
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into fluoride's role in MFI zeolites: unveiling the link between location and synthesis conditions†
Joaquín
Martinez-Ortigosa
 ab,
Reisel
Millán
a,
Jorge
Simancas
ac,
J. Alejandro
Vidal-Moya
a,
Charlotte
Martineau-Corcos‡
d,
Fernando
Rey
*a and
Teresa
Blasco
*a
ab,
Reisel
Millán
a,
Jorge
Simancas
ac,
J. Alejandro
Vidal-Moya
a,
Charlotte
Martineau-Corcos‡
d,
Fernando
Rey
*a and
Teresa
Blasco
*a
aInstituto de Tecnología Química, Universitat Politècnica de València – Consejo Superior de Investigaciones Científicas (UPV-CSIC), Avda. de los Naranjos s/n, 46022, Valencia, Spain. E-mail: tblasco@itq.upv.es; frey@itq.upv.es
bLaboratorio de Nanotecnología Molecular, Departamento de Química Inorgánica, Universidad de Alicante, 03690, Alican-te, Spain
cInstitute of Advanced Materials (INAM), Universitat Jaume I (UJI), Castelló de la Plana, 12071, Spain
dUniversité Paris-Saclay, UVSQ, CNRS, Institut Lavoisier de Versailles, 78000, Versailles, France
First published on 24th January 2025
Abstract
This study examines the fluoride distribution in pure silica MFI zeolite synthesized in fluoride medium using various tetra-alkyl ammonium and phosphonium cations as organic structure directing agents (OSDAs). The 19F NMR spectra show a resonance at ∼−65 ppm, related with the presence of fluoride atoms in the t-mel cage bonded to the silicon at T9 site, and another signal at ∼−80 ppm. Through DFT calculations, the ∼−80 ppm 19F NMR resonance is attributed to fluoride within the t-mel cage bonded to silicon at the T12 site. Thus, the results reported here reveal that, contrary to previous reports, there is more than one position for fluoride in the zeolite structure. The location of fluoride at T12 is stabilized by using bulky OSDAs and by increasing the concentration of defects in the MFI framework. Moreover, it is shown that the OSDA used in the synthesis determines the content and distribution of siloxy–silanol defects and fluoride in pure silica MFI zeolite. These findings highlight the impact of synthesis parameters in the charge distribution in zeolites, which ultimately governs key structural properties and potential applications of zeolite materials in catalysis and adsorption processes.
1. Introduction
Zeolites are microporous materials widely used in industrial processes, especially in separations and catalysis. To illustrate their importance, the global consumption of synthetic zeolites in 2017 was estimated at ∼1.8 million metric tons per year, with 17% used as heterogeneous catalysts and 10% as adsorbents/desiccants.1 Zeolites possess unique crystalline frameworks, primarily composed of [SiO4] tetrahedra sharing oxygen atoms, forming channels and cavities of molecular dimensions.2,3 Their regular microporous structure and diverse chemical compositions (commonly silicates and aluminosilicates, but also borosilicates, titanosilicates, stannosilicates and others) support a broad range of catalytic processes.4–6 Zeolites are also valuable in selective adsorption based on its pore size, shape, and polarity, making them essential in industrial applications, such as petrochemical, gas separation, and environmental remediation processes.7–9 The wide range of zeolite structures and the tunability of their physicochemical properties, such as hydrophilicity and hydrophobicity, further enhances their versatility in both traditional and emerging applications.10,11In the field of catalytic materials, charge matching between cationic species within zeolite voids and the negatively charged framework—typically aluminosilicate—plays a crucial role in the location of active sites, significantly influencing the selectivity of zeolites in catalytic reactions. A deeper understanding of how charge balance dictates active site positioning during synthesis enables the design of zeolites with enhanced catalytic properties, resulting in more efficient and selective catalysts for industrial applications.12,13
A critical performance factor in the use of zeolites for selective separation processes, especially in alcohol recovery and water treatment, is the concentration of framework defects.14,15 High defect concentrations increase hydrophilicity, essential for the selective adsorption of polar molecules like water or small alcohols.16,17 These defects are associated with (i) isolated silanol groups (Si–OH) within pores or at crystal surfaces and (ii) H-bonded silanol networks, formed from framework vacancies or Si–O–Si bond breakage.11,16 In addition, silanol clusters in zeolites are prone to incorporate other elements suitable for catalytic applications,18 and can serve as active sites for specific reactions.19,20 Thus, understanding the nature of zeolitic silanol is not only academically significant but also crucial for optimizing the performance of zeolites in catalytic and adsorption applications. Charge distribution in the synthesis medium is widely recognized as a crucial factor in determining the hydrophobic or hydrophilic nature of zeolites, prompting extensive research into their crystallization mechanisms.21–24
Zeolites are typically synthesized using organic cations as organic structure-directing agents (OSDAs), which occupy the voids and, in the case of aluminosilicate zeolites, are charge-balanced by Al3+ substituting for framework Si4+.25,26 However, in high-silica zeolites, charge compensation for OSDAs strongly depends on the synthesis conditions. When synthesis occurs in basic media (high pH), OSDA cations are compensated by structural defects based on siloxy species associated with silanol groups (SiO−⋯HOSi), which have been the subject of numerous investigations.27–30 Additionally, some studies suggest that the type of OSDA used in synthesis can influence the location of silanol defects,31 similar to how it affects Al3+ siting in aluminosilicate materials.32 In fluoride-mediated synthesis, OSDA cations are charge-balanced by fluoride anions that occupy small zeolite cages,33,34 resulting in defect-free or low-defect hydrophobic materials. In all-silica MFI zeolite (Silicalite-1), fluoride ions are located in t-mel cages, forming a Si–F bond at the T9 site,35 which, to date, remains the only position with a Si–F linkage in the extensively studied MFI structure.
Here, we investigate the influence of N- and P-based OSDAs on the concentration of silanol defects and the fluoride location in pure silica zeolite MFI by applying Magic Angle Spinning Nuclear Magnetic Resonance (MAS-NMR) spectroscopy and DFT calculations. Our findings reveal that, contrary to previous reports, fluoride occupies different sites depending on the OSDA type and synthesis conditions, offering a promising approach to tailoring MFI material properties by fine-tuning synthesis parameters.
2. Experimental
2.1. Silicalite-1 synthesis
The synthesis of Silicalite-1 zeolites (IZA code: MFI) was performed using the sol–gel methodology, consisting in the preparation of a suspension by mixing the different reactants that constitute the synthesis gel in polypropylene beakers. As a general procedure, a given amount of tetraethylorthosilicate (TEOS, 99% Aldrich) was added to an aqueous solution containing the quaternary ammonium or phosphonium cation or a mixture of both in their hydroxide form. The mixture was kept under stirring for 4 hours to let the TEOS to be hydrolysed and to evaporate the ethanol and the necessary amount of water to yield a synthesis gel with molar ratio of 1 SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :0.4 OSDA(OH):10 H2O. Then, fluorhydric acid (48 wt% in water, Aldrich) was added in the same molar amount than the number of OH− groups of the OSDA and the mixture is further stirred for homogenization, which usually took 15 min. After that, the resulting gels were transferred into Teflon lined stainless-steel autoclaves and heated at 175 °C at its autogenous pressure under tumbling (60 rpm) for times ranging from 5 hours to 30 days. The solids were recovered by filtration, washed exhaustively with distilled boiling water and dried at 100 °C overnight.
:0.4 OSDA(OH):10 H2O. Then, fluorhydric acid (48 wt% in water, Aldrich) was added in the same molar amount than the number of OH− groups of the OSDA and the mixture is further stirred for homogenization, which usually took 15 min. After that, the resulting gels were transferred into Teflon lined stainless-steel autoclaves and heated at 175 °C at its autogenous pressure under tumbling (60 rpm) for times ranging from 5 hours to 30 days. The solids were recovered by filtration, washed exhaustively with distilled boiling water and dried at 100 °C overnight.
A large variety of ammonium or phosphonium-based OSDA cations were employed in this work. The OSDAs employed are listed next: tetraethyl ammonium (TEA) and tetraethyl phosphonium (TEP), tetrapropyl ammonium (TPA) and tetrapropyl phosphonium (TPP), tetrabutyl ammonium (TBA) and tetrabutyl phosphonium (TBP), tributylmethyl ammonium (TBMA) and tributylmethyl phosphonium (TBMP); butane-1,4-diylbis(triethylphosphonium) (EPPE) and butane-1,4-diylbis(tripropylphosphonium) (BPPB). The sample name will be stablished by the letter code of the OSDA and in case of using a mixture, the subindex number will indicate the P-OSDA proportion.
2.2. General characterization
The powder X-ray diffractograms were obtained by using a PANalytical X'Pert PRO instrument equipped with a graphite monochromator, operating at 45 kV and 40 mA, and using Cu Kα radiation (λ = 1.542 Å). The chemical composition of the solids was determined by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) in a Varian 710-ES device or by the employ of the EuroEA3000 of Eurovector for the C and N elemental analysis. A Bruker Avance III HD 400 MHz spectrometer was used to record the following MAS-NMR spectra. 19F NMR spectra at ν0(19F) = 376.5 MHz were acquired with a 3.2 mm probe spinning the sample at 20 kHz, using a π/2 pulse length of 5.5 μs and 60 s recycle delay. 1H NMR spectra at ν0(1H) = 400.1 MHz were acquired with a 3.2 mm probe at 20 kHz as a spinning speed, π/2 pulse length of 2.5 μs and 15 s as recycle delay. 29Si NMR measurements at ν0(29Si) = 79.5 MHz were carried out in a 7.0 mm probe with 3.5 μs pulse length corresponding to 60° flip angle, spinal proton decoupling, and 180 s as recycle delay. The 29Si low temperature (LT) cross-polarization (CP) MAS NMR spectra were recorded on a 18.8 T Avance III WB spectrometer using a LT-MAS 3.2 mm probe at temperature range of 100 K. MAS frequency was set to 10 kHz. 1H–29Si contact time for the CP is 3 ms, radiofrequency field on 1H and 29Si was set to 50 kHz. Initial 90° pulse duration for 1H is 2.3 μs. Recycle delay was 5 s for TEP-MFI and 7 s for TBP-MFI, and ∼8000 transients were accumulated for each sample. NMR spectra were referenced to TMS, CFCl3 and water for 29Si, 19F and 1H respectively. Na2SiF6 (δ19F = −152.46 ppm) was used as a secondary reference for 19F.2.3. DFT simulations
Periodic calculations were carried out with the VASP code.36 Energy minimizations were performed with the Perdew–Burke–Ernzerhof generalized gradient approximation (PBE).37,38 In all calculations, the projected augmented wave (PAW)39 pseudopotentials were used to describe the interaction of the valence electrons with the nuclei and core electrons. An energy cutoff of 600 eV was used to expand the plane wave basis set. The Brillouin zone was sampled at the gamma point. The structures were considered converged when the forces acting on atoms were lower than 0.01 eV Å−1. Dispersion energies were evaluated using the D3 Grimme's method40,41 with the Becke–Johnson42 as implemented in VASP. The NMR absolute shielding tensors were computed with the GIPAW approach implemented in VASP. The same energy cut-off as in the optimizations was used but with a tighter convergence criterion of 10–10 eV for the electronic energy. The underestimation of band gaps with GGA functionals leads to a slope that deviates from unity when predicting the chemical shift of alkaline fluorides from linear regression models.43,44 The modified Becke–Johnson exchange potential (TB-mBJ)45 shows a better relation between experimental chemical shifts (δiso) and absolute isotropic shieldings (σiso). The linear regression parameters were taken from ref. 46 and are also shown in ESI†3. Results
In this study, we used as OSDA tetraalkyl-ammonium and phosphonium cations with alkyl chains of different lengths displayed in Fig. 1. Although this work is mainly focused on pure silica MFI zeolites synthesized by the classical route, we have also studied MFI zeolite synthesized using the dual template route with TEA/TEP, TPA/TPP47,48 and TBA/TBP (see Materials and method section of the ESI† for detailed description of the zeolite synthesis procedures). The XRD patterns of the samples, typical of the MFI-type structure, are shown in Fig. S1.† The unit cell parameters (Table S1†) exhibit a slight increase with the elongation of the alkyl chain, reaching a maximum value for the butyl-, tributylmethyl- and diquats OSDA families. | ||
| Fig. 1 OSDAs used for the synthesis of the silicalite-1 zeolites (X refers to N or P, depending on whether it is a tetraalkylammonium or tetraalkylphosphonium ion, respectively). | ||
The chemical compositions of MFI zeolites synthesized using single OSDAs (i.e. phosphonium or ammonium cations, but not their mixtures) are detailed in Table 1 (Table S1† shows the chemical compositions of all samples reported in this study). The materials synthesized with tetraethyl- and tetrapropyl- ammonium/phosphonium and the non-symmetric tributylmethyl- ammonium/phosphonium cations contain approximately four OSDAs per unit cell.35,49 Meanwhile, the zeolites obtained with the bulkier tetrabutyl-ammonium/phosphonium, or with the di-phosphonium (EPPE and BPPB) cations contain fewer than four OSDAs per unit cell, likely due to steric hindrance. The relatively low F content in these samples cannot be attributed to a lack of crystallinity, as their XRD patterns demonstrate that the samples exhibit very high crystallinity (Fig. S1†). Table 1 shows the fluorine content (expressed as F atoms per u.c. and in mmol g−1) and the F/OSDA molar ratio, which represents the fraction of organic cations balanced by fluoride anions. From these results, the remaining OSDAs (i.e., [1 − F/OSDA]) must be compensated by SiO−⋯HOSi defects in the zeolite structure.27,28 Only the TEA-MFI zeolite contains nearly four (3.9) F− per unit cell and a F/OSDA ratio close to one (0.9), indicating that this material contains very few SiO−⋯SiOH defects. The rest of samples have fewer than four F− per unit cell and F/OSDA molar ratios significantly below 1.
| Sample | OSDA/u.ca | F/u.c.a,b | F TOT (mmol g−1) | F/OSDAc | % F−80d |
|---|---|---|---|---|---|
| a Unit cell Si96O192: determined by 19F MAS-NMR by using an external standard. b Expressed as molar ratio. c Relative intensity of signal at −80 ppm. d %F−65 = 100 − %F−80. | |||||
| TEA | 4.4 | 3.9 | 0.59 | 0.91 | 1.0 |
| TEP | 4.1 | 2.7 | 0.43 | 0.66 | 2.9 |
| TPA | 4.5 | 3.3 | 0.48 | 0.74 | 6.9 |
| TPP | 4.3 | 2.4 | 0.34 | 0.56 | 18.8 |
| TBA | 3.6 | 1.8 | 0.29 | 0.50 | 42.6 |
| TBP | 3.0 | 1.5 | 0.24 | 0.49 | 46.8 |
| TBMA | 4.2 | 2.4 | 0.36 | 0.56 | 18.9 |
| TBMP | 4.2 | 1.5 | 0.23 | 0.36 | 54.2 |
| EPPE | 3.8 | 1.9 | 0.32 | 0.49 | 19.1 |
| BPPB | 3.2 | 1.9 | 0.27 | 0.59 | 45.2 |
Inspection of Table 1 shows that both the fluoride content and the F/OSDA ratio in the MFI zeolite decrease as the length of the alkyl chains of the OSDA increases or when using the phosphonium counterpart or the dual template route in the synthesis (see Table S1†). This trend suggests that bulkier OSDAs lead to a reduction in fluoride incorporation, which in turn results in a higher prevalence of structural defects.27 The presence of defects in the zeolite structure is confirmed by the appearance of a 1H NMR signal at δ1H ≈ 10.5 ppm attributed to silanol groups involved in SiO−⋯HOSi defect types (see Fig. S2a†).27,28,47 These structural defects are also evidenced by the broadening of 29Si NMR signals and the presence of a resonance at δ29Si ≈ −103 ppm corresponding to Q3 sites (Fig. S2b†).34 This is illustrated in Fig. S2c† for selected samples highlighting the impact of the OSDAs on the structural defects in the zeolite.
The 19F ss-NMR spectra of the as-synthesized MFI samples are displayed in Fig. 2. The spectrum of the TEA-MFI zeoliteconsists of a single signal at δ19F ≈ −65 ppm attributed to fluoride within the t-mel cage of the MFI structure.35 The 19F NMR spectra of the TEP-MFI and TPA-MFI zeolite show, in addition to the signal at δ19F ≈ −65 ppm, another weak one at δ19F ≈ −80 ppm. The relative intensity of the signal at δ19F ≈ −80 ppm increases for the TPP-MFI sample (Fig. 2) and very sharply for the TBA-MFI and TBP-MFI zeolites, where it accounts for almost half of the total fluoride in these samples. The 19F NMR spectra of the EPPE-MFI and BPPB-MFI zeolites, synthesized with di-cationic OSDAs, closely resemble those of the TEP-MFI and TBP-MFI samples, respectively. In particular, the 19F NMR signals at δ19F ≈ −65 ppm and δ19F ≈ −80 ppm exhibit similar intensities for the zeolites synthesized with butyl-OSDAs. However, for the samples prepared with ethyl-OSDAs, the signal at δ19F ≈ −65 ppm is noticeably more intense. This similarity suggests that the distribution of fluoride in the MFI zeolite synthesized with dicationic OSDAs depends on the length of the side alkyl groups, rather than on the alkyl chain connecting the two phosphorus atoms.
 | ||
| Fig. 2 19F MAS-NMR spectra of the as-made zeolites synthesized with the OSDA indicated in the figure. | ||
The 19F NMR resonance at δ19F ≈ −80 ppm has been already reported for MFI type zeolites, but its nature and location are still under debate.50–54 It has been attributed either to fluoride associated with nearby siloxy defects, or to fluoride interacting with di-cationic imidazolium OSDA. As shown in Fig. 3a, the amount of fluoride contributing to the signal at δ19F ≈ −65 ppm, increases linearly with the total fluoride content in the MFI zeolite, whereas the opposite trend is observed for that giving the signal at δ19F ≈ −80 ppm (Fig. 3b). Our results indicate that the fraction of fluoride responsible of the NMR signal at δ19F ≈ −80 ppm (Table 1), is higher in MFI zeolites synthesized with larger OSDAs, which also correspond to an increase in structural defects. Inspection of Table 1 and S1† (summarized in Fig. 3) indicates that the δ19F ≈ −80 ppm signal accounts for half or more of total fluoride in samples with an F/OSDA molar ratio around 0.5 or lower. Therefore, it is difficult to unambiguously stablish whether the development of the signal at δ19F ≈ −80 ppm is primarily driven by the size of the OSDA, the presence of structural defects, or both, as these factors are closely interconnected.
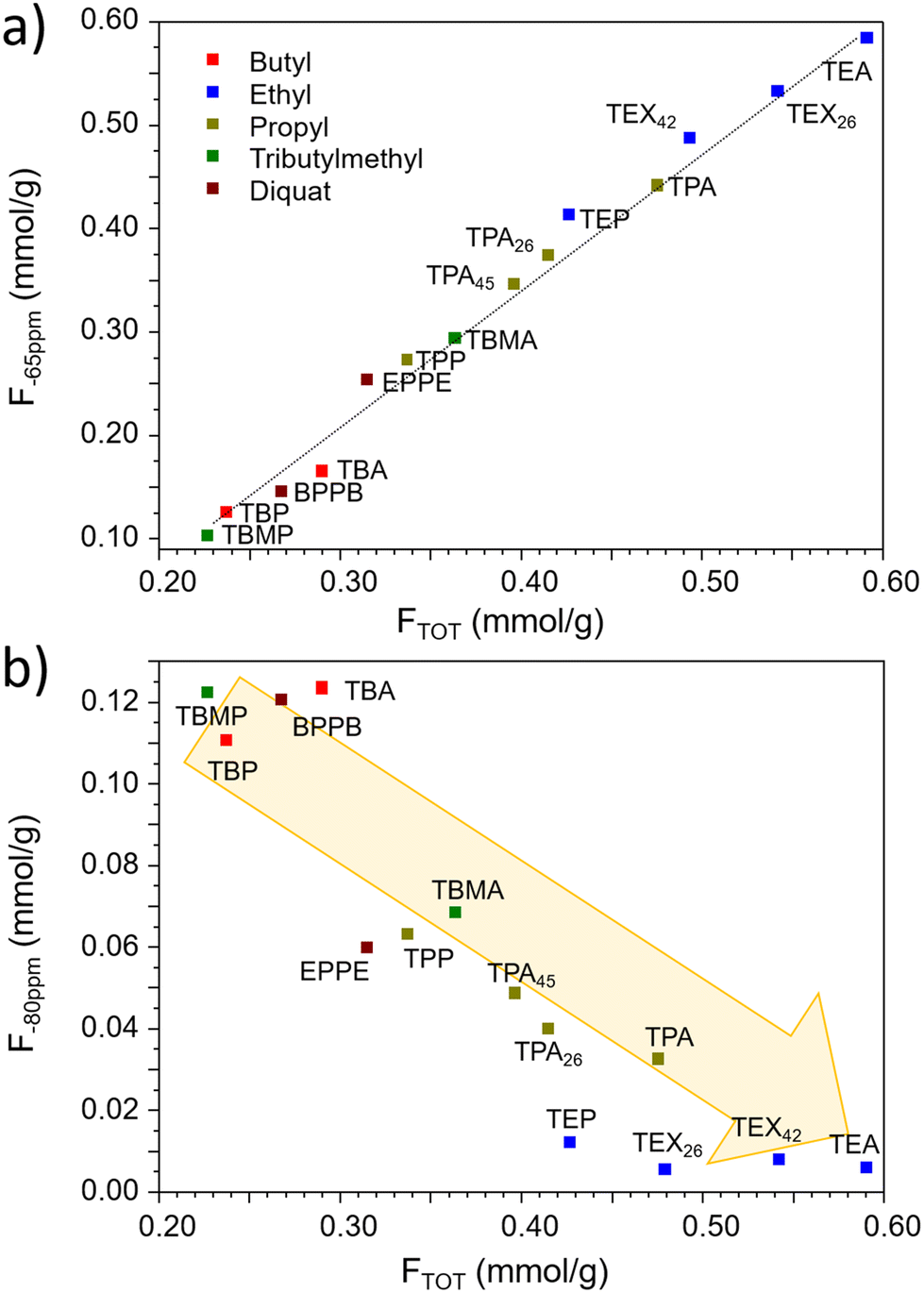 | ||
| Fig. 3 Correlation between the amount of fluoride producing the NMR signals at −65 ppm (a) and at −80 ppm (b) and the total fluoride content for the samples indicated in the figure. The samples synthesized by the dual template route are included in the plots (see ESI†). | ||
To better understand the role of defects in generating the 19F NMR signal at −80 ppm, we studied a set of fully crystalline MFI samples synthesized using TPA as the OSDA, with synthesis times ranging from 5 hours to 720 hours (see ESI†). Our findings show that as synthesis time increases, more fluoride is incorporated into the zeolite, leading to less defective crystals, supported also by the disappearance of the Q3 signal in the 29Si MAS-NMR spectra (Fig. S3†). After 5 hours of synthesis, the intensity of the 19F NMR resonance at −80 ppm accounts for about 30% of the total fluoride, but this intensity sharply decreases with longer synthesis times, becoming almost undetectable after 720 hours (see Fig. S3†). This result provides strong evidence of a direct relationship between the intensity of the NMR signal at δ19F ≈ −80 ppm and the quantity of structural defects present in the zeolite. 19F NMR signal at δ19F ≈ −65 ppm is assigned to fluoride atoms placed within the t-mel cage of the MFI zeolite bonding to silicon atom at the T9 site, jumping between neighboring Si at this position.35 Due to this dynamic behavior, the silicon coordination changes continuously between four and five giving a large 29Si NMR signal in the region between SiO4 and SiO4-F. This is the case for the majority of the MFI samples studied here (see Fig. S2b†). When the 29Si NMR is recorded at low temperature, the fluoride becomes static and the signal of five coordinated Si becomes evident at a chemical shift of approximately −150 ppm.49,55,56 In the particular case of using TBMA as OSDA the fluoride is static also at room temperature and it is possible to detect the pentacoordinated silicon (SiO4-F) in the 29Si MAS-NMR spectrum without cooling the sample (Fig. S2b†).55,56 Theoretical calculations have pointed out that the dynamic behaviour of fluoride within smaller d4r cages is influenced by both the framework composition and the type of OSDA used.57
To shed light on the environment of the two fluoride anions, we recorded the 1H–29Si CP MAS NMR spectra at 100 K for the TEP-MFI and TBP-MFI zeolites (see Fig. 4), which were selected due to their different 19F NMR spectra (Fig. 2). The 19F NMR spectrum of the TEP-MFI is dominated by the signal at −65 ppm. The corresponding 29Si NMR spectrum (at 100 K), shown in Fig. 4, contains only a signal at δ29Si = −147 ppm, which is typical for five-coordinate silicon (F–SiO4). Meanwhile, the 19F NMR spectrum of the TBP-MFI sample contains two signals at ≈−65 ppm and ≈−80 ppm with practically the same intensity. The corresponding 29Si NMR spectrum, shown in Fig. 4, presents besides the peak at δ29Si = −147 ppm, another one at δ29Si ≈ −151 ppm. Both signals have similar intensity and are assigned to five coordinate silicon.35,49 It must be noted that these 29Si NMR signals do not show the doublet of the scalar coupling JF–Si, typically around 150 kHz, because their width (240–550 Hz) is larger likely due to the presence of defects in the structure.
 | ||
| Fig. 4 Low Temperature (100 K) 1H–29Si CP MAS-NMR spectra of the TEP-MFI and TBP-MFI zeolites. | ||
Therefore, the 29Si NMR signal at δ29Si ≈ −151 ppm is connected with the 19F signal at −80 ppm, which is consistently attributed to fluoride within the t-mel cage bonded to a Si in a crystallographic site different to T9.
To assist in the elucidation of the specific fluoride location in the t-mel cage we performed calculations of 19F chemical shielding. Several models were created by bonding fluoride to the five Si positions of the t-mel cage (Fig. 5). The MFI framework structure was modeled using a lattice of 288 atoms and optimized lattice parameters a = 19.97 Å, b = 19.75 Å, c = 13.20 Å, α = β = γ = 90° for the models with TEA and TPA and a = 21.57 Å, b = 21.33 Å, c = 14.26 Å, α = β = γ = 90° for TBA. The models contained four fluoride anions compensating the four OSDA cations (TEA, TPA and TBA) per unit cell, with the following chemical compositions: Si96O192(NC8H10)4F4, Si96O192(NC12H28)4F4 and Si96O192(NC16H36)4F4, respectively. Further details are available in the Theoretical Calculation section of the ESI.†
 | ||
| Fig. 5 Fluoride locations in t-mel cage along the b direction (upper panel) and the c direction (lower panel). (a) Representation of the five fluoride positions. (b) Optimized structure with F− in T9. (c) Optimized structure with F− in T12. | ||
Table 2 reports the relative energies and the calculated δ19F of the 15 models, five for each of the three OSDAs used: TEA, TPA and TBA. The calculated average δiso19F values for fluoride bonded to the T9 and T10 sites, which range from −54 to −70 ppm, closely match with the experimental 19F NMR signal observed at −65 ppm. In contrast, fluoride associated with T8, T11, and T12 sites exhibits average δiso19F values between −76 and −84 ppm, corresponding to the experimental 19F NMR signal at −80 ppm. However, the T8 and T11 sites, with higher relative energies (96–130 kJ mol−1 for TEA and TPA; 20 kJ mol−1 for TBA), are less stable, making them unlikely locations for fluoride. For TEA and TPA OSDAs, fluoride bonded to the T9 site emerges as the most stable configuration. When the bulkier TBA is used as OSDA, the T12 site becomes the most stable fluoride location, though the T9 and T10 sites remain relatively competitive, being only 8 kJ mol−1 and 2 kJ mol−1 less stable, respectively. The findings summarized in Table 2 show a strong correlation between the OSDA size, fluoride location and structural stability. It can be concluded that larger OSDAs like TBA stabilizes fluoride at T12 site, while smaller OSDAs like TEA and TPA stabilizes fluoride at T9 site in agreement with the literature data.35 Further analysis of the models reveals that the relative stability of fluoride coordinated to silicon atoms at the different T sites within the t-mel cage is strongly influenced by the symmetry of the trigonal bipyramidal geometry surrounding the five-coordinate F-SiO4 sites. Fig. S7† shows that the environment of the T9 site, the most stable location for fluoride in the models with TEA and TPA OSDAs, closely resembles an ideal trigonal bipyramidal geometry. Meanwhile, fluoride species at other T sites exhibit greater distortion, leading to lower stability. In contrast, the bulkier TBA OSDA enforces a nearly perfect trigonal bipyramidal geometry at all positions within the t-mel cage. Consequently, the stabilization energies across different positions become nearly equivalent, explaining the occupancy of fluoride at multiple crystallographic sites (T12 and T9, as supported by our calculations). Fig. 5 and Fig. S4† illustrate both fluoride locations within the MFI zeolite structure, highlighting the impact of OSDA size on the distribution and stability of fluoride in the zeolite framework.
| Structure | OSDA | E rel | δ iso 19F (ppm) | Average δiso19F (ppm) |
|---|---|---|---|---|
| T8 | TEA | 96 | −76, −77, −77, −78 | −77 |
| T9 | TEA | 0 | −60, −59, −58, −61 | −60 |
| T10 | TEA | 25 | −58, −60, −60, −60 | −60 |
| T11 | TEA | 129 | −81, −81, −84, −83 | −82 |
| T12 | TEA | 42 | −78, −78, −78, −78 | −78 |
| T8 | TPA | 104 | −76, −76, −77, −76 | −76 |
| T9 | TPA | 0 | −61, −62, −63, −62 | −62 |
| T10 | TPA | 79 | −51, −55, −54, −56 | −54 |
| T11 | TPA | 126 | −81, −82, −82, −83 | −82 |
| T12 | TPA | 68 | −74, −76, −74, −74 | −75 |
| T8 | TBA | 20 | −82, −83, −82, −82 | −82 |
| T9 | TBA | 8 | −64, −63, −67, −63 | −64 |
| T10 | TBA | 2 | −70, −71, −68, −71 | −70 |
| T11 | TBA | 56 | −82, −82, −82, −83 | −82 |
| T12 | TBA | 0 | −78, −76, −77, −75 | −77 |
We performed additional calculations to verify whether the presence of defects could shift the NMR resonance of fluoride linked to the T9 site at δ19F ≈ −65 ppm to δ19F ≈ −80 ppm. We used the five models with TPA OSDA, incorporating three F− anions and one SiO− defect per unit cell (see Table S5†), which is very close to the experimental composition of TPA-MFI sample (3F/u.c., see Table 1). The predicted average δiso19F are very similar to those reported in Table 2, suggesting that the presence of defects does not significantly impact the δ19F value. The relative energies are generally lower but follow the same trend as those in Table 2. These results confirm the assignment of the 19F NMR signal at −80 ppm to fluoride in a location different to T9, more specifically in the T12 site.
4. Conclusions
In summary, the combination of NMR spectroscopy and theoretical calculations confirms the previous assignment of the 19F NMR signal at −65 ppm to fluoride bonded to Si at the T9 sites and allows to assign the resonance at δ19F ≈ −80 ppm to fluoride linked to silicon at the T12 sites. Both T9 and T12 sites form part of the t-mel cage of the MFI zeolite. Our results suggest that the stability of the fluoride at the T12 site is noticeably enhanced either by using bulkier OSDAs or the presence of defects. We have determined that the stability of fluoride positioning within the t-mel cage is directly linked to the distortion of the trigonal bipyramidal geometry around the five-coordinate silicon site, which is influenced by the choice of OSDA. Alternatively, the fluoride at the T12 position can be stabilized by increasing the concentration of SiO−⋯HOSi defects above a certain level (about 1.9 or fewer F/u.c.). The presence of defects is favored at shorter synthesis times and when using bulky OSDAs. Therefore, the distribution of fluoride within the structure can be modified by varying the synthesis conditions.These findings highlight the significant influence of the OSDA size and the presence of defects on the distribution of fluoride anions within the MFI structure. Larger OSDAs and higher concentration of defects promote the incorporation of fluoride bonded to silicon at the T12 site. Changes on the content and location of fluoride in the zeolite sites must be accompanied by changes in the distribution of silanols. Our findings are crucial for understanding the charge-matching mechanism in zeolites, which ultimately impacts their structural properties and potential applications.
Author contributions
Investigation: J. M.-O., R. M., J. S., J.-A. V.-M. and C. M.-C. Methodology: J. M.-O. and R. M. Supervision: T. B. and F. R. Visualization: J. M.-O. and R. M. Writing – original draft: J. M.-O., T. B. and R. M. Writing – review and editing: T. B., J. M.-O., R. M., C. M.-C. and F. R. Project administration: F. R. and T. B. Funding acquisition: F.R.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Spanish Ministry of Science and Innovation (CEX2021-001230-S and PID2022-136934OB-I00 grants funded by MCIN/AEI/10.13039/501100011033 funded by “ERDF A way of making Europe” and TED2021–130191B-C41 grant funded by the European Union NextGenerationEU/PRTR) are gratefully acknowledged. Authors thank also the financial support by the Generalitat Valenciana (Prometeo 2021/077). This study forms part of the Advanced Materials programme and was supported by MCIN with partial funding from European Union Next Generation EU (PRTR-C17. I1) and by Generalitat Valenciana (MFA/2022/012 and MFA/2022/047). Financial support from the IR INFRANALYTICS FR2054 for conducting the research is gratefully acknowledged. J. M.-O. acknowledges the financial support from the European Union – NextGeneration EU through the Universitat Politècnica de València, Ministerio de Universidades, Plan de Recuperación, Transformación y Resiliencia for the Margarita Salas grant. Authors thank funding for open access charge: CRUE-Universitat Politècnica de València.References
- R. Millini and G. Bellussi, Zeolites in Catalysis: Properties and Applications, ed. J. Čejka, R. E. Morris and P. Nachtigall, The Royal Society of Chemistry, 2017, ch. 1, pp. 1–36 Search PubMed.
- R. F. Lobo, S. I. Zones and M. E. Davis, Structure-direction in zeolite synthesis, J. Inclusion Phenom. Macrocyclic Chem., 1995, 21, 47–78 CrossRef CAS.
- C. S. Cundy and P. A. Cox, The hydrothermal synthesis of zeolites: History and development from the earliest days to the present time, Chem. Rev., 2003, 103, 663–701 CrossRef CAS PubMed.
- N. Y. Chen and W. E. Garwood, Industrial Application of Shape-Selective Catalysis, Catal. Rev., 1986, 28, 185–264 CrossRef CAS.
- B. Yilmaz and U. Müller, Catalytic applications of zeolites in chemical industry., Top. Catal., 2009, 52, 888–895 CrossRef CAS.
- M. Shamzhy, M. Opanasenko, P. Concepción and A. Martínez, New trends in tailoring active sites in zeolite-based catalysts., Chem. Soc. Rev., 2019, 48, 1095–1149 RSC.
- Y. Li, L. Li and J. Yu, Applications of Zeolites in Sustainable Chemistry, Chem, 2017, 3, 928–949 CAS.
- Y. Li and J. Yu, Emerging applications of zeolites in catalysis, separation and host–guest assembly., Nat. Rev. Mater., 2021, 6, 1156–1174 CrossRef CAS.
- E. Pérez-Botella, S. Valencia and F. Rey, Zeolites in Adsorption Processes: State of the Art and Future Prospects., Chem. Rev., 2022, 122, 17647–17695 CrossRef PubMed.
- W. Yan, Y. Li, F. S. Xiao, Z. Liu and J. Yu, The Future of Zeolites, Chem. Mater., 2024, 36, 7103–7105 CrossRef CAS.
- E. Dib, E. B. Clatworthy, H. Cruchade, I. C. Medeiros-Costa, N. Nesterenko, J. P. Gilson and S. Mintova, Exploration, explanation and exploitation of hydroxyls in zeolites, Natl. Sci. Rev., 2022, 9, nwac081 CrossRef PubMed.
- H. Windeck, F. Berger and J. Sauer, Spectroscopic Signatures of Internal Hydrogen Bonds of Brønsted-Acid Sites in the Zeolite H-MFI., Angew. Chem., Int. Ed., 2023, 62, e202303204 CrossRef CAS PubMed.
- E. M. Gallego, M. T. Portilla, C. Paris, A. León-Escamilla, M. Boronat, M. Moliner and A. Corma, “Ab initio” synthesis of zeolites for preestablished catalytic reactions, Science, 2017, 355, 1051–1054 CrossRef CAS PubMed.
- M. Gao, H. Li, Y. Tian, J. Yu, M. Ye and Z. Liu, Surface Diffusion Barriers and Catalytic Activity Driven by Terminal Groups at Zeolite Catalysts, ACS Catal., 2023, 13, 11598–11609 CrossRef CAS.
- S. Radhakrishnan, C. Lejaegere, K. Duerinckx, W. S. Lo, A. F. Morais, D. Dom, C. V. Chandran, I. Hermans, J. A. Martens and E. Breynaert, Hydrogen bonding to oxygen in siloxane bonds drives liquid phase adsorption of primary alcohols in high-silica zeolites, Mater. Horiz., 2023, 10, 3702 RSC.
- I. C. Medeiros-Costa, E. Dib, N. Nesterenko, J. P. Dath, J. P. Gilson and S. Mintova, Silanol defect engineering and healing in zeolites: opportunities to fine-tune their properties and performances, Chem. Soc. Rev., 2021, 50, 11156–11179 RSC.
- G. Hou, T. Fu, X. Li, Q. Ma and Z. Li, Creation of silanol nests on HZSM-5 catalyst to boost the alkylation of toluene with methanol for PX synthesis., Appl. Catal., A, 2022, 642, 118713 CrossRef CAS.
- S. C. Purdy, G. Collinge, J. Zhang, S. N. Borate, K. A. Unocic, Q. Wu, E. C. Wegener, A. J. Kropf, N. R. Samad, S. F. Yuk, D. Zhang, S. Habas, T. R. Krause, J. W. Harris, M.-S. Lee, V.-A. Glezakou, R. Rousseau, A. D. Sutton and Z. Li, Dynamic Copper Site Redispersion through Atom Trapping in Zeolite Defects, J. Am. Chem. Soc., 2024, 146, 8280–8297 CrossRef CAS PubMed.
- G. P. Heitmann, G. Dahlhoff and W. F. Hölderich, Catalytically Active Sites for the Beckmann Rearrangement of Cyclohexanone Oxime to ε-Caprolactam., J. Catal., 1999, 186, 12–19 CrossRef CAS.
- I. Lezcano-Gonzalez, M. Boronat and T. Blasco, Investigation on the Beckmann rearrangement reaction catalyzed by porous solids: MAS NMR and theoretical calculations, Solid State Nucl. Magn. Reson., 2009, 35, 120–129 CrossRef CAS PubMed.
- J. D. Epping and B. F. Chmelka, Nucleation and growth of zeolites and inorganic mesoporous solids: Molecular insights from magnetic resonance spectroscopy, Curr. Opin. Colloid Interface Sci., 2006, 11, 81–117 CrossRef CAS.
- T. Wakihara and T. Okubo, Hydrothermal synthesis and characterization of zeolites, Chem. Lett., 2005, 34, 276–281 CrossRef CAS.
- M. Moliner, F. Rey and A. Corma, Towards the rational design of efficient organic structure-directing agents for zeolite synthesis, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef CAS PubMed.
- D. Liang, Y. Liu, R. Zhang, Q. Xie and L. Zhang, A Review on the Influence Factors in the Synthesis of Zeolites and the Transformation Behavior of Silicon and Aluminum During the Process, Comments Inorg. Chem., 2024, 44(6), 461–497 CrossRef CAS.
- R. M. Barrer, Zeolites and Clay Minerals as Sorbents and Molecular Sieves, Academic Press, London., 1978 Search PubMed.
- D. W. Breck, Zeolite Molecular Sieves: Structure, Chemistry and Use, John Wiley & Sons Inc., New York, 1984 Search PubMed.
- H. Koller, R. F. Lobo, S. L. Burkett and M. E. Davis, SiO−⋯HOSi Hydrogen Bonds in As-Synthesized High-Silica Zeolites, J. Phys. Chem., 1995, 99, 12588–12596 CrossRef CAS.
- G. Brunklaus, H. Koller and S. I. Zones, Defect Models of As-Made High-Silica Zeolites: Clusters of Hydrogen-Bonds and Their Interaction with the Organic Structure-Directing Agents Determined from 1H Double and Triple Quantum NMR Spectroscopy, Angew. Chem., Int. Ed., 2016, 55, 14459 CrossRef CAS PubMed.
- E. Dib, J. Grand, S. Mintova and C. Fernandez, Structure-Directing Agent Governs the Location of Silanol Defects in Zeolites, Chem. Mater., 2015, 27, 7577–7579 CrossRef CAS.
- E. Dib, I. Medeiros-Costa, G. N. Vayssilov, H. A. Aleksandrov and S. Mintova, Complex H-bonded silanol network in zeolites revealed by IR and NMR spectroscopy combined with DFT calculations, J. Mater. Chem. A, 2021, 9, 27347 RSC.
- E. Dib, J. Grand, A. Gedeon, S. Mintova and C. Fernandez, Control the position of framework defects in zeolites by changing the symmetry of organic structure directing agents, Microporous Mesoporous Mater., 2021, 315, 110899 CrossRef CAS.
- J. Li, M. Gao, W. Yan and J. Yu, Regulation of the Si/Al ratios and Al distributions of zeolites and their impact on properties, Chem. Sci., 2023, 14, 1935–1959 RSC.
- J. L. Guth, H. Kessler, J. M. Higel, J. M. Lamblin, J. Patarin, A. Seive, J. M. Chezeau and R. Wey, Zeolite Synthesis in the Presence of Fluoride Ions: A Comparison with Conventional Synthesis Methods, Zeolite Synth., 1989, 13, 176–195 Search PubMed.
- M. A. Camblor, L. A. Villaescusa and M. J. Díaz-Cabañas, Synthesis of all-silica and high-silica molecular sieves in fluoride media, Top. Catal., 1999, 9, 59–76 CrossRef CAS.
- C. A. Fyfe, D. A. Brouwer, A. R. Lewis and J. M. Chezeau, Location of the Fluoride Ion in Tetrapropylammonium FluorideSilicalite-1 Determined by 1 H/19F/29Si Triple Resonance CP, REDOR,and TEDOR NMR Experiments, J. Am. Chem. Soc., 2001, 123, 6882–6891 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew and Y. Wang, Accurate and simple analytic representation of the electron-gas correlation energy, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 16955487 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 21370243 Search PubMed.
- R. Laskowski, P. Blaha and F. Tran, Assessment of DFT functionals with NMR chemical shifts, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 195130 CrossRef.
- R. Laskowski and P. Blaha, Calculations of NMR chemical shifts with APW-based methods, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 035132 CrossRef.
- F. Tran and P. Blaha, Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential, Phys. Rev. Lett., 2009, 102, 22641 Search PubMed.
- J. Martinez-Ortigosa, R. Millán, J. Simancas, M. Hernández-Rodríguez, J. A. Vidal-Moya, J. L. Jordá, C. Martineau-Corcos, V. Sarou-Kanian, M. Boronat, T. Blasco and F. Rey, Crystalline phase transition in as-synthesized pure silica zeolite RTH containing tetra-alkyl phosphonium as organic structure directing agent, J. Mater. Chem. A, 2024,12, 876–891 Search PubMed.
- J. Martinez-Ortigosa, J. Simancas, J. A. Vidal-Moya, P. Gaveau, F. Rey, B. Alonso and T. Blasco, Host–Guest and Guest–Guest Interactions of P- and N-Containing Structure Directing Agents Entrapped inside MFI-Type Zeolite by Multinuclear NMR Spectroscopy, J. Phys. Chem. C, 2019, 123, 22324–22334 CrossRef CAS.
- J. Martinez-Ortigosa, J. Simancas, J. A. Vidal-Moya, F. Rey, S. Valencia and T. Blasco, A Multi-Nuclear MAS-NMR Study on the Structural Properties of Silicalite-1 Zeolite Synthesized Using N- and P-Based Organic Structure Directing Agents, Appl. Sci., 2021, 11, 6850 CrossRef CAS.
- M. Fabbiani, S. Al-Nahari, L. Piveteau, E. Dib, V. Veremeienko, A. Gaje, D. G. Dumitrescu, P. Gaveau, T. Mineva, D. Massiot, A. van der Lee and J. Haines, Alonso, Host–Guest Silicalite-1 Zeolites: Correlated Disorder and Phase Transition Inhibition by a Small Guest Modification, Chem. Mater., 2022, 34, 366–387 CrossRef CAS.
- P. Lu, L. Gómez-Hortigüela and M. A. Camblor, Synthesis of pure silica MFI zeolites using imidazolium-based long dications. A comparative study of structure-directing effects derived from a further spacer length increase, Dalton Trans., 2018, 47, 7498–7504 RSC.
- E. Dib, A. Gimenez, T. Mineva and B. Alonso, Preferential orientations of structure directing agents in zeolites, Dalton Trans., 2015, 44, 16680–16683 RSC.
- A. Rojas, L. Gómez-Hortigüela and M. A. Camblor, Zeolite Structure Direction by Simple Bis(methylimidazolium)Cations: The Effect of the Spacer Length on Structure Direction andof the Imidazolium Ring Orientation on the 19F NMR Resonances, J. Am. Chem. Soc., 2012, 134, 3845–3856 CrossRef CAS PubMed.
- L. Delmotte, M. Soulard, F. Guth, A. Seive, A. Lopez and J. L. Guth, 19F MAS n.m.r, studies of crystalline microporous solids synthesized in the fluoride medium, Zeolites, 1990, 10, 778–783 CrossRef CAS.
- P. Losch, A. B. Pinar, M. G. Willinger, K. Soukup, S. Chavan, B. Vincent, P. Pale and B. Louis, H-ZSM-, 5 zeolite model crystals: Structure-diffusion-activity relationship in methanol-to-olefins catalysis, J. Catal., 2017, 345, 11–23 CrossRef CAS.
- S. L. Brace, P. Wormald and R. J. Darton, The effect of structure directing agents on the ordering of fluoride ions in pure silica MFI zeolites, Phys. Chem. Chem. Phys., 2015, 17, 11950–11953 RSC.
- H. Koller, A. Wölker, L. A. Villaescusa, M. J. Diaz-Cabañas, S. Valencia and M. A. Camblor, Five-Coordinate Silicon in High-Silica Zeolites, J. Am. Chem. Soc., 1999, 121, 3368–3376 CrossRef CAS.
- M. Fischer and L. Freymann, Local Distortions in a Prototypical Zeolite Framework Containing Double Four-Ring Cages: The Role of Framework Composition and Organic Guests, ChemPhysChem, 2021, 22, 40 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and methods; XRD patterns; 1H & 29Si MAS-NMR spectra; synthesis of the MFI zeolite using TPA as OSDA and varying the synthesis time; Simulation of 19F chemical shift. Simulation of connectivity defects. See DOI: https://doi.org/10.1039/d4qi03001j |
| ‡ Present address: CortecNet, 7 avenue du Hoggar, 91940 Les Ulis, France. |
| This journal is © the Partner Organisations 2025 |