
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe side-arm-effect controlled stereodivergent polymerization of 1-butene
Guangyu
Zhu†
ab,
Liang
Wang†
ab,
Wenjie
Tao
*abc,
Hongbin
Hou
ab,
Guangqiang
Xu
 abc,
Bo
Wang
ab and
Qinggang
Wang
*abc
abc,
Bo
Wang
ab and
Qinggang
Wang
*abc
aKey Laboratory of Photoelectric Conversion and Utilization of Solar Energy, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao, 266101, China. E-mail: taowj@qibebt.ac.cn; wangqg@qibebt.ac.cn
bShandong Energy Institute, Qingdao, 266101, China
cCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, 100049, China
First published on 1st August 2025
Abstract
The stereocontrolled synthesis of poly(1-butene) has been established using metallocene catalysts in the field of homogeneous catalysis. However, there are few reports on the efficient and controllable polymerization of 1-butene using non-metallocene catalysts, possibly due to the limited strategies for generating stereospecific active species specifically for high-carbon α-olefins. In this study, a series of novel tridentate [O−NX] titanium complexes were designed and used to synthesize poly(1-butene) materials with high activity (up to 3.41 × 106 g mol−1 h−1), ranging from atactic to isotactic (up to 91% mmmm). The physical properties of the obtained poly(1-butene) materials were highly correlated with their isotacticity, and highly isotactic poly(1-butene) possessed good toughness.
Introduction
Isotactic poly(1-butene) is a semi-crystalline polyolefin with exceptional properties, including a high heat distortion temperature, high creep resistance, good impact resistance and superior toughness, and it has been successfully industrialized and applied in food packing and hot-water pipes.1–3 Meanwhile, atactic and moderately isotactic poly(1-butene) have shown potential applications as adhesives and elastomers.4,5 Poly(1-butene) was initially produced by Natta in 1955,6,7 and its controllable synthesis has received more attention since the 1980s thanks to the remarkable development of single-site metallocene catalysts.8–27 In 1985, Kaminsky used ansa-zirconocene 1 to furnish poly(1-butene) with activity of 2.64 × 106 g mol−1 h−1 with a high isotacticity index determined by IR spectroscopy (Chart 1, 1).28 In 2006, Resconi developed a series of ansa-zirconocenes 2,29 which produced poly(1-butene) with activity of over 108 g mol−1 h−1 with isotacticities of 78–96% mmmm (Chart 1, 2). Though catalyst performance is no longer a question regarding the industrialization of metallocene-catalyzed poly(1-butene),30 commercially available products are still rare. Non-metallocene31–38 catalysts can be prepared with diverse ligand skeletons, however, their application in 1-butene polymerization has not concurrently shown good polymerization activity and isotacticity. In 2003, Miyatake utilized thiobis(phenoxy)titanium 3 and water/C6F5OH-modified MMAO to yield poly(1-butene) with good activity (1.26 × 106 g mol−1 h−1) and mediocre isoselectivity (30% mmmm) (Chart 1, 3).39 In 2005, Pellecchia achieved 1-butene polymerization by employing salen zirconium 4, which exhibited low activity (7.40 × 102 g mol−1 h−1) and moderate isoselectivity (60% mmmm) (Chart 1, 4).40 Subsequently, Pellecchia developed the tridentate [O−NN] zirconium complex 5 for the polymerization of 1-butene, and this increased the isotacticity of the resulting polymer to 87% mmmm, albeit with low activity (9.70 × 102 g mol−1 h−1) (Chart 1, 5).41 Mechanistic studies prove that the ligand of the active species becomes dianionic from dehydrochlorination, constructing a covalent bond between sp3-N and the Zr center. Thus, there are still significant challenges in developing non-metallocene catalysts to achieve the efficient, low-cost, and controllable production of poly(1-butene).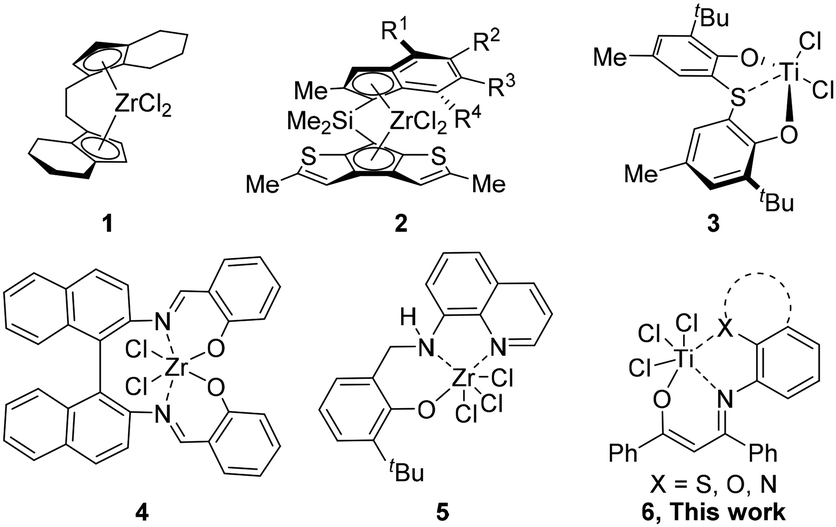 | ||
| Chart 1 Representative single-site catalysts investigated for the synthesis of isotactic poly(1-butene). | ||
To this end, a mechanistic understanding of stereoregularity control when using single-site catalysts8–27,31–38,42 is important. In an ansa-metallocene catalyst system, the C2-symmetric active species achieves isotactic polymerization through the alternation of polymer chain growth between the two identical face-selective sites.43 Meanwhile, the isotactic preference of C1-symmetric species originates from the site epimerization of the polymer chain and the face selectivity of the unique coordination site.44,45 Although these stereochemical principles are also applicable to group-IV non-metallocene catalysts,31–38,46,47 most of them exhibit low activity towards high-carbon α-olefin polymerization due to their congested tetracoordinate environment in C2- or C1-symmetric systems. In contrast, tridentate group-IV complexes, such as (pyridylamido)Hf discovered by Dow and Symyx in 2003, exhibit excellent activity and isoselectivity during α-olefin polymerization.48–57 Coates proposed that the isoselectivity of these Cs-symmetric tridentate hafnium complexes was caused by unexpected in situ monomer insertion into the Hf–Caryl bond, leading to C1-symmetric active species.58 Similarly, a C1-symmetric [(N−NN−)Zr(μ-H)nAliBu2]+ species was proposed to affect stereoselectivity in Pellecchia's tridentate Cs-symmetric[N−NN−] zirconium complexes,59,60 which was also supported by a computational study by Talarico.61 A rare tridentate hafnium catalyst reported by Voskoboynikov in 2021 showcased high isoselectivity with its C2 symmetry formed by the rigid ligand skeleton.62 Notably, most tridentate non-metallocene catalysts involved in the stereoselective polymerization of α-olefins employed dianionic ligands. To the best of our knowledge, the field of α-olefin polymerization remains unexplored in terms of tridentate non-metallocene catalysts with a monoanionic ligand,63–80 particularly regarding their potential for stereoregularity control.
Herein, we report that a tridentate titanium complex family with a [O−NX] ligand can catalyze stereoselective 1-butene polymerization to produce poly(1-butene), from atactic to isotactic (up to 91% mmmm), with high activity (up to 3.41 × 106 g mol−1 h−1) and with different side-arm donors (Chart 1, 6). Compared with previous reports,48–62,83,84 this is the first report on the application of group-IV non-metallocene catalysts with a monoanionic ligand for the stereoselective polymerization of α-olefins.
Results and discussion
Tridentate [O−NX] titanium complexes, originally developed by Tang's group, have shown remarkable applications in the regulation of polyethylene structures and properties by employing different side-arm donors.70–82 We assume that tridentate [O−NX] titanium complexes with more open catalytic space are good candidates to catalyse the polymerization of 1-butene with high activity. In this research, a series of novel tridentate [O−NX] titanium complexes is designed and thoroughly investigated for 1-butene polymerization.Based on reported methods,75L6 ligands were synthesized via the condensation of 1,3-diphenylpropane-1,3-dione and different substituted anilines. L6 ligands were treated with titanium chloride to afford complexes 6a–6e with side arms bearing S, O and N (Scheme 1), which were characterized carefully by 1H NMR, 13C NMR and elemental analysis. Single crystals of complexes 6a, 6c, 6d and 6e were grown by the slow vapor diffusion of n-hexane into their toluene solutions. Single-crystal X-ray diffraction studies confirmed their structures.
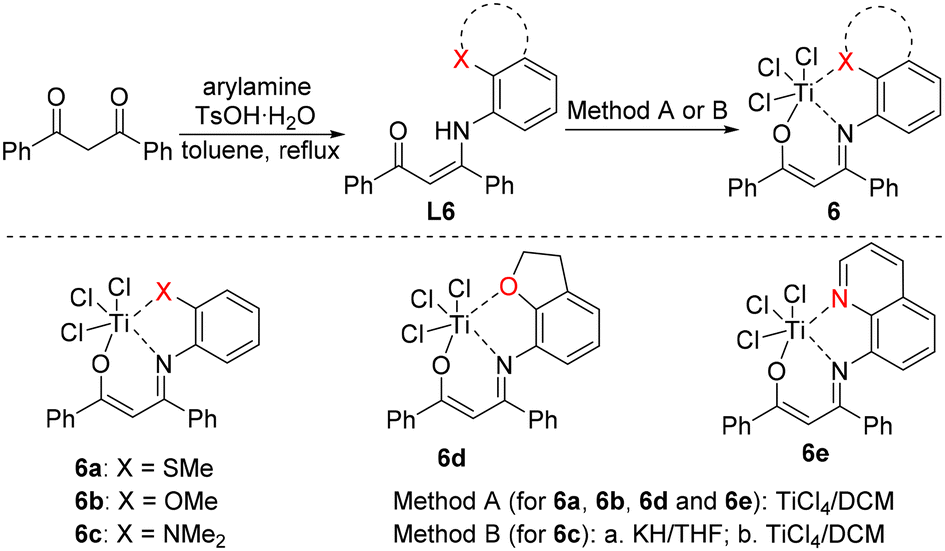 | ||
| Scheme 1 Synthetic pathways to the tridentate [O−NX] titanium complexes 6a–6e. | ||
As shown in Fig. 1,85 complexes 6a, 6c, 6d and 6e feature distorted octahedral coordination at the titanium center, with three chlorine ligands in a mer arrangement. The N, O, Ti, X (side-arm heteroatom) and equatorial Cl are essentially in the same plane. Slight distortion was discovered between an X-containing five-membered ring (N1–C10–C15–S1–Ti1 in 6a) and an O-containing six-membered ring (O1–C1–C2–C3–N1–Ti1 in 6a). Interestingly, the single crystal of 6a is a mixture of enantiomers at the chiral S atom, indicating that 6a is actually C1-symmetric (see SI, crystal data). The bond angle sum of C15–S1–C16 (102.9°), C15–S1–Ti1 (95.0°) and C16–S1–Ti1 (108.6°) is 306.5°, suggesting that the S atom in 6a is sp3-hybridized. The bond angle sum of C22–O2–C21 (104.9°), Ti1–O2–C22 (130.7°) and Ti1–O2–C21 (110.7°) in 6d is nearly 360°, revealing the near-sp2-hybridization of the O2 atom. The different hybridizations of S and O in the side arms are consistent with previous reports.73 Both 6d and 6e are nearly planar, with similar Ti–X bond lengths (2.17 Å vs. 2.16 Å), which may lead to similar catalytic polymerization performance.
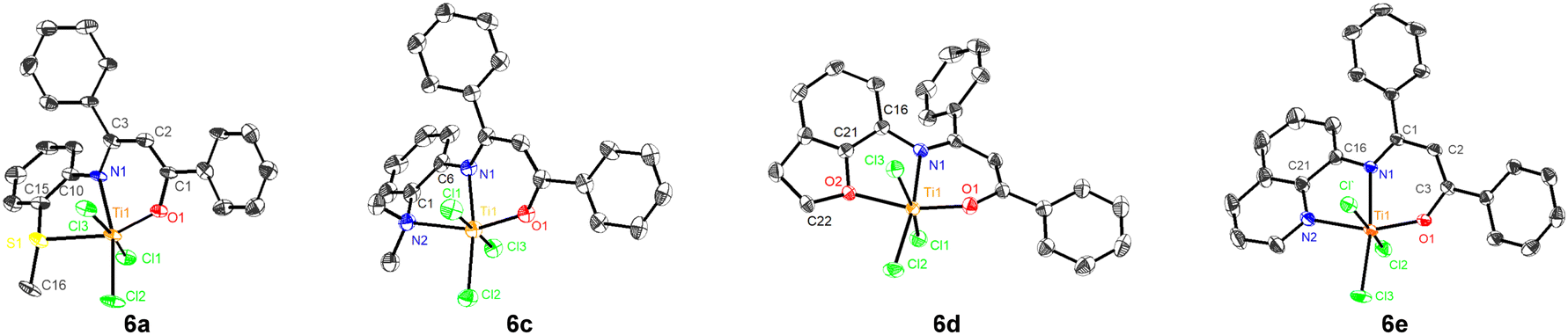 | ||
| Fig. 1 Molecular structures of 6a, 6c, 6d and 6e. Selected bond lengths (Å) and angles (°): 6a: Ti1–O1 1.833(6), Ti1–N1 2.137(7), Ti1–S1 2.595(2); S1–Ti1–N1 75.29(19), S1–Ti1–O1 159.2(2), N1–Ti1–O1 84.9(3); 6c: Ti1–O1 1.852(5), Ti1–N1 2.102(6), Ti1–N2 2.306(6); N2–Ti1–N1 74.3(2), N2–Ti1–O1 157.5(2), N1–Ti1–O1 83.2(2); 6d: Ti1–O1 1.8252(15), Ti1–N1 2.1551(16), Ti1–O2 2.1684(14); O2–Ti1–N1 74.40(6), O2–Ti1–O1 160.87(6), N1–Ti1–O1 84.49(6); 6e: Ti1–O1 1.8262(14), Ti1–N1 2.1713(15), Ti1–N2 2.1616(18); N2–Ti1–N1 75.34(6), N2–Ti1–O1 159.72(6), N1–Ti1–O1 84.50(6). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 30% probability. | ||
Complex 6a with a pendant –SMe group was chosen as the model catalyst to optimize the 1-butene polymerization conditions, inspired by the excellent performance of S-containing tridentate [O−NX] titanium complexes for ethylene polymerization.70–82 Some representative results are summarized in Table 1. When using 500 equivalents of MMAO as the co-catalyst, the activity of 1-butene polymerization was 4.8 × 104 g mol−1 h−1 (Table 1, entry 1), which is much lower than that of reported ethylene polymerization. Increasing the Al/Ti ratio from 500 to 1000 improved the activity obviously (Table 1, entry 2). However, further increasing the Al/Ti ratio to 2000 had a negative effect on activity (Table 1, entry 3). MAO as the cocatalyst resulted in low activity (5.7 × 104 g mol−1 h−1) when the Al/Ti ratio was 1000 (Table 1, entry 4). To our delight, it was found that employing alkyl aluminum and [Ph3C][B(C6F5)4] as co-catalysts resulted in higher activity (Table 1, entries 5–7), and the Al/Ti ratio can be reduced to 50. AlEt3 facilitated complete monomer conversion within 120 min with an activity of 1.25 × 105 g mol−1 h−1 (Table 1, entry 7). By shortening the reaction time (Table 1, entry 8) and increasing the monomer loading (Table 1, entry 9), the activity can reach 7.36 × 105 g mol−1 h−1. Reducing the Al/Ti ratio from 50 to 25 (Table 1, entry 10) resulted in a higher activity (7.52 × 105 g mol−1 h−1) and molecular weight (38 kDa). However, further reducing the Al/Ti ratio to 10 (Table 1, entry 11) resulted in a loss of activity (4.64 × 105 g mol−1 h−1) with a higher molecular weight (163 kDa). Of note, an induction period was needed, since sticky reaction mixtures and an exothermic phenomenon will not be observed until 15 min after the addition of [Ph3C][B(C6F5)4]. In summary, we used model complex 6a and successfully optimized the 1-butene polymerization conditions. Interestingly, we can employ alkyl aluminum (25 eq.) and [Ph3C][B(C6F5)4] (1.0 eq.) as cocatalysts to replace costly MAO or MMAO and produce poly(1-butene) efficiently.
| Entry | Co-cat. | [Al]/[Ti]/[B] | Time (min) | Yield (g) | Act.b |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (kDa) c (kDa) |
Đ |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1-butene (2.4–2.5 g), 6a (0.01 mmol in 2 mL of toluene), co-catalyst, 25 °C, 30–120 min, in a sealed tube, quenched by acidified ethanol at the set time. b Activity is in units of 105 g mol−1 h−1. c Determined via GPC. d 4.8–4.9 g of 1-butene was used. | |||||||
| 1 | MMAO | 500/1/0 | 120 | 0.96 | 0.48 | 11 | 2.4 |
| 2 | MMAO | 1000/1/0 | 120 | 1.63 | 0.82 | 24 | 2.1 |
| 3 | MMAO | 2000/1/0 | 120 | 0.98 | 0.49 | 27 | 2.2 |
| 4 | MAO | 1000/1/0 | 120 | 1.13 | 0.57 | 9 | 3.2 |
| 5 | AlMe3/[Ph3C][B(C6F5)4] | 50/1/1 | 120 | 1.50 | 0.75 | 49 | 2.1 |
| 6 | AliBu3/[Ph3C][B(C6F5)4] | 50/1/1 | 120 | 1.13 | 0.57 | 37 | 2.5 |
| 7 | AlEt3/[Ph3C][B(C6F5)4] | 50/1/1 | 120 | 2.50 | 1.25 | 7 | 2.5 |
| 8 | AlEt3/[Ph3C][B(C6F5)4] | 50/1/1 | 30 | 2.43 | 4.86 | 8 | 2.4 |
| 9d | AlEt3/[Ph3C][B(C6F5)4] | 50/1/1 | 30 | 3.68 | 7.36 | 14 | 2.2 |
| 10 | AlEt 3 /[Ph 3 C][B(C 6 F 5 ) 4 ] | 25/1/1 | 30 | 3.76 | 7.52 | 38 | 2.3 |
| 11d | AlEt3/[Ph3C][B(C6F5)4] | 10/1/1 | 30 | 2.32 | 4.64 | 163 | 2.5 |
With the best polymerization conditions in hand (Table 1, entry 10), 1-butene polymerizations using catalysts 6 with different side-arm donors were further investigated, as shown in Table 2. Atactic poly(1-butene) was obtained by using 6a (Table 2, entry 1) in the form of a viscous oil. Complex 6b bearing an –OMe group gave a slightly lower activity (3.92 × 105 g mol−1 h−1) and moderate isotacticity (44% mmmm) (Table 2, entry 2), producing poly(1-butene) with elasticity. 6c, with an sp3-N-donor pendant group, did not yield observable polymers (Table 2, entries 3 and 4), whether using AlEt3/[Ph3C][B(C6F5)4] or MMAO as cocatalysts, which agreed with previous reports regarding ethylene polymerization.69 We speculate that an increase in poly(1-butene) isotacticity is possibly attributed to smaller steric hindrance from the –OMe group. As a result, we designed complex 6d with an alkyl-constrained dihydrofuran structure. Interestingly, 6d exhibited better isoselectivity (60% mmmm) and higher activity (6.00 × 105 g mol−1 h−1) compared with 6b (Table 2, entry 5 vs. entry 2). Notably, lowering the reaction temperature to −20 °C further increased the isotacticity (80% mmmm) and polymerization activity, with the corresponding product taking the form of hard plastic (Table 2, entry 6). Given that a significant quantity of emitted heat was observed during the polymerization process, we speculate that at a lower temperature, such as −20 °C, the thermal decomposition of active species was avoided so that the polymerization activity was improved (Table 2, entry 6 vs. entry 5). When MMAO was used as a co-catalyst, the isotacticity was improved to 75% mmmm, although the activity (1.50 × 105 g mol−1 h−1) was reduced quite a lot (Table 2, entry 7 vs. entry 5). The performance of 6d confirmed our hypothesis that smaller side-arms lead to better isoselectivity and activity. Next, we turned to modifying sp3-N in 6c to sp2-N. Surprisingly, when –NMe2 was replaced with pyridine to yield the complex 6e, there was a remarkable increase in polymerization activity (2.60 × 106 g mol−1 h−1) and isotacticity (82% mmmm) (Table 2, entry 8 vs. entry 5). Boiling of the reaction mixture within a few seconds of the addition of [Ph3C][B(C6F5)4] was observed. Consequently, the magnetic stirrer stopped stirring after 5 min due to the increased viscosity of the reaction solution. The poor stirring and high reaction viscosity of the mixtures may have led to the relatively wide PDI values (Table 2, entry 8: PDI = 2.8; entry 9: PDI = 3.2). Encouraged by the above findings, we tried 1-butene polymerization using catalyst 6e at −20 °C and produced poly(1-butene) with increased activity (3.41 × 106 g mol−1 h−1) and isotacticity (85% mmmm) (Table 2, entry 9). Of note, isotacticity can be further improved to 91% mmmm by using MMAO as the co-catalyst at room temperature, with reduced activity (1.82 × 105 g mol−1 h−1) (Table 2, entry 10). To the best of our knowledge, the above experiment results represent the highest level of activity and isoselectivity obtained in non-metallocene-catalyzed 1-butene polymerization. 6e avoids the use of expensive MAO and has a simple synthesis process; however, it still shows unsatisfactory activity and isoselectivity in 1-butene polymerization compared with metallocene catalysts.29 More structural modifications of 6e to enhance its practicality are currently underway in our lab.
| Entry | Side-arm (cat.) | Time (min) | Yield (g) | Act.b |
M
nc (kDa) |
Đ | mmmm |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 1-butene (4.8–4.9 g), 6 (0.01 mmol in 2 mL of toluene), AlEt3 (0.25 mmol, 1 M in hexane), [Ph3C][B(C6F5)4] (0.01 mmol in 2 mL of toluene), 25 °C, 5–30 min, in a sealed tube, quenched by acidified ethanol at the set time. b Activity is in units of 105 g mol−1 h−1. c Determined via GPC. d Determined via quantitative 13C NMR spectrum analysis. e 10 mmol of MMAO was used instead of AlEt3/[Ph3C][B(C6F5)4]. f The reaction is run at −20 °C. | |||||||
| 1 | SMe (6a) | 30 | 3.76 | 7.52 | 38 | 2.3 | <10 |
| 2 | OMe (6b) | 30 | 1.96 | 3.92 | 57 | 2.0 | 44 |
| 3 | NMe2 (6c) | 120 | n.r. | n.p. | — | — | — |
| 4e | NMe2 (6c) | 120 | n.r. | n.p. | — | — | — |
| 5 | Dihydrofuran (6d) | 30 | 3.00 | 6.00 | 41 | 2.4 | 60 |
| 6f | Dihydrofuran (6d) | 30 | 3.28 | 6.56 | 129 | 2.1 | 80 |
| 7e | Dihydrofuran (6d) | 30 | 0.75 | 1.50 | 38 | 1.9 | 75 |
| 8 | Pyridine (6e) | 5 | 2.17 | 26.0 | 41 | 2.8 | 82 |
| 9f | Pyridine (6e) | 5 | 2.84 | 34.1 | 52 | 3.2 | 85 |
| 10e | Pyridine (6e) | 30 | 0.91 | 1.82 | 42 | 2.1 | 91 |
The quantitative 13C NMR spectra of corresponding poly(1-butene) samples from Table 2 are shown in Fig. 2, ranging from isotactic to atactic. The rrrr pentad cannot be observed, implying that the resulting polymers are not hemi-isotactic. This suggests that consecutive migratory insertions between the axial site and the equatorial site are not feasible. On the other hand, mmmr, mmrr, and mrrm pentad peaks retain clear signals with increased tacticity, suggesting the prompt correction of stereoirregularity via an ESC mechanism19 and that site epimerization is likely operative. In addition, regioerror and chain-end peaks were not observed in any of the polymers.
 | ||
| Fig. 2 Quantitative 13C NMR (298 K, CDCl3) spectra analysis of the methylene carbon in the side chains of the obtained poly(1-butene) samples. | ||
To correlate poly(1-butene) isotacticities with physical properties, the thermal and mechanical properties of representative polymers were investigated. To minimize polymer molecular-weight effects on the physical properties, the samples fall within a comparable molecular weight range of 38–57 kDa (Table 2, entries 1, 2, 5, 8 and 10). Interestingly, good correlation was discovered between the glass-transition temperature (Tg) and the isotacticity, i.e., the higher the isotacticity, the lower the Tg value (varying from −15.5 to −32.1 °C, Fig. 3a, a–e), which suggested that the highly isotactic poly(1-butene) may have applications in a wide temperature range. This trend may because the atactic amorphous structure reduced the mobility of the chain end, slowing down the transition kinetics. No Tm was found for low-isotacticity poly(1-butene) (Fig. 3b, a and b), indicating the low crystallinity of these polymers. More than one Tm peak was observed in moderately to highly isotactic poly(1-butene). The highest Tm values were positively correlated with the isotacticity of the corresponding polymers (varying from 71.8 to 81.8 °C, Fig. 3b, c–e). Moreover, an exothermic peak was discovered for 91% mmmm poly(1-butene) (Fig. 3b, e) at 23.9 °C with heating, indicating the occurrence of cold crystallization. We believe that highly isotactic poly(1-butene) does not crystallize during a direct cooling process; however, cold crystallization occurs during subsequent heating.
 | ||
| Fig. 3 Characterization of obtained poly(1-butene) samples. (a) DSC thermograms from the first heating cycle showing Tg. (b) DSC thermograms from the second heating cycle showing Tc and Tm. (c) Wide-angle X-ray diffraction (WAXD) profiles. (d) Stress–strain curves measured at a speed of 50 mm min−1 for all specimens after aging for 24 h. | ||
Fig. 3c shows 1D WAXD profiles of the above five examples aged at room temperature for 30 days. For low-isotacticity poly(1-butene) (Fig. 3c, a and b), crystalline peaks cannot be observed clearly. The low-crystalline nature of these polymers was also supported by DSC analysis (Fig. 3b, a and b). As expected, when the isotacticity of poly(1-butene) is increased (Fig. 3c, c–e), the crystallization peaks of the corresponding poly(1-butene) samples become clear and sharp in the WAXD spectra, which indicated that the thermal properties were greatly affected by the isotacticity of the materials. In addition, the distinct diffraction peaks observed at 2θ values of 10.1°, 17.5°, and 20.2° corresponded to the (110), (300), and (220) + (211) crystallographic planes of hexagonal86 form I, while peaks of other crystal forms are invisible, suggesting that a complete phase transition from tetragonal form II to hexagonal form I has been achieved.
Subsequently, we attempted to investigate the mechanical characteristics of poly(1-butene) with different isotacticities through tensile tests. It is noteworthy that only poly(1-butene) materials with medium to high isotacticity (Fig. 3d, c–e) could be successfully fashioned into tensile-test specimens, while sample a and b failed due to their excessive viscosity. 91% mmmm poly(1-butene) (Fig. 3d, e) showed the highest yield strength (σY = 9.5 MPa) but the lowest elongation at break (εB = 513%). Sample d with 82% mmmm isotacticity (Fig. 3d, d) was softer, with a yield strength of σY = 8.3 MPa and an elongation at break of εB = 837%. Sample c with 60% mmmm (Fig. 3d, c) showed a marked decrease in yield strength (σY = 2.9 MPa) accompanied by the highest elongation at break value (εB = 946%). Compared to commercial LDPE (Fig. 3d, f; σY = 12.1 MPa, εB = 703%), poly(1-butene) with 82% mmmm isotacticity possesses much higher tensile strength (26.0 MPa vs. 15.1 MPa) and a larger area under the stress–strain curve, indicating its good toughness and potential applications as a plastic film and packaging material.
To rationalize the remarkable effects of side-arm donors on stereocontrol, we first studied the steric hindrance effects and generated steric maps of complexes 6a, 6b and 6e and their possible active species Et-6a, Et-6b and Et-6e using the SambVca 2.0 program87 (Fig. 4). The structures of complexes were optimized via DFT computation at the level of the B3LYP functional with D3 dispersion and the 6-31G(d, p) basis set. The buried volume (%Vbur) was used to measure the steric environment of the side-arm donors around the Ti active site. The order of %Vbur was 6a (43.1%) > 6b (41.8%) > 6e (41.5%), which is opposite to the isotacticities of the corresponding polymers. The –SMe group in 6a caused obvious steric congestion at the north poles. Considering that atactic poly(1-butene) is generated using 6a, while an isotactic polymer is preferentially produced by 6e, it can be preliminarily inferred that the steric hindrance of side-arm donors may be a negative factor affecting isoselectivity. The order of %Vbur for possible active species was Et-6a (58.8%) > Et-6b (56.9%) > Et-6e (56.7%), which is consistent with the order of the catalyst precursors.
 | ||
| Fig. 4 A comparison of steric hindrance. Steric maps of 6a, 6b and 6e, and their corresponding active species Et-6a, Et-6b and Et-6e. The titanium center is placed at the center of the xyz coordinate system. The Ti–N bond coincides with the z-axis. The y-axis represents the axial position of the xz-plane containing the titanium center. The chloride atoms are omitted in 6a, 6b and 6e, and the ethyl groups are retained in Et-6a, Et-6b and Et-6e. | ||
Conclusions
In summary, an elegant system to generate isospecific C1-symmetric active species from tridentate [O−NX] titanium complexes has been discovered, leading to the efficient synthesis of a range of poly(1-butene) samples, from atactic to isotactic (up to 91% mmmm), with good polymerization activity (up to 3.41 × 106 g mol−1 h−1). To the best of our knowledge, no non-metallocene titanium complex catalysts have demonstrated such high activity and isoselectivity in catalyzing the polymerization of α-olefins. Interestingly, isoselectivity can be effectively regulated by modifying one functional group or even one heteroatom of the ligands in this system. In addition, the material properties of poly(1-butene) samples with different isotacticities have been preliminarily investigated, indicating the much better toughness of isotactic poly(1-butene) compared to commercial LDPE. These newly synthesized tridentate [O−NX] titanium complexes bearing different side-arm heteroatoms showed slightly C1-symmetric structures in steric maps and single-crystal diffraction analysis. However, this is not sufficient to explain their differential isotactic selectivities. 13C NMR microstructural analysis of poly(1-butene) suggested that the isospecificity results from a mechanism of steric control based on enantiomorphic sites. Due to the fact that these trichloride precursors will grow two polymer chains after activation,88 explaining their polymerization behavior on the basis of generally accepted symmetry rules42 is complicated at present. Experimental and theoretical investigations are currently in progress in order to address the origin of the isoselectivity of these tridentate [O−NX] titanium complexes in our lab.Author contributions
Conceptualization: Wenjie Tao, Qinggang Wang; experimentation: Guangyu Zhu, Liang Wang, Bo Wang; supervision: Wenjie Tao, Hongbin Hou, Guangqiang Xu, Qinggang Wang; writing: Guangyu Zhu, Wenjie Tao.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the SI.Supplementary information is available and includes full synthetic sequences, experimental procedures, characterization data and NMR spectra. See DOI: https://doi.org/10.1039/d5py00583c.
CCDC 2412497, 2412499, 2412500 and 2412502 contains the supplementary crystallographic data for this paper.85a–d
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22471282, W. T.), Major Science and Technology Innovation Program of Shandong Province (2022CXGC020604, Q. W.), Taishan Scholars Program of Shandong Province (tstp20240520, Q. W.), Natural Science Foundation of Shandong Province (ZR2023ME008 for G. X., and ZR2023QB156 for G. Z), and the Scientific Research and Innovation Fund Project of Shandong Energy Research Institute (SEI I202004, G. X.). We also thank Dr Yanshan Gao for helpful discussion and suggestions.References
- Y. Men, J. Rieger and J. Homeyer, Macromolecules, 2004, 37, 9481–9488 CrossRef CAS.
- U. W. Gedde, J. Viebke, H. Leijstrom and M. Ifwarson, Polym. Eng. Sci., 1994, 34, 1773–1787 CrossRef CAS.
- L. Luciani, J. Seppälä and B. Löfgren, Prog. Polym. Sci., 1988, 13, 37–62 CrossRef CAS.
- Q. Huang, Q. Wu, F. Zhu and S. Lin, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4068–4073 CrossRef.
- S. Abedi and N. Sharifi-Sanjani, J. Appl. Polym. Sci., 2000, 78, 2533–2539 CrossRef.
- G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708–1710 CrossRef.
- G. Natta, J. Polym. Sci., 1955, 16, 143–154 CrossRef.
- Z. Chen, Y. Mao, Y. Cao, S. Liang, S. Song, C. Ni, Z. Liu, X. Ye, A. Shen and H. Zhu, Chin. J. Org. Chem., 2018, 38, 2937–2992 CrossRef.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef.
- B. Wang, Coord. Chem. Rev., 2006, 250, 242–258 CrossRef.
- C. Cobzaru, S. Hild, A. Boger, C. Troll and B. Rieger, Coord. Chem. Rev., 2006, 250, 189–211 CrossRef.
- F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170–188 CrossRef CAS.
- A. Razavi and U. Thewalt, Coord. Chem. Rev., 2006, 250, 155–169 CrossRef CAS.
- S. Prashar, A. Antiñolo and A. Otero, Coord. Chem. Rev., 2006, 250, 133–154 CrossRef CAS.
- J.-Y. Dong and Y. Hu, Coord. Chem. Rev., 2006, 250, 47–65 CrossRef CAS.
- P. C. Möhring and N. J. Coville, Coord. Chem. Rev., 2006, 250, 18–35 CrossRef.
- H. Braunschweig and F. M. Breitling, Coord. Chem. Rev., 2006, 250, 2691–2720 CrossRef CAS.
- S. Lin and R. M. Waymouth, Acc. Chem. Res., 2002, 35, 765–773 CrossRef CAS PubMed.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- G. Fink, B. Steinmetz, J. Zechlin, C. Przybyla and B. Tesche, Chem. Rev., 2000, 100, 1377–1390 CrossRef CAS.
- G. G. Hlatky, Chem. Rev., 2000, 100, 1347–1376 CrossRef CAS.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1346 CrossRef CAS PubMed.
- G. W. Coates, Chem. Rev., 2000, 100, 1223–1252 CrossRef CAS.
- H. G. Alt and A. Köppl, Chem. Rev., 2000, 100, 1205–1222 CrossRef CAS.
- A. L. McKnight and R. M. Waymouth, Chem. Rev., 1998, 98, 2587–2598 CrossRef CAS.
- W. Kaminsky, Macromol. Chem. Phys., 1996, 197, 3907–3945 CrossRef CAS.
- G. M. Diamond, R. F. Jordan and J. Petersen, J. Am. Chem. Soc., 1996, 118, 8024–8033 CrossRef CAS.
- W. Kaminsky, K. Külper, H. H. Brintzinger and F. R. W. P. Wild, Angew. Chem., Int. Ed. Engl., 1985, 24, 507–508 CrossRef.
- L. Resconi, I. Camurati and F. Malizia, Macromol. Chem. Phys., 2006, 207, 2257–2279 CrossRef CAS.
- C. De Rosa, F. Auriemma and L. Resconi, Angew. Chem., Int. Ed., 2009, 48, 9871–9874 CrossRef CAS PubMed.
- Z. Wen, C. Wu, J. Chen, S. Qu, X. Li and W. Wang, Polymers, 2024, 16, 406–424 CrossRef CAS PubMed.
- C. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed.
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722–9244 CrossRef CAS PubMed.
- C. Redshaw and Y. Tang, Chem. Soc. Rev., 2012, 41, 4484–4510 RSC.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS.
- M. Mitani, J. Saito, S. Ishii, Y. Nakayama, H. Makio, N. Matsukawa, S. Matsui, J. Mohri, R. Furuyama, H. Terao, H. Bando, H. Tanaka and T. Fujita, Chem. Rec., 2004, 4, 137–158 CrossRef CAS.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–315 CrossRef CAS PubMed.
- H. Kawamura-Kuribayashi and T. Miyatake, J. Organomet. Chem., 2003, 674, 73–85 CrossRef CAS.
- M. Lamberti, M. Consolmagno, M. Mazzeo and C. Pellecchia, Macromol. Rapid. Commun., 2005, 26, 1866–1871 CrossRef CAS.
- M. Lamberti, M. Bortoluzzi, G. Paolucci and C. Pellecchia, J. Mol. Catal. A: Chem., 2011, 351, 112–119 CrossRef.
- J. A. Ewen, J. Mol. Catal. A: Chem., 1998, 128, 103–109 CrossRef.
- G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217–219 CrossRef PubMed.
- W. Kaminsky, O. Rabe, A.-M. Schauwienold, G. U. Schupfner, J. Hanss and J. Kopf, J. Organomet. Chem., 1995, 497, 181–193 CrossRef.
- J. A. Ewen, Macromol. Symp., 1995, 89, 181–196 CrossRef.
- K. Press, A. Cohen, I. Goldberg, V. Venditto, M. Mazzeo and M. Kol, Angew. Chem., Int. Ed., 2011, 50, 3529–3532 CrossRef.
- A. Cohen, J. Kopilov, I. Goldberg and M. Kol, Organometallics, 2009, 28, 1391–1405 CrossRef.
- Y. Gong, L. Jiang, J. Zhang, S. Liu, P. Braunstein and Z. Li, ACS Catal., 2025, 15, 8442–8453 CrossRef.
- S. Kanesato, K. Yasoshima, K. Matsumoto, N. Misawa, Y. Suzuki, N. Koga and M. Nagaoka, J. Phys. Chem. B, 2024, 128, 6178–6188 CrossRef CAS PubMed.
- Y. Kang, H. Wang, X. Li, F. Meng, H. Liu, Y. Xiao, Z. Jiang, H. Gao, C. Liu, F. Wang, Li Pan and Y. Li, Macromolecules, 2024, 57, 4208–4219 CrossRef CAS.
- Y. Xing, S. Liu and Z. Li, Polym. Chem., 2024, 15, 4141–4150 RSC.
- J. M. Eagan, J. Xu, R. Di Girolamo, C. M. Thurber, C. W. Macosko, A. M. LaPointe, F. S. Bates and G. W. Coates, Science, 2017, 355, 814–816 CrossRef CAS PubMed.
- K. A. Frazier, R. D. Froese, Y. He, J. Klosin, C. N. Theriault, P. C. Vosejpka, Z. Zhou and K. A. Abboud, Organometallics, 2011, 30, 3318–3329 CrossRef CAS.
- P. S. Chum and K. W. Swogger, Prog. Polym. Sci., 2008, 33, 797–819 CrossRef CAS.
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714–719 CrossRef CAS PubMed.
- T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. K. Leclerc, V. Murphy, J. A. W. Shoemaker, H. Turner, R. K. Rosen, J. C. Stevens, F. Alfano, V. Busico, R. Cipullo and G. Talarico, Angew. Chem., Int. Ed., 2006, 45, 3278–3283 CrossRef CAS.
- T. R. Boussie, G. M. Diamond, C. Goh, K. A. Hall, A. M. LaPointe, M. Leclerc, C. Lund, V. Murphy, J. A. W. Shoemaker, U. Tracht, H. Turner, J. Zhang, T. Uno, R. K. Rosen and J. C. Stevens, J. Am. Chem. Soc., 2003, 125, 4306–4317 CrossRef CAS PubMed.
- G. J. Domski, E. B. Lobkovsky and G. W. Coates, Macromolecules, 2007, 40, 3510–3513 CrossRef CAS.
- L. Annunziata, D. Pappalardo, C. Tedesco and C. Pellecchia, Macromolecules, 2009, 42, 5572–5578 CrossRef CAS.
- G. Li, C. Zuccaccia, C. Tedesco, I. D'Auria, A. Macchioni and C. Pellecchia, Chem. – Eur. J., 2014, 20, 232–244 CrossRef CAS.
- C. De Rosa, R. Di Girolamo, A. B. Muñoz-García, M. Pavone and G. Talarico, Macromolecules, 2020, 53, 2959–2964 CrossRef CAS.
- G. P. Goryunov, M. I. Sharikov, A. N. Iashin, J. A. M. Canich, S. J. Mattler, J. R. Hagadorn, D. V. Uborsky and A. Z. Voskoboynikov, ACS Catal., 2021, 11, 8079–8086 CrossRef.
- J. Tian, Z. Gao, Y. Liu, P. Braunstein, S. Liu and Z. Li, Inorg. Chem. Front., 2024, 11, 613–623 RSC.
- Y. Chen, S. Zhou, W. Yang and S. Liu, Organometallics, 2022, 41, 3724–3731 CrossRef CAS.
- A. C. Pinheiro, S. M. da Silva, T. Roisnel, E. Kirilov, J. F. Carpentier and O. L. Casagrande, New J. Chem., 2018, 42, 1477–1483 RSC.
- L. A. Wright, E. G. Hope, G. A. Solan, W. B. Cross and K. Singh, Organometallics, 2016, 35, 1183–1191 CrossRef CAS.
- R. Zhao, T. Liu, L. Wang and H. Ma, Dalton Trans., 2014, 43, 12663–12677 RSC.
- Y. Wang, W. Zhang, W. Huang, L. Wang, C. Redshaw and W.-H. Sun, Polymer, 2011, 52, 3732–3737 CrossRef CAS.
- W. Huang, W. Zhang, S. Liu, T. Liang and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1887–1894 CrossRef CAS.
- M. Xue, L. Lei, S. Ren, T. Li, Q. You and G. Xie, Polymer, 2023, 277, 125995 CrossRef CAS.
- L. Ji, J.-S. Liu, X.-Y. Wang, J.-F. Li, Z. Chen, S. Liao, X.-L. Sun and Y. Tang, Polym. Chem., 2019, 10, 3604–3609 RSC.
- C. Xu, Z. Chen, Q. Shen, X.-L. Sun and Y. Tang, J. Organomet. Chem., 2014, 761, 142–146 CrossRef CAS.
- Z. Wang, A.-Q. Peng, X.-L. Sun and Y. Tang, Sci. China: Chem., 2014, 57, 1144–1149 CrossRef CAS.
- D.-W. Wan, Z. Chen, Y.-S. Gao, Q. Shen, X.-L. Sun and Y. Tang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2495–2503 CrossRef CAS.
- Z. Chen, J.-F. Li, W.-J. Tao, X.-L. Sun, X.-H. Yang and Y. Tang, Macromolecules, 2013, 46, 2870–2875 CrossRef CAS.
- P. Tao, X.-Y. Tang, B.-X. Li, J.-Y. Liu and Y.-S. Li, Dalton Trans., 2012, 41, 7390–7398 RSC.
- X. Wang, M.-M. Sit, J. Sun, Y. Tang and Z. Xie, Acta Chim. Sin., 2012, 70, 1909–1916 CrossRef.
- X.-H. Yang, Z. Wang, X.-L. Sun and Y. Tang, Dalton Trans., 2009, 8945–8954 RSC.
- X.-H. Yang, C.-R. Liu, C. Wang, X.-L. Sun, Y.-H. Guo, X.-K. Wang, Z. Wang, Z. Xie and Y. Tang, Angew. Chem., Int. Ed., 2009, 48, 8099–8102 CrossRef PubMed.
- X.-H. Yang, X.-L. Sun, F.-B. Han, B. Liu, Y. Tang, Z. Wang, M.-L. Gao, Z.-W. Xie and S.-Z. Bu, Organometallics, 2008, 27, 4618–4624 CrossRef.
- C. Wang, Z. Ma, X.-L. Sun, Y. Gao, Y.-H. Guo, Y. Tang and L.-P. Shi, Organometallics, 2006, 25, 3259–3266 CrossRef.
- W.-Q. Hu, X.-L. Sun, C. Wang, Y. Gao, Y. Tang, L.-P. Shi, W. Xia, J. Sun, H.-L. Dai, X.-Q. Li, X.-L. Yao and X.-R. Wang, Organometallics, 2004, 23, 1684–1688 CrossRef.
- A. Cicolella, E. Romano, V. Barone, C. De Rosa and G. Talarico, Organometallics, 2022, 41, 3872–3883 CrossRef.
- C. De Rosa, R. Di Girolamo and G. Talarico, ACS Catal., 2016, 6, 3767–3770 CrossRef.
- (a) ; CCDC 2412497 (for 6a): Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2lzdhh; (b) CCDC 2412499 (for 6c): Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2lzdkk; (c) CCDC 2412500 (for 6d): Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2lzdll; (d) CCDC 2412502 (for 6e): Experimental Crystal Structure Determination, DOI:10.5517/ccdc.csd.cc2lzdnn.
- A. C. K. Tashiro, J. Hu, H. Wang, M. Hanesaka and A. Saiani, Macromolecules, 2016, 49, 1392–1404 CrossRef.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef PubMed.
- Y. Suzuki, S. Kinoshita, A. Shibahara, S. Ishii, K. Kawamura, Y. Inoue and T. Fujita, Organometallics, 2010, 29, 2394–2396 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |