
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel benzothiazole-1,2,3-triazole-based arene osmium(II) complex as an effective rhabdomyosarcoma cancer stem cell agent†
Sofia
Sharkawy‡
 ab,
Alba
Hernández-García‡
c,
Hana
Kostrhunova
a,
Delia
Bautista
d,
Lenka
Markova
a,
María Dolores
Santana
c,
Jana
Kasparkova
ae,
Viktor
Brabec
*ae and
José
Ruiz
*c
ab,
Alba
Hernández-García‡
c,
Hana
Kostrhunova
a,
Delia
Bautista
d,
Lenka
Markova
a,
María Dolores
Santana
c,
Jana
Kasparkova
ae,
Viktor
Brabec
*ae and
José
Ruiz
*c
aCzech Academy of Sciences, Institute of Biophysics, CZ-61200 Brno, Czech Republic. E-mail: vbrabec44@gmail.com
bFaculty of Science, Department of Biochemistry, Masaryk University, CZ-62500 Brno, Czech Republic
cDepartamento de Química Inorgánica, Universidad de Murcia, and Murcia BioHealth Research; Institute (IMIB-Arrixaca), E-30100 Murcia, Spain. E-mail: jruiz@um.es
dACTI, Universidad de Murcia, Murcia E-30100, Spain
eDepartment of Biophysics, Faculty of Science, Palacky University, Slechtitelu 27, 783 71 Olomouc, Czech Republic
First published on 10th January 2025
Abstract
We designed a series of pseudo-octahedral arene Os(II) complexes (Os1–Os5) with the general formula [(η6-p-cym)Os(BTAT)Cl]+, where BTAT represents chelating N^N′ ligands based on the 1-aryl-4-benzothiazolyl-1,2,3-triazole scaffold. The structures of Os3 and Os5 were confirmed by X-ray diffraction, and Os5 exhibits a bathochromic shift in its absorption band compared to the other complexes, likely due to the electron-donating properties of the substituent NMe2. Os5 also hydrolyzed without losing its BTAT ligand and exhibited the highest cellular accumulation in Rhabdomyosarcoma (RD) cancer cells. The investigated Os(II) complexes demonstrated moderate antiproliferative activity across six cancer cell lines, with Os5 being the most potent, showing activity comparable to or better than conventional cisplatin. Cellular accumulation was a key factor influencing their antiproliferative effect, though binding to human serum albumin did not play a significant role. Further studies with Os5 in RD cells, the most responsive cell line, revealed that its mechanism of action includes mitochondrial dysfunction, apoptosis via a caspase-dependent pathway, and cell cycle arrest at the G1 phase. Os5 also increased the production/generation of reactive oxygen species (ROS) in RD cells, implicating ROS production as a contributor to its activity. Importantly, Os5 was effective against cancer stem cells (CSCs) in 3D spheroid models, marking the first report of an osmium-based compound targeting CSC-enriched RD cells. This highlights the potential of Os5 as a CSC-targeted therapy, addressing the need for treatments that prevent relapse and metastasis. The study underscores the promising role of metal-based complexes in cancer stem cell chemotherapy.
Introduction
Cancer is the second leading cause of mortality worldwide. According to data provided by the International Agency for Research on Cancer (IARC), it is estimated that by 2022, approximately 20 million new cancer cases and 9.7 million cancer-related deaths occurred.1,2Over the last few decades, the prognosis for children with localized Rhabdomyosarcoma (RMS) has significantly improved, with a 5-year overall survival rate of >70%.3 At the same time, despite aggressive combination therapy, no further significant improvements have been made for the treatment of children with high-risk disease or recurrent disease (5-year survival <30% and 17%, respectively).4 Conventional therapies usually aim to attack rapidly dividing non-stem (differentiated) cancer cells, ultimately forming the bulk of a tumor. However, by adopting slow proliferation states, cancer stem cells (CSCs) can evade antiproliferative treatments due to their stem cell-like properties.5 The survival of a small number of CSCs within the tumor population can regenerate the original tumor and/or produce invasive cancer cells that can colonize distant organs.6 Therefore, CSCs are thought to be responsible for cancer relapse and metastasis.7–10 To overcome this clinical problem, novel therapeutics and modalities for treatments that can eradicate both fast-growing cancer cells and quiescent CSCs are urgently needed.
The discovery of anticancer properties of some ruthenium compounds led to the development and research of osmium analogs. The application of osmium compounds as therapeutic agents was initially met with hesitation because osmium complexes have been generally considered either toxic11 or substitution-inert,12 which may explain the limited exploration of their therapeutic potential. Nevertheless, subsequent research revealed that these compounds present significant anticancer potential. The first osmium compounds considered as potential organometallic anticancer drugs were closely related congeners of RuII-arene-pta compounds (pta = phosphine 1,3,5-triaza-7-phosphatricyclo[3.3.1.1]decane).13 Other categories of organometallic Os complexes that show promising in vitro activity against cancer cells and adjustable reactivity in aqueous environments include arene complexes containing a series of different bidentate nitrogen chelators,14 picolinate derivatives,15 or paullone-derived ligands,16 and also osmium analogs of ruthenium-based antimetastatic agent NAMI-A type compound [(NAMI-A) = (ImH)[trans-RuCl4(dmso-S)(Im)], Im = imidazole].17 Lippard et al. reported the first osmium compound [osmium(VI) nitrido complex] to exhibit selective toxicity against breast CSC-enriched HMLERtax cell populations.18 A selection of reported Os(II) arene anticancer complexes are shown in Scheme 1. The one synthesized by Sadler et al., known as FY26 (OsA in Scheme 1A), is particularly noteworthy.19 FY26 demonstrated substantially greater potential than cisplatin, and its ruthenium analog, RuA, across various cell lines, and recently, in vivo studies were conducted in mice to ascertain its pharmacokinetics and tolerability in the treatment of liver cancer.20–22
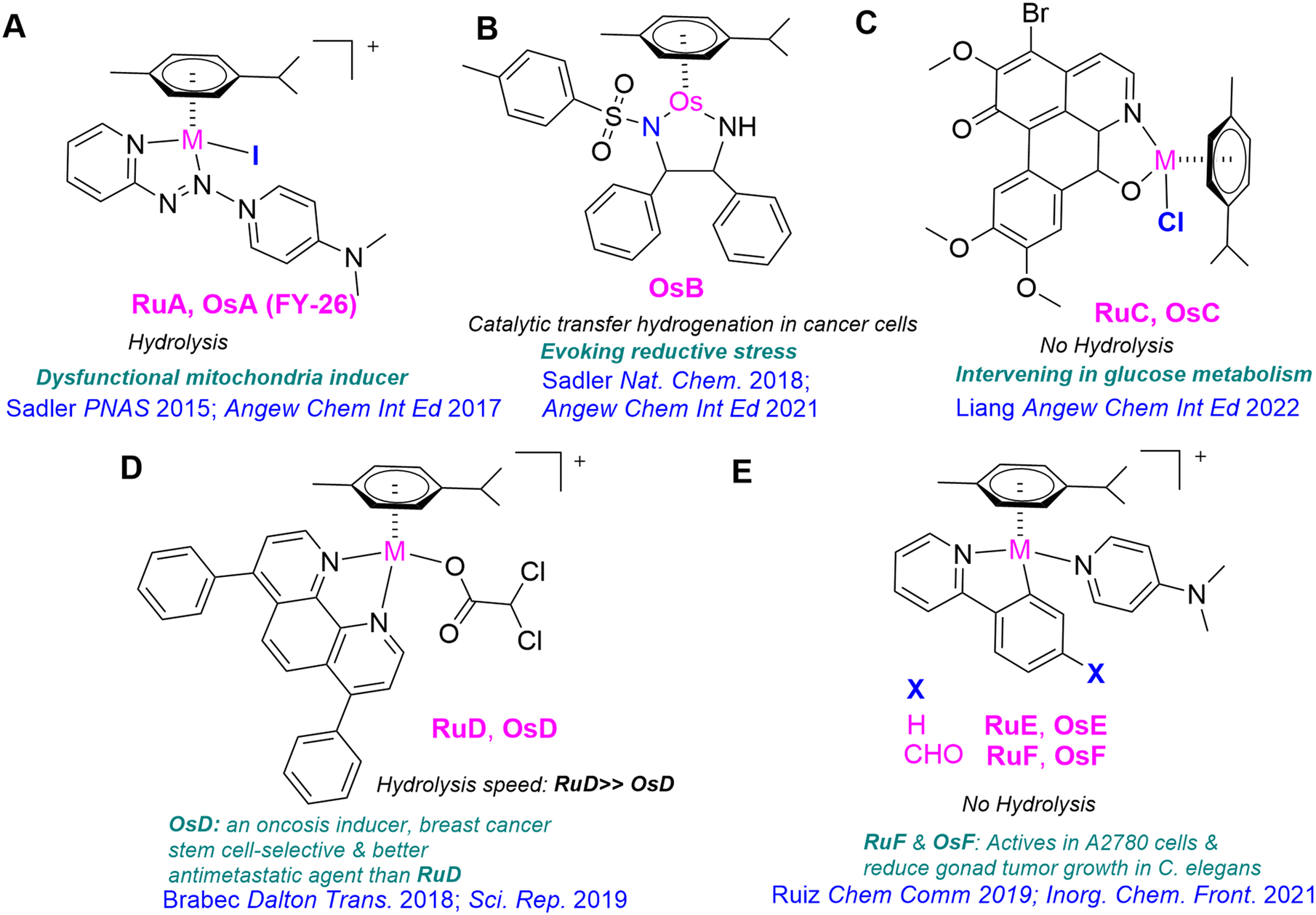 | ||
| Scheme 1 A selection of reported examples of Ru(II) and Os(II) arene anticancer complexes by Sadler's lab (A and B), Liang's lab (C) Trávníček's lab (D) and Ruiz's lab (E), highlighting their biological properties. | ||
Sadler et al. have recently reported a few interesting half-sandwich sixteen electron Os(II) transfer hydrogenation catalysts (OsBScheme 1) to achieve in-cell pyruvate and quinone reduction as a novel anticancer mechanism.23,24 Os(II) complexes bearing a pH-sensitive reversible tether between the η6 ligand and the monodentate coordination site have been shown to improve aqueous solution stability. They can catalyze the in-cell reduction of pyruvate to lactate.25 Recently, it has been shown that a family of arene osmium(II) complexes with oxoglaucine (OsC in Scheme 1) can induce glucose metabolism reprogramming and exhibit good anti-tumor activity in vivo with low side effects under hypoglycemia.26
Some of us have synthesized other noteworthy osmium half-sandwich complexes.27–29 Brabec et al. have developed an Os(II) compound incorporating bathophenanthroline, OsD (Scheme 1), which exhibits antimetastatic properties against triple-negative breast cancer.27 Furthermore, recent studies have demonstrated its superior efficacy against human breast cancer stem cells compared to salinomycin and RuD (Scheme 1).28 Importantly, RuD hydrolyzed very fast, whereas OsD did not. Additionally, Ruiz et al. synthesized the Os(II) compounds [(η6-p-cym)Os(C^N)(4-NMe2-py)]+ (C^N = ppy OsE and ppy-CHO OsF, Scheme 1), which have shown activity against ovarian cancer in vivo in Caenorhabditis elegans in the tumoral strain JK1466.29 In the last series, the activity of OsF and the analogous RuF was similar, with no hydrolysis observed for either of them.29,30
On the other hand, benzothiazole and 1,2,3-triazole heterocycles are chemical structures found in various pharmaceuticals and biologically active molecules. BLZ945 (sotuletinib), a derivative of 2-aminobenzothiazole, is currently undergoing clinical trials.31 Additionally, they are components of ligands in metal complexes with biological activity, as exemplified by certain Ru(II) compounds.32–34 Notably, both a series of benzothiazolyl-1,2,3-triazole (BTAT) derivatives with push–pull architecture and bioactive properties35 and a family of photoactivatable BTAT-based octahedral ruthenium(II) complexes of general formula [Ru(phen)2(BTAT)]2+ has been recently developed by some of us (Ru1-Ru5 in Scheme 2A).36 It is noteworthy that, as reported, the cytotoxicity of these octahedral heteroleptic BTAT Ru complexes towards several differentiated human cancer cells under dark conditions was very low, even at high concentrations. These complexes exhibited significant phototoxicity indexes toward several cancer cells only after exposure to blue light irradiation. It was further shown by HPLC-MS spectrometry and UV–vis spectroscopy that these heteroleptic Ru complexes photoreleased the BTAT ligand upon irradiation in water with light (as illustrated in Scheme 2A).
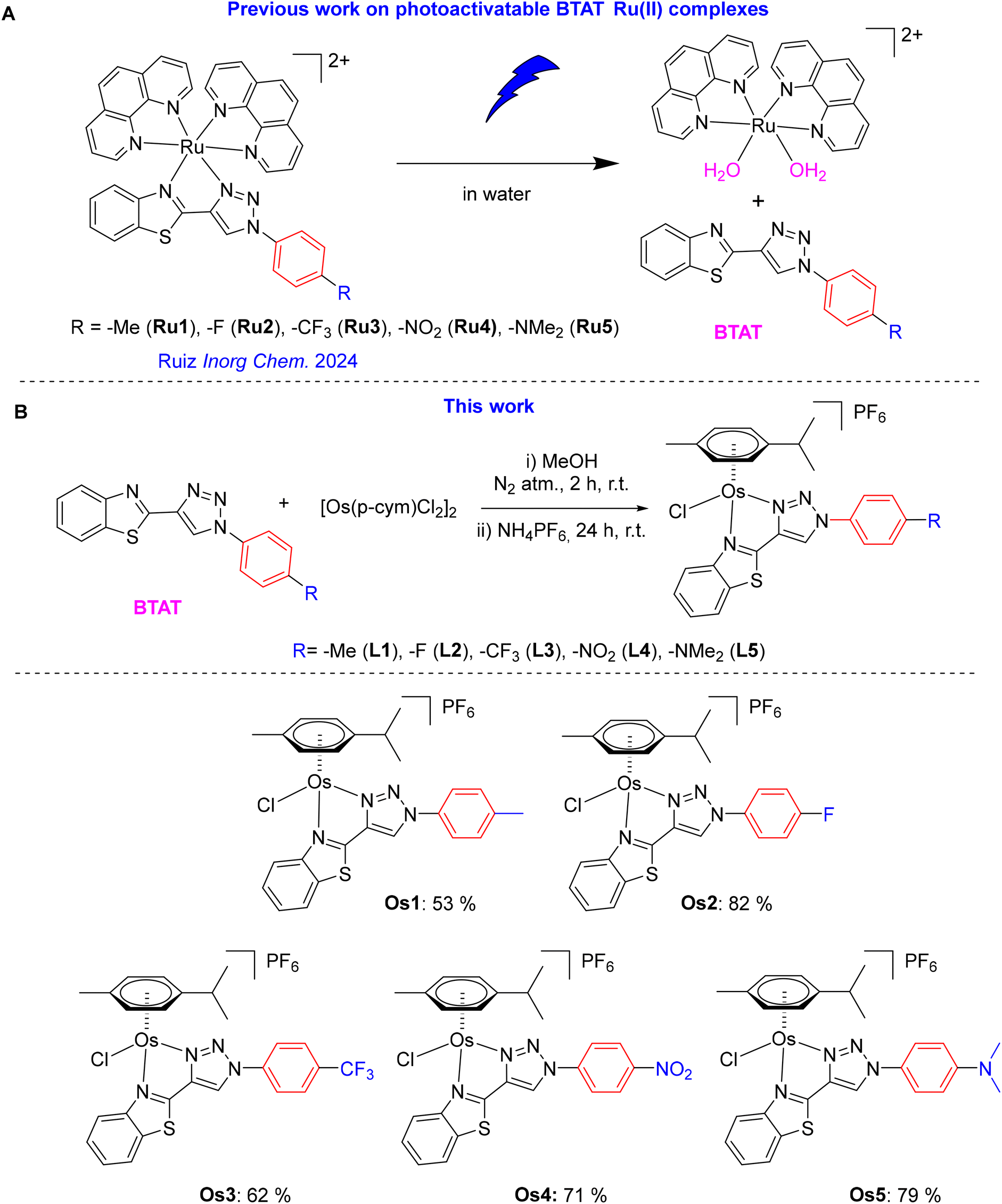 | ||
| Scheme 2 (A) Previously reported photoactivable octahedral Ru BTAT complexes (B) synthesis of new osmium arene BTAT complexes Os1–Os5 investigated in this work. | ||
Herein, we present the synthesis and characterization of novel osmium(II) arene compounds of the type [Os(η6-p-cymene)(N^N′)Cl]PF6, where the N^N′ ligands are BTAT ligands with different substituents in the R4 position of the phenyl ring (Scheme 2B). Our goal was to develop potential osmium-based anticancer drugs that can inhibit both the proliferation of differentiated tumor cells and reduce the tumorigenic potential of cancer stem cells (CSCs). Our research demonstrates the antiproliferative effects of these compounds in both monolayer and 3D spheroid assays across a variety of cancer cell lines under dark conditions. This work contributes to understanding the structure–activity relationship and key aspects of the mechanism of action of these novel osmium compounds, particularly in rhabdomyosarcoma (RD) cells, which are associated with one of the most challenging and hard-to-treat pediatric tumors.
Results and discussion
Synthesis and characterization of osmium complexes (Os1–Os5)
The BTAT ligands L1–L5 were obtained following a synthetic three-steps procedure recently reported (see Schemes S1 and S2 in ESI† for further details).35 Yellow solids Os1–Os5 were synthesized from the reaction between the dimeric precursor [{Os(η6-p-cymene)Cl(μ-Cl)}2] and the corresponding BTAT chelating ligand, following similar procedures to those reported for other half-sandwich arene-complexes (Scheme 2B).19,25,37 Osmium half-sandwich complexes were isolated as pure hexafluorophosphate salts and fully characterized by multinuclear NMR spectroscopy (Fig. S1–S16†). The purities of complexes were assessed by elemental analysis of C, H, N, and S, ESI-HRMS (positive mode, Fig. S17–S21†), and it was also confirmed that the purities of complexes were higher than 95% through RP-HPLC/MS in ACN/H2O (Table S1 and Fig. S22, S23†). ESI-MS spectra from HPLC-MS displayed a peak due to the ion [M − PF6]+ with the expected isotopic distribution.The 1H NMR spectra were recorded in CD3CN, and in all cases, they show resonances due to the aromatic and aliphatic protons of the p-cymene ligand. Thus, the three characteristic resonances of the isopropyl group (two doublets at 0.9 and 1 ppm, and a septuplet around 2.5 ppm), the characteristic resonance of the methyl group appearing as a singlet around 2.3 ppm, and the expected ABCD spin system for the aromatic resonances were observed in the p-cymene derivatives. Due to the coordination of the N,N′ chelating BTAT ligands L1–L5, some characteristic changes in the splitting pattern of the p-cymene ring protons were observed. The C2v symmetry of the complexes was disrupted, and the peaks corresponding to the four aromatic protons of the p-cymene ring were observed as four distinct doublets between 6.2 and 6.5 ppm. Moreover, the frequencies of the H6+7, H8+9, and H5 proton resonances (doublets and singlet, respectively) depend on the substituent (–R), resulting in lower frequencies for Os5 (containing the –NMe2 group) and higher frequencies for Os3 and Os4 (whose substituents are –CF3 and –NO2, with electron-withdrawing nature) than the other osmium complexes, Os1 and Os2. All other proton resonances of the complexes Os1–Os5 were assigned based on their coupling profiles in the 1H–1H COSY and 1H–1H-NOESY spectra obtained and by comparing these profiles between different complexes within the series (Fig. 1 and Fig. S8–S16†).
 | ||
| Fig. 1 1H-NMR stacking spectra (aromatic region) for compounds Os1–Os5 in acetonitrile-d3 along with proton assignment. | ||
Crystal structure by X-Ray diffraction
Suitable single crystals of Os3 and Os5 for X-ray diffraction analysis were obtained by slow diffusion of diethyl ether into a saturated acetonitrile solution at 5 °C for 3 days. The molecular structures of both complexes are shown in Fig. 2.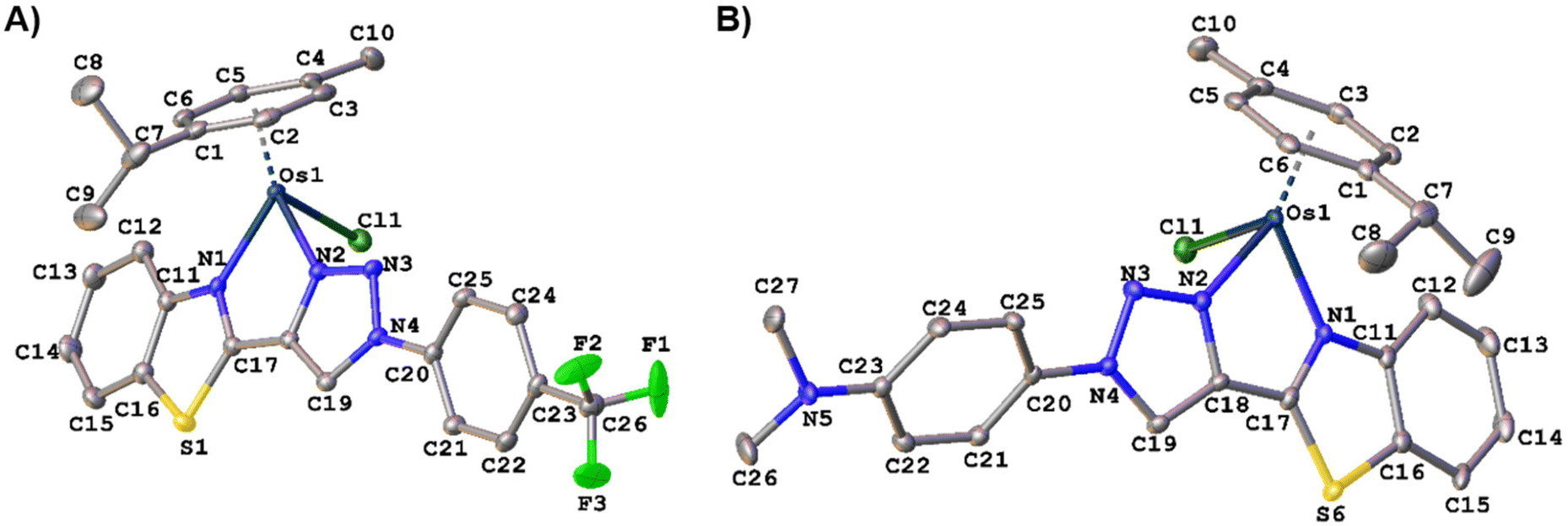 | ||
| Fig. 2 (A) ORTEP plot of the cation of complex Os3. Selected bond lengths (Å) and angles (°) for Os3: Os–N2: 2.064(2), Os–N1: 2.112(2), Os–Cl: 2.3949(7), Os–Cg(p-cymene): 1.6808(12), N2–Os1–N1: 75.15(8), N1–Os–Cl: 81.96(6), N2–Os–Cl: 83.98(6). CCDC reference number is: 2393537. (B) ORTEP plot of the cation of complex Os5. Selected bond lengths (Å) and angles (°) for Os5: Os–N2: 2.072 (2), Os–N1: 2.114(2), Os–Cl: 2.3958(6), Os–Cg(p-cymene): 1.6774(11), N2–Os1–N1: 75.29(8), N1–Os–Cl: 82.61 (6), N2–Os–Cl: 83.05(6). CCDC reference number is: 2393536. Cg = the centroid of the p-cymene ring defined by C1–C6 atoms. | ||
Crystallographic data are given in Tables S2 and S5.† The structures of complexes were unambiguously confirmed by X-ray diffraction, supporting our predicted molecular structure. Both complexes present the pseudo-octahedral “three-legged piano-stool” geometry, with the metal atom π-bonded to the η6-p-cymene ligand, whereas the benzothiazole-1,2,3-triazole ligands assume a bidentate chelate coordination mode (κ2-N,N′), occupying two coordination positions. The Os–N bond distances (2.064–2.112 Å) and Os–Cl (2.3949 Å) are within the range reported for osmium half sandwich complexes.19,37–39 Apart from the cation-hexafluorophosphate anion coulombic interaction, the packing in Os3 is organized by intermolecular interactions C–H⋯F (Table S3 and Fig. S24†) and C–H⋯π interactions (Table S4 and Fig. S25†). However, the packing in Os5 is organized not only by intermolecular interactions C–H⋯X (X = F and Cl, Table S6 and Fig. S26†) and C–H⋯π interactions (Table S8 and Fig. S28†), but also by π–π interactions (Table S7 and Fig. S27†).
Photophysical characterization of the compounds
The UV/Vis absorption spectra of complexes Os1–Os5 (50 μM) were recorded in DMSO and acetonitrile (Fig. 3A, B and Table S9†) at room temperature. As can be observed in Fig. 3A and B, all osmium half-sandwich complexes show intense sharp bands between 260 and 330 nm, with extinction coefficients around 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 in both solvents, that can be mainly assigned to the 1π–π* transition aromatic system of the ligands, and other less intense broad bands around 380 nm, due to metal–ligand charge transfer transitions (1MLCT), with extinction coefficients around 5000 M−1 cm−1. Noteworthy, the Os5 complex undergoes bathochromic displacement of that band compared to the rest of the osmium complexes, possibly due to the electron donor nature of its substituent (–NMe2).
000 M−1 cm−1 in both solvents, that can be mainly assigned to the 1π–π* transition aromatic system of the ligands, and other less intense broad bands around 380 nm, due to metal–ligand charge transfer transitions (1MLCT), with extinction coefficients around 5000 M−1 cm−1. Noteworthy, the Os5 complex undergoes bathochromic displacement of that band compared to the rest of the osmium complexes, possibly due to the electron donor nature of its substituent (–NMe2).
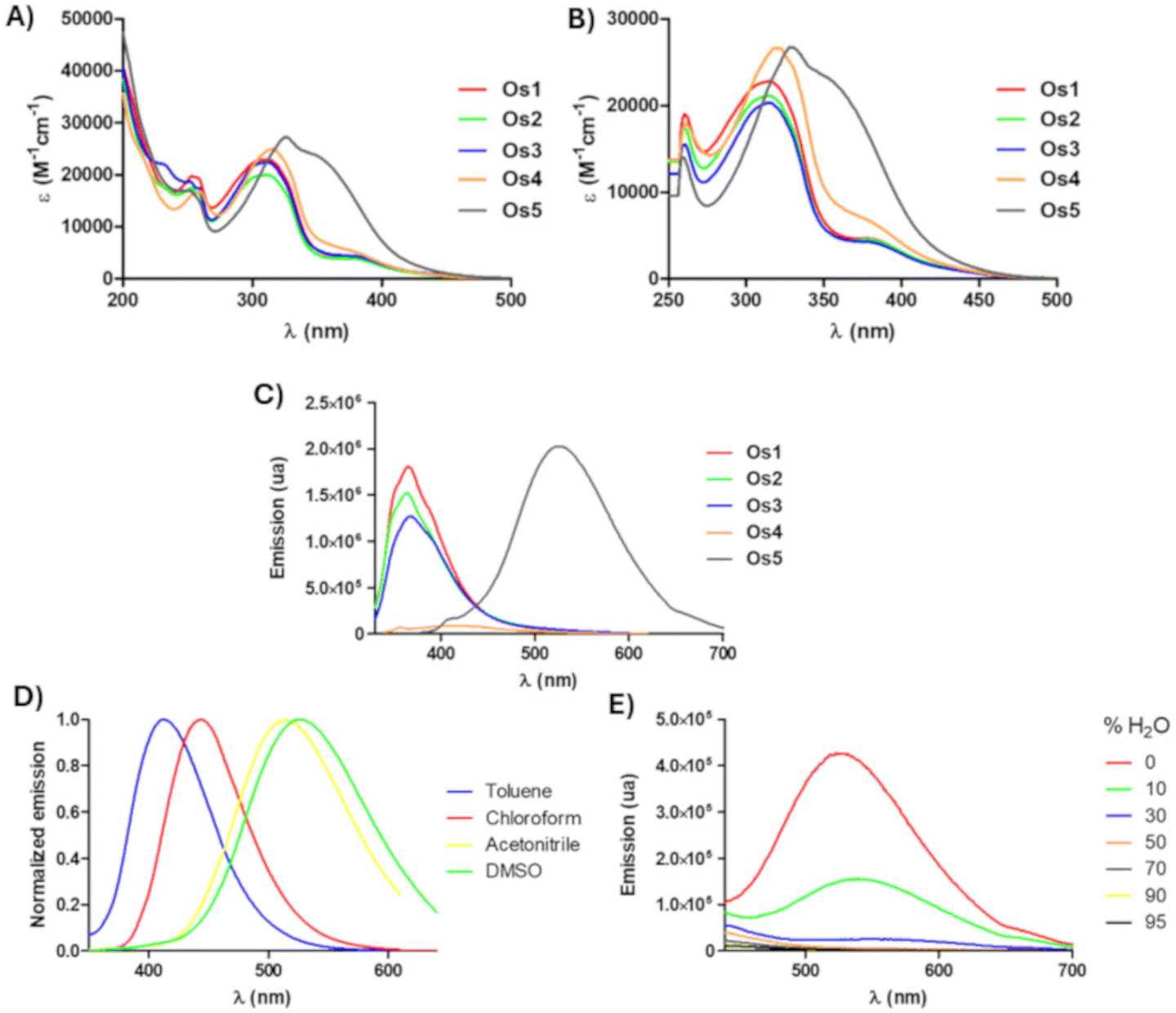 | ||
| Fig. 3 UV/Vis absorption spectra of Os1–Os5 (50 μM) in acetonitrile (A) and DMSO (B) at room temperature. (C) Emission spectra of complexes Os1–Os3 (λex = 310 nm), Os4 (λex = 320 nm), and Os5 (λex = 360 nm) in aerated DMSO at room temperature. Normalized emission spectra of Os5 in different solvents (D). Emission spectra of Os5 (λex = 360 nm, 50 μM) in DMSO/water mixtures with different fw (E). | ||
The emission spectra of Os1–Os5 (50 μM) compounds were also recorded in DMSO (Fig. 3C). Except for Os4, all the complexes were found to be emitters in DMSO, with compound Os5 highlighted for presenting an intense and broad emission band, whose maximum is shifted to longer wavelengths (529 nm). These emission results agree with those obtained for the free BTAT ligands.35 To understand the intramolecular charge transfer (ICT) character of the excited state, the emission spectra of Os5 were recorded in different solvents at room temperature (Fig. 3D). Large and positive solvatofluorochromism indicates considerable ICT characteristics of the compound.40Os5 shows strong solvatochromic behavior; as solvent polarity increases (e.g., toluene, chloroform, acetonitrile, and DMSO), the emission maxima are red-shifted (λem 413, 445, 515, and 529 nm, respectively). On the other hand, the aggregation-caused quenching (ACQ) effect of Os5 was next evaluated in DMSO/water mixtures with varied water volumetric fractions (fw). As shown in Fig. 3E, Os5 shows classic ACQ properties, as at 30% water, there has been complete emission extinction.41,42
Stability and hydrolysis
We next investigated the stability and hydrolysis behavior of the new BTAT-based osmium(II) since this is one of the possible mechanisms for activating halide osmium arene complexes in their interaction with biological targets.43 Therefore, we first evaluated the stabilities of Os1–Os5 in dimethylsulfoxide (DMSO), a frequently used solvent, to ensure the solubility of compounds in biological media. Stabilities in DMSO were checked by UV/Visible spectra, which remained unchanged after 48 h. Stability in aqueous media was then evaluated by 1H-NMR spectroscopy by dissolving each Os complex (1 mM) in 1:2 D2O:DMSO-d6 (v/v) and recording spectra at different times. The 1H-NMR spectra of complexes Os1–Os5 remained almost unaltered after 24 h, except for the disappearance of H5 of the corresponding BTAT ligand, which was attributed to substituting a proton with deuterium (Fig. S30–S34†). It is worth mentioning that the ESI-MS spectrum of Os5 in 1:2 D2O:DMSO-d6 mixture displayed a peak corresponding to the ion [M + 1–PF6]+ (Fig. S35†). To further confirm that the disappearance of the proton in the aforementioned NMR spectra is due to the substitution by deuterium, a 1H-NMR experiment was performed for Os3, showing the reversibility of the process, with details provided in the ESI (Fig. S36†). Based on these results, we can conclude that all new half-sandwich Os(II) complexes remained stable and did not hydrolyze over that period under those conditions. Furthermore, hydrolysis of the Os complexes was also studied by HPLC-MS in mixtures containing a higher percentage of water than previously used. For this purpose, solutions of Os1–Os5 were prepared in water (5% DMSO), and HPLC-MS spectra were recorded at the initial time and after 24 h of incubation at room temperature. The results reveal that compounds Os1–Os4 are hardly hydrolyzed (Fig. S37–S40†), whereas Os5 (Fig. S41†) hydrolyzes readily, achieving up to 76% hydrolysis at 24 h under these conditions. This is observed by MS from the peak of interest extracted from HPLC. However, the formation of adducts with any other biological nucleophiles cannot be discarded in the cell culture medium. To clarify this point a bit further, we have performed the HPLC-MS studies of the Os complexes (Fig. S42–S46†) dissolved in RPMI culture medium (5% DMSO). As shown in Fig. S46,† complex Os5 hydrolyzed to a lesser extent in these conditions, likely due to the high concentration of sodium chloride (6 g L−1) present in the RPMI-1640 medium. Os1–Os4 remained unaltered also in these conditions. Notably, Os5, which undergoes faster hydrolysis, turned out to be the best performer of the series for biological applications (vide infra), so it can be concluded that the hydrolysis process could play a crucial role in its cancer treatment properties. Os5 is the complex that incorporates the electron donor and steric bulk NMe2 group in the BTAT ligand.
Interaction of half-sandwich osmium complexes with human serum albumin
As proteins are present in biological membranes, our next investigations focused on assessing the impact of the investigated Os compounds on proteins, with Human Serum Albumin (HSA) being the most abundant protein in blood serum.44 The fluorescence of HSA is mainly due to tryptophan residues and, to a lesser extent, tyrosine and phenylalanine residues. Changes in the conformation of HSA, resulting in a noticeable change in fluorescence emission intensity, can occur upon binding with the interacting compounds. Given the high lipophilicity of the investigated Os compounds, it is reasonable to anticipate that hydrophobic interactions would be predominant in protein-compound interactions. Previous studies have revealed that the surface of HSA contains both hydrophobic and hydrophilic domains, rendering it a suitable model for investigating compound interactions.To determine the possible interaction of the complexes Os1–Os5 with HSA, luminescence quenching experiments were carried out by exciting at 295 nm and adding variable amounts (0–27 μM) of complexes Os1–Os5 and HSA (2.7 μM) at room temperature (Fig. 4B for Os3 and Fig. S47† for the rest) and following the decrease in luminescence intensity, which usually fits the Stern Volmer (SV) equation (Fig. S48†). The Ksv values, obtained from the slope of the representation of F0/F against [C], are collected in Table 1. The linear fit obtained for the SV quenching analysis for all complexes indicates a single quenching mechanism. To establish whether the extinction mechanism is due to a static or dynamic process, the variation in the absorption intensity of HSA in the presence of the complexes was studied, as can be seen in Fig. 4A for Os3 and Fig. S49† for the rest. The absorption intensity of HSA decreased drastically in the presence of complexes Os1–Os5, suggesting a disturbance in the secondary structure of HSA. Thus, the addition of the complexes to the HSA produces changes in the excited state of the fluorophore and also induces changes in the absorption spectrum, so the possible quenching mechanism induced by the complexes would be a static mechanism that has been previously described in the literature.45 When the static mechanism takes place, it is assumed that the complex interacts independently with a set of equivalent binding sites in the HSA.46 The binding parameters can be determined according to the Scatchard equation, where KB is the binding constant and “n” is the number of binding sites per HSA molecule. The linear fit of the plot of log(F0 − F/F) versus log[C] (Fig. S50†) at room temperature allows obtaining KB for the complexes and the values of n (0.9–1.3) indicating that there is only one binding site accessible in HSA for complexes Os1–Os5.47–49
 | ||
| Fig. 4 Interaction of Os3 with HSA. (A) UV/Visible absorption spectrum of HSA (1 μM) with the addition of different equivalents of Os3 (0–2 eq.). (B) Emission spectrum of HSA (2.7 μM) in the presence of increasing amounts of Os3 (0–27 μM) (top to bottom gradual increments). | ||
| Complex | K SV × 105 (M−1) | K B (M−1) × 105 | n |
|---|---|---|---|
| Os1 | 0.96 | 28.58 | 1.33 |
| Os2 | 1.13 | 0.77 | 0.97 |
| Os3 | 1.24 | 1.39 | 1.01 |
| Os4 | 1.76 | 15.09 | 1.21 |
| Os5 | 2.08 | 23.85 | 1.09 |
As shown in Table 1, complexes Os1 and Os5, which contain the donor substituents Me and NMe2, respectively, in the R4 position of the phenyl ring of the BTAT ligands, exhibit the highest binding constants to HSA.
Site-selective binding of complexes on HSA
To improve the future design of new drugs, there is growing interest in understanding the mechanism of action of metallodrugs, including the nature of the binding of metallodrugs to biomolecules. To identify the location of the binding site of new osmium compounds in the HSA region, competitive binding experiments were carried out. Albumins have two main drug-binding sites characterized as sites I and II.50 These sites bind drugs selectively. Site I is located in the subdomain IIA of HSA and involves the lone tryptophan of the protein (214Trp), whereas site II is positioned in subdomain IIIA and primarily involves the individual amino acid residues 410Arg and 411Tyr.51 We used warfarin as a marker substance for the specific binding of the investigated Os complexes on the HSA molecule site I52 and ibuprofen on-site II.53Warfarin fluorescence at λ = 320 nm increases considerably upon binding to serum albumin due to its interaction with Trp214. This fluorescence decreases if a second compound competes for the same site. Thus, the binding of the complexes is assessed by monitoring the changes in the fluorescence spectrum when increasing amounts of each of the complexes are added (0–25 μM) (Fig. 5A for Os3 and Fig. S51† for the rest). Similarly, a decrease in the fluorescence of the [HSA-IBU] complex upon the addition of increasing amounts of each osmium compound indicates the binding of the complexes to site II of HSA (Fig. 5B and Fig. S52† for the rest). As observed in Table 2 and Fig. S53,† compounds Os1–Os4 prefer binding to site II of HSA, as indicated by the higher SV fluorescence quenching constants in all instances. In contrast, compound Os5 binds indiscriminately to both binding sites, displaying no preference for either.
 | ||
| Fig. 5 Emission spectra of HSA-WF (A) and HSA-IBU (B) in the presence of increasing amounts of complex Os3. λex = 320 nm, [HSA–WF] = 1:1 μM; λex = 280 nm, [HSA–IBU] = 1:1 μM. [Complex Os3]: 0–25 μM (top to bottom gradual increments). | ||
| Complex | K SV × 105 (M−1) | ||
|---|---|---|---|
| Interaction with HSA | WF displacement | IBU displacement | |
| Os1 | 0.96 | 0.38 | 0.69 |
| Os2 | 1.13 | 0.49 | 0.59 |
| Os3 | 1.24 | 0.48 | 0.68 |
| Os4 | 1.76 | 0.82 | 0.94 |
| Os5 | 2.08 | 1.15 | 1.11 |
Effect on growth and viability of cancer cells
A panel of six cancer cell lines was employed to examine the antiproliferative potential of the new half-sandwich Os compounds. The anticancer activity was determined with MTT assay after a 72 h treatment and expressed as IC50 values shown in Table 3. The osmium complexes Os1–Os4 manifested medium cytotoxicity with IC50 ranging from 15 to 66 μM. The best compound was Os1, carrying the methyl group with IC50 of 15 μM in rhabdomyosarcoma (RD) cells. It has shown that the presence of the dimethylamine substituent in the molecule of Os5 is the most advantageous and that this compound displays the highest inhibitory effect in the cancer cells tested. With the IC50 values ranging from 4.3 to 8.3 μM, its activity is comparable or, in some cases, better than that of the established anticancer drug cisplatin. Representative dose–response curves are shown in Fig. S54.†a values (μM) determined with MTT assay
| RD | HCT116 | PSN1 | MCF7 | MDA-231 | HeLa | MRC5 | |
|---|---|---|---|---|---|---|---|
| a Data represents MEAN ± SD from at least three independent measurements. The cells were treated for 72 h. | |||||||
| Os1 | 15 ± 5 | 25 ± 2 | 27 ± 4 | 27 ± 8 | 27 ± 5 | 23 ± 1 | 91 ± 18 |
| Os2 | 38 ± 3 | 53 ± 8 | 66 ± 5 | 42 ± 5 | 60 ± 16 | 46 ± 10 | >100 |
| Os3 | 24 ± 6 | 34 ± 3 | 47 ± 2 | 45 ± 9 | 40 ± 1 | 43 ± 7 | 76 ± 12 |
| Os4 | 43 ± 8 | 45 ± 9 | 43 ± 9 | 45 ± 4 | 44 ± 7 | 36 ± 7 | 69 ± 4 |
| Os5 | 4 ± 1 | 6.6 ± 0.6 | 7 ± 2 | 6 ± 1 | 6 ± 1 | 8 ± 2 | 45 ± 3 |
| Cisplatin | 3.1 ± 0.2 | 8.8 ± 0.6 | 3.3 ± 0.4 | 21 ± 2 | 19 ± 3 | 15 ± 3 | 9 ± 2 |
Besides the tumor cell lines, the human lung fibroblast MRC5, a non-cancerous cell line, was included in the experiment. Selectivity indexes (SIs) for the osmium complexes were calculated and are shown in Table 4. The SIs determined for Os5 were noticeably higher than those calculated for Os1–Os4, except Os1 in RD cells. This result indicates that the selectivity of Os5 for the chosen cancer cell lines over MRC5 is better than that of the remaining agents, including clinically used cisplatin. Nevertheless, the SIs of all the compounds and all cell lines are higher than 1, i.e., the complexes inhibit the proliferation of the chosen cancer cells to a greater extent than the non-cancerous lung fibroblasts. Hence, exceptional SI values obtained especially for Os5 (SI = 5.4–10.4) reveal its remarkable selectivity for cancer over noncancerous cells, thereby alleviating possible toxic side effects – a foundational prerequisite in developing anticancer therapies.
| RD | HCT116 | PSN1 | MCF7 | MDA-231 | HeLa | |
|---|---|---|---|---|---|---|
| a The SIs were calculated as IC50 (MRC5)/IC50 (indicated cancer cell line). | ||||||
| Os1 | 6.07 | 3.64 | 3.37 | 3.37 | 3.37 | 3.96 |
| Os2 | >2.63 | >1.89 | >1.52 | >2.38 | >1.67 | >2.17 |
| Os3 | 3.17 | 2.24 | 1.62 | 1.69 | 1.90 | 1.77 |
| Os4 | 1.60 | 1.53 | 1.60 | 1.53 | 1.57 | 1.92 |
| Os5 | 10.47 | 6.82 | 6.34 | 7.03 | 7.38 | 5.42 |
| Cisplatin | 2.90 | 1.02 | 2.73 | 0.43 | 0.47 | 0.60 |
Cellular accumulation
The series of processes leading to cell death commences with the cellular uptake of a compound and its intracellular accumulation. The amounts of osmium localized inside RD cells following a 5 h treatment with the new Os complexes were determined. The results are shown in Table 5, and the data inspection confirms that the compounds are accumulated in RD cells with various effectivity. The most abundant intracellular osmium was found in the cells exposed to Os5. According to Os uptake, the agents may be organized in the order Os5 > Os1 > Os3 > Os2 > Os4. A comparison of the results from Table 3 (RD column) and Table 5 discloses a strong correlation between cellular accumulation of the osmium complexes and their antiproliferative activity. It is, therefore, reasonable to suggest that cellular accumulation of the investigated osmium complexes plays a significant role in the mechanism of their antiproliferative activity.Transport into cells and accumulation in cells of several known drugs are mediated by their binding to HSA.44 The comparison of the binding constants of the investigated Os complexes when they bind to HSA (Table 1) and their accumulation in RD cells (Table 5) shows no correlation. This comparison may, therefore, be interpreted to mean that the interaction of the investigated Os complexes with HSA does not play a fundamental role in their transport and accumulation in tumor cells.
It should also be noted that according to HPLC (Fig. S22†), Os5 is one of the more lipophilic complexes of the series; Os5 is the complex that incorporates the electron donor and steric bulk NMe2 group in the BTAT ligand.
Mechanism of action
Since Os5 proved to be the most active compound of the new group of osmium(II) half-sandwich complexes, we chose this agent to study the mechanism of action in cancer cells. Rhabdomyosarcoma (RD) was selected as a model cell line for two reasons. First, rhabdomyosarcomas are one of the most frequent sarcomas in childhood and adolescence; they are highly aggressive and belong to poorly treatable cancers with survival rates of metastatic and relapsed cases of less than 20%.54,55 Therefore, it is of high importance to find a satisfactory cure for this kind of cancer. Second, from the selected panel of cancer cells, the RD cell line was the most sensitive to the treatment with the new group of Os(II) complexes, with the highest sensitivity to Os5.Localization of Os5 in rhabdomyosarcoma cells
We took advantage of the fact that Os5 possesses fluorescence properties to monitor its cellular accumulation and localization (Fig. 6). The cells were treated with 12.9 μM (3 × IC50) Os5 for 3 h, and then confocal imaging was performed using UV laser (Ex/Em = 355 nm/400–485 nm). Brightfield images were taken to provide information on cell morphology. As shown in Fig. 6, the fluorescence signal from Os5 was localized mainly in the cytoplasm, partly as a weak blue background and partly as discrete puncta emanating a strong signal. Control non-treated cells produced no fluorescence signal under the same laser and detector settings. Thus, the observed fluorescence may be attributed to Os5, not cellular autofluorescence. In the confocal images, some cells gradually adopted different morphology (Fig. 6(C)). They lost stretched, adhered shape and became rounded with apparent blebbing membranes. At the same time, the discrete blue puncta disappeared, and only a weak blue background remained.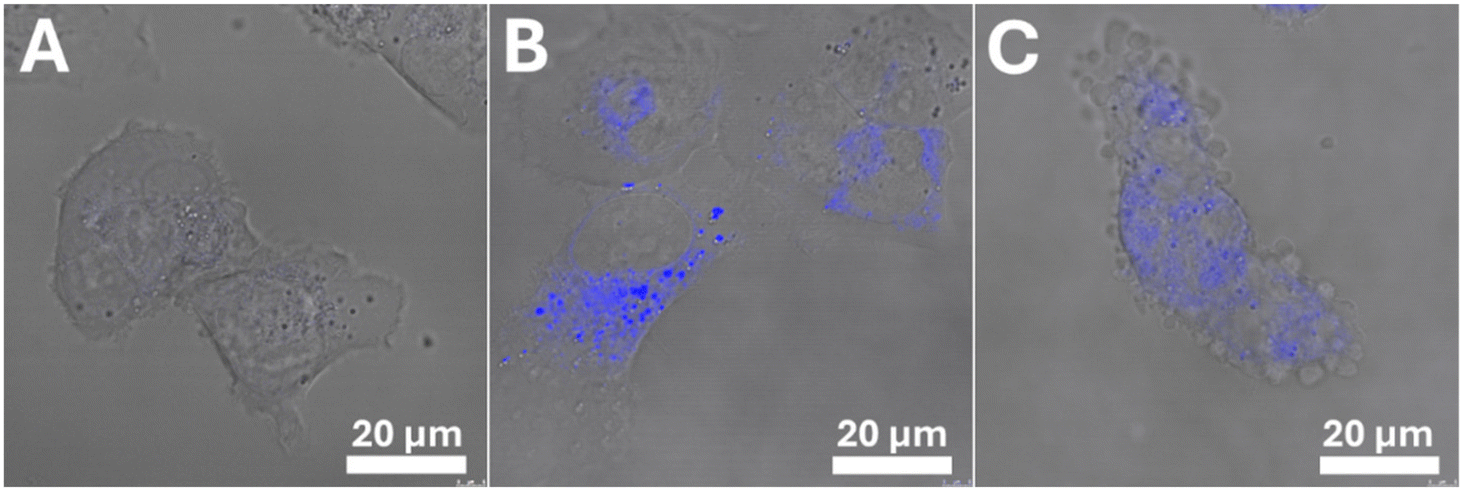 | ||
| Fig. 6 Localization of Os5 in Rhabdomyosarcoma cells. The cells were exposed to Os5 (12.9 μM) for 3 h, and then confocal images were taken using a UV laser (λex/λem = 355/400–485 nm). The signal from Os5 (blue) is merged with the brightfield channel. (A) – control (non-treated cells); (B) and (C) – cells were treated with Os5. | ||
To better reveal the mechanism of action of Os5, we aimed to detect the specific cellular component with which Os5 associates. Understanding the intracellular distribution of a drug is essential for elucidating its mechanism of action, as the drug must localize within the specific intracellular compartment containing its target to exert its therapeutic effect. Therefore, a colocalization assay with Lyso-Tracker and ER-Tracker dyes was performed. The results are shown in Fig. S55.† Pearson correlation coefficients were calculated from 30–40 images using ImageJ software and are shown in Fig. S56.† The correlation coefficient for Os5 with Lyso-Tracker (0.65 ± 0.09) was the highest found. Even lower correlation coefficient values (0.57 ± 0.07) were found for colocalization of Os5 with endoplasmic reticulum. Both findings indicate a moderate/weak correlation.56 The fact that the fluorescence signal associated with the Os5 complex does not strongly correlate with any of the trackers used (Fig. S55 and S56†) shows that the complex is not selectively accumulated in any particular cell organelles. Still, it is distributed among different organelles, including partial localization freely in the cytoplasm (Fig. 6). This suggests that Os5 could affect multiple cellular targets simultaneously.
In the attempt to determine the degree of colocalization of Os5 with mitochondria, we obtained poor results since, upon the addition of Os5 to Mito-Tracker-stained cells, the Mito-Tracker signal gradually disappeared (not shown). We repeated the attempt with TMRE-stained cells. The strong TMRE signal enabled us to capture better the mitochondria in cells exposed to Os5 (Fig. 7). The correlation coefficient for Os5 and TMRE calculated from the images showed only very weak (0.35 ± 0.05) colocalization of Os5 with mitochondria. However, a closer look at Fig. 7, especially at the detailed images, discloses that mitochondria undergo significant changes in the presence of Os5. The characteristic tubular network visible in non-treated cells (yellow arrow) rapidly disappears upon the addition of Os5, and punctate spheres appear (red arrow), which finally disappear in time (not shown).
 | ||
| Fig. 7 Colocalization of Os5 and TMRE-stained mitochondria in RD cells. The cells were prestained with TMRE and then exposed to Os5 (12.9 μM) for 1 h. Os5 (blue) – the signal of Os5; TMRE (green) – the signal of TMRE stained mitochondria; brightfield – brightfield channel; merge – the merged signal of the three channels; detail – the magnified image of merged channels, the cropped area is marked with a square in merge images. (A) – cells exposed to Os5 only; (B) – the cells prestained with TMRE, non-treated; (C) – the TMRE prestained cells treated with Os5. | ||
To determine which biomolecules Os5 is associated with, the cells treated with Os5 were fractionated using a commercial FractionPREP™ Cell Fractionation Kit. The amount of Os in each fraction was determined by ICP-MS. Most Os (71 ± 6%) was associated with a membrane/particulate fraction containing phospholipids and membrane proteins. Cytosolic fraction contained 21 ± 4% of Os, whereas 6.3 ± 0.9% was found in the fraction comprising nuclear proteins and nuclear membrane. Only a negligible portion of Os was found in the cytoskeleton and nuclear DNA containing fraction (1.7 ± 0.3%) (Table S10†). The results are in agreement with the data from confocal microscopy and indicate that the antiproliferative effect of Os5 can be mainly caused by damage to cell membranes (plasma membrane or membranes of intracellular organelles). However, interactions with cytosolic biomolecules (presumably proteins) may also play a significant role.
In pursuit of revealing target biomolecule(s), further experiments were performed using cell-free media. Since the vast majority of Os is located in membranes and the cytoplasm of the cell, the binding ability of Os5 to biomolecules present in these cellular compartments, i.e., phospholipids (in membranes), RNA (in the cytoplasm), and proteins (in both membrane and cytosol) was assessed as described in Experimental section. Model molecules HSA, t-RNA, and 18:0 DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine) were used as models for proteins, RNA, and phospholipids, respectively. We found that Os5 binds most rapidly to proteins (57.6% of total Os5 present in the reaction was bound within 24 h) and phospholipids (54.5% of total Os5 was bound within 24 h), whereas binding to RNA was significantly slower, though still notable (18% of total Os5 was bound within 24 h). These results further support the view that the biological action of Os5 may likely be related to its binding to proteins and phospholipids and damage to these molecules.
Loss of mitochondrial membrane potential
The results from colocalization experiments suggest that mitochondria in RD cells are affected by the presence of Os5. Therefore, we decided to analyze the changes in mitochondrial membrane potential (MMP) inflicted by Os5. In one experiment (Fig. 8A), the cells were prestained with TMRE and then analyzed with flow cytometry immediately after adding Os5 or staurosporine (for comparison). The analysis was repeated several times within an hour. The graph (Fig. 8A) shows that the presence of Os5 rapidly causes the loss of mitochondrial membrane potential, while the effect of staurosporine is much lower (or slower). In the second experiment, the RD cells were incubated with a series of Os5 concentrations for periods of 1, 2, 5.5, and 24 h and then stained with TMRE. TMRE fluorescence was analyzed with the flow cytometer. The results shown in Fig. 8B reveal that even at concentrations lower than IC50, there is a significant loss of mitochondrial membrane potential. Moreover, the graph shows that the MMP decreases within the first two hours following the treatment and then remains steady for up to 24 h, and that the MMP decrease is proportional to the Os5 concentration applied. These results, together with the information provided by confocal microscopy, indicate that following the RD cells’ exposure to Os5, mitochondria in some cells lose their potential and characteristic morphology. | ||
| Fig. 8 The loss of mitochondrial membrane potential. (A) RD cells were prestained with TMRE (100 nM; 30 min) and then aliquoted into three parts. The first part remained untreated, staurosporine (2 μM) was added to the second part, and Os5 (20 μM) was added to the third part. Flow cytometry analysis of TMRE fluorescence was performed immediately, and further analyses were performed several times within an hour. The graph (A) displays relative TMRE fluorescence depending on the treatment time. (B) RD cells were treated with several concentrations of Os5 (indicated in the graph) for 1, 2.5, 5.5, and 24 h, stained with TMRE, and analyzed using flow cytometry. The graph (B) displays relative TMRE fluorescence depending on the treatment time. | ||
Comparison of cytotoxicity determination with MTT and neutral red (NR)
The knowledge that mitochondria are affected by the presence of Os5 prompted us to assess whether there is a difference in cytotoxicity evaluation by MTT (a method based on the activity of mitochondrial succinate dehydrogenase) and a mitochondria-independent assay based on the uptake of neutral red dye. The results of both assays are shown in Table 6 and Fig. S57,† and they reveal that there is no significant difference between the IC50 values determined by either method after 24- and 72 h treatment with Os5.| 24 h | 72 h | |
|---|---|---|
| a Data represents MEAN ± SD from at least three independent measurements. | ||
| MTT | 13 ± 2 | 4.3 ± 0.6 |
| NR | 13 ± 3 | 4.0 ± 0.2 |
Detection of cell death
 | ||
| Fig. 9 Mode of cell death. (A–D) Flow cytometry analysis of Annexin V/Propidium iodide dual staining of rhabdomyosarcoma cells treated with Os5 complex. The cells were treated with several concentrations of Os5 for 24 h. X- and Y-axes indicate Annexin V negative/positive (A−/A+) and Propidium iodide negative/positive (PI−/PI+) populations. Left bottom quadrant (A−/PI−) – living cells; right bottom quadrant (A+/PI−) – early apoptotic cells; left upper quadrant (A−/PI+) – necrotic cells and right upper quadrant (A+/PI+) – late apoptotic/necrotic cells. (A) control non-treated cells; (B–D) Os5 treated cells; (B) 5 μM; (C) 10 μM (D) 20 μM. (E) RealTime Annexin V Apoptosis and Necrosis Assay. RD cells were seeded in a 96-well plate, and the next day, EtOH (10%), Staurosporine (2 μM), or Os5 (at indicated concentrations) were added to the wells immediately followed by the addition of the kit components. Luminescence (apoptosis) and Fluorescence (necrosis) were measured in a kinetic mode for cca 1000 min at 37 °C. | ||
From the results obtained, it is evident that apoptosis is the leading cause of death induced in RD cells with Os5. Confocal images of TMRE-stained mitochondria (Fig. 7) demonstrate notable changes in mitochondrial morphology. Kinetic studies of the loss of mitochondrial membrane potential (MMP) (Fig. 8) showed that the mitochondrial membrane integrity is disrupted within minutes following the addition of Os5. This phenomenon is strictly compound dose-dependent. The experiments based on phosphatidylserine exposure on the outer leaflet of the plasma membrane (Fig. 9) reveal that apoptosis progression to this step is slower than in the case of staurosporine, which is again dose-dependent. These findings suggest that Os5 triggers apoptosis in RD cells by mitochondrial damage.
Generation of reactive oxygen species (ROS)
We examined the generation of ROS in RD cells exposed to the Os5 complex. The level of ROS was detected with CellROX Green reagent. The method is based on the probe emitting fluorescence after ROS oxidation. The results are shown in Fig. 10. The RD cells treated with Os5 exhibited elevated fluorescence compared to the untreated control. As shown in the graph, the production of ROS induced with Os5 was concentration-dependent. Os5 at a concentration corresponding to its 3 × IC50 value induced ROS generation approximately twice the level of untreated control, whereas menadione used as positive control triggered ROS to a significantly higher level (cca 6-fold of control level). Overall, this result indicates a relatively low ROS production induced by Os5 in RD cells that might, to a certain extent, contribute to its anticancer activity.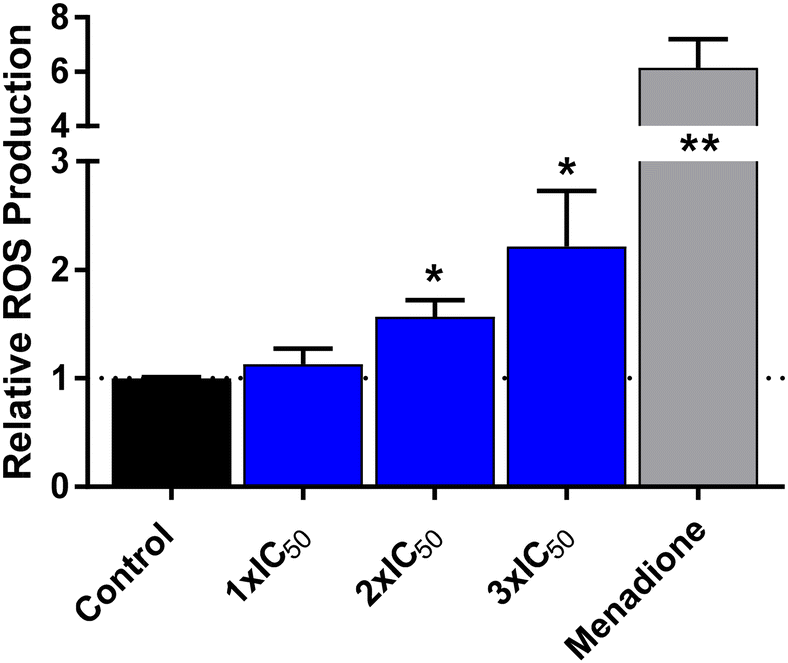 | ||
| Fig. 10 ROS production. Quantification of ROS produced in RD cells exposed to Os5 and menadione (positive control). The cells were treated with three concentrations of Os5 – 4.3 μM (1 × IC50), 8.6 μM (2 × IC50), and 12.9 μM (3 × IC50) for 3 h or with 100 μM menadione for 1 h. The ROS production was evaluated with CellRox®-green reagent on BD FACS Verse flow cytometer. The bars in the graph show relative fluorescence (vs. untreated control) and represent MEAN ± SD from three independent experiments. The results were subjected to statistical analysis using an unpaired Student t-test, and statistically significant differences from untreated control are marked with asterisks with *p ≤ 0.05 or **p ≤ 0.01. | ||
The effect of Os5 on cell cycle
Drug treatment often induces modulation of cell cycle progression. Hence, we analyzed the cell cycle perturbation in RD cells exposed to Os5 (Fig. 11). The cells were treated with Os5 at concentrations corresponding to 1× and 2 × IC50 values for 24 h. A close inspection of the distribution of the treated RD cells into individual cell cycle phases indicates that a significant cell count is arrested in the G1 phase compared to untreated control cells. While 40% of untreated cells are in the G1 phase, 69 and 72% of cells treated with Os5 at the two concentrations are in the G1 phase, respectively. The increase in the G1 population occurs at the expense of both the S and G2/M phases but mostly the S phase, where there are only 18 and 13% cells (vs. 41% of untreated control), respectively. The results reveal that Os5 arrests RD cells in the G1 phase and significantly reduces cell portion (vs. control) in the S phase. | ||
| Fig. 11 Cell cycle distribution. RD cells were treated with Os5 at concentrations corresponding to 1× and 2 × IC50 for 24 h. Propidium iodide was used as a nuclear staining. (A) nontreated control; (B) cells treated with 4.3 μM Os5; (C) cells treated with 8.6 μM Os5; (D) bar graph of cell populations (%) in individual cell cycle phases. Red – G1; blue striped – S; green – G2/M. (A–C) representative images, (D) – the graph shows the results of 3 measurements (MEAN ± SD). The results were subjected to statistical analysis using an unpaired Student t-test, and statistically significant differences from untreated control are marked with asterisks with *p ≤ 0.05 or **p ≤ 0.01. | ||
In the mechanistic section (vide supra), we focused on clarifying the mechanism of action of Os5. At the reviewer's request, we clarify why we did not use cisplatin for positive control in the mechanistic studies in the following paragraph.
The results described in the present study clearly show that the mechanism of action of the investigated Os complexes is markedly different from that of cisplatin. The major mechanism of cisplatin activity is generally accepted to be related to damage to nuclear DNA, to which cisplatin binds covalently (coordinatively). The resulting adducts then inhibit replication and especially transcription.57 We did not observe this mechanism for Os complexes; on the contrary, it is clear that the action is related to damage to mitochondrial membranes, thus impacting mitochondrial function in cancer cells. This mechanism was not observed for cisplatin.
Cytotoxic/antiproliferative effect in RD·CD133− and RD·CD133+ spheroids
In cancer treatment, a population of cells called cancer stem cells (CSC) represents a small but dangerous cell fraction.58 Whereas the bulk of tumor cells is effectively eradicated, CSCs escape the treatment and serve as a reservoir of resistant cancer cells that reinitiate secondary or tertiary tumors. The inefficiency of current anticancer drugs to successfully and permanently eliminate tumors is, to a major extent, due to their failure to eliminate CSC. It is, therefore, worth the effort to detect compounds that can kill CSCs to the same or even higher extent than the non-stem cells.59–63 In this work, we showed that the Os5 complex significantly damages mitochondria (Fig. 7 and 8). The ability of the complexes to effectively damage mitochondria suggests that they might also be effective in CSCs since mitochondria are the determinant of the function and fate of CSCs, playing core roles in the regulation of CSC stemness maintenance and differentiation,64 likely due to the higher mitochondrial load. Therefore, we decided to examine the efficacy of the new osmium compounds in inhibiting the growth and viability of the sarcoma stem cell subpopulation. The isolation of sarcoma CSCs was based on the CSC's expression of the CD133 marker. CD133 is a transmembrane protein known as Prominin1 and has been previously used as a distinct marker for selecting CSCs in rhabdomyosarcoma.63 We sorted the RD cells into CD133+ (expressing CD133) and CD133− (nonexpressing CD133) groups. Both populations were cultured for 96 h until rhabdospheres of the desired size (211 ± 23 nm in diameter) were grown. The spheroids were then treated with the new osmium complexes for another 72 h. Viability at the end of the incubation period was determined with Cell TiterGlo 3D assay. Representative dose–response curves are shown in Fig. S59.†Cyclophosphamide, salinomycin, and cisplatin were included in the assessment for comparative purposes. The results expressed as IC50 values in Fig. 12 and Table S10† indicate that both RD populations (CD133+ and CD133−) were sensitive to the newly synthesized Os compounds to a similar extent. In some cases (Os1 and Os3), the agent was more potent in the RD CD133+ fraction. Similar results were obtained for salinomycin and cyclophosphamide. Although salinomycin was effective at lower concentrations than the tested Os complexes, this finding does not necessarily diminish the potential importance of Os5 for treating tumor cells, including CSCs.65–67 Furthermore, it should be pointed out that Os5 is about two orders of magnitude more effective than cyclophosphamide, a drug used to treat rhabdomyosarcoma in clinic.68,69 Moreover, cancer stem cells are known to be inherently resistant to cisplatin.70,71 The finding that osmium complexes demonstrated comparable activity in both CSC-depleted and CSC-enriched RD cells suggests their potential to target both differentiated cancer cells and cancer stem cells concurrently. This characteristic could represent a significant advantage in chemotherapy, as it may reduce the reliance on chemotherapeutics tailored to specific tumor cell subtypes when used in combination.
 | ||
| Fig. 12 Anticancer activity in RD·CD133+ and RD·CD133− cells expressed as IC50 values (μM). 96-Hour-old spheroids were incubated with the Os compounds for 72 h, and cell viability was then determined with Cell TiterGlo 3D assay. SAL – salinomycin; CF – cyclophosphamide. The asterisks denote significant differences as determined with the Student t-test (p < 0.05). | ||
Confocal microscopy of RD·CD133+ spheroids
To visualize the effect of Os5 on RD·CD133+ spheroids, we stained untreated and treated representative spheroids with Calcein AM (live-cells label), propidium iodide (stains cells with disturbed membrane integrity) and Hoechst 33258 (stains all cell nuclei). Representative images are shown in Fig. 13. The images reveal that spheroids exposed to Os5 differ from untreated control in size, integrity, and propidium iodide intensity. The treated samples are smaller than the control, and spheroid disintegration is apparent in the case of the 10 μM concentration applied. Meanwhile, in untreated spheroid, a small portion of cells displays propidium iodide positivity, and markedly increased cell count with compromised membrane integrity (PI) is present in treated spheroids. The quantification of the ratios of Calcein AM fluorescence intensity to PI fluorescence intensity is shown in Fig. S60.† The graph shows that while the green and red channel intensity ratio for untreated control is 3.2, it is only cca 1.3 for treated spheroids. Significantly more dead cells are present in treated spheroids than in control, and the treated spheroids are smaller, and their integrity is impaired. | ||
| Fig. 13 Confocal microscopy of RD spheroids exposed to Os5. The spheroids were stained with Hoechst 33258 dye (Hoechst), Calcein AM, and propidium iodide. Merge – all three channels merged. (A) – Untreated control, (B) – spheroid treated with Os5 (5 μM, 72 h), (C and D) – spheroids treated with Os5 (10 μM, 72 h). Individual pictures were obtained as the maximal projection from 10 z-stacks. Representative images. | ||
Conclusions
In conclusion, we have designed a series of pseudo-octahedral arene Os(II) complexes, Os1–Os5, with the general formula [(η6-p-cym)Os(BTAT)Cl]+, where BTAT represents various chelating N^N′ ligands based on the 1-aryl-4-benzothiazolyl-1,2,3-triazole scaffold. The molecular structures of Os3 and Os5 were confirmed via X-ray diffraction. Notably, Os5 exhibits a bathochromic shift in its absorption band compared to the other osmium complexes, likely due to the electron-donating properties of its substituent (–NMe2). Additionally, HPLC-MS studies show that Os5 undergoes hydrolysis without losing its BTAT ligand and has the highest cellular accumulation in RD cancer cells.The new Os(II) half-sandwich complexes demonstrated moderate antiproliferative activity across six cancer cell lines, with greater potency compared to non-cancerous lung fibroblasts. Os5, which contains a dimethylamine substituent, was the most active compound, showing comparable or superior activity to cisplatin, a widely used anticancer drug. Our findings also highlight that cellular accumulation is a crucial factor influencing the antiproliferative activity of these Os complexes. Although it is known that several drugs rely on binding to HSA for cellular uptake and accumulation, our data suggest that HSA does not play a significant role in the transport of these Os complexes into tumor cells.
Further investigations focused on the mechanism of action (MoA) of the investigated Os complexes, with Os5 serving as the representative molecule. We selected RD cells for these studies, as they were the most responsive to Os complexes, and rhabdomyosarcoma is a highly aggressive cancer with a poor prognosis for metastatic and relapsed cases, with survival rates below 20%.54,55
Our initial goal was to identify the specific cellular component with which Os5 interacts. Colocalization assays revealed that Os5 may target multiple cellular components simultaneously, potentially providing an advantage in its biological effects by making it more difficult for cells to develop resistance. However, these experiments also indicated that Os5 impacts mitochondrial function in RD cells, causing loss of membrane potential and changes in morphology.
Annexin V/propidium iodide staining of RD cells treated with Os5 confirmed that apoptosis, driven by a caspase-dependent pathway, is the primary mode of cell death induced by Os5. Additionally, cell cycle analysis in RD cells revealed that Os5 increases the G1 phase population while reducing the S and G2/M phase populations, indicating that Os5 may inhibit cancer cell proliferation by preventing progression through the DNA synthesis and mitosis phases.
Previous studies on osmium-based anticancer complexes have shown their antiproliferative effects can be mediated through pathways involving reactive oxygen species (ROS) production. In our study, elevated ROS levels were detected in RD cells treated with Os5, suggesting that ROS production may contribute to its mechanism of action.
The activity of these Os complexes was further demonstrated in both 2D monolayer cultures and 3D spheroids, the latter of which more closely resemble solid tumors and serve as a promising in vitro model. One challenge with conventional anticancer metallopharmaceuticals is their inefficacy against cancer stem cells (CSCs), which often survive chemotherapy, leading to relapse and metastasis. For this reason, we prepared 3D spheroids from CSC-enriched RD cells and found that the Os complexes were not only effective against differentiated RD cells but also against RD CSCs. This may be related to the ability of the investigated Os complexes to increase intracellular ROS levels. CSCs have been shown to be hypersensitive to ROS, as they maintain lower ROS levels than bulk differentiated cancer cells,72 which helps them elude therapeutic interventions. Targeting the redox state within CSCs with drugs capable of elevating the ROS level and thus disrupting their survival advantage is, therefore, considered a promising strategy for cancer treatment.73,74
Importantly, Os5 exhibited anti-CSC properties, marking the first report of an osmium-based compound showing activity against CSC-enriched RD cells. Notably, the CSC-targeting potency of Os5 rivals some of the most CSC-selective compounds identified thus far. Given the critical need for CSC-specific therapies to prevent cancer relapse and metastasis, the preclinical potential of Os5 is highly promising. This study underscores the largely untapped potential of metal-based complexes for CSC-targeted chemotherapy and offers insights into the mechanisms and targets involved in osmium-induced toxicity.
Experimental section
Reagents and chemicals
Synthesis-grade solvents were employed in all cases. Deuterated solvents were purchased from Euriso-top. OsCl3·3H2O, α-terpinene, and ammonium hexafluorophosphate were obtained from Merck (Madrid, Spain). [Os(η6-p-cymene)Cl2]2 was prepared as previously reported,75 and benzothiazolyl-1,2,3-triazole derivatives (L1–L5) were synthesized following a synthetic procedure published by some of us (Scheme S1†).29 The purities ≥95% of the synthesized complexes used for biological evaluation were determined by RP-HPLC.[Os(η6-p-cymene)L1Cl]PF6 (Os1). Yellow solid. Isolated yield: 53%. 1H NMR (401 MHz, Acetonitrile-d3) δ 9.26 (s, 1H), 8.30–8.23 (m, 2H), 7.92–7.83 (m, 3H), 7.79 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.56–7.51 (m, 2H), 6.55–6.50 (m, 1H), 6.47 (dd, J = 5.8, 1.1 Hz, 1H), 6.35–6.28 (m, 1H), 6.25–6.18 (m, 1H), 2.49 (m, 4H), 2.29 (s, 3H), 1.01 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CD3CN) δ 160.3, 150.2, 144.6, 142.9, 134.7, 134.5, 131.7, 130.3, 129.4, 126.3, 125.2, 124.1, 122.2, 98.6, 97.5, 79.8, 76.0, 74.0, 73.0, 32.0, 23.0, 22.1, 21.3, 19.2. ESI-MS (positive ion. mode): m/z = 653.1226 [M − PF6]+, calcd m/z: 653.1182. Anal. calcd for C26H26ClF6N4OsPS: C, 39.17; H, 3.29; N, 7.03; S, 4.02. Found: C, 39.24; H, 3.11; N, 6.96; S, 4.09 (%).
[Os(η6-p-cymene)L2Cl]PF6 (Os2). Yellow solid. Isolated yield: 82%. 1H NMR (600 MHz, Acetonitrile-d3) δ 9.25 (s, 1H), 8.28–8.24 (m, 2H), 8.04–7.97 (m, 2H), 7.89 (ddd, J = 8.5, 7.1, 1.2 Hz, 1H), 7.81–7.76 (m, 1H), 7.51–7.44 (m, 2H), 6.53 (d, J = 5.7 Hz, 1H), 6.47 (d, J = 6.0 Hz, 1H), 6.31 (d, J = 5.7 Hz, 1H), 6.22 (d, J = 5.8 Hz, 1H), 2.50 (sept, J = 6.9 Hz, 1H), 2.28 (s, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H). 13C NMR (151 MHz, CD3CN) δ 165.5, 163.8, 160.2, 150.2, 144.8, 134.7, 133.2, 133.1, 130.3, 129.4, 126.4, 125.2, 125.0, 124.9, 124.1, 98.6, 97.7, 79.8, 76.1, 74.1, 73.2, 32.1, 23.0, 22.1, 19.2. ESI-MS (positive ion mode, CH3CN): m/z = 657.0977 [M − PF6]+, calcd m/z: 657.0931. Anal. calcd for C25H23ClF7N4OsPS: C, 37.48; H, 2.89; N, 6.99; S, 4.00. Found: C, 37.43; H, 2.70; N, 6.67; S, 3.93 (%).
[Os(η6-p-cymene)L3Cl]PF6 (Os3). Yellow solid. Isolated yield: 62%. 1H NMR (300 MHz, Acetonitrile-d3) δ 9.40 (s, 1H), 8.31–8.28 (m, 1H), 8.28–8.24 (m, 1H), 8.20 (d, J = 8.6 Hz, 2H), 8.11–8.04 (m, 2H), 7.90 (ddd, J = 8.5, 7.2, 1.3 Hz, 1H), 7.80 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 6.54 (d, J = 5.7 Hz, 1H), 6.49 (d, J = 5.8 Hz, 1H), 6.33 (d, J = 5.5 Hz, 1H), 6.24 (d, J = 5.8 Hz, 1H), 2.50 (sept, J = 6.8 Hz, 1H), 2.29 (s, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CD3CN) δ 156.0, 150.2, 145.0, 139.4, 134.7, 133.0, 132.6, 130.4, 129.5, 128.7, 128.6, 128.6, 128.5, 126.4, 125.2, 124.1, 122.9, 98.7, 97.8, 79.8, 76.2, 74.2, 73.2, 32.0, 23.0, 22.1, 19.1. ESI-MS (positive ion mode, CH3CN): m/z = 707.0938 [M − PF6]+, calcd m/z: 707.0899. Anal. calcd for C26H23ClF9N4OsPS: C, 36.69; H, 2.72; N, 6.58; S, 3.77 (%). Found: C, 36.54; H, 2.58; N, 6.67; S, 3.68 (%).
[Os(η6-p-cymene)L4Cl]PF6 (Os4). Yellow solid. Isolated yield: 71%. 1H NMR (401 MHz, Acetonitrile-d3) δ 9.43 (s, 1H), 8.63–8.49 (m, 2H), 8.31–8.25 (m, 2H), 8.25–8.21 (m, 2H), 7.91 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 7.81 (ddd, J = 8.3, 7.2, 1.1 Hz, 1H), 6.55 (d, J = 5.7 Hz, 1H), 6.49 (d, J = 5.9 Hz, 1H), 6.33 (d, J = 5.7 Hz, 1H), 6.25 (d, J = 5.8 Hz, 1H), 2.51 (sept, J = 6.9 Hz, 1H), 2.29 (s, 3H), 1.03 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H). 13C NMR (101 MHz, CD3CN) δ 159.9, 150.2, 149.8, 145.2, 140.7, 134.8, 130.4, 129.6, 126.8, 126.5, 125.3, 124.2, 123.2, 98.8, 98.0, 79.9, 76.3, 74.3, 73.4, 32.1, 23.0, 22.1, 19.2. ESI-MS (positive ion mode, CH3CN): m/z = 684.0900 [M − PF6]+, calcd m/z: 684.0876. Anal. calcd for C25H23ClF6N5O2OsPS: C, 36.26; H, 2.80; N, 8.46; S, 3.87 (%). Found: C, 36.42; H, 2.68; N, 8.47; S, 3.74 (%).
[Os(η6-p-cymene)L5Cl]PF6 (Os5). Yellow solid. Isolated yield: 79%. 1H NMR (401 MHz, Acetonitrile-d3) δ 9.11 (s, 1H), 8.29–8.21 (m, 2H), 7.89 (ddd, J = 8.6, 7.3, 1.2 Hz, 1H), 7.81–7.73 (m, 3H), 6.96–6.90 (m, 2H), 6.51 (d, J = 5.7 Hz, 1H), 6.47 (d, J = 5.8, 1H), 6.30 (d, J = 5.7, 1H), 6.19 (d, J = 5.7, 1H), 3.07 (s, 6H), 2.49 (sept, J = 6.8 Hz, 1H), 2.29 (s, 3H), 1.00 (d, J = 6.9 Hz, 3H), 0.92 (d, J = 6.9 Hz, 3H).
13C NMR (151 MHz, CD3CN) δ 160.5, 153.0, 150.2, 144.3, 134.5, 130.2, 129.3, 125.5, 125.2, 125.0, 124.0, 123.2, 113.1, 98.6, 97.3, 79.7, 75.9, 73.9, 72.9, 40.5, 32.1, 23.0, 22.2, 19.2. ESI-MS (positive ion mode, CH3CN): m/z = 682.1460 [M − PF6]+, calcd m/z: 682.1447. Anal. calcd for C25H23ClF6N5O2OsPS: C, 39.25; H, 3.46; N, 8.48; S, 3.88. Found: C, 39.26; H, 3.46; N, 8.36; S, 3.82 (%).
Methods and instrumentation
:56%.
Interaction with biomacromolecules
000 M−1 cm−1.78 Quantitative analyses of the interaction between Os1–Os5 complexes and HSA were performed by fluorimetric titration. A 3.0 mL portion of aqueous protein solution (2.5 μM) was titrated by successive additions of complex solution (0–25 μM). The mixing solution was stirred for each addition and allowed to stand for 5 min. Fluorescence intensities were then measured at excitation wavelengths of λ = 295 nm. The width of the excitation and emission slit was set at 5 nm, and the emission fluorescence spectra were recorded in the wavelength range λ = 305–570 nm.
Cell lines, culture conditions, and stock solution
Human rhabdomyosarcoma cells (RD) were obtained from the ATCC (Manassas, VA, USA), human colorectal carcinoma cells (HCT116) and human breast cancer cells (MCF7) were kindly provided by Prof. B. Keppler from University of Vienna (Austria), human pancreatic adenocarcinoma cells (PSN1), human highly invasive breast carcinoma cells (MDA-MB-231), human cervix carcinoma cells (HeLa) and human cells derived from normal lung tissue (MRC5) were purchased from ECACC (Salisbury, UK). RD and PSN1 cells were cultured in RPMI 1640 medium (Biosera, Boussens, France) and the other cells were grown in DMEM medium (high glucose, 4.5 g L−1, PAA, Pasching, Austria); both media were supplemented with gentamycin (50 mg mL−1, Serva, Heidelberg, Germany) and 10% heat-inactivated fetal bovine serum (PAA, Pasching, Austria). Medium for MRC5 cells was further supplemented with 1% nonessential amino acids (Sigma-Aldrich, Prague, Czech Republic). The cells were cultured in a humidified atmosphere (37 °C; 5% CO2) and subcultured twice a week. For in cellulo experiments, the stock solutions of Os complexes were freshly prepared in DMSO and subsequently diluted to the respective cell-culture medium. The final concentration of DMSO in biological experiments did not exceed 0.5% (v/v). For the negative controls, a matched DMSO concentration was used.Cytotoxic/antiproliferative efficiency
The cells were seeded at a proper density – 2 × 103 cells per well (RD, HCT116, PSN1), 3 × 103 cells per well (MCF7, MDA-MB-231, HeLa), and 5 × 103 cells per well (MRC5) in 96-well plates one day before the treatment. The cells were treated with a series of concentrations of the tested compounds in DMEM or RPMI medium. After 72 h of treatment, an MTT assay was performed to determine the inhibition effect of the compounds (0.125 mg mL−1 MTT, 3–4 h; the medium was then removed and replaced with 100 μL DMSO). Absorbance was read at 570 nm (vs. 620 nm). IC50 values refer to compound concentrations corresponding to 50% absorbance (vs. control).Cellular accumulation
Rhabdomyosarcoma cells were seeded in Petri dishes at 2 × 106 cells per dish density and incubated overnight. Cells were treated with 50 μM compounds for 5 h. Following the treatment, the cells were harvested, washed, counted, and pelleted. Cell pellets were lyzed with the microwave acid (HCl) digestion system (CEM Mars®). The metal content was measured with the ICP-MS system.Localization of Os5 in rhabdomyosarcoma cells
RD cells were seeded in confocal dishes (35 mm, MatTek (Ashland, MA, USA)) at 1.5 × 105 cells per dish density and incubated overnight. Os5 or vehicle (DMSO) was added to the indicated concentration, and the cells were visualized with a Leica CM SP5 confocal microscope. Fluorescence of Os5 was obtained using a UV laser (λex/λem = 355/400–485 nm).Colocalization studies were conducted under similar conditions. The cells were prestained with LysoTracker (LysoTracker™ Green DND-26, Invitrogen; 75 nM, 37 °C, 2 h), ER-Tracker (ER-Tracker™ Red, Invitrogen; 1 μM/HBSS, 37 °C, 30 min), or Tetramethylrhodamine ethyl ester (TMRE, Invitrogen; 0.1 μM, 37 °C, 30 min) and then Os5 was added to indicated concentrations and the cells were subjected to confocal microscopy with Leica CM SP5 confocal microscope. Sequential scanning was applied (Os5 – λex/λem = 355/400–485 nm; LysoTracker – λex/λem = 504/511–585 nm; ER-Tracker (λex/λem = 587/615–660 nm); or TMRE – λex/λem = 549/574–620 nm).
Fractionation of rhabdomyosarcoma cells
RD cells were seeded in Petri dishes (2 × 06 cells per dish), incubated overnight, and exposed to the tested compound for 5 h. The cells were then harvested, washed, and pelleted. The pellets were consequently processed with a FractionPREP™ Cell Fractionation kit (BioVision, CA, USA) following the manufacturer′s instructions. The individual fractions were lyzed with HCl, and osmium content was measured using the ICP-MS system.Cell-free determination of Os5 binding to HSA, tRNA, and phospholipid
The experiment was performed in PBS. Os5 (200 μM) was incubated with HSA (1:1), t-RNA (Os:nucleotide ratio = 0.1) and 18:0 DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine) (1 mg mL−1) at 37 °C for 24 h. Os5 (200 μM) sample was used as a control. Control, HSA, and t-RNA samples were then filtered through an Amicon® Ultra centrifugal filter device (3 K). DSPC from the DSPC sample was extracted with chloroform. Os5 content in the filtrates and water phase was determined spectrophotometrically. Os5 bound to HSA, t-RNA, or DSPC was calculated as A327 nm (control) − A327 nm (HSA, t-RNA, or DSPC)/A327 nm (control) × 100.
Loss of mitochondrial membrane potential
Neutral red assay
The cells were cultured and treated in the same way as for the MTT assay mentioned above. The final volume at the treatment was 200 μL per well. Following 24 or 72 h of cell incubation with the tested compounds, 20 μL of Neutral red reagent (Sigma, Prague, Czech Republic) was added, and the cells were incubated for another 3 h. The medium was then removed, the cells were briefly washed with PBS, and 100 μL solubilization solution (1% Acetic acid in 50% Ethanol) was added. The resulting absorbance was read at 540 nm (vs. 690 nm) on an absorbance reader (Tekan Spark, Switzerland).Detection of cell death
:20 dilution) and Propidium iodide (10 μg mL−1). The cells were analyzed on BD FACS Verse Flow Cytometer and evaluated with FCS Express 6 (DeNovo Software, Glendale, CA).
RealTime-Glo™ Annexin V apoptosis and necrosis assay
RD cells were grown in a 96-well plate (Corning, ME, USA) and the next day, EtOH (10%), Staurosporine (2 μM) or Os5 (at indicated concentrations) were added to the wells immediately followed by the addition of the kit (RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay, Promega, Madison, WI, USA) components following the manufacturer's instructions. Luminescence (apoptosis) and Fluorescence (necrosis) were measured in a kinetic mode for cca 1000 min at 37 °C with a Fluorescence reader (Tecan Spark, Switzerland).Activation of caspase-3
Caspase-3 activation was detected with CellEvent® Caspase3/7 Green Detection Reagent (Invitrogen). RD cells were treated with vehicle or Os5 at indicated concentrations for 24 h. STAU – cells were treated with staurosporine (2 μM) for 3 h. Following the staining with the green reagent (10 μM, 37 °C, 30 min), the cells were analyzed with flow cytometry. 2 × 104 cells were analyzed per run; the experiment was performed twice.Detection of ROS
RD cells were seeded on 6-well plates at 2 × 105 cells per well density and incubated for 16 h. The cells were then exposed to the tested compounds for 24 h at given concentrations. 5 μM CellROX®Green reagent was added to the cells, and the cells were incubated for another 30 min at 37 °C. The cells were then harvested, washed in PBS, and analyzed with a flow cytometer (BD FACS Verse). Data were analyzed with FCS Express 6 (DeNovo software, Glendale, CA). 100 μM Menadione was used as a positive control.Cell cycle
RD cells were seeded in 6-well plates (3 × 105 cells per well) and incubated for 24 h. The cells were treated with the tested compounds at given concentrations for 24 h. After harvesting and washing, the cells were pelleted (250 g, 3 min), and the pellets were resuspended in 70% ethanol. The next day, the cells were rinsed twice with PBS and stained with propidium iodide (50 μg mL−1 with 100 μg mL−1 RNase A) in Vindel's solution (10 mM Tris-Cl (pH = 8.0), 10 mM NaCl, 0.1% Triton X-100) for 30 min at 37 °C. Cell cycle profiles were gathered with FACS Verse flow cytometer, and data were analyzed with FCS Express 6 (DeNovo Software, Glendale, CA).Preparation of CSC-enriched rhabdomyosarcoma Cells (RD·CD133+)
CSC marker antibodies with CD133/1-APC (Miltenyi Biotec, Reutlingen, Germany) were used to stain RD CD133-positive cells (10 min, 4 °C). The cells were then washed and coupled with anti-APC microbeads (15 min, 4 °C). Following another washing, the cells were magnetically sorted on an LS column using magnets on a MACS stand. CD133− and CD133+ fractions were consequently eluted.3D rhabdospheres
Sorted RD·CD133− and RD·CD133+ cells were seeded into ultralow attachment U-shape 96-well plates (Corning, NY, USA) at 800 cells per well density. The cells were grown for 4 days in a non-differentiation medium DMEM/F12 Ham (Merck, Sigma-Aldrich, Germany) supplemented with 2% B27 (Invitrogen, Thermofisher Scientific, Inc., MA, USA), fibroblast growth factor (FGF, 10 ng mL−1), epidermal growth factor (EGF, 20 ng mL−1) and 0.4% bovine serum albumin (BSA).Cytotoxic/antiproliferative effect in RD·CD133− and RD·CD133+ spheroids
The spheroids were treated with a series of concentrations of the tested compounds for 72 h. After this period, CellTiter-Glo® Cell Viability Assay (Promega) was performed. The resulting luminescence was measured with a SPARK® microplate reader. IC50 values correspond to a 50% inhibition effect (vs. control).Confocal microscopy of RD·CD133+ spheroids
Representative RD·CD133+ spheroids were transferred to confocal dishes (MatTek) and stained with Calcein AM (Invitrogen, 2 μM) and propidium iodide (PI, Invitrogen, 8 μg mL−1) at 37 °C for 2 h. Hoechst 33342 (Enzo, Farmingdale, NY, USA; 20 μg mL−1) was added to help define the shape of the spheroids. Confocal images were recorded with a Leica SP8 SMD laser scanning confocal microscope (Leica Microsystems, Wetzlar, Germany). Individual pictures were obtained as the maximal projection from 10 z-stacks. The fluorescence intensity of the Calcein AM channel and PI channel was measured with ImageJ software.Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for Os5 and Os3 has been deposited at the CCDC under accession numbers 2393536 and 2393537,† respectively.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research of S. S., H. K., L. M., J. K. and V. B. was supported by the Czech Science Foundation (grant 23-06316S). The research of A. H-G., M. D. S., J. R. was supported by the Spanish Ministerio de Ciencia e Innovación-Agencia Estatal de Investigación (grant PID2021-122850NB-I00) and Fundación Séneca-CARM (project 21989/PI/22). A.H.-G. thanks Fundación Séneca-CARM for a grant (project 21426/FPI/20). The graphical abstract was created with BioRender.com.References
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Cancer statistics, 2022, CA Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- R. Dasgupta, J. Fuchs and D. Rodeberg, Rhabdomyosarcoma, Semin. Pediatr. Surg., 2016, 25, 276–283 CrossRef PubMed.
- J. F. Shern, M. E. Yohe and J. Khan, Pediatric Rhabdomyosarcoma, Crit. Rev. Oncog., 2015, 20, 227–243 CrossRef PubMed.
- A. Mitra, L. Mishra and S. Li, EMT, CTCs and CSCs in tumor relapse and drug-resistance, Oncotarget, 2015, 6, 10697–10711 CrossRef PubMed.
- E. Batlle and H. Clevers, Cancer stem cells revisited, Nat. Med., 2017, 23, 1124–1134 CrossRef CAS PubMed.
- Z. Xiao, A. Johnson, K. Singh and K. Suntharalingam, The discrete breast cancer stem cell mammosphere activity of group 10-bis(azadiphosphine) metal complexes, Angew. Chem., Int. Ed., 2021, 60, 6704–6709 CrossRef CAS PubMed.
- U. Das, S. Shanavas, A. H. Nagendra, B. Kar, N. Roy, S. Vardhan, S. K. Sahoo, D. Panda, B. Bose and P. Paira, Luminescent 11-{naphthalen-1-yl}dipyrido[3,2-a:2′,3′-c]phenazine-based Ru(II)/Ir(III)/Re(I) complexes for HCT-116 colorectal cancer stem cell therapy, ACS Appl. Bio. Mater., 2023, 6, 410–424 CrossRef CAS PubMed.
- P. Kumari, S. Ghosh, S. Acharya, P. Mitra, S. Roy, S. Ghosh, M. Maji, S. Singh and A. Mukherjee, Cytotoxic imidazolyl-mesalazine ester-based Ru(II) complexes reduce expression of stemness genes and induce differentiation of oral squamous cell carcinoma, J. Med. Chem., 2023, 66, 14061–14079 CrossRef CAS PubMed.
- M. R. Chang, D. A. Rusanov, J. Arakelyan, M. Alshehri, A. V. Asaturova, G. S. Kireeva, M. V. Babak and W. H. Ang, Targeting emerging cancer hallmarks by transition metal complexes: Cancer stem cells and tumor microbiome. Part I, Coord. Chem. Rev., 2023, 477, 214923 CrossRef CAS.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Anticancer activity of complexes of the third row transition metals, rhenium, osmium, and iridium, Dalton Trans., 2018, 47, 9934–9974 RSC.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Designing organometallic compounds for catalysis and therapy, Chem. Commun., 2012, 48, 5219–5246 RSC.
- A. Dorcier, W. H. Ang, S. Bolaño, L. Gonsalvi, L. Juillerat-Jeannerat, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, In vitro evaluation of rhodium and osmium RAPTA analogues: The case for organometallic anticancer drugs not based on ruthenium, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- A. F. A. Peacock, A. Habtemariam, S. A. Moggach, A. Prescimone, S. Parsons and P. J. Sadler, Chloro half-sandwich osmium(II) complexes: Influence of chelated N,N-ligands on hydrolysis, guanine binding and cytotoxicity, Inorg. Chem., 2007, 46, 4049–4059 CrossRef CAS PubMed.
- S. H. van Rijt, A. Mukherjee, A. M. Pizarro and P. J. Sadler, Cytotoxicity, Hydrophobicity, uptake, and distribution of osmium(II) anticancer complexes in ovarian cancer cells, J. Med. Chem., 2010, 53, 840–849 CrossRef CAS PubMed.
- W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Highly antiproliferative ruthenium(II) and osmium(II) arene complexes with paullone-derived ligands, Organometallics, 2007, 26, 6643–6652 CrossRef CAS.
- B. Cebrián-Losantos, A. A. Krokhin, I. N. Stepanenko, R. Eichinger, M. A. Jakupec, V. B. Arion and B. K. Keppler, Osmium NAMI-A analogues: synthesis, structural and spectroscopic characterization, and antiproliferative properties, Inorg. Chem., 2007, 46, 5023–5033 CrossRef PubMed.
- K. Suntharalingam, T. C. Johnstone, P. M. Bruno, W. Lin, M. T. Hemann and S. J. Lippard, Bidentate ligands on osmium(VI) nitrido complexes control intracellular targeting and cell death pathways, J. Am. Chem. Soc., 2013, 135, 14060–14063 CrossRef CAS PubMed.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. van Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, Organometallic osmium arene complexes with potent cancer cell cytotoxicity, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS PubMed.
- S. A. Kumar, R. J. Needham, K. Abraham, H. E. Bridgewater, L. A. Garbutt, H. Xandri-Monje, R. Dallmann, S. Perrier, P. J. Sadler and F. Lévi, Dose- and time-dependent tolerability and efficacy of organo-osmium complex FY26 and its tissue pharmacokinetics in hepatocarcinoma-bearing mice, Metallomics, 2020, 13, mfaa003 CrossRef PubMed.
- J. M. Hearn, I. Romero-Canelón, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Potent organo-osmium compound shifts metabolism in epithelial ovarian cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3800–E3805 CrossRef CAS PubMed.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelón, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, In-cell activation of organo-osmium(II) anticancer complexes, Angew. Chem., Int. Ed., 2017, 56, 1017–1020 CrossRef CAS PubMed.
- E. M. Bolitho, J. P. C. Coverdale, H. E. Bridgewater, G. J. Clarkson, P. D. Quinn, C. Sanchez-Cano and P. J. Sadler, Tracking reactions of asymmetric organo-osmium transfer hydrogenation catalysts in cancer cells, Angew. Chem., Int. Ed., 2021, 60, 6462–6472 CrossRef CAS PubMed.
- J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- S. Infante-Tadeo, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Osmium(II) tethered half-sandwich complexes: pH-dependent aqueous speciation and transfer hydrogenation in cells, Chem. Sci., 2021, 12, 9287–9297 RSC.
- Q.-Y. Yang, R. Ma, Y.-Q. Gu, X.-F. Xu, Z.-F. Chen and H. Liang, Arene-ruthenium(II)/osmium(II) complexes potentiate the anticancer efficacy of metformin via glucose metabolism reprogramming, Angew. Chem., Int. Ed., 2022, e202208570 CAS.
- J. Pracharova, V. Novohradsky, H. Kostrhunova, P. Starha, Z. Travnicek, J. Kasparkova and V. Brabec, Half-sandwich Os(II) and Ru(II) bathophenanthroline complexes: anticancer drug candidates with unusual potency and cellular activity profile in highly invasive triple-negative breast cancer cells, Dalton Trans., 2018, 47, 12197–12208 RSC.
- V. Novohradsky, L. Markova, H. Kostrhunova, Z. Trávníček, V. Brabec and J. Kasparkova, An anticancer Os(II) bathophenanthroline complex as a human breast cancer stem cell selective, mammosphere potent agent that kills cells by necroptosis, Sci. Rep., 2019, 9, 13327 CrossRef PubMed.
- E. Ortega, F. J. Ballester, A. Hernández-García, S. Hernández-García, M. A. Guerrero-Rubio, D. Bautista, M. D. Santana, F. Gandía-Herrero and J. Ruiz, Novel organo-osmium(II) proteosynthesis inhibitors active against human ovarian cancer cells reduce gonad tumor growth in Caenorhabditis elegans, Inorg. Chem. Front., 2021, 8, 141–155 RSC.
- F. J. Ballester, E. Ortega, V. Porto, H. Kostrhunova, N. Davila-Ferreira, D. Bautista, V. Brabec, F. Domínguez, M. D. Santana and J. Ruiz, New half-sandwich ruthenium(II) complexes as proteosynthesis inhibitors in cancer cells, Chem. Commun., 2019, 55, 1140–1143 RSC.
- C. R. G. D. Alcazar, V. Xu, C.-c. Lin, M. Gil-Martin, A. Naing, L. Fan, F. Yang, D. Quinn, J. Wu, C. Quadt and J. Mataraza, Abstract CT094: Targeting CSF1R with BLZ945 results in effective peripheral and tumor immune microenvironment modulation in advanced solid tumors and may be associated with limited efficacy in recurrent non-mesenchymal glioblastoma, Cancer Res., 2024, 84, CT094–CT094 CrossRef.
- M. Lari, M. Martínez-Alonso, N. Busto, B. R. Manzano, A. M. Rodríguez, M. I. Acuña, F. Domínguez, J. L. Albasanz, J. M. Leal, G. Espino and B. García, Strong influence of ancillary ligands containing benzothiazole or benzimidazole rings on cytotoxicity and photoactivation of Ru(II) arene complexesc, Inorg. Chem., 2018, 57, 14322–14336 CrossRef CAS PubMed.
- O. A. Lenis-Rojas, R. Cabral, B. Carvalho, S. Friães, C. Roma-Rodrigues, J. A. A. Fernández, S. F. Vila, L. Sanchez, C. S. B. Gomes, A. R. Fernandes and B. Royo, Triazole-based half-sandwich ruthenium(II) compounds: From in vitro antiproliferative potential to in vivo toxicity evaluation, Inorg. Chem., 2021, 60, 8011–8026 CrossRef CAS PubMed.
- P. R. Florindo, D. M. Pereira, P. M. Borralho, P. J. Costa, M. F. M. Piedade, C. M. P. Rodrigues and A. C. Fernandes, New [(η5-C5H5)Ru(N–N)(PPh3)][PF6] compounds: colon anticancer activity and GLUT-mediated cellular uptake of carbohydrate-appended complexes, Dalton Trans., 2016, 45, 11926–11930 RSC.
- F. J. Ballester, E. Ortega-Forte, D. Bautista, M. D. Santana, G. Barone and J. Ruiz, Newly synthesized benzothiazolyl-1,2,3-triazole derivatives: Intramolecular charge transfer tuning, solvatofluorochromism and antiproliferative properties, Dyes Pigm., 2023, 209, 110905 CrossRef CAS.
- F. J. Ballester, A. Hernández-García, M. D. Santana, D. Bautista, P. Ashoo, E. Ortega-Forte, G. Barone and J. Ruiz, Photoactivatable ruthenium complexes containing minimal straining benzothiazolyl-1,2,3-triazole chelators for cancer treatment, Inorg. Chem., 2024, 63, 6202–6216 CrossRef CAS PubMed.
- S. Infante-Tadeo, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Osmium(II) tethered half-sandwich complexes: pH-dependent aqueous speciation and transfer hydrogenation in cells, Chem. Sci., 2021, 12, 9287–9297 RSC.
- Y. Fu, A. Habtemariam, A. M. B. H. Basri, D. Braddick, G. J. Clarkson and P. J. Sadler, Structure–activity relationships for organometallic osmium arene phenylazopyridine complexes with potent anticancer activity, Dalton Trans., 2011, 40, 10553–10562 RSC.
- P. Starha, Z. Travnicek, R. Herchel, P. Jewula and Z. Dvorak, A potential method to improve the in vitro cytotoxicity of half-sandwich Os(II) complexes against A2780 cells, Dalton Trans., 2018, 47, 5714–5724 RSC.
- A. Slodek, D. Zych, A. Maroń, R. Gawecki, A. Mrozek-Wilczkiewicz, K. Malarz and R. Musioł, Phenothiazine derivatives - synthesis, characterization, and theoretical studies with an emphasis on the solvatochromic properties, J. Mol. Liq., 2019, 285, 515–525 CrossRef CAS.
- L. Di, Y. Xing, Z. Yang, C. Qiao and Z. Xia, Photostable aggregation-induced emission of iridium(III) complex realizing robust and high-resolution imaging of latent fingerprints, Sens. Actuators, B, 2023, 375, 132898 CrossRef CAS.
- J. Zhuang, B. Wang, H. Chen, K. Zhang, N. Li, N. Zhao and B. Z. Tang, Efficient NIR-II type-I AIE photosensitizer for mitochondria-targeted photodynamic therapy through synergistic apoptosis–ferroptosis, ACS Nano, 2023, 17, 9110–9125 CrossRef CAS PubMed.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, Tuning the hydrolytic aqueous chemistry of osmium arene complexes with N,O-chelating ligands to achieve cancer cell cytotoxicity, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef CAS PubMed.
- D. A. Belinskaia, P. A. Voronina, V. I. Shmurak, R. O. Jenkins and N. V. Goncharov, Serum albumin in health and disease: Esterase, antioxidant, transporting and signaling properties, Int. J. Mol. Sci., 2021, 22, 10318 CrossRef CAS PubMed.
- H. Y. Liu, Z. H. Xu, X. H. Liu, P. X. Xi and Z. Z. Zeng, Analysis of binding interaction between bovine serum albumin and the cobalt(II) complex with salicylaldehyde-2-phenylquinoline-4-carboylhydrazone, Chem. Pharm. Bull., 2009, 57, 1237–1242 CrossRef CAS PubMed.
- G. Devagi, F. Dallemer, P. Kalaivani and R. Prabhakaran, Organometallic ruthenium(II) complexes containing NS donor Schiff bases: Synthesis, structure, electrochemistry, DNA/BSA binding, DNA cleavage, radical scavenging and antibacterial activities, J. Organomet. Chem., 2018, 854, 1–14 CrossRef CAS.
- P. Kalaivani, R. Prabhakaran, E. Ramachandran, F. Dallemer, G. Paramaguru, R. Renganathan, P. Poornima, V. Vijaya Padma and K. Natarajan, Influence of terminal substitution on structural, DNA, Protein binding, anticancer and antibacterial activities of palladium(II) complexes containing 3-methoxy salicylaldehyde-4(N) substituted thiosemicarbazones, Dalton Trans., 2012, 41, 2486–2499 RSC.
- J. Ruiz, C. Vicente, C. de Haro and D. Bautista, Novel bis-C,N-cyclometalated iridium(III) thiosemicarbazide antitumor complexes: Interactions with human serum albumin and DNA, and inhibition of cathepsin B, Inorg. Chem., 2013, 52, 974–982 CrossRef CAS PubMed.
- H. Zhang, X. Huang and M. Zhang, Spectral diagnostics of the interaction between pyridoxine hydrochloride and bovine serum albumin in vitro, Mol. Biol. Rep., 2008, 35, 699–705 CrossRef CAS PubMed.
- G. Sudlow, D. J. Birkett and D. N. Wade, The characterization of two specific drug binding sites on human serum albumin, Mol. Pharmacol., 1975, 11, 824–832 CAS.
- U. Kragh-Hansen, V. T. G. Chuang and M. Otagiri, Practical aspects of the ligand-binding and enzymatic properties of human serum albumin, Biol. Pharm. Bull., 2002, 25, 695–704 CrossRef CAS PubMed.
- J. Pablo Villamor and A. M. a. L. Zatón, Data plotting of warfarin binding to human serum albumin, J. Biochem. Biophys. Methods, 2001, 48, 33–41 CrossRef CAS PubMed.
- S. Baroni, M. Mattu, A. Vannini, R. Cipollone, S. Aime, P. Ascenzi and M. Fasano, Effect of ibuprofen and warfarin on the allosteric properties of haem-human serum albumin. A spectroscopic study, Eur. J. Biochem., 2001, 268, 6214–6620 CrossRef CAS PubMed.
- D. Egas-Bejar and W. W. Huh, Rhabdomyosarcoma in adolescent and young adult patients: current perspectives, Adolesc. Health, Med. Ther., 2014, 5, 115–125 Search PubMed.
- F. Dela Cruz, Cancer stem cells in pediatric sarcomas, Front. Oncol., 2013, 3, 168 Search PubMed.
- D. S. Moore, W. Notz and M. A. Fligner, The Basic Practice of Statistics 6th Ed, W.H. Freeman and Co., New York, 6th edn, 2013 Search PubMed.
- R. C. Todd and S. J. Lippard, Inhibition of transcription by platinum antitumor compounds, Metallomics, 2009, 1, 280–291 CrossRef CAS PubMed.
- J. Marx, Cancer's perpetual source?, Science, 2007, 317, 1029–1031 CrossRef CAS PubMed.
- P. B. Gupta, C. L. Chaffer and R. A. Weinberg, Cancer stem cells: mirage or reality?, Nat. Med., 2009, 15, 1010 CrossRef CAS PubMed.
- S. P. McDermott and M. S. Wicha, Targeting breast cancer stem cells, Mol. Oncol., 2010, 4, 404–419 CrossRef CAS PubMed.
- H. Zhang, H. Wu, J. Zheng, P. Yu, L. Xu, P. Jiang, J. Gao, H. Wang and Y. Zhang, Transforming growth factor β1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma, Stem Cells, 2013, 31, 433–446 CrossRef PubMed.
- J. Kaiser, The cancer stem cell gamble, Science, 2015, 347, 226–229 CrossRef CAS PubMed.
- M. Hreusova, V. Novohradsky, L. Markova, H. Kostrhunova, I. Potočňák, V. Brabec and J. Kasparkova, Gallium(III) complex with cloxyquin ligands induces ferroptosis in cancer cells and is a potent agent against both differentiated and tumorigenic cancer stem Rhabdomyosarcoma cells, Bioinorg. Chem. Appl., 2022, 2022, 3095749 CrossRef PubMed.
- X.-x. Zheng, J.-j. Chen, Y.-b. Sun, T.-q. Chen, J. Wang and S.-c. Yu, Mitochondria in cancer stem cells: Achilles heel or hard armor, Trends Cell Biol., 2023, 33, 708–727 CrossRef CAS PubMed.
- K. Suntharalingam, W. Lin, T. C. Johnstone, P. M. Bruno, Y.-R. Zheng, M. T. Hemann and S. J. Lippard, A breast cancer stem cell-selective, mammospheres-potent osmium(VI) nitrido complex, J. Am. Chem. Soc., 2014, 136, 14413–14416 CrossRef CAS PubMed.
- K. Laws, G. Bineva-Todd, A. Eskandari, C. Lu, N. O'Reilly and K. Suntharalingam, A Copper(II) Phenanthroline Metallopeptide That Targets and Disrupts Mitochondrial Function in Breast Cancer Stem Cells, Angew. Chem., Int. Ed., 2018, 57, 287–291 CrossRef CAS PubMed.
- K. Laws, A. Eskandari, C. Lu and K. Suntharalingam, Highly charged, cytotoxic, cyclometalated iridium(III) complexes as cancer stem cell mitochondriotropics, Chem. – Eur. J., 2018, 24, 15205–15210 CrossRef CAS PubMed.
- P. A. Carpenter, L. White, G. B. McCowage, V. Nayanar, I. Toogood, P. J. Shaw, L. Lockwood, K. Tiedemann, f. t. A. Group and N. Z. C. s. C. Study, A dose-intensive, cyclophosphamide-based regimen for the treatment of recurrent/Progressive or advanced solid tumors of childhood, Cancer, 1997, 80, 489–496 CrossRef CAS.
- C. A. S. Arndt, J. A. Stoner, D. S. Hawkins, D. A. Rodeberg, A. A. Hayes-Jordan, C. N. Paidas, D. M. Parham, L. A. Teot, M. D. Wharam, J. C. Breneman, S. S. Donaldson, J. R. Anderson and W. H. Meyer, Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk Rhabdomyosarcoma: Children's oncology group study D9803, J. Clin. Oncol., 2009, 27, 5182–5188 CrossRef PubMed.
- B. Thakur and P. Ray, Cisplatin triggers cancer stem cell enrichment in platinum-resistant cells through NF-κB-TNFα-PIK3CA loop, J. Exp. Clin. Cancer Res., 2017, 36, 164 CrossRef PubMed.
- Y. Li, Z. Wang, J. A. Ajani and S. Song, Drug resistance and Cancer stem cells, Cell Commun. Signaling, 2021, 19, 19 CrossRef CAS PubMed.
- P. L. de Sá Junior, D. A. D. Câmara, A. S. Porcacchia, P. M. M. Fonseca, S. D. Jorge, R. P. Araldi and A. K. Ferreira, The roles of ROS in cancer heterogeneity and therapy, Oxid. Med. Cell. Longevity, 2017, 2017, 2467940 CrossRef PubMed.
- J. N. Boodram, I. J. Mcgregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Breast cancer stem cell potent copper(II)–non-steroidal anti-inflammatory drug complexes, Angew. Chem., Int. Ed., 2016, 55, 2845–2850 CrossRef CAS PubMed.
- X. Shi, Y. Zhang, J. Zheng and J. Pan, Reactive oxygen species in cancer stem cells, Antioxid. Redox Signal., 2012, 16, 1215–1228 CrossRef CAS PubMed.
- S. Stahl and H. Werner, A new family of (arene)osmium(0) and -osmium(II) complexes, Organometallics, 1990, 9, 1876–1881 CrossRef CAS.
- Bruker, SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2001 Search PubMed.
- G. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- J. Yellol, S. A. Perez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, Novel C,N-cyclometalated benzimidazole ruthenium(II) and iridium(III) complexes as antitumor and antiangiogenic agents: A structure-activity relationship study, J. Med. Chem., 2015, 58, 7310–7327 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2393536 and 2393537 for Os5 and Os3. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02737j |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |