DOI:
10.1039/D5PY00401B
(Paper)
Polym. Chem., 2025,
16, 4590-4598
Designing catechol-poly(ethylene terephthalate) as a depolymerizable adhesive for PET packaging
Received
23rd April 2025
, Accepted 2nd September 2025
First published on 9th September 2025
Abstract
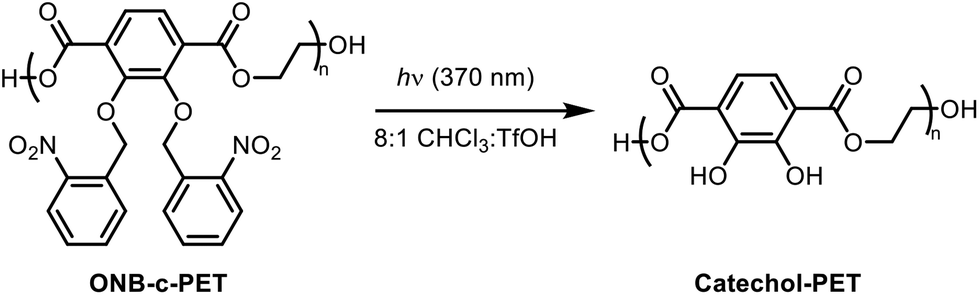
2,3-Dihydroxyterephthalate, a catechol-functionalized derivative of the conventional poly(ethylene terephthalate) monomer, was converted into a reactive monomer that polymerized into polyesters that exhibit adhesive properties. The monomer synthesis required two steps and proceeded with an overall yield of 95%, incorporating photoprotecting groups, ortho-nitrobenzyl (ONB), on the catechol to prevent side reactions. Polymerization with ethylene glycol produced a catechol-protected material, poly(ONB-2,3-dihydroxyterephthalate) (ONB-c-PET), and by varying the polymerization conditions, molecular weights ranged from 3000 to 8000 g mol−1 with dispersity values ranging from 1.13 to 1.72. These polymers were deprotected using a UV lamp (370 nm), producing poly(2,3-dihydroxyterephthalate), c-PET. The glass transition temperatures (Tg) measured for ONB-c-PET and c-PET were 34 °C and 56 °C, respectively. Adhesion lap-shear testing of c-PET (ASTM D1002) showed adhesion strengths of 0.77 ± 0.09 MPa and 0.38 ± 0.11 MPa on a PET substrate. Degradation studies showed complete conversion to monomers via methanolysis over 16 hours. The resulting monomers, 2,3-dihydroxyterephthalic acid and 2,3-dihydroxyterephthalate, can be separated from ethylene terephthalate due to their water solubility. These results highlight the potential of this adhesive to facilitate the recycling of plastic materials contaminated with residual adhesives.
Introduction
Poly(ethylene terephthalate) (PET) is an aromatic polyester polymer widely used in beverage bottles and textiles and is labeled as #1 for U.S. recycling facilities. PET is the third most produced plastic material, behind polyethylene (PE) and polypropylene (PP).1 PET is often produced from the polycondensation reaction of dimethyl terephthalate (DMT) with ethylene glycol (EG) in the presence of a metal catalyst. Valued for its thermal and mechanical properties, its industrial uses are extensive, including in bottling and textiles. Additionally, PET has thermal stability up to 80 °C without losing any mechanical properties, making it an ideal material for electronic components, i.e., computer microchips.2 The good mechanical properties of PET allow it to hold in heat and serve as a gas barrier against other chemicals, such as oxygen and carbon dioxide. While the advantages of PET are numerous, end-of-life PET presents a global challenge. Research on organocatalysts,3–5 enzymatic degradation,6,7 and chemical recycling8,9 has made great advances in end-of-life processing. However, additional challenges, especially within industrial processes, must also be addressed.
The production of PET is roughly 60 million tons per year, of which only 30% is recycled, and the rest ends up in the environment, both on land and in the sea.1 It is estimated that roughly 1.5 billion plastic bottles are discarded daily in nature, totaling more than 500 billion plastic bottles per year.10 While PET bottles are recyclable via glycolysis,11 methanolysis,12 hydrolysis,13 or aminolysis,14 sorting and separating PET bottles from other mixed plastics is a practical challenge at an industrial scale.15 For example, Eastman Chemicals®, located in Kingsport, Tennessee, has opened the largest PET recycling plant in the United States to recycle PET bottles via methanolysis.16 Once at a plant, the bottles go through a complex process to remove labels, adhesives, dirt, and food residues.17 The Association of Plastic Recyclers’ (APR) critical guidance testing for PET has determined that haze- and color-impacted PET cannot be recycled into clear plastic bottles again.18 The discoloration comes from the contaminants, including adhesives. According to the APR, using labels and adhesives designed for compatibility with PET recycling is one of the most important steps package designers can take to improve the recycling process.19 Specifically, wash-off capabilities to cleanly remove adhesives need to be developed for each type of plastic.
Adhesives have become integral to society through their numerous uses, such as food storage, acrylic in drywall constructions, and formaldehyde in concrete.20 Moreover, adhesion is not solely a human innovation; nature also relies on adhesive mechanisms. These examples include barnacles and mussels on boats and rocks, human cell adhesion, and ice on roads.21 One widely studied and mimicked motif is catechol (1,2-dihydroxybenzene). This natural motif is found in mussels’ protein chains. This catechol motif allows mussels to form a sticky adhesive to wet surfaces such as rocks, even under extreme weather conditions.22 In addition, these catechol motifs also have biological significance, e.g., Parkinson's disease, due to the presence of the amino acid L-3,4-dihydroxyphenylalanine (L-DOPA).23 Mimetic materials have been well explored in a large number of applications, including adhesives,24 coatings,25 elastomers,26 hydrogels,27 and other materials.28,29 Focusing on thermoplastic catechol polymer adhesives, many additive properties have been reported such as high strength underwater,30 applications in specialty adhesives,31 and enhancement to common adhesives such as polysiloxane.32 Catechol-based adhesives exemplify how nature-inspired designs can drive innovation across diverse material applications, including in nature-inspired adhesives.33,34
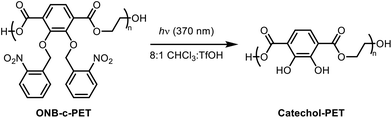
To begin to address the adhesive issue in recycling, we explored the use of a catechol motif in a poly(ethylene terephthalate) derivative. The starting monomer (2,3-bishydroxyterephthalic acid) was protected and polymerized to achieve polyesters with reasonable molecular weights upon optimization of the conditions. Then, the thermal and mechanical properties of c-PET were analysed for its potential as a replacement for the sticky labels on current beverage bottles (Fig. 1).
 |
| | Fig. 1 Dimethyl-2,3-dihydroxyterephthalate was chemically modified to enable polymerization. Following deprotection, adhesive properties were revealed, suggesting its potential as a wash-off adhesive. | |
Experimental
Materials and methods
All chemicals and solvents were purchased from Oakwood Chemicals®, Acros®, AmBeed®, or Fisher Scientific® and used as received. Unless otherwise noted, all reactions were conducted under a nitrogen atmosphere.
Fourier transform infrared (FTIR) spectroscopy.
FTIR spectra were recorded on a Nicolet iS10 infrared spectrometer using an ATR attachment equipped with a diamond crystal. Spectra were analyzed using the OMNIC software package and processed with OriginLab 2024b.
Nuclear magnetic resonance (NMR) experiments.
1H NMR and 13C-NMR spectra were recorded with chloroform-d (CDCl3), dimethyl sulfoxide (DMSO-d6), or water (D2O) on a JOEL ECS 400 MH1 spectrometer and analyzed using MestReNova® software.
Size exclusion chromatography (SEC).
SEC was performed on an Agilent 1260 Infinity II with a PL gel 5 μm MIXED-C 300 × 7.5 mm column equipped with light scattering (LS) and a refractive index (RI) detector. The polymerized material was dissolved in HPLC grade DMF with 0.025 M LiBr and then filtered with a 0.2 μm PTFE filter. Values were calculated using polystyrene as a standard. Data were processed using OriginLab 2024b.
Thermal characterization.
Thermogravimetric analysis (TGA) was performed under a nitrogen environment using a Shimadzu DTG-60H. Samples were first dried at 120 °C to remove all residual solvents. Samples (5–10 mg) with a heating rate of 10 °C min−1 were measured at 5% degradation (Td5%), and first derivative measurements were recorded as well. Differential scanning calorimetry (DSC) measurements were performed on a Shimadzu DSC-60 Plus with samples (4–8 mg) using alumina pans. The experiment was conducted from 25 to 300 °C at a rate of 10 °C min−1 under a nitrogen environment. The glass transition temperature (Tg) was determined by taking the midpoint of the reversible exotherm on the 3rd heating cycle.
Adhesion lap-shear testing.
Lap shear adhesive testing was used to measure the mechanical response of adhesive polymers using a TA.XTPlus Connect instrument (Stable Micro Systems, USA). Specimens were prepared according to ASTM D1002, with the adhesive applied to a 25.4 mm × 12.7 mm overlapping area on 127 mm × 25.4 mm aluminium substrates. Samples underwent uniaxial tensile loading at 0.25 mm min−1 until failure. For the PET substrates, it was first cleaned with a dilute sulfuric acid wash, followed by methanol. Next, the polymer was heated to 100 °C to soften the material. The adhesive was placed between the two PET coupons. Next, the sample was placed in an oven at 50 °C with a weight on top overnight. Once the sample was removed from the oven and cooled, it was taken directly to testing.
All samples failed adhesively. Force and displacement measurements were collected to calculate stress and strain with 4–5 samples tested for each set of conditions.
Synthesis of monomers and polymers
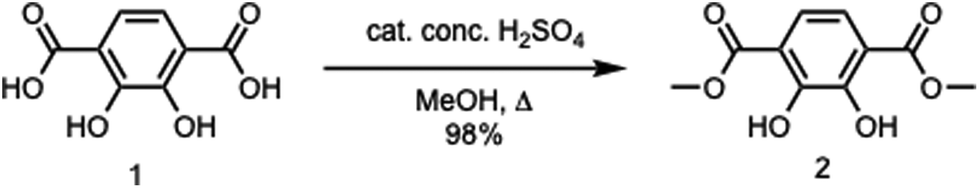
Dimethyl-2,3-dihydroxyterephthalate.
To a round bottom flask were added 2,3-dihydroxyterephthalic acid (2.42 g, 12.2 mmol), methanol (250 mL), and concentrated sulfuric acid (5 mL). The reaction mixture was then allowed to heat under reflux overnight. The reaction mixture was then cooled down to room temperature, followed by the removal of the solvent in vacuo. A saturated sodium bicarbonate solution (50 mL) was added, followed by extraction with dichloromethane (DCM) (2 × 50 mL). The organic layer was then dried with magnesium sulfate, filtered, and then concentrated under vacuum to afford a light yellow solid (2.70 g, 98%). 1H NMR (CDCl3, 400 MHz): δ 3.98 (s, 6H), 7.33 (s, 2H), 10.93 (s, 2H). 13C NMR (CDCl3, 400 MHz): δ 53.18, 116.27, 118.73, 151.94, 170.55. IR (neat) νmax: 3147, 2960, 1683, 1625, 1435, 1322, 1235, 1191, 1142, 1034, 983, 912, 828, 810, 788, 742, 695, 639, 556, 536, 474, 449 cm−1 (Scheme 1).
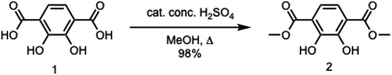 |
| | Scheme 1 Synthesis of dimethyl-2,3-dihydroxyterephthalate. | |
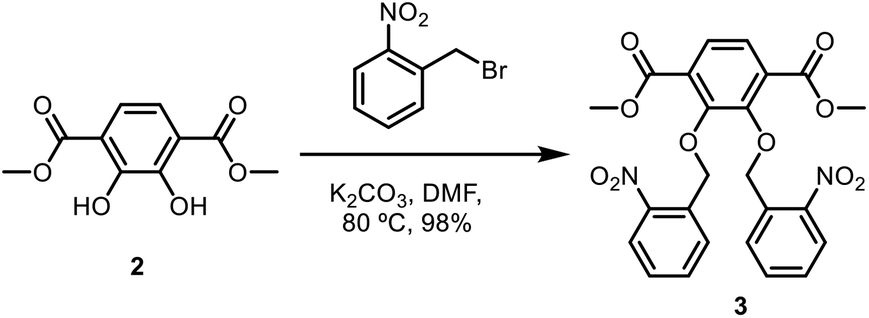
Dimethyl-2,3-bis(nitrobenzyl(oxo))terephthalate.
To a round-bottom flask was added dimethyl-2,3-dihydroxyterephthalate (2.70 g, 11.94 mmol) in N,N-dimethylformamide (DMF) (82 mL). Then, ortho-nitrobenzyl bromide (5.67 g, 26.25 mmol) and potassium carbonate (6.93 g, 50.1 mmol) were added to the flask. The reaction mixture was then heated to 80 °C and stirred overnight. The reaction mixture was then diluted with water (500 mL) to precipitate out dimethyl-2,3-bis(nitrobenzyl(oxo))terephthalate. The product was collected by vacuum filtration, followed by washing with methanol (50 mL) and dried under vacuum to obtain a yellow solid (5.86 g, 98%). 1H NMR (CDCl3, 400 MHz): δ 3.84 (s, 6H), 5.46 (s, 4H), 7.32–7.36 (dt, 2H), 7.51–7.55 (dt, 2H), 7.68 (s, 2H), 7.90–7.92 (dd, 2H), 7.98–8.01 (dd, 2H). 13C NMR (CDCl3, 400 MHz): δ 52.55, 72.75, 124.43, 126.28, 127.93, 129.82, 133.82, 146.11, 152.46, 162.55, 165.24. IR (neat) νmax: 2951, 1724, 1613, 1578, 1522, 1480, 1427, 1365, 1335, 1281, 1240, 1193, 1147, 1083, 1042, 1021, 998, 902, 858, 838, 814, 804, 791, 758, 725, 686, 656, 478 cm−1 (Scheme 2).
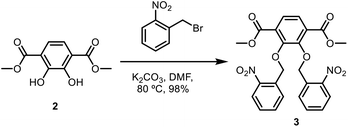 |
| | Scheme 2 Synthesis of dimethyl-2,3-bis(nitrobenzyl-(oxo))terephthalate. | |
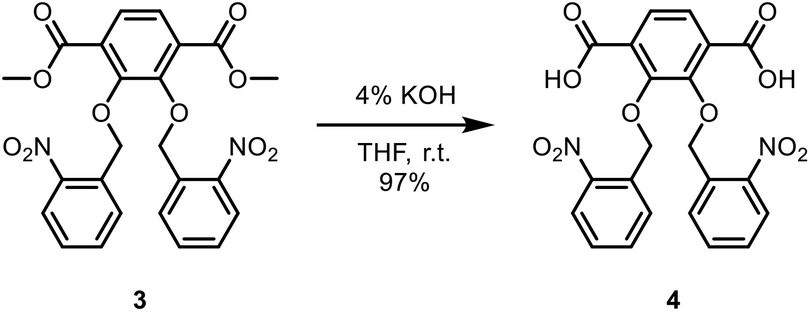
2,3-Bis(nitrobenzyl(oxo))terephthalic acid.
To a round bottom flask was added dimethyl-2,3-bis(nitrobenzyl(oxo))terephthalate (5.86 g, 11.8 mmol). A solution of tetrahydrofuran (THF) (80 mL) and 4% potassium hydroxide solution (KOH) (80 mL) was then added, and the mixture was stirred overnight at room temperature. THF was then evaporated. Diethyl ether (50 mL) was then added to the round-bottom flask, and the aqueous layer was extracted once. The aqueous layer was acidified to pH 1 with concentrated hydrochloric acid (HCl) (6 mL). The precipitate was then filtered and washed with water (50 mL). 2,3-Bis(nitrobenzyl(oxo))terephthalic acid was then collected and vacuum dried overnight to obtain a light yellow solid (5.35 g, 97%). 1H NMR (DMSO-D6, 400 MHz): δ 5.36 (s, 4H), 7.48–7.52 (t, 2H), 7.58 (s, 2H), 7.61–7.63 (t, 2H), 7.79–7.81 (d, 2H), 8.00–8.02 (d, 2H). 13C NMR (DMSO-D6, 400 MHz): δ 166.82, 151.65, 146.85, 134.40, 133.41, 131.16, 129.14, 129.07, 126.13, 124.92, 72.77. IR (neat) νmax: 3854, 3752, 3676, 3650, 3629, 2956, 1810, 1718, 1616, 1579, 1519, 1482, 1428, 1337, 1283, 1231, 1191, 1146, 1085, 1046, 1023, 1000, 908, 859, 806, 790, 758, 724, 687, 476, 432, 419 cm−1 (Scheme 3).
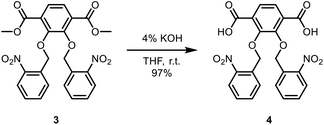 |
| | Scheme 3 Synthesis of 2,3-bis(nitrobenzyl(oxo))terephthalic acid. | |
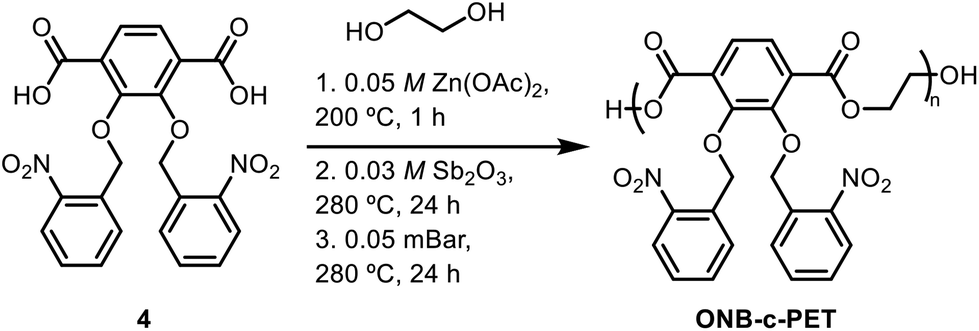
Poly(2,3-bis(nitrobenzyl(oxo))terephthalic acid).
To a round bottom flask was added 2,3-bis(nitrobenzyl(oxo))terephthalic acid (200 mg, 0.427 mmol), followed by ethylene glycol (EG) (33 μL, 0.59 mmol) and 0.05 M zinc acetate [Zn(OAc)2] in EG (427 μL). A distillation reaction was then carried out, and the reaction mixture was heated to 200 °C for 1 hour. 0.03 M antimony(III) oxide (Sb2O3) in EG (1.33 mL) was then added, and the reaction mixture was heated to 280 °C overnight. The excess EG was removed the next day in vacuo, while the reaction mixture was stirred at 280 °C for 24 hours. After cooling down to room temperature, a solution of chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trifluoroacetic acid (CHCl3:TfOH) (8:1) (10 mL) was then added and stirred for thirty minutes. Next, cold methanol (50 mL) was poured into the solution and stirred for an additional 30 minutes. The solid material was then filtered and dried in vacuo for two hours. 1H NMR (CDCl3, 400 MHz): δ 3.87 (s, 4H), 4.00 (s, 4H), 4.47 (s, 3H), 4.64 (s, 1H), 7.87 (s, 10H), 8.56 (s, 1H) (Scheme 4).
:trifluoroacetic acid (CHCl3:TfOH) (8:1) (10 mL) was then added and stirred for thirty minutes. Next, cold methanol (50 mL) was poured into the solution and stirred for an additional 30 minutes. The solid material was then filtered and dried in vacuo for two hours. 1H NMR (CDCl3, 400 MHz): δ 3.87 (s, 4H), 4.00 (s, 4H), 4.47 (s, 3H), 4.64 (s, 1H), 7.87 (s, 10H), 8.56 (s, 1H) (Scheme 4).
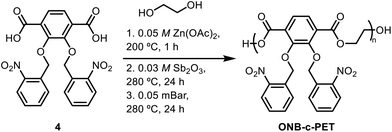 |
| | Scheme 4 Synthesis of poly(2,3-bis(nitrobenzyl(oxo))-terephthalic acid) (ONB-c-PET). | |
Photodeprotection of poly(2,3-bis(nitrobenzyl(oxo))terephthalic acid).
To a round bottom flask containing poly(2,3-bis(nitrobenzyl(oxo))terephthalic acid) (1.5 g), 8:1 CHCl3:TfOH (50 mL) was added. The round bottom flask was then placed on a stir plate about two centimeters away from a UV lamp (370 nm), and the solution was irradiated for 48 hours. The solvent was then removed in vacuo, and excess hexane was added to precipitate out poly(2,3-bishydroxyterephthalic acid) (catechol-PET) as a sticky, black, and sticky solid material. Further drying was done at 150 °C to remove leftover solvents or water residues. Heating the catechol-PET polymer above this temperature will turn the polymer into a hard, solid, cross-linked network without any adhesive properties. 1H NMR (CDCl3, 400 MHz): δ 3.66–3.74 (d, 2H), 4.24 (s, 2H), 4.52 (s, 2H), 7.15–7.43 (d, 2H). 13C NMR (DMSO-D6, 400 MHz): δ 60.80, 60.84, 63.36, 70.33, 72.82, 72.88, 144.10 (Scheme 5).
 |
| | Scheme 5 Synthesis of the catechol-PET homopolymer. | |
Poly[(2,3-bis(nitrobenzyl(oxo))terephthalic acid)-co-terephthalic acid] (ONB-c-PET-80).
To a round bottom flask were added 2,3-bis(nitrobenzyl(oxo))terephthalic acid (375 mg, 0.80 mmol) and dimethyl terephthalate (DMT) (39 mg, 0.20 mmol), followed by ethylene glycol (EG) (90 μL, 1.62 mmol) and 0.05 M zinc acetate [Zn(OAc)2] in EG (0.80 mL). A distillation reaction was then carried out, and the reaction mixture was heated to 200 °C for 1 hour. 0.03 M antimony(III) oxide (Sb2O3) in EG (1.33 mL) was then added, and the reaction mixture was heated to 280 °C overnight. The excess EG was removed the next day in vacuo, while the reaction mixture was stirred at 280 °C for 24 hours. After cooling down to room temperature, a solution of chloroform:trifluoroacetic acid (CHCl3:TfOH) (8:1) (10 mL) was then added and stirred for thirty minutes. Next, cold methanol (50 mL) was poured into the solution and stirred for an additional 30 minutes. The solid material was then filtered and dried in vacuo for two hours.
Depolymerization of poly[(2,3-bis(nitrobenzyl(oxo))terephthalic acid)].
The homopolymer (2.5 g) was refluxed in methanol (250 mL) overnight with 10 mg of zinc acetate [Zn(OAc)2]. After cooling to room temperature, the methanol was evaporated using a rotary evaporator. Then, 50 mL of saturated sodium bicarbonate (NaHCO3) and 50 mL of dichloromethane (DCM) were added to the round-bottom flask. The organic layer was separated, dried with magnesium sulfate, and evaporated to afford 0.26 g of dimethyl-2,3-dihydroxyterephthalate. The aqueous layer was acidified with 1 M HCl (5 mL) and then distilled at 80 °C under vacuum to remove ethylene glycol and water, recovering 0.20 g of 2,3-dihydroxy-4-(methoxycarbonyl)-benzoic acid. The distillate was then redistilled at 80 °C under vacuum to recover 0.10 g of ethylene glycol.
Results and discussion
Monomer and polymer synthesis
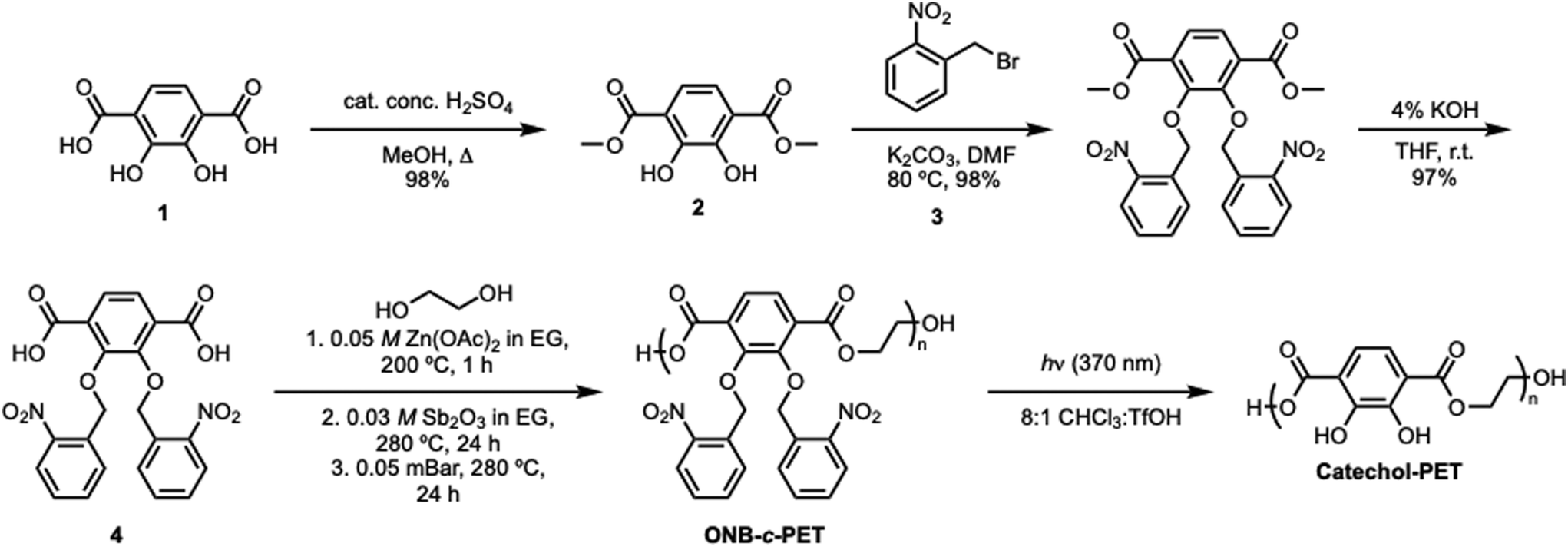
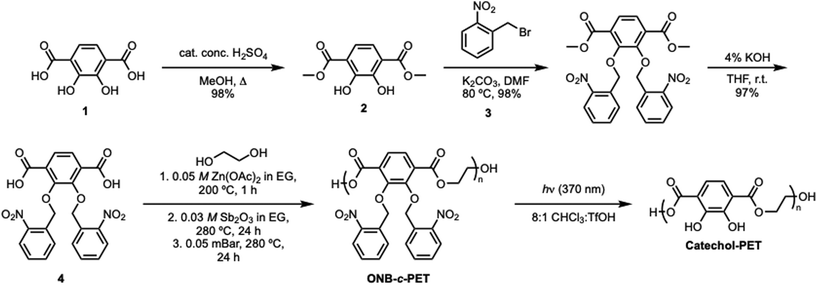
The synthesis of catechol-PET (c-PET) began with the preparation of the monomer, 2,3-bis(nitrobenzyl(oxo))terephthalic acid (Scheme 6). First, an esterification reaction of 2,3-dihydroxyterephthalic acid using a catalytic amount of sulfuric acid in refluxing methanol produced dimethyl-2,3-dihydroxyterephthalate in 98% yield, similar to the method of Cohen and co-workers.35 The catechol functionality was then protected using ortho-nitrobenzyl bromide and potassium carbonate in N,N-dimethylformamide (DMF) at 80 °C, affording dimethyl-2,3-bis(nitrobenzyl(oxo))terephthalate in 98% yield. The protecting group was necessary prior to polymerization with EG; otherwise, hyperbranched polymers could form. Specifically, the use of the ortho-nitrobenzyl functionality as a photoprotective group allowed the ease of removal for adhesive properties via an external stimulus such as light, which is highly advantageous for an adhesive. The final step of the monomer synthesis was completed by base-promoted ester hydrolysis using a 1:1 ratio of 4% potassium hydroxide and tetrahydrofuran (THF), followed by acidification to pH = 1 with concentrated hydrochloric acid (HCl) to afford 2,3-bis(nitrobenzyl(oxo)terephthalic acid) in 97% yield. All small-molecule precursors and monomers were characterized by 1H and 13C NMR (Fig. S2–S13), HRMS (Fig. S14–S16), and IR (Fig. S17–S20).
 |
| | Scheme 6 Synthetic scheme for the protected catechol-PET homopolymer. | |
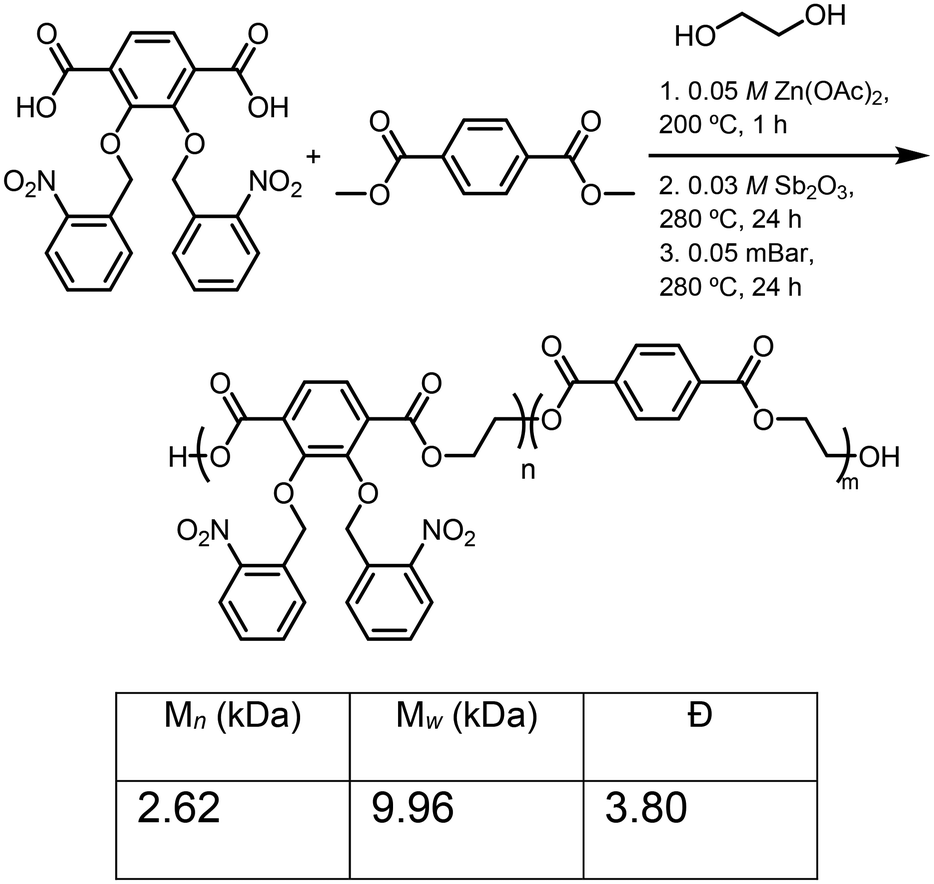
Early polymerization methods were performed by using various catalysts previously used for PET synthesis to afford poly(ONB-2,3-dihydroxyterephthalate) (ONB-c-PET). These included antimony(III) oxide (Sb2O3),36 titanium(IV) butoxide [Ti(OCH2CH2CH2CH3)4],37 antimony oxide (Sb2O3) or zinc acetate dihydrate [Zn(OAc)2-2H2O],38 4-dimethylaminopyridine (DMAP) and triflic acid (TfOH) (1:1),39 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) and benzoic acid.40 The use of these catalysts resulted in the production of oligomers of poly(2,3-bisnitrobenzyl(oxo)-terephthalic acid), rather than higher molecular weight polymers. Therefore, after exploring these catalysts, we were delighted to discover that the dual catalyst action of Sb2O3 and Zn(OAc)2 could catalyze the step-growth polymerization of 2,3-bis(nitrobenzyl(oxo))terephthalic acid and ethylene glycol (EG). Specifically, the polymerization process of 2,3-bis(nitrobenzyl(oxo)terephthalic acid) with 2.02 equivalents of ethylene glycol using dual catalysts of zinc acetate (Zn(OAc)2) and antimony(III) oxide (Sb2O3) was chosen to mimic similar polymerization conditions of PET in the literature.41 However, the excess ethylene glycol capped the polymer, limiting the molecular weight, and the end cap protons were observed as shown in Fig. S7. Three stages were used in each polymerization process. First, 5 mol% of 0.05 M zinc acetate (Zn(OAc)2) in EG was added and heated to 200 °C. Next, 5 mol% of 0.03 M antimony(III) oxide (Sb2O3) in EG was added, and the temperature was then raised to 280 °C. The final stage allowed the reaction to proceed under vacuum heating. The time was altered for each stage to understand the effects on the molecular weight and Đ. All polymers were characterized by SEC (Fig. S29–S38). The initial condition attempted gave Mn = 4.04 kDa (Table 1, entry 1). Then, increasing the time for the second and third stages did not increase the molecular weight but did greatly broaden the dispersity (Table 1, entry 2). The most significant rise in molecular weight was observed when the heating duration of the second stage was extended to 24 hours. Traditionally, condensation polymerization exhibits exponential growth in molecular weight over time, and the highest molecular weight was obtained under these conditions (Table 1, entry 3). Ethylene glycol is difficult to remove, and therefore, the 24-hour mark on the vacuum removed it completely. In an effort to reduce the time used for the polymerization, stages 1 and 2 were altered. Notably, increasing stage 1 anywhere between 3 and 24 h did not greatly increase the molecular weight. This indicated that the activation from Zn(OAc)2 was complete in 1 h (Table 1, entries 4–9). The excess EG was removed the next day under vacuum (0.05 mBar) for 24 h (Table 1, entries 2–9). The dispersity values of poly(2,3-bisnitrobenzyl(oxo)terephthalic acid) ranged from 1.1 to 1.7. Current PET polymers have dispersity values that are much broader, ranging from 1.5 to 7, which is most likely due to the extra aromatic groups used as a protecting group that decrease solubility.
Table 1 Synthetic conditions and SEC data for homopolymerization
| Entry |
Stage 1 |
Stage 2 |
M
n (kDa) |
M
w (kDa) |
Đ
|
| 1 |
0.05 M Zn(OAc)2, 1 h |
0.03 M Sb2O3, 2 h, 1 h vacuum |
4.04 |
4.55 |
1.13 |
| 2 |
0.05 M Zn(OAc)2, 1 h |
0.03 M Sb2O3, 10 h, 24 h vacuum |
2.48 |
3.96 |
1.60 |
| 3 |
0.05 M Zn(OAc)2, 1 h |
0.03 M Sb2O3, 24 h, 24 h vacuum |
8.19 |
10.14 |
1.24 |
| 4 |
0.05 M Zn(OAc)2, 2 h |
0.03 M Sb2O3, 8 h, 24 h vacuum |
5.39 |
7.34 |
1.36 |
| 5 |
0.05 M Zn(OAc)2, 3 h |
0.03 M Sb2O3, 8 h, 24 h vacuum |
2.58 |
4.29 |
1.66 |
| 6 |
0.05 M Zn(OAc)2, 3 h |
0.03 M Sb2O3, 3 h, 24 h vacuum |
2.62 |
4.50 |
1.72 |
| 7 |
0.05 M Zn(OAc)2, 5 h |
0.03 M Sb2O3, 5 h, 24 h vacuum |
5.18 |
6.44 |
1.25 |
| 8 |
0.05 M Zn(OAc)2, 12 h |
0.03 M Sb2O3, 5 h, 24 h vacuum |
2.87 |
3.09 |
1.33 |
| 9 |
0.05 M Zn(OAc)2, 24 h |
0.03 M Sb2O3, 5 h, 24 h vacuum |
2.82 |
3.25 |
1.15 |
After the polymerization heating stages, the reaction mixture was then cooled down to room temperature and deprotected using a solution of 8:1 (v/v) chloroform:trifluoroacetic acid (CHCl3:TfOH). First, a beaker of cold methanol was added to the reaction flask. The solution was then filtered and dried to afford the poly(2,3-bis(nitro-benzyl(oxo))terephthalic acid) polymer as a black, viscous liquid. The solution of 8:1 (v/v) chloroform:trifluoroacetic acid (TfOH) was then added, and the solution was irradiated under a 370 nm UV lamp for 48 h. The solvent was then evaporated, followed by precipitation into hexanes, which afforded the catechol-poly(ethylene terephthalate) polymer (c-PET) as a black, sticky, hard solid material.
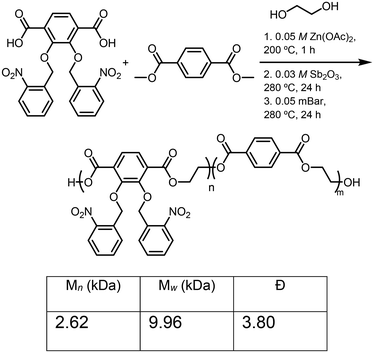
The synthesis of the catechol-PET copolymer was performed using a similar strategy to homopolymerization to incorporate a cheap, commercially available monomer to decrease the reliance on the synthesized monomer, which included several steps and is more costly than terephthalic acid ($0.092 vs. $2.90, Sigma-Aldrich 2025). To test the effect on the thermal and mechanical properties, 20% terephthalic acid comonomer was incorporated. Dimethyl terephthalate (DMT) (Fig. S7) was synthesized starting with terephthalic acid, refluxing in methanol. Then, thionyl chloride (SOCl2) was slowly added dropwise into the solution and refluxed overnight to afford the ester monomer.42 The reaction mixture was then cooled down to room temperature and extracted. After removing the solvent in vacuo, DMT was produced in >90% yields. Next, 2,3-bis(nitrobenzyl(oxo)terephthalic acid) to DMT were combined at a ratio of 8:2, polymerized, and then photodeprotected to afford the catechol-PET copolymer (Scheme 7) using the optimized conditions for homopolymerization, resulting in Mn = 2.62 kDa and Đ = 3.80 for the protected polymerization (ONB-c-PET-80). The deprotection was confirmed by FTIR, resulting in c-PET-80. While the degree of incorporation was investigated, the overlapping peaks in the 1H NMR did not allow for the ratio to be determined.
 |
| | Scheme 7 Synthesis of the protected catechol-PET copolymer. | |
Thermal characterization
The DSC thermogram results showed that poly(2,3-bisnitrobenzyl(oxo)terephthalic acid) had a glass transition temperature (Tg) of 34 °C. After the photodeprotection reaction, the Tg increased to 56 °C, attributed to the hydrogen bonding that occurs upon deprotection. TGA studies (Table 2) showed that ONB-c-PET had a degradation temperature of 284 °C and when deprotected the degradation temperature was 317 °C. We proposed that the deprotected polymer had a higher degradation temperature of approximately 30 °C due to the hydrogen bonding that is revealed when the nitrobenzyl groups are removed. The copolymer had a higher T5% at 370 °C due to the incorporation of the terephthalic acid monomer. PET undergoes isothermal degradation in the temperature range of 270–370 °C.43 Together, these thermal analyses demonstrate that photodeprotection significantly alters the polymer's thermal behavior, enhancing Tg and Td5% due to hydrogen bonding.
Table 2 TGA and DSC data for the protected and deprotected homopolymer and copolymer
| Polymer |
TGA |
DSC |
|
T
5% (°C) |
T
g (°C) |
C
p (J g−1 °C−1) |
|
c-PET
|
317 |
56 |
0.17 |
|
ONB-c-PET
|
284 |
34 |
0.082 |
|
c-PET-80
|
370 |
45 |
0.0018 |
Adhesion testing
Lap shear adhesion measurements were performed on the c-PET homopolymer and copolymers, similar to other catechol-based materials.44,45 The configuration was lap shear using an aluminium based on ASTM International D1002-05.46 The polymers were studied at room temperature to understand the adhesion under conditions for the intended use. Lap-shear results for all polymers are provided in Fig. 2A. c-PET and c-PET-80 have lap-shear adhesion strengths of 0.77 ± 0.09 MPa and 0.55 ± 0.16 MPa, respectively. The decrease in adhesion strength by 29% is approximately proportional to the decrease in catechol groups in the backbone of the copolymer due to the incorporation of the comonomer without catechol groups.
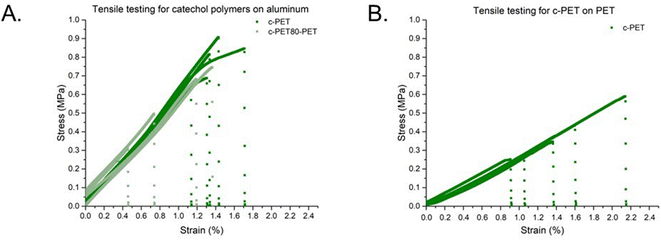 |
| | Fig. 2 Lap shear adhesion on aluminium plates using c-PET and c-PET80-PET (A) and c-PET on PET (B). Adhesion strengths were calculated from an average of 4–5 replicates and reported with standard errors. | |
Next, adhesion lap-shear testing on PET for the homopolymer was performed using a modified procedure. Coupons of PET from ULINE plastic juice bottles (model number: S-21727W) were cut to the same size as the aluminium substrate from ASTM International D1002-05. The polymer was heated to 100 °C to allow for the polymer to soften and then placed between the PET coupons. The sample was then placed in an oven at 50 °C with a weight on it overnight. Once removed from the oven, the sample was cooled to room temperature and taken directly to testing. c-PET resulted in 0.38 ± 0.11 MPa (Fig. 2B). The reported literature values showed that casein adhesives, the most commonly used adhesive in PET bottling, have a range between 5.28 and 5.40 MPa on PET.47 The c-PET material exhibits a lower adhesion lap-shear strength relative to standard casein; however, its degradability and potential to improve recycling provide a viable alternative for adhesives.
Degradation studies
The recycling of PET bottles alleviates part of the plastic pollution problem that persists in the environment by having a closed-loop process. However, chemical recycling strategies such as hydrolysis48 require high heat and pressure (>400 °C and 400 atm) under neutral conditions. Here, we chose methanolysis49 as our strategy to produce starting monomers because it allows for shorter times, lower pressure, and lower temperatures. Conditions used for the methanolysis were 200–280 °C and <1 atm. As shown in Fig. 3, the homopolymer was refluxed in methanol overnight with zinc acetate [Zn(OAc)2], producing a mixture of monomers. After evaporation of methanol, a solution of saturated sodium bicarbonate (NaHCO3) and dichloromethane (DCM) was added. The organic layer was evaporated to recover dimethyl-2,3-dihydroxyterephthalate. The aqueous layer was collected and acidified with 1 M HCl, followed by distillation to recover 2,3-dihydroxy-4-(methoxycarbonyl)-benzoic acid. The remaining materials were oligomers. The distillate was then redistilled to recover ethylene glycol (EG). Overall, 23% of the EG and 30% of the terephthalate derivatives were recovered (Fig. S39–S41), which have the potential to be used again for polymerization.
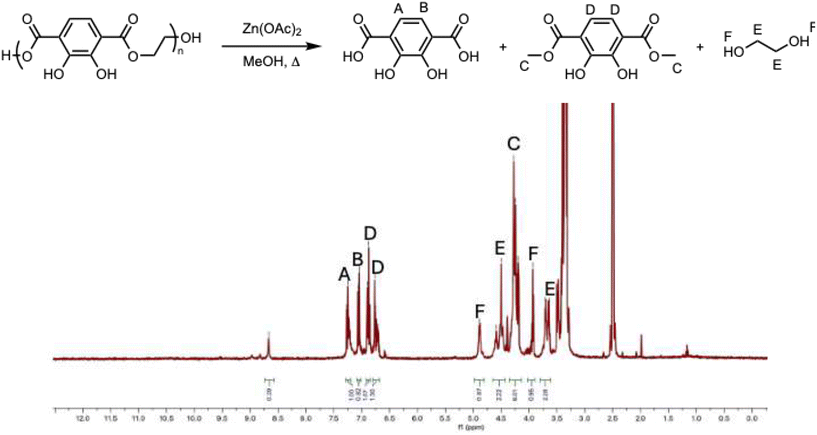 |
| | Fig. 3
1H NMR of the degradation of homopolymer via methanolysis in DMSO-D6. | |
Conclusions
The catechol motif was integrated into the common polyester PET as a terephthalic acid derivative. The polymerization of 2,3-bis((2-nitrobenzyl)oxy)terephthalic acid with ethylene glycol was performed by using a dual catalyst system of zinc acetate and antimony(III) oxide. Polymerization produced polymers (Mn = 3000–8000 g mol−1) with dispersity values (1.1–1.7). The photodeprotection step was performed using a UV lamp (370 nm), which allowed access to the catechol-PET polymer and copolymer when combined with DMT. Both catechol-PET and catechol-PET copolymers exhibited adhesive properties on both aluminum and PET. This catechol-PET also showed degradability by returning to starting monomers when heated with methanol. Potential future applications of this material include use as a substitute adhesive material for beverage bottles to increase recyclability due to the ease of washing off the labels.
Author contributions
S. L. K. conceptualized the project and interpreted the data. T. N. and C. T. C. synthesized the monomers and polymers. F. O. interpreted the data. W. R. and P. E. performed the adhesion testing.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the SI. Supplementary information: 1H NMR, TGA, and DSC thermograms. See DOI: https://doi.org/10.1039/d5py00401b.
Acknowledgements
We would like to thank Texas Tech University for the start-up funding that supported this work.
References
-
S. Thomas, A. V. Rane, K. Kanny, V. Abitha and M. G. Thomas, Recycling of polyethylene terephthalate bottles, William Andrew, 2018 Search PubMed.
- T. M. Joseph, S. Azat, Z. Ahmadi, O. M. Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Polyethylene terephthalate (PET) recycling: a review, Case Stud. Chem. Environ. Eng., 2024, 100673 CrossRef.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Organocatalysis for depolymerisation, Polym. Chem., 2019, 10(2), 172–186 RSC.
- K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Dual-catalytic depolymerization of polyethylene terephthalate (PET), Polym. Chem., 2020, 11(8), 1450–1453 RSC.
- I. Olazabal, E. J. Luna Barrios, S. De Meester, C. Jehanno and H. Sardon, Overcoming the Limitations of Organocatalyzed Glycolysis of Poly(ethylene terephthalate) to Facilitate the Recycling of Complex Waste Under Mild Conditions, ACS Appl. Polym. Mater., 2024, 6(7), 4226–4232 CrossRef CAS.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guemard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. Andre, S. Duquesne and A. Marty, An engineered PET depolymerase to break down and recycle plastic bottles, Nature, 2020, 580(7802), 216–219 CrossRef CAS PubMed.
- Z. Chen, R. Duan, Y. Xiao, Y. Wei, H. Zhang, X. Sun, S. Wang, Y. Cheng, X. Wang, S. Tong, Y. Yao, C. Zhu, H. Yang, Y. Wang and Z. Wang, Biodegradation of highly crystallized poly(ethylene terephthalate) through cell surface codisplay of bacterial PETase and hydrophobin, Nat. Commun., 2022, 13(1), 7138 CrossRef CAS PubMed.
- M. Babaei, M. Jalilian and K. Shahbaz, Chemical recycling of Polyethylene terephthalate: A mini-review, J. Environ. Chem. Eng., 2024, 12(3), 112507 CrossRef CAS.
- Z. Guo, J. Wu and J. Wang, Chemical degradation and recycling of polyethylene terephthalate (PET): a review, RSC Sustainability, 2025, 3, 2111–2133 RSC.
- M. T. Hossain, M. A. Shahid, N. Mahmud, M. A. Darda and A. B. Samad, Techniques, applications, and prospects of recycled polyethylene terephthalate bottle: a review, J. Thermoplast. Compos. Mater., 2024, 37(3), 1268–1286 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. De Rivas, S. Arnaiz and J. I. Gutierrez-Ortiz, Chemical recycling of post-consumer PET wastes by glycolysis in the presence of metal salts, Polym. Degrad. Stab., 2010, 95(6), 1022–1028 CrossRef.
- D. D. Pham and J. Cho, Low-energy catalytic methanolysis of poly (ethyleneterephthalate), Green Chem., 2021, 23(1), 511–525 RSC.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis, Green Chem., 2020, 22(16), 5376–5394 RSC.
- D. A. Mersha, T. N. Gesese, Z. B. Sendekie, A. T. Admase and A. J. Bezie, Operating conditions, products and sustainable recycling routes of aminolysis of polyethylene terephthalate (PET)–a review, Polym. Bull., 2024, 1–17 Search PubMed.
- J. Payne and M. D. Jones, The chemical recycling of polyesters for a circular plastics economy: challenges and emerging opportunities, ChemSusChem, 2021, 14(19), 4041–4070 CrossRef CAS.
-
R. D. Allen and M. I. James, Chemical recycling of PET, in Circular economy of polymers: Topics in recycling technologies, ACS Publications, 2021, pp. 61–80 Search PubMed.
- T. Muringayil Joseph, S. Azat, Z. Ahmadi, O. Moini Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Polyethylene terephthalate (PET) recycling: A review, Case Stud. Chem. Environ. Eng., 2024, 9, 100673 CrossRef CAS.
- . Recyclers, A. o. P. Critical Guidance Protocol for Clear PET Articles with Labels and Closures Document number: PET-CG-02 2024.
- . Recyclers, A. o. P. PET Thermoform Packaging Design Resource Document Document Number RES-PET-01 2024.
-
D. J. Dunn, Adhesives and sealants: technology, applications and markets, iSmithers Rapra Publishing, 2003 Search PubMed.
- M. Li, A. Mao, Q. Guan and E. Saiz, Nature-inspired adhesive systems, Chem. Soc. Rev., 2024, 8240–8305 RSC.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, The chemistry behind catechol–based adhesion, Angew. Chem., Int. Ed., 2019, 58(3), 696–714 CrossRef CAS.
- S. Espinoza, F. Managò, D. Leo, T. D. Sotnikova and R. R. Gainetdinov, Role of catechol-O-methyltransferase (COMT)-dependent processes in Parkinson's disease and L-DOPA treatment, CNS Neurol. Disord.: Drug Targets, 2012, 11(3), 251–263 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, A reversible wet/dry adhesive inspired by mussels and geckos, Nature, 2007, 448(7151), 338–341 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Mussel-Inspired Surface Chemistry for Multifunctional Coatings, Science, 2007, 318(5849), 426–430 CrossRef CAS.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. H. Waite, J. N. Israelachvili, B. K. Ahn and M. T. Valentine, Toughening elastomers using mussel-inspired iron-catechol complexes, Science, 2017, 358(6362), 502–505 CrossRef CAS.
- W. Zhang, R. Wang, Z. Sun, X. Zhu, Q. Zhao, T. Zhang, A. Cholewinski, F. K. Yang, B. Zhao, R. Pinnaratip, P. K. Forooshani and B. P. Lee, Catechol-functionalized hydrogels: biomimetic design, adhesion mechanism, and biomedical applications, Chem. Soc. Rev., 2020, 49(2), 433–464 RSC.
- M. A. Rahim, S. L. Kristufek, S. Pan, J. J. Richardson and F. Caruso, Phenolic Building Blocks for the Assembly of Functional Materials, Angew. Chem., Int. Ed., 2019, 58(7), 1904–1927 CrossRef CAS.
- J. Yang, M. A. Cohen Stuart and M. Kamperman, Jack of all trades: versatile catechol crosslinking mechanisms, Chem. Soc. Rev., 2014, 43(24), 8271–8298 RSC.
- M. A. North, C. A. Del Grosso and J. J. Wilker, High Strength Underwater Bonding with Polymer Mimics of Mussel Adhesive Proteins, ACS Appl. Mater. Interfaces, 2017, 9(8), 7866–7872 CrossRef CAS PubMed.
- T. S. Pal, S. K. Raut and N. K. Singha, Mussel-Inspired Catechol-Functionalized EVA Elastomers for Specialty Adhesives; Based on Triple Dynamic Network, Chem. Mater., 2025, 37(7), 2516–2534 CrossRef CAS.
- A. Fukuyoshi, S. Oshiro, Y. Seino, Y. Uchida, T. Aketa, T. Ozai, H. Nakagawa and Y. Kaneko, Enhancement of the adhesion of polysiloxane-based adhesives through catechol functionalization, Polym. Chem., 2025, 16(28), 3237–3243 RSC.
- M. Li, A. Mao, Q. Guan and E. Saiz, Nature-inspired adhesive systems, Chem. Soc. Rev., 2024, 53(16), 8240–8305 RSC.
- H. Montazerian, E. Davoodi, A. Baidya, M. Badv, R. Haghniaz, A. Dalili, A. S. Milani, M. Hoorfar, N. Annabi, A. Khademhosseini and P. S. Weiss, Bio-macromolecular design roadmap towards tough bioadhesives, Chem. Soc. Rev., 2022, 51(21), 9127–9173 RSC.
- K. K. Tanabe, C. A. Allen and S. M. Cohen, Photochemical activation of a metal-organic framework to reveal functionality, Angew. Chem., Int. Ed., 2010, 49(50), 9730–9733 CrossRef CAS PubMed.
- T. W. Son, H. S. Son, W. K. Kim, D. W. Lee, K. I. Kim and J. H. Jeong, Preparation of PET using homogeneous catalysts. II. Effect of BHPP, NPG and PD in Sb 2 O 3 glycol solution catalysts, Fibers Polym., 2000, 1, 6–11 CrossRef CAS.
- L. Finelli, C. Lorenzetti, M. Messori, L. Sisti and M. Vannini, Comparison between titanium tetrabutoxide and a new commercial titanium dioxide based catalyst used for the synthesis of poly (ethylene terephthalate), J. Appl. Polym. Sci., 2004, 92(3), 1887–1892 CrossRef CAS.
- S. Mohammadi and M. Enayati, Dual catalytic activity of antimony(III) oxide: The polymerization catalyst for synthesis of polyethylene terephthalate also catalyze depolymerization, Polym. Degrad. Stab., 2022, 206, 110180 CrossRef CAS.
- S. Kaiho, A. A. R. Hmayed, K. Delle Chiaie, J. Worch and A. Dove, Designing Thermally Stable Organocatalysts for Poly (ethylene terephthalate) Synthesis, Macromolecules, 2022, 10628–10639 CrossRef CAS.
- I. Flores, J. Demarteau, A. Müller, A. Etxeberria, L. Irusta, F. Bergman, C. Koning and H. Sardon, Screening of different organocatalysts for the sustainable synthesis of PET, Eur. Polym. J., 2018, 104, 170–176 CrossRef CAS.
- R. M. Gohil, Designing polyethylene terephthalate–based barrier resins, J. Appl. Polym. Sci., 2012, 126(1), 260–272 CrossRef CAS.
- A. A. Khan, F. Kamena, M. S. Timmer and B. L. Stocker, Development of a benzophenone and alkyne functionalised trehalose probe to study trehalose dimycolate binding proteins, Org. Biomol. Chem., 2013, 11(6), 881–885 RSC.
- F. Samperi, C. Puglisi, R. Alicata and G. Montaudo, Thermal degradation of poly(ethylene terephthalate) at the processing temperature, Polym. Degrad. Stab., 2004, 83(1), 3–10 CrossRef CAS.
- J. Zhou, A. P. Defante, F. Lin, Y. Xu, J. Yu, Y. Gao, E. Childers, A. Dhinojwala and M. L. Becker, Adhesion properties of catechol-based biodegradable amino acid-based poly(ester urea) copolymers inspired from mussel proteins, Biomacromolecules, 2015, 16(1), 266–274 CrossRef CAS.
- C. R. Matos-Perez, J. D. White and J. J. Wilker, Polymer composition and substrate influences on the adhesive bonding of a biomimetic, cross-linking polymer, J. Am. Chem. Soc., 2012, 134(22), 9498–9505 CrossRef CAS PubMed.
-
International, A., Standard Test Method for Apparent Shear Strength of Single-Lap-Joint Adhesively Bonded Metal Specimens by Tension Loading(Metal-to-Metal), in ASTM International D1002–05, West Conshocken, PA, 2005 Search PubMed.
- A. Cherkashina, A. Rassokha, I. Ryshchenko and O. Komarova, Development and research of a label caseine adhesive for packaging the industrial and household products, East.-Eur. J. Enterp. Technol., 2020, 2(6), 104 Search PubMed.
- G. P. Karayannidis and D. S. Achilias, Chemical recycling of poly (ethylene terephthalate), Macromol. Mater. Eng., 2007, 292(2), 128–146 CrossRef CAS.
-
M. Crippa and B. Morico, PET depolymerization: a novel process for plastic waste chemical recycling, in Studies in Surface Science and Catalysis, Elsevier, 2020, vol. 179, pp. 215–229 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Corey T.
Chiu
a,
William
Renter
b,
Ferley
Orozco
a,
Paul F.
Egan
b and
Samantha L.
Kristufek
a,
Corey T.
Chiu
a,
William
Renter
b,
Ferley
Orozco
a,
Paul F.
Egan
b and
Samantha L.
Kristufek