium complexes bearing dianionic [C(PPh2S)2] carbene pincer-type ligands revealed Sc–C distances as short as 2.200(3) Å,38 and non-chelated phosphoniomethylidene complex (PNP)Sc(μ2-CPPh3)(AlMe3) feature the shortest Sc–C distance (2.036(5) Å) recorded to date.36,39
Reactivity studies of methylidenes CpRSc(CH2)(AlMe3)2 and [Cp′Sc(CH2)2AlMe]3
Monomeric methylidenes 8R eliminate methane at 80 °C, yielding unidentified products. Trimeric 11′, in contrast, is stable at 100 °C. Compounds 8R methylenate fluorenone instantly at ambient temperature, whereas the reaction of 11′ with fluorenone takes 3 h at 80 °C until completion (cf., ESI†). Full conversion of methylidene complexes 8R is indicated by 45Sc NMR spectroscopy, revealing chemical shifts to higher field by ca. 50 ppm, likely ascribed to the symmetric oxo species CpRSc(O)(AlMe3)2 (cf., ESI†). Since the reactions were conducted with two equivalents of fluorenone, methylenation is competing with or followed by methylation.9c,12c Hence, comparatively low yields of dibenzofulvene were obtained (8*, 45%; 8′, 46%; 11′, 19%). It should be noted that DFT calculations on the methylenation of ketones by the trinuclear cluster [PhC(NC6H3iPr2-2,6)2]3Sc3(μ3-CH2)(μ3-Me)(μ2-Me)3 showed that methylene transfer rather than methyl transfer applies.40 Reactivities of the methylidenes toward multiple donors were tested as well. Compound 8* was treated with pyridine, leading to an immediate color change from colorless to yellow. After two hours at ambient temperature, a dark-brown solution had evolved. From this dark-brown solution, colorless crystals of AlMe3(pyr) could be obtained at −40 °C (cf., ESI†). Compound 11′ does not react with pyridine, THF nor diethyl ether even at 100 °C. Compounds 8R and 11′ oligomerize δ-valerolactone instead of promoting methylenation (cf., ESI†). This suits the known reactivities of rare-earth-metal methylidenes in contrast to Tebbe's reagent.11 A preliminary evaluation of the oligomerization/polymerization performance of complexes 8R reveal an activity comparable to [Cp′ScMe2]2 (ref. 22) but significantly higher than Cp*2YMe(thf) or commercially available yttrium isopropoxide (conditions: toluene as a solvent, ambient temperature, 1 h; Table S5†).Conclusions
Cyclopentadienyl-supported discrete methylidenes CpRSc(CH2)(AlMe3)2 (CpR = C5Me5, C5Me4SiMe3) are accessible directly by a tandem metathesis reaction/thermally induced methane elimination by treatment of CpRScCl2(μ-Cl)Li(thf)3 with the alkylating mix LiAlMe4/AlMe3. These reactions proceed via isolable intermediates as evidenced for [CpRSc(AlMe4)Cl]2 and [Cp′Sc(AlMe4)Me]. Here, in situ45Sc NMR spectroscopy provides a valuable tool to assess the progress of such one-pot reactions. The mixed methyl/tetramethylaluminato complex [Cp′Sc(AlMe4)Me] reacts with GaMe3 to yield the first example of a mixed Sc/Al/Ga methylidene, Cp′Sc(CH2)(AlMe3)(GaMe3). Thermal treatment of Cp′Sc(CH2)(AlMe3)(GaMe3) generates the new trimeric scandium aluminum bis(methylidene) cluster [Cp′Sc(CH2)2AlMe]3, under release of GaMe3 and CH4. The {Sc3Al3} hexametallic cluster is closing the gap between methylidenes solely bound to rare-earth-metal centers of the {Sc4} cubane-type, [Cp*Sc(CH2)]4 (ref. 13), and {ScAl2}-type methylidenes bound to only one rare-earth-metal center and two stabilizing Lewis acids, like CpRSc(CH2)(AlMe3)2. X-ray diffraction and NMR analyses revealed enhanced rigidity of the thermally quite stable {Sc3Al3} cluster in contrast to the fluxional methylidenes CpRSc(CH2)(AlMe3)2. Distinct reactivity of the {ScAl2} and {Sc3Al3} methylidenes is found toward donors and for ketone methylenation. The {ScAl2}-type methylidene converts fluorenone immediately at ambient temperature, whereas the {Sc3Al3} cluster needs prolonged reaction time at elevated temperatures. Finally, taking the earlier reported orange oil, putative [Cp′Sc(AlMe4)2], under close scrutiny led to the development of this new line of methylidenes with commercially available ligands Cp* and Cp′.Author contributions
GTLZ, synthesis and characterization of compounds, writing and editing original draft; SAZ, synthesis and characterization of compound 11′; JR, synthesis and characterization of compound 8*; HS, crystallography of compound 8′; CM-M, crystallography, editing original draft; RA, conceptualization, supervision, writing and editing original draft, project administration, funding acquisition.Data availability
Experimental, spectroscopic and structural data supporting this article have been uploaded as part of the ESI.† Crystallographic data for all compounds have been deposited at the CCDC under 2410874–2410889,† and can be obtained from https://www.ccdc.cam.ac.uk/structures/.Conflicts of interest
There are no conflicts to declare.References
- (a) R. Schrock, The First Isolable Transition Metal Methylene Complex and Analogs. Characterization, Mode of Decomposition, and Some Simple Reactions, J. Am. Chem. Soc., 1975, 97, 6577–6578 CrossRef CAS; (b) R. R. Schrock, Alkylidene Complexes of Niobium and Tantalum, Acc. Chem. Res., 1979, 12, 98–104 CrossRef CAS.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, Olefin Homologation with Titanium Methylene-Compounds, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS.
- Olefination: (a) S. H. Pine, G. S. Shen and H. Hoang, Ketone Methylenation Using the Tebbe and Wittig Reagents - A Comparison, Synthesis, 1991, 165–167 CrossRef CAS; (b) Modern Carbonyl Olefination, ed. T. Takeda, Wiley-VCH, 2004 Search PubMed.
- (a) T. R. Howard, J. B. Lee and R. H. Grubbs, Titanium metallacarbene-metallacyclobutane reactions: stepwise metathesis, J. Am. Chem. Soc., 1980, 102, 6876–6878 CrossRef CAS; (b) K. C. Ott, E. J. M. De Boer and R. H. Grubbs, An investigation of the reaction of bis(cyclopentadienyl)titanium dichlorides with trimethylaluminum. Mechanism of an α-hydrogen abstraction reaction, Organometallics, 1984, 3, 223–230 CrossRef CAS; (c) J. D. Meinhart, E. V. Anslyn and R. H. Grubbs, Synthesis, reactivity and kinetic studies of bis(η5-cyclopentadienyl)titanium methylidene phosphine complexes, Organometallics, 1989, 8, 583–589 CrossRef CAS; (d) R. Thompson, E. Nakamaru-Ogiso, C.-H. Chen, M. Pink and D. J. Mindiola, Structural Elucidation of the Illustrious Tebbe Reagent, Organometallics, 2013, 33, 429–432 CrossRef.
- J. Scott and D. J. Mindiola, A tribute to Frederick Nye Tebbe. Lewis acid stabilized alkylidyne, alkylidene, and imides of 3d early transition metals, Dalton Trans., 2009, 8463–8472 RSC and references therein.
- M. Kamitani, B. Pinter, C.-H. Chen, M. Pink and D. J. Mindiola, Mononuclear and Terminal Zirconium and Hafnium Methylidenes, Angew. Chem., Int. Ed., 2014, 53, 10913–10915 CrossRef CAS PubMed.
- S. Senthil, D. Fehn, M. R. Gau, A. M. Bacon, P. J. Carroll, K. Meyer and D. J. Mindiola, A Vanadium Methylidene, J. Am. Chem. Soc., 2024, 146, 15666–15671 CrossRef CAS PubMed.
- J. Scott, H. J. Fan, B. F. Wicker, A. R. Fout, M.-H. Baik and D. J. Mindiola, Lewis Acid Stabilized Methylidene and Oxoscandium Complexes, J. Am. Chem. Soc., 2008, 130, 14438–14439 CrossRef CAS PubMed.
- (a) R. Litlabø, M. Zimmermann, K. Saliu, J. Takats, K. W. Törnroos and R. Anwander, A Rare-Earth Metal Variant of the Tebbe Reagent, Angew. Chem., Int. Ed., 2008, 47, 9560–9564 CrossRef PubMed; (b) T. E. Rieser, R. Thim-Spöring, D. Schädle, P. Sirsch, R. Litlabø, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, Open-Shell Early Lanthanide Terminal Imides, J. Am. Chem. Soc., 2022, 144, 4102–4113 CrossRef CAS PubMed; (c) D. Schädle, R. Litlabø, M. Meermann-Zimmermann, R. Thim-Spöring, C. Schädle, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Rare-Earth-Metal Methyl and Methylidene Complexes Stabilized by TpR,R’-Scorpionato Ligands−Size Matters, Inorg. Chem., 2024, 63, 9624–9637 CrossRef PubMed.
- Similar Lewis acid stabilized complexes were obtained by introducing neutral donors to homoleptic tris(tetramethyl)aluminates Ln(AlMe4)3: (a) L. C. H. Gerber, E. LeRoux, K. W. Törnroos and R. Anwander, Elusive trimethyllanthanum: snapshots of extensive methyl group degradation in La–Al heterobimetallic complexes, Chem. – Eur. J., 2008, 14, 9555–9564 CrossRef CAS PubMed; (b) A. Venugopal, I. Kamps, D. Bojer, R. J. F. Berger, A. Mix, A. Willner, B. Neumann, H.-G. Stammler and N. W. Mitzel, Neutral ligand induced methane elimination from rare-earth metal tetramethylaluminates up to the six-coordinate carbide state, Dalton Trans., 2009, 5755–5765 RSC; (c) D. Bojer, A. Venugopal, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, C–H Activation versus Yttrium–Methyl Cation Formation from [Y(AlMe4)3] Induced by Cyclic Polynitrogen Bases: Solvent and Substituent-Size Effects, Chem. – Eur. J., 2011, 17, 6248–6255 CrossRef CAS PubMed.
- (a) H. M. Dietrich, K. W. Törnroos and R. Anwander, “Ionic carbenes”: Synthesis, structural characterization, and reactivity of rare-earth metal methylidene complexes, J. Am. Chem. Soc., 2006, 128, 9298–9299 CrossRef CAS PubMed; (b) V. M. Birkelbach, F. Kracht, H. M. Dietrich, C. Stuhl, C. Maichle-Mössmer and R. Anwander, A Rare-Earth-Metal Ensemble of the Tebbe Reagent: Scope of Coligands and Carbonyl Olefination, Organometallics, 2020, 39, 3490–3504 CrossRef CAS; (c) D. A. Buschmann, L. Schumacher and R. Anwander, Rare-earth-metal half-sandwich complexes incorporating methyl, methylidene, and hydrido ligands, Chem. Commun., 2022, 58, 9132–9135 RSC.
- Motif III is the most frequently one encountered for rare-earth-metal methylidenes: (a) M. Zimmermann, D. Rauschmaier, K. Eichele, K. W. Törnroos and R. Anwander, Amido-stabilized rare-earth metal mixed methyl methylidene complexes, Chem. Commun., 2010, 46, 5346–5348 RSC; (b) J. Kratsch and P. W. Roesky, Rare-Earth-metal methylidene complexes, Angew. Chem., Int. Ed., 2014, 53, 376–383 CrossRef CAS PubMed; (c) D. Schädle, M. Meermann-Zimmermann, C. Maichle-Mössmer, C. Schädle, K. W. Törnroos and R. Anwander, Rare-earth metal methylidene complexes with Ln3(μ3-CH2)(μ3-Me)(μ2-Me)3 core structure, Dalton Trans., 2015, 44, 18101–18110 RSC; (d) J. Hong, Z. Li, Z. Chen, L. Weng, X. Zhou and L. Zhang, Small molecule activation by mixed methyl/methylidene rare earth metal complexes, Dalton Trans., 2016, 45, 6641–6649 RSC.
- P. Deng, X. Shi, X. Gong and J. Cheng, Trinuclear scandium methylidyne complexes stabilized by pentamethylcyclopentadienyl ligands, Chem. Commun., 2021, 57, 6436–6439 RSC.
- W.-X. Zhang, Z. Wang, M. Nishiura, Z. Xi and Z. Hou, Ln4(CH2)4 Cubane-Type Rare-Earth Methylidene Complexes Consisting of “(C5Me4SiMe3)LnCH2” Units (Ln = Tm, Lu), J. Am. Chem. Soc., 2011, 133, 5712–5715 CrossRef CAS PubMed.
- J. Hong, L. Zhang, X. Yu, M. Li, Z. Zhang, P. Zheng, M. Nishiura, Z. Hou and X. Zhou, Syntheses, Structures, and Reactivities of Homometallic Rare-Earth-Metal Multimethyl Methylidene and Oxo Complexes, Chem. – Eur. J., 2011, 17, 2130–2137 CrossRef CAS PubMed.
- D. S. Levine, T. D. Tilley and R. A. Andersen, Evidence for the Existence of Group 3 Terminal Methylidene Complexes, Organometallics, 2017, 36, 80–88 CrossRef CAS.
- J. Zhou, T. Li, L. Maron, X. Leng and Y. Chen, A Scandium Complex Bearing Both Methylidene and Phosphinidene Ligands: Synthesis, Structure, and Reactivity, Organometallics, 2015, 34, 470–476 CrossRef CAS.
- D. Barisic, D. Diether, C. Maichle-Mössmer and R. Anwander, Trimethylscandium, J. Am. Chem. Soc., 2019, 141, 13931–13940 CrossRef CAS PubMed.
- N. A. Petasis and E. I. Bzowej, Titanium-mediated carbonyl olefinations. 1. Methylenations of carbonyl compounds with dimethyltitanocene, J. Am. Chem. Soc., 1990, 112, 6392–6394 CrossRef CAS.
- A. Mortis, C. Maichle-Mössmer and R. Anwander, Distinct Reactivity of Trimethylytterbium toward AlMe3 and GaMe3. Synthesis of Donor-Stabilized Dimethylytterbium, Chem. – Eur. J., 2023, 29, e202203824 CrossRef CAS PubMed.
- (a) J. Hitzbleck, K. Beckerle and J. Okuda, Half-sandwich dibenzyl complexes of scandium: Synthesis, structure, and styrene polymerization activity, J. Organomet. Chem., 2007, 692, 4702–4707 CrossRef CAS; (b) A. Fridrichová, A. Růžička, M. Lamač and M. Horáček, Structural differences of half-sandwich complexes of scandium and yttrium containing bulky substituents, Inorg. Chem. Commun., 2017, 76, 62–66 CrossRef.
- D. Robert, T. P. Spaniol and J. Okuda, Neutral and monocationic half-sandwich methyl rare-earth metal complexes: Synthesis, structure, and 1,3-butadiene polymerization catalysis, Eur. J. Inorg. Chem., 2008, 2801–2809 CrossRef CAS.
- P. L. Watson, Ziegler-Natta Polymerization: The Lanthanide Model, J. Am. Chem. Soc., 1982, 104, 337–339 CrossRef CAS.
- K. A. Tupper and T. D. Tilley, Synthesis and characterization of scandium complexes with reduced ligands: Crystal structures of Cp*ScI2, [Cp*ScI(bpy)]2, and [Cp*ScCl(bpy)]2, J. Organomet. Chem., 2005, 690, 1689–1698 CrossRef CAS.
- H. M. Dietrich, O. Schuster, C. Maichle-Mössmer and R. Anwander, Heterobimetallic Halflanthanidocene Clusters: Novel Mixed Tetramethylaluminato Chloro Coordination, Angew. Chem., Int. Ed., 2006, 45, 4858–4863 CrossRef CAS PubMed.
- J. J. Byers, W. T. Pennington and G. H. Robinson, Bis-Adducts of Trimethylaluminum and Trimethylgallium with N,N,N′,N’-Tetramethylethylenediamine, Acta Crystallogr., 1992, C48, 2023–2025 CAS.
- F. Thomas, T. Bauer, S. Schulz and M. Nieger, Comparative structural studies on 4-dimethylaminopyridine-adducts, Z. Anorg. Allg. Chem., 2003, 629, 2018–2027 CrossRef CAS.
- H. Schumann, I. Albrecht, J. Pickardt and E. Hahn, Metallorganische Verbindungen der Lanthanoide. XXIII. Synthese und Struktur von [Li(tmed)2][C5Me5Lu(CH3)3], J. Organomet. Chem., 1984, 276, C5–C9 CrossRef CAS.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- M. Zimmermann, R. Litlabø, K. W. Törnroos and R. Anwander, “Metastable” Lu(GaMe4)3 Reacts Like Masked [LuMe3]: Synthesis of an Unsolvated Lanthanide Dimethyl Complex, Organometallics, 2009, 28, 6646–6649 CrossRef CAS.
- H. M. Dietrich, H. Drove, K. W. Törnroos and R. Anwander, Multiple C–H Bond Activation in Group 3 Chemistry: Synthesis, structural characterization of an Yttrium–Aluminum–Methine Cluster, J. Am. Chem. Soc., 2006, 128, 1458–1459 CrossRef CAS PubMed.
- G. Spiridopoulos, M. Kramer, F. Kracht, C. Maichle-Mössmer and R. Anwander, [(CH3)Al(CH2)]12: Methylaluminomethylene (MAM-12), Chem. – Eur. J., 2022, 28, e202200823 CrossRef CAS PubMed.
- (a) M. Cesari, G. Perego, G. Delpiero, S. Cucinella and E. Cernia, Chemistry and Stereochemistry of Poly(N-Alkyl-Iminoalanes): II. Crystal and Molecular Structure of Hexamer, (HAlN-i-Pr)6, Organomet. Chem., 1974, 78, 203–213 CrossRef CAS; (b) G. Delpiero, M. Cesari, G. Perego, S. Cucinella and E. Cernia, Chemistry and Stereochemistry of Poly(N-Alkyliminoalanes): XI. Crystal and Molecular Structure of Hexamer (HAlN-n-Pr)6 and Octamer (HAlN-n-Pr)8, J. Organomet. Chem., 1977, 129, 289–298 CrossRef CAS; (c) G. Delpiero, S. Cucinella and M. Cesari, Chemistry and the Stereochemistry of Poly(N-Alkyliminoalanes): XVI. Crystal and Molecular Structure of the Compound [HAlNCH(CH3)C6H5]6·1/3 C6H14, J. Organomet. Chem., 1979, 173, 263–268 CrossRef CAS; (d) A. A. I. Alwassil, P. B. Hitchcock, S. Sarisaban, J. D. Smith and C. L. Wilson, Complexes of Organoaluminium Compounds. Part 13. Preparation and Nuclear Magnetic Resonance Spectra of the Arylamido-Compounds AlMe2(NHR’)(R’= Ph, C6H4Me-o, C6H4Me-p, C6H3Me2-2,6) and the Imido-Compounds AlMe(NR’). Crystal and Molecular Structures of [{AlMe2(NHC6H4Me-o,)}2] and [{AlMe(NPh)}6], J. Chem. Soc., Dalton Trans., 1985, 1929–1934 RSC; (e) C. Schnitter, S. D. Waezsada, H. W. Roesky, M. Teichert, I. Uson and E. Parisini, Synthesis and characterization of (4-fluorophenyl)amino-based amino- and iminometallanes of group 13. Crystal structures of (MeAlNRf)4, (MeMNRf)6·nTHF (M = Al, n = 2; M = Ga, n = 7), and (MeIn(THF)NRf)4 (Rf = 4-C6H4F), Organometallics, 1997, 16, 1197–1202 CrossRef CAS; (f) J. E. Park, B.-J. Bae, Y. Kim, J. T. Park and I.-H. Suh, Reactions of AlR3 (R = Me, Et) with H2NCH2CH2NMe2: Synthesis and characterization of amido- and imidoalanes, Organometallics, 1999, 18, 1059–1067 CrossRef CAS; (g) N. D. Reddy, H. W. Roesky, M. Noltemeyer and H. G. Schmidt, Reactions of AIH3·NMe3 with nitriles: Structural characterization and substitution reactions of hexameric aluminum imides, Inorg. Chem., 2002, 41, 2374–2378 CrossRef CAS PubMed; (h) E. K. Styron, C. H. Lake, D. H. Powell, L. K. Krannich and C. L. Watkins, Reactivity of Me3M (M = Al, Ga, In) with benzylamine to form the dimers, [Me2MN(H)CH2Ph]2, and the hexamers, [MeAlNCH2Ph]6 and [MeGaNCH2Ph]6: molecular structures of trans-[Me2MN(H)CH2Ph]2 and [MeAlNCH2Ph]6, J. Organomet. Chem., 2002, 649, 78–85 CrossRef CAS; (i) N. D. Reddy, S. S. Kumar, H. W. Roesky, D. Vidovic, J. Magull, M. Noltemeyer and H. G. Schmidt, Synthesis of a hexadentate hexameric aluminum imide and its metathesis reactions, Eur. J. Inorg. Chem., 2003, 442–448 CrossRef CAS.
- C. O. Hollfelder, L. N. Jende, H. M. Dietrich, K. Eichele, C. Maichle-Mössmer and R. Anwander, 1,3-Diene Polymerization Promoted by Half-Sandwich Rare-Earth-Metal Dimethyl Complexes: Active Species Clustering and Cationization/Deactivation Processes, Chem. – Eur. J., 2019, 25, 7298–7302 CrossRef CAS PubMed.
- W. Huang, C. T. Carver and P. L. Diaconescu, Transmetalation Reactions of a Scandium Complex Supported by a Ferrocene Diamide Ligand, Inorg. Chem., 2011, 50, 978–984 CrossRef CAS PubMed.
- P. Zatsepin, E. Lee, J. Gu, M. R. Gau, P. J. Carroll, M.-H. Baik and D. J. Mindiola, Tebbe-like and Phosphonioalkylidene and -alkylidyne Complexes of Scandium, J. Am. Chem. Soc., 2020, 142, 10143–10152 CrossRef CAS PubMed.
- T. Shima, T. Yanagi and Z. Hou, Rare-earth-iridium heterobimetallic complexes with bridging imido and silylmethyl ligands: synthesis, structure and reactivity, New J. Chem., 2015, 39, 7608–7616 RSC.
- (a) M. Fustier, X. F. Le Goff, P. Le Floch and N. Mézailles, Nucleophilic Scandium Carbene Complexes, J. Am. Chem. Soc., 2010, 132, 13108–13110 CrossRef CAS PubMed; (b) M. Fustier, X. F. Le Goff, M. Lutz, J. C. Slootweg and N. Mézailles, Scandium Carbene Complexes: Synthesis of Mixed Alkyl, Amido, and Phosphido Derivatives, Organometallics, 2015, 34, 63–72 CrossRef CAS.
- (a) C. Wang, J. Zhou, X. Zhao, L. Maron, X. Leng and Y. Chen, Non-Pincer-Type Mononuclear Scandium Alkylidene Complexes: Synthesis, Bonding, and Reactivity, Chem. – Eur. J., 2016, 22, 1258–1261 CrossRef CAS PubMed; (b) W. Mao, L. Xiang, C. A. Lamsfus, L. Maron, X. Leng and Y. Chen, Highly Reactive Scandium Phosphinoalkylidene Complex: C–H and H–H Bonds Activation, J. Am. Chem. Soc., 2017, 139, 1081–1084 CrossRef CAS PubMed; (c) W. Mao, L. Xiang, L. Maron, X. Leng and Y. Chen, Nonchelated Phosphinomethylidene Complexes of Scandium and Lutetium, J. Am. Chem. Soc., 2017, 139, 17759–17762 CrossRef CAS PubMed.
- G. Luo, Y. Luo, J. Qu and Z. Hou, Mechanistic Insights into the Methylenation of Ketone by a Trinuclear Rare-Earth-Metal Methylidene Complex, Organometallics, 2015, 34, 366–372 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, detailed crystallographic data, spectroscopic data (NMR, IR), analytical details. CCDC 2410874–2410889. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00152h |
| This journal is © the Partner Organisations 2025 |