DOI:
10.1039/D5PY00255A
(Paper)
Polym. Chem., 2025,
16, 3119-3128
Furfural-derived 5-alkoxy-2(5H)-furanones as cationically copolymerizable cyclic hemiacetal esters†
Received
12th March 2025
, Accepted 10th June 2025
First published on 11th June 2025
Abstract
We have focused on cyclic hemiacetal esters as monomers that undergo cationic ring-opening copolymerization. In this study, we investigated the cationic ring-opening copolymerization of 5-alkoxy-2(5H)-furanones (ROFOs), which are cyclic hemiacetal esters synthesized from a plant-derived compound (furfural) via photochemical reaction. ROFOs did not undergo cationic homopolymerization, while copolymerization with oxiranes successfully proceeded to yield polymers with ester and acetal moieties in the main chain. In addition, cationic terpolymerization of vinyl ether (VE), oxirane, and ROFO proceeded via highly selective crossover reactions, resulting in sequence-controlled terpolymers with VEn–oxiranem–ROFO-type periodic structures. The co- and terpolymers could be degraded by acid hydrolysis via the scission of the acetal moieties in the main chain. The copolymers obtained from ROFO have β-substituted acrylate structures in the main chain; hence, the polymers could be modified by the thiol–ene reaction.
Introduction
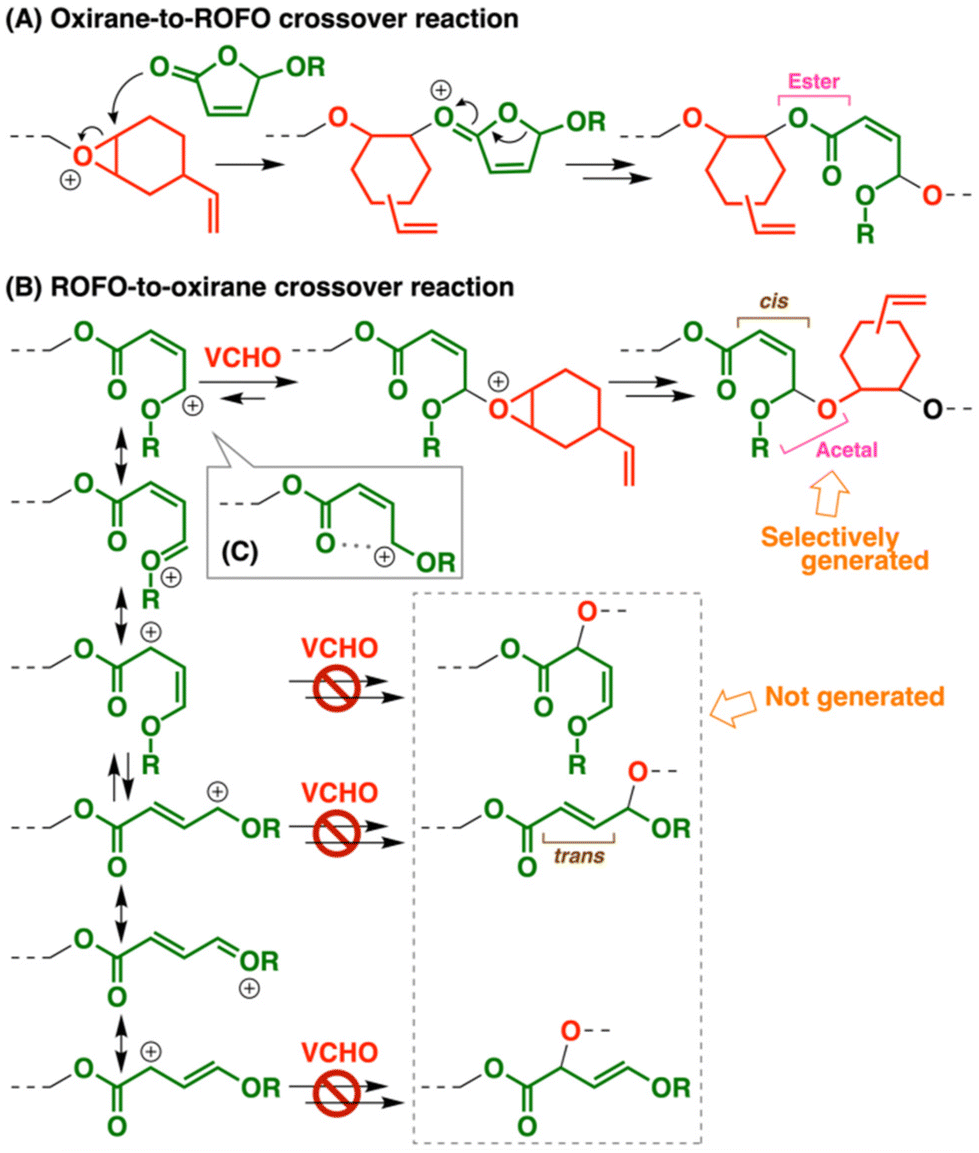
Polymerizability of cyclic monomers in ring-opening polymerization (ROP) is governed by several factors, such as ring strain, reactivity of propagating species, and stability of the structures resulting from propagation reactions.1 Cyclic ethers including oxiranes and oxetanes undergo cationic ROP via the oxonium ion-mediated propagation reactions, which relies on the liberation of ring strain as a driving force.2–4 In addition, cyclic monomers that do not exhibit homopolymerizability potentially undergo copolymerization when appropriate comonomers are used. For example, nonhomopolymerizable cyclic acetals5 undergo cationic copolymerization with vinyl monomers such as vinyl ethers (VEs) and styrene derivatives.6,7 The crossover reaction from a cyclic acetal to a vinyl monomer occurs via the carbocation (oxocarbenium ion) generation through the ring-opening of the cyclic acetal-derived oxonium ion.
Cyclic hemiacetal esters have a carbon atom adjacent to both ester and alkoxy groups in a ring, which is responsible for the characteristic polymerizability. Coordination–insertion-type ROP of 1,3-dioxolan-4-ones (DOLOs), which are five-membered cyclic hemiacetal esters, was reported to proceed by a metal complex catalyst, while a carbonyl compound eliminates from the propagating end, resulting in not poly(DOLO)s but poly(α-hydroxy acid)s, such as polylactide.8,9 Copolymerization of DOLOs and cyclic esters was also reported to proceed.10,11 We previously reported that DOLOs did not undergo cationic homopolymerization but underwent cationic copolymerization with oxiranes.12 An alkoxy group-adjacent carbocation (oxocarbenium ion), which has a structure similar to that of a VE-derived carbocation, is most likely generated via the ring-opening reaction that occurred after the reaction between an oxirane-derived oxonium ion and the carbonyl group of a DOLO (Scheme 1A). In addition, cationic copolymerization of VE and DOLO did not proceed, while terpolymerization proceeded when VE, oxirane, and DOLO were combined. Moreover, we demonstrated that 3-alkoxyphthalides (ROPTs; Scheme 1A), which have a fused aryl ring in a five-membered cyclic hemiacetal ester structure, underwent cationic copolymerization with oxiranes via more frequent crossover reactions than the DOLO case.13 ROPTs also did not undergo cationic homopolymerization. Unlike these five-membered cyclic hemiacetal esters, six- and seven-membered counterparts were reported to undergo cationic homopolymerization.14,15 Bicyclic compounds with a five-membered cyclic hemiacetal structure also undergo cationic homopolymerization.16
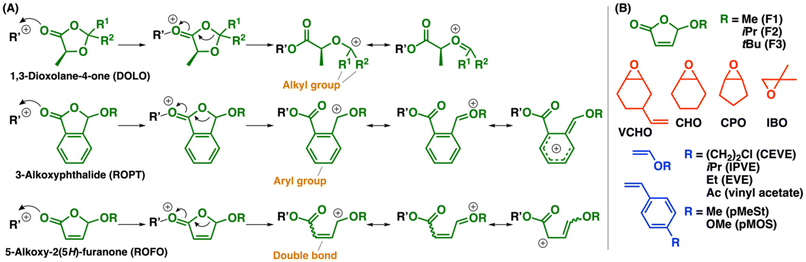 |
| | Scheme 1 (A) Propagating species generated from DOLO, ROPT, or ROFO. (B) Monomers used in this study. | |
As potentially polymerizable cyclic hemiacetal esters, we focused on 5-alkoxy-2(5H)-furanones (ROFOs), which are synthesized from furfural by the photochemical reaction.17–20 ROFOs have a five-membered cyclic hemiacetal structure with a carbon–carbon double bond in the ring. This structure corresponds to a β-substituted acrylate. Indeed, ROFOs were reported to undergo radical copolymerization with vinyl monomers through the reaction of a radical species and the double bond.21,22 However, the use of ROFOs as cyclic monomers for ROP was not previously reported as far as we know. When the carbonyl group of an ROFO reacts with a cationic species, subsequent ring-opening reaction is expected to result in a carbocation adjacent to both alkoxy group and carbon–carbon double bond (Scheme 1A). This carbocation has a structure different from those of the DOLO-derived, alkoxy group-adjacent carbocation and the ROPT-derived, alkoxy group- and aryl group-adjacent carbocation;12,13 hence, ROFOs are expected to exhibit polymerizability different from those of DOLOs and ROPTs.
In this study, we examined the cationic polymerization of ROFOs with a methoxy, isopropoxy, or tert-butoxy group (Scheme 1B). Cationic homopolymerization of ROFOs did not proceed at all, whereas cationic copolymerization with oxiranes successfully proceeded to yield copolymers with acetal, ester, and carbon–carbon double bond moieties in the main chain. Interestingly, the ROFO-derived carbocation underwent highly selective reaction with an oxirane monomer, resulting in a single structure among the four possible structures associated with carbocation conjugation and cis–trans configuration. Cationic terpolymerization of ROFOs with VEs and oxiranes also proceeded efficiently. In addition, the obtained copolymers could be modified by the thiol–ene reaction of the ROFO-derived β-substituted acrylate moieties in the main chain.
Results and discussion
Synthesis of ROFOs
ROFOs were synthesized from furfural.17–20 A solution of furfural in methanol was bubbled with air under irradiation with a white LED floodlight in the presence of rose bengal, resulting in 5-hydroxy-2(5H)-furanone. 5-Methoxy-2(5H)-furanone (F1) and 5-isopropoxy-2(5H)-furanone (F2) were synthesized by the reaction of 5-hydroxy-2(5H)-furanone in methanol and 2-propanol, respectively. 5-tert-Butoxy-2(5H)-furanone (F3) was synthesized from 5-hydroxy-2(5H)-furanone by the reported method.22 See the Experimental section in the ESI† for the details of monomer synthesis.
Cationic homopolymerization of ROFOs
Cationic homopolymerization of ROFOs was examined with B(C6F5)3 or GaCl3 as Lewis acid catalysts at different monomer concentrations and temperatures. However, ROFOs were not consumed at all under any conditions examined (entries 1–5 in Table 1). DOLOs and ROPTs also did not undergo homopolymerization in our previous studies.12,13 The inertness of the five-membered cyclic hemiacetal esters in cationic homopolymerization is likely derived from both low ring-strain and ineffective generation of a hemiacetal ester moiety via the homopropagation reaction (Scheme 2A).
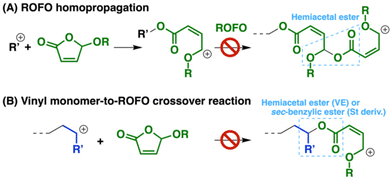 |
| | Scheme 2 (A) Homopropagation reaction of ROFO and (B) crossover reaction from a vinyl monomer to ROFO. | |
Table 1 Cationic homopolymerization of ROFOs and copolymerization with vinyl monomersa
| Entry |
ROFO |
Vinyl monomer |
Time (h) |
Conv. (%) |
| ROFO |
Vinyl |
|
[ROFO]0 = 0.75 M, [vinyl monomer]0 = 0 or 0.75 M, [B(C6F5)3]0 = 3.0 mM (except for entry 3), in dichloromethane at −78 °C (except for entry 2).
At 0 °C.
GaCl3 (4.0 mM) was used instead of B(C6F5)3.
|
| 1 |
F1 |
— |
166 |
0 |
— |
| 2b |
F1 |
— |
96 |
0 |
— |
| 3c |
F1 |
— |
96 |
0 |
— |
| 4 |
F2 |
— |
120 |
0 |
— |
| 5 |
F3 |
— |
72 |
0 |
— |
| 6 |
F1 |
CEVE |
166 |
0 |
0 |
| 7 |
F1 |
pMeSt |
24 |
0 |
0 |
| 8 |
F1 |
pMOS |
24 |
0 |
56 |
| 9 |
F2 |
CEVE |
120 |
0 |
7 |
| 10 |
F3 |
CEVE |
72 |
0 |
2 |
Cationic copolymerization
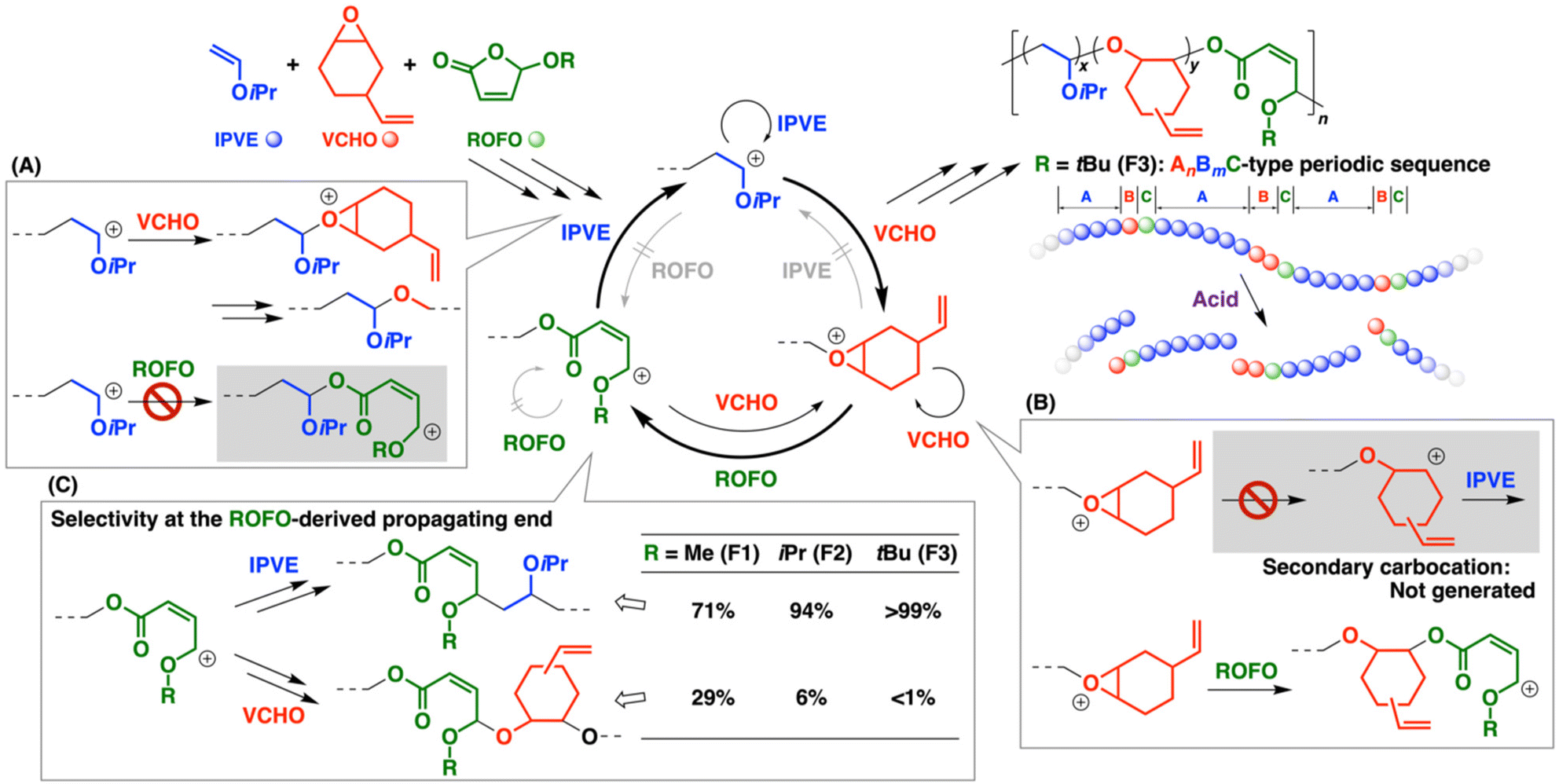
Cationic copolymerization of 2-chloroethyl VE (CEVE) and ROFOs was ineffective (entries 6, 9, and 10 in Table 1), which is similar to the DOLO case.12 Inefficient generation of a hemiacetal ester moiety by the crossover reaction from VE to ROFO is likely responsible for the inertness of ROFOs in the copolymerization with VEs (Scheme 2B). Styrene derivatives [p-methylstyrene (pMeSt) and p-methoxystyrene (pMOS)] were also ineffective as comonomers for copolymerization with ROFOs (entries 7 and 8).
Unlike unsuccessful homopolymerization and copolymerization with VEs, ROFOs were demonstrated to undergo cationic copolymerization with oxiranes. When F1 was copolymerized with VCHO using B(C6F5)3 as a catalyst, both monomers were consumed to reach F1 and VCHO conversion of 57% and 11%, respectively, in 1 min (entry 1 in Table 2). An obtained polymer had an Mn value of 17.8 × 103 (Fig. 1A, black). 1H NMR analysis suggested that the copolymer was produced by the crossover reactions between F1 and VCHO (Fig. 2A). An acetal moiety was generated via the crossover reaction from F1 to VCHO although the acetal peak (peak 11) partly overlapped with the peak of the double bonds in the main chain (peak 10) at 6.0–6.3 ppm. An ester moiety was generated via the crossover reaction from VCHO to F1 although the ester-adjacent methine peak (peak 8) also overlapped with the peaks of the VCHO-derived vinyl groups (peaks 6 and 7). The assignments of peaks shown in Fig. 2A were confirmed by 13C and 2D NMR analyses (Fig. S1–S4†). From the integral ratios of the peaks in the 1H NMR spectrum, the average number of VCHO/F1 units per block was calculated to be 5.4/1.0, respectively. The number of VCHO units per block decreased as the polymerization proceeded (entry 2, 3.6/1.0 for VCHO/R1) because of the faster decrease in the amount of VCHO than that of F1 during polymerization.
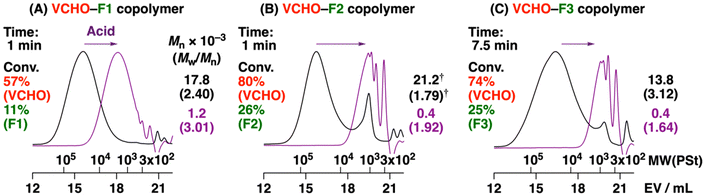 |
| | Fig. 1 MWD curves of (A) VCHO–F1 (entry 1 in Table 2), (B) VCHO–F2 (entry 3), and (C) VCHO–F3 (entry 4) copolymers (black) and their acid hydrolysis products (purple). See Table 2 for the polymerization conditions.† Values for a main peak. | |
 |
| | Fig. 2
1H NMR spectra of (A) VCHO–F1 copolymer (entry 1 in Table 2; Fig. 1A), (B) its acid hydrolysis product (purple curve in Fig. 1A), (C) VCHO–F2 copolymer (entry 3 in Table 2; Fig. 1B), and (D) VCHO–F3 copolymer (entry 4 in Table 2; Fig. 1C, after reprecipitation in methanol). In CDCl3 at 30 °C. The integral ratios of unnecessary peaks (water, residual monomer, etc.) were subtracted. * CHCl3, residual monomer, 1,2-dimethoxyethane, etc. | |
Table 2 Cationic copolymerization of VCHO and ROFOsa
The VCHO–F1 copolymer has acetal moieties in the main chain; hence, the copolymer was degraded into oligomers by acid hydrolysis with HCl. The Mn value decreased from 17.8 × 103 to 1.2 × 103, while keeping a unimodal distribution (Fig. 1A, purple), indicating that acetal moieties were statistically located in the original copolymer. In the 1H NMR spectrum (Fig. 2B), the peaks of the acetal and carbon–carbon double bond moieties at 6.0–6.3 ppm disappeared and instead peaks assigned to an alkenal structure23 appeared (peaks 12–14), which is consistent with the scission of the acetal moiety adjacent to the double bond.
The copolymerization mechanism is explained as follows: the polymerization starts from the interaction between B(C6F5)3 and VCHO or ROFO and/or a proton derived from the reaction between adventitious water and B(C6F5)3. The homopropagation reaction of VCHO proceeds via the oxonium ion-mediated cationic ring-opening mechanism. An ROFO monomer reacts with the VCHO-derived oxonium ion through the attack of the carbonyl oxygen on the carbon atom of the three-membered ring moiety in the oxonium ion (Scheme 3A) in a manner similar to the cases of other cyclic hemiacetal esters (DOLOs and ROPTs).12,13 Subsequently, the ring-opening of the ROFO-derived oxonium ion occurs to generate a carbocation adjacent to the alkoxy group, which has a structure of an oxocarbenium ion. This crossover reaction results in an ester moiety in the main chain (Scheme 3B). The homopropagation reaction of ROFOs does not proceed (Scheme 2A); hence, the carbocation reacts with a VCHO monomer, which is followed by the VCHO homopropagation. An acetal moiety is generated in the main chain by the crossover reaction from ROFO to VCHO. The monomer reactivity ratios of VCHO and F1 were determined to be 4.7 and 0, respectively, by the Meyer–Lowry method (Fig. S5 and Table S1†).24,25 The values are consistent with the average number of monomer units per block in the copolymer chains.
 |
| | Scheme 3 (A) Crossover reaction from VCHO to ROFO, (B) crossover reaction from ROFO to VCHO, and (C) possible interaction of a carbocation with the carbonyl group. | |
Four different ROFO-derived structures are potentially generated in the main chain because the ROFO-derived carbocation has an adjacent carbon–carbon double bond (Scheme 3B). As expected from the resonance structure and the cis–trans configurations, this carbocation potentially reacts with VCHO at not only the alkoxy group-adjacent carbon atom but also the ester-adjacent carbon atom. In addition, cis- and trans-carbon–carbon double bonds are potentially generated after the reaction with VCHO. Among the four possible structures, the copolymer chain was found to exclusively have cis-double bonds derived from the reaction of the alkoxy group-adjacent carbon atom and VCHO, which was confirmed by the comparison of the 1H NMR spectrum with those of the corresponding compounds26 (also confirmed by cis–trans isomerization during the radical reaction; see below). The superiority of an alkoxy group-adjacent carbocation (oxocarbenium ion) in the resonance structures and the interaction between the carbonyl group with a cationic center (Scheme 3C) are possible causes of the highly selective reaction.
F2 and F3, which are isopropoxy and tert-butoxy counterparts of F1, respectively, also underwent cationic copolymerization with VCHO, resulting in copolymers with Mn values over 104 (entries 3 and 4 in Table 2; Fig. 1B and C). The average number of VCHO units per block of the copolymers, which was calculated from 1H NMR spectra (Fig. 2C and D; see Fig. S6–S9† for the 13C and 2D NMR spectra of the VCHO–F2 copolymer), was comparable among the three ROFOs when the samples of similar VCHO conversion were compared (entries 2–4 in Table 2). Small peaks were detected in the MWD curves of the F2- and F3-derived copolymers (Fig. 1B and C). These peaks are likely assigned to cyclic oligomers from the results of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) analysis (Fig. S10†).
Other oxiranes than VCHO were also copolymerized with ROFOs. Copolymers were generated by the copolymerization of CHO with F1, F2, or F3 (entries 1–3 in Table 3, Fig. S11 and S12†), while the MWs of the polymers were lower and the average number of oxirane units per block was larger than the VCHO counterparts. The copolymerization of CPO with ROFOs proceeded at much slower rates than those of the copolymerization with VCHO or CHO (entries 4–6 in Table 3, Fig. S11 and S13†). The MWs and the crossover frequency were comparable to those of the copolymerization with CHO. Unlike these two oxiranes, the use of IBO as an oxirane comonomer resulted in oligomers in the copolymerization with F1 or F2 (entries 7 and 8 in Table 3, Fig. S11†).
Table 3 Cationic copolymerization of ROFOs with CHO, CPO, or IBOa
| Entry |
Oxirane |
ROFO |
Time |
Conv. (%) |
M
n × 10−3b |
M
w/Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Average no. of monomer units per blockc |
| Oxirane |
ROFO |
Oxirane |
ROFO |
|
[Oxirane]0 = 0.75 M, [ROFO]0 = 0.75 M, [B(C6F5)3]0 = 3.0 (except for entries 1 and 2) or 1.0 (entries 1 and 2) mM, in dichloromethane at −78 °C.
Determined by GPC (polystyrene calibration).
Calculated by 1H NMR.
|
| 1 |
CHO |
F1 |
30 s |
90 |
11 |
10.6 |
2.06 |
8.3 |
1.0 |
| 2 |
|
F2 |
1 min |
43 |
9 |
7.9 |
2.55 |
6.0 |
1.0 |
| 3 |
|
F3 |
5 min |
93 |
21 |
2.1 |
2.61 |
4.4 |
1.0 |
| 4 |
CPO |
F1 |
20 h |
59 |
8 |
6.0 |
1.96 |
7.0 |
1.0 |
| 5 |
|
F2 |
68 h |
35 |
5 |
4.3 |
1.35 |
6.2 |
1.0 |
| 6 |
|
F3 |
50 h |
30 |
5 |
4.3 |
1.59 |
7.2 |
1.0 |
| 7 |
IBO |
F1 |
45 h |
2 |
<1 |
0.8 |
1.71 |
— |
— |
| 8 |
|
F2 |
144 h |
4 |
1 |
0.4 |
1.32 |
— |
— |
Comparison of cyclic hemiacetal esters
ROFOs exhibited higher and smaller reactivity than ROPTs and DOLOs did, respectively,12,13 as indicated by the number of oxirane units per block in the copolymers (Fig. 3). The nucleophilicity of cyclic hemiacetal esters (paths i and ii in Scheme 4; VCHO–ROFO copolymerization as an example), the ring-opening rate of the oxonium ion (path iii), and/or the reaction rate of the carbocation and an oxirane (path iv and v) are likely responsible for the difference in the incorporated ratios of cyclic hemiacetal esters in copolymers. This is considered assuming that the cyclic hemiacetal ester-derived oxonium ion formation (path ii in Scheme 4), the ring-opening reaction (path iii), and the oxirane-derived oxonium ion formation by the reaction of the carbocation and oxirane (path iv) are reversible processes. The difference of the carbocation stability (Scheme 1A) potentially affects the frequency of carbocation generation, the reverse reaction from the carbocation to the oxonium ion, and the reaction of a carbocation with an oxirane (paths iii and iv).
 |
| | Fig. 3 Comparison of the Mn values and the average number of monomer units per block of the copolymers obtained by the copolymerization of oxiranes and cyclic hemiacetal esters ([oxirane]0 = 0.75 M, [cyclic hemiacetal ester]0 = 0.75 M, [B(C6F5)3]0 = 1.0 or 3.0 mM), in dichloromethane at −78 °C. Ref. 12 and 13 for the DOLO and ROPT data, respectively (except for the DOLO–CPO copolymer [unpublished data]). | |
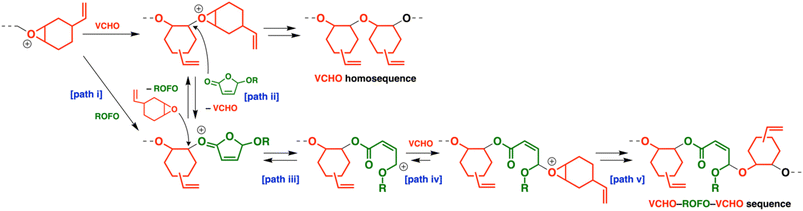 |
| | Scheme 4 Plausible processes of the propagation reactions. | |
ROFOs and ROPTs exhibited higher reactivity than DOLO did, while the copolymerization of ROFOs or ROPTs with IBO was not effective unlike that of DOLO (Fig. 3).12 The ROFO- and ROPT-derived carbocations, which have double bond- and aryl group-conjugated structures, respectively (Scheme 1A), are likely not suited for the reaction with IBO.
Cationic terpolymerization
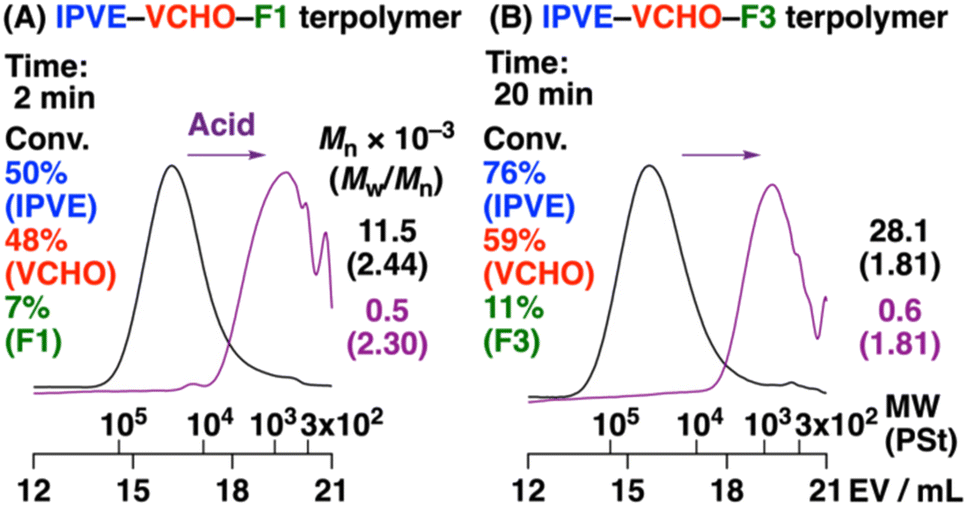
Cationic copolymerization of VEs and ROFOs did not proceed as explained above, while ROFOs potentially undergo cationic terpolymerization with VEs and oxiranes, like the DOLO and ROPT cases,12,13 because the ROFO-derived carbocation (oxocarbenium ion), which is generated by the reaction with an oxirane, is expected to react with a VE monomer. We therefore conducted terpolymerization of IPVE, VCHO, and ROFOs under similar conditions to those for the copolymerization of VCHO and ROFOs. A higher concentration of IPVE than that of VCHO was used to facilitate crossover reactions from VCHO to ROFO. As a result, polymers with Mn values of 11.5, 8.0, or 28.1 × 103 were obtained from F1, F2, or F3, respectively (entries 1–3 in Table 4; Fig. 4). 1H NMR analysis of the products suggested that IPVE, VCHO, and ROFOs were incorporated into polymer chains (Fig. 5). From the integral ratios of key peaks (explained below), the average number of IPVE/VCHO/ROFO units was calculated to be 7.1/1.8/1.0, 5.1/1.9/1.0, and 6.7/1.3/1.0 for F1, F2, and F3, respectively, indicating that terpolymers were successfully produced by the crossover reactions among the three monomers. The terpolymers could be degraded into low-MW compounds by acid via the cleavage of the acetal moieties in the main chain (Fig. 4, purple).
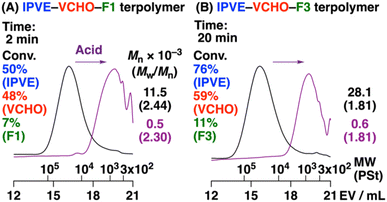 |
| | Fig. 4 MWD curves of (A) IPVE–VCHO–F1 and (B) IPVE–VCHO–F3 terpolymers (black) and their acid hydrolysis products (purple). See Table 4 for the polymerization conditions. | |
 |
| | Fig. 5
1H NMR spectra of (A) IPVE–VCHO–F1 terpolymer (entry 1 in Table 3; Fig. 4A), (B) its acid hydrolysis product (purple curve in Fig. 4A), (C) IPVE–VCHO–F3 terpolymer (entry 3 in Table 3; Fig. 4B), and (D) its acid hydrolysis product (purple curve in Fig. 4B). In CDCl3 at 30 °C. *CHCl3, water, 1,2-dimethoxyethane, etc. | |
Table 4 Cationic terpolymerization of VE, VCHO, and ROFOa
The terpolymerization most likely proceeded via selective crossover reactions at the VE- and VCHO-derived propagating ends (Scheme 5). The VE-derived carbocation reacted with VE or VCHO, while it did not react with ROFO as expected by ineffective copolymerization of VE and ROFO (Scheme 5A). At the VCHO-derived end, a carbocation was not generated by the ring-opening reaction of the VCHO-derived oxonium ion, resulting in the absence of the reaction with VE (Scheme 5B), as demonstrated by our previous studies.12,13,27 Accordingly, the VCHO-derived oxonium ion reacted with VCHO or ROFO (Scheme 5B).
 |
| | Scheme 5 Cationic terpolymerization of IPVE, VCHO, and ROFO: Propagation reactions at (A) VE-, (B) VCHO, and (C) ROFO-derived propagating ends. | |
We revealed the reaction selectivity at the ROFO-derived propagating end by elaborate analysis of the 1H NMR spectra (Fig. 5). The amounts of homosequence units (IPVE and VCHO) and crossover reaction-derived units (IPVE-to-VCHO, ROFO-to-IPVE, and ROFO-to-VCHO crossover reactions) were calculated by assuming that the amounts of the ROFO-to-IPVE and IPVE-to-VCHO crossover reaction-derived units are the same. For example, from the integral ratios of peaks at 5.6–6.3 (peaks 9, 15, 16, and 18), 4.6–5.4 (peaks 10–12, 14, and 17), 3.0–4.1 (peaks 2, 3, 5, 13, and a), and 2.1–2.5 (peak 8) ppm (Fig. 5A), we concluded that 71% and 29% of the F1-derived propagating species reacted with IPVE and VCHO, respectively (entry 1 in Table 4). Interestingly, this ratio varied among ROFOs (Scheme 5C) and it is most likely that the F3-derived propagating species exclusively reacted with IPVE (>99%/<1% for IPVE/VCHO; entry 3; Fig. 5C; see Fig. S14–S17† for the 13C and 2D NMR spectra). Indeed, peaks assigned to the structures resulting from the degradation of the VCHO–ROFO–VCHO sequence (peaks 25–27) were absent in the 1H NMR spectrum of the degradation product (Fig. 5D), which is in contrast to the F1 counterpart (Fig. 5B). The difference likely stemmed from the stabilities of the acetal moieties resulting from the reaction of the ROFO-derived carbocation and an oxirane. Methoxy, isopropoxy, or tert-butoxy group-containing acetal moieties are generated from F1, F2, or F3, respectively. The stability of acetals decreases with an increase in bulkiness of alkoxy groups. Indeed, the acid hydrolysis rates of acetals were reported to increase in the order of methoxy < isopropoxy < tert-butoxy group-containing acetal.28 In the terpolymerization, the F3-derived acetal moieties were less favorably generated by the reaction of the F3-derived carbocation with VCHO, resulting in preferential reactions with IPVE, compared to the F1 case. Similar exclusive reactions for VEs rather than oxiranes were observed in the terpolymerization of VE, oxirane, and ROPT in our previous study.13 The use of ethyl VE, which is a less reactive VE than IPVE, resulted in a terpolymer with shorter VE homosequences (entry 4 in Table 4), while keeping the very high selectivity of the crossover reactions at the F3-derived propagating end (98%/2% for EVE/VCHO). We tried to analyze the terpolymer by MALDI-TOF-MS; however, it was difficult to detect clear peaks because the product is composed of many polymer chains consisting of the different number of the three monomers (Fig. S18†). Sequences consisting of VCHO–ROFO–IPVE were detected by electrospray ionization mass spectrometry (ESI-MS) analysis of the products obtained by acid hydrolysis of the terpolymers (Fig. S19†).
Cationic alternating copolymerization of vinyl acetate and ROFO
We previously reported that vinyl acetate, which is a cationically nonhomopolymerizable monomer, undergoes cationic alternating copolymerization with DOLOs or ROPTs.29,30 To examine the potential of ROFOs as comonomers for vinyl acetate, we conducted cationic copolymerization of vinyl acetate and F1 with GaCl3 as a catalyst in dichloromethane at −40 °C. As a result, copolymerization successfully proceeded to yield an alternating copolymer [Mn(GPC) = 5.4 × 103; Fig. S20†]. MALDI-TOF-MS analysis also suggested that the copolymer chains had alternating sequences (Fig. S21†).
Thiol–ene reaction of ROFO-derived copolymer
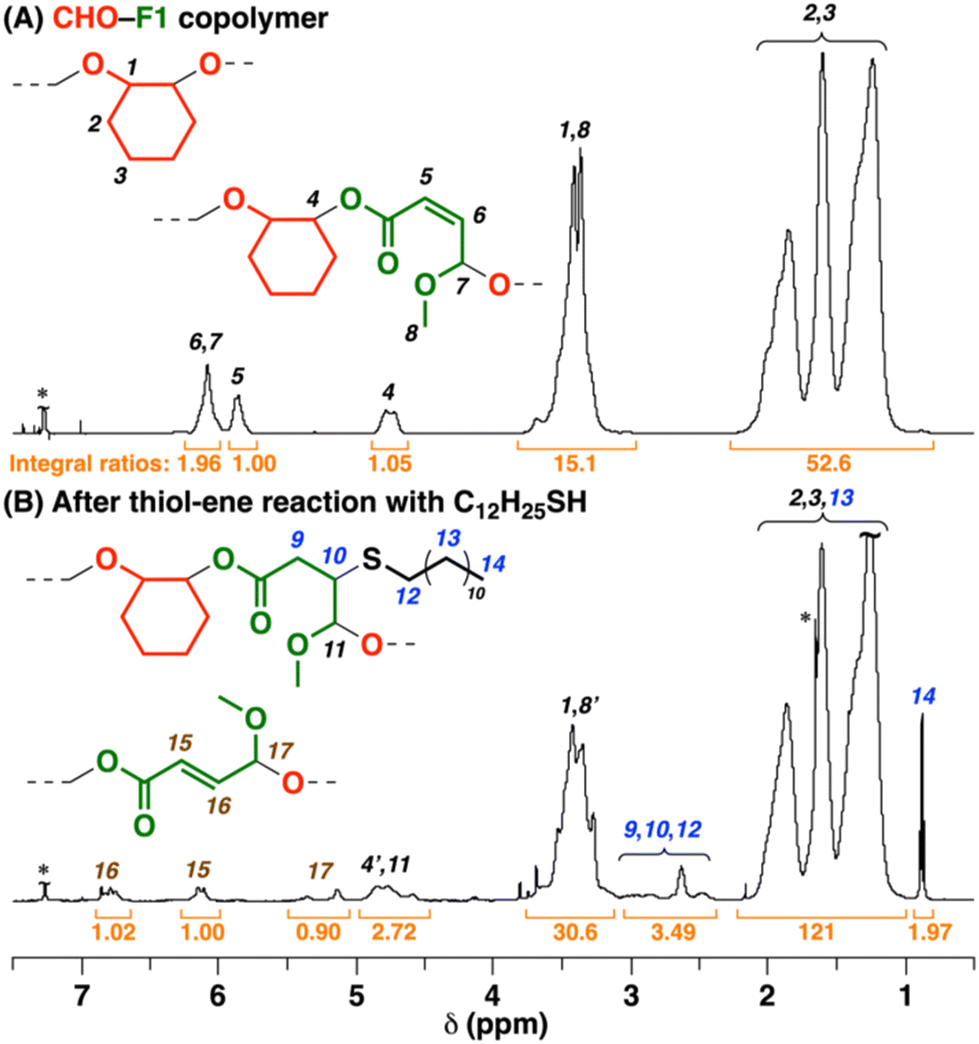
The copolymers obtained from ROFOs have β-substituted acrylate (isocrotonate) moieties in the main chain. To demonstrate functionalization of the β-substituted acrylate moieties, we conducted the thiol–ene reaction31,32 using 1-dodecanethiol as a model thiol. After the reaction of a CHO–F1 copolymer using 2,2′-azobis(isobutyronitrile) (AIBN) as a radical generator for 24 h, a peak assigned to the methyl group of the 1-dodecanethiol moieties was observed at 0.9 ppm (peak 14 in Fig. 6B) in the 1H NMR spectrum of the product, which indicates the successful incorporation of the thiol by the thiol–ene reaction (incorporated ratio = 40%).
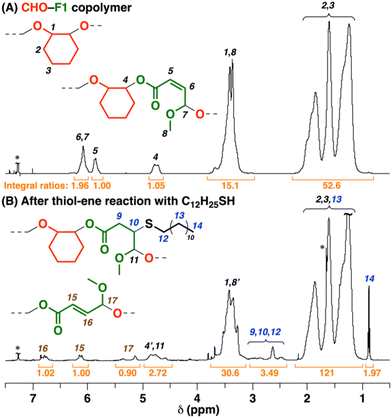 |
| | Fig. 6
1H NMR spectra of (A) CHO–F1 copolymer (obtained under similar conditions to those for entry 1 in Table 3; the average number of CHO/F1 units per block = 6.6/1.0) and (B) the product obtained by the thiol–ene reaction of the copolymer ([double bonds in copolymer]0 = 0.062 M, [C12H25SH]0 = 0.31 M, [AIBN]0 = 0.031 M, in toluene at 80 °C, 24 h). See Fig. S22† for the MWD curves. * CHCl3 or water. | |
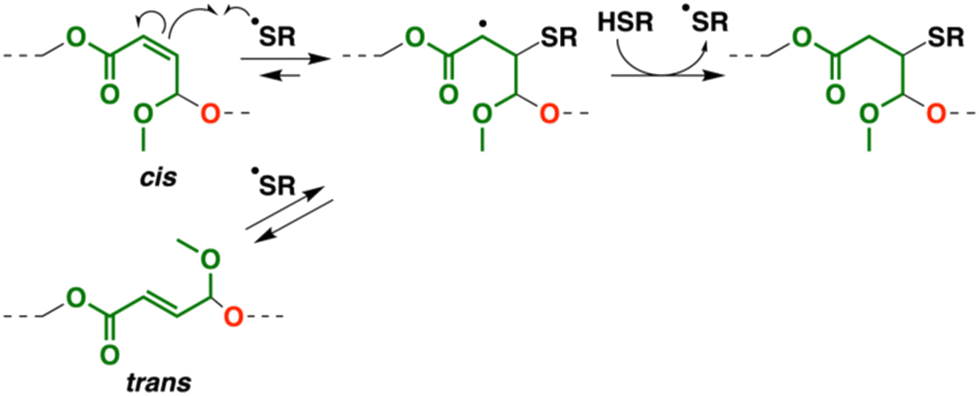
Interestingly, in the 1H NMR spectra, the peaks of the cis-double bonds in the original copolymer (peaks 5 and 6 in Fig. 6A) disappeared, while new peaks emerged at 6.8, 6.1, and 5.1–5.4 ppm (peaks 15–17 in Fig. 6B). These peaks were assigned to trans-double bonds,26 which indicates that the intermediate radical species generated by the reaction of the cis-double bond and a thiyl radical underwent the thiyl-radical elimination reaction into not the original cis-double bond but a trans-double bond during the thiol–ene reaction (Scheme 6).33
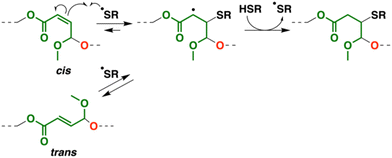 |
| | Scheme 6 Thiol–ene reaction and isomerization into trans-double bond. | |
Thermal properties
The thermal properties of the copolymers consisting of oxiranes and ROFOs were examined by differential scanning calorimetry (DSC). VCHO–F1, VCHO–F2, and VCHO–F3 copolymers exhibited glass transition temperatures (Tgs) of 60, 45, and 66 °C, respectively (entries 1–3 in Table 5, Fig. S23†), which were higher or comparable to the Tg of a VCHO homopolymer (50 °C, entry 4). The difference of the alkoxy groups derived from ROFOs affected the thermal properties of the copolymers. The balance of flexibility and bulkiness of the alkyl substituents likely affected the Tgs. Indeed, the order of the Tgs (substituent: tBu > Me > iPr) is similar to those of poly(alkyl acrylate)s and poly(alkyl methacrylate)s.34
Table 5
T
g values of VCHO–ROFO copolymers and VCHO homopolymera
| Entry |
Polymer |
Average no. of monomer units per blockb |
M
n × 10−3c |
T
g (°C) |
| VCHO |
ROFO |
|
By DSC (the second heating scan, 10 °C min−1; Fig. S23†).
Calculated by 1H NMR. After purification by preparative GPC (entries 1 and 2) or reprecipitation in methanol (entry 3).
Determined by GPC (polystyrene calibration).
From ref. 13.
|
| 1 |
VCHO–F1 |
3.9 |
1.0 |
17.8 |
60 |
| 2 |
VCHO–F2 |
3.3 |
1.0 |
17.8 |
45 |
| 3 |
VCHO–F3 |
3.0 |
1.0 |
13.8 |
66 |
| 4d |
VCHO homo |
— |
— |
45.1 |
50 |
Conclusion
In conclusion, ROFOs successfully underwent cationic copolymerization with oxiranes and terpolymerization with VEs and oxiranes using B(C6F5)3 as a catalyst, resulting in acidically degradable polymers. Homopolymerization of ROFOs did not proceed, while copolymerization with oxiranes smoothly proceeded because of the efficient generation of ester and acetal moieties by the crossover reactions between oxiranes and ROFOs. One of the four possible structures, which are related to the conjugated structure and cis–trans configuration of the ROFO-derived carbocation, was selectively generated by the crossover reactions from ROFO. The terpolymerization also proceeded via the highly selective crossover reactions particularly when F3 was used, resulting in a terpolymer with repetitive IPVEn–VCHOm–F3 sequences. In addition, we could use the ROFO-derived β-substituted acrylate moieties as reaction sites for polymer modification by the thiol–ene reaction. The results obtained in this study will contribute to both understanding the design strategy of nonhomopolymerizable comonomers for cationic copolymerization and developing new degradable polymers from bio-derived monomers.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the ESI.†
Acknowledgements
This work was partially supported by JSPS KAKENHI Grant 23H02010.
References
-
S. Penczek and K. Kaluzynski, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier B.V., Amsterdam, 2012, Vol. 4.02 Search PubMed.
-
P. Kubisa, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier B.V., Amsterdam, 2012, Vol. 4.08 Search PubMed.
- S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
-
P. Kubisa and J. P. Vairon, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier B.V., Amsterdam, 2012, Vol. 4.10 Search PubMed.
- K. Maruyama, A. Kanazawa and S. Aoshima, Polym. Chem., 2019, 10, 5304–5314 RSC.
- K. Maruyama, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 4034–4045 CrossRef CAS.
- S. A. Cairns, A. Schultheiss and M. P. Shaver, Polym. Chem., 2017, 8, 2990–2996 RSC.
- Y. Xu, M. R. Perry, S. A. Cairns and M. P. Shaver, Polym. Chem., 2019, 10, 3048–3054 RSC.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- T. Şucu, M. Wang and M. P. Shaver, Macromolecules, 2023, 56, 1625–1632 CrossRef PubMed.
- K. Hyoi, A. Kanazawa and S. Aoshima, ACS Macro Lett., 2019, 8, 128–133 CrossRef CAS PubMed.
- Y. Takahashi, A. Kanazawa and S. Aoshima, Macromolecules, 2023, 56, 4198–4207 CrossRef CAS.
- A. E. Neitzel, T. J. Haversang and M. A. Hillmyer, Ind. Eng. Chem. Res., 2016, 55, 11747–11755 CrossRef CAS.
- A. E. Neitzel, L. Barreda, J. T. Trotta, G. W. Fahnhorst, T. J. Haversang, T. R. Hoye, B. P. Fors and M. A. Hillmyer, Polym. Chem., 2019, 10, 4573–4583 RSC.
- M. Okada, H. Sumitomo and Y. Yamamoto, Makromol. Chem., 1974, 175, 3023–3028 CrossRef.
- G. O. Schenck, Justus Liebigs Ann. Chem., 1953, 584, 156–176 CrossRef CAS.
- J. C. de Jong, F. van Bolhus and B. L. Feringa, Tetrahedron: Asymmetry, 1991, 2, 1247–1262 CrossRef CAS.
- P. Esser, B. Pohlmann and H.-D. Scharf, Angew. Chem., Int. Ed. Engl., 1994, 33, 2009–2023 CrossRef.
- Y. Morita, H. Tokuyama and T. Fukuyama, Org. Lett., 2005, 7, 4337–4340 CrossRef CAS PubMed.
- J. G. H. Hermens, T. Freese, K. J. van den Berg, R. van Gemert and B. L. Feringa, Sci. Adv., 2020, 6, eabe0026 CrossRef CAS PubMed.
- M. L. Lepage, G. Alachouzos, J. G. H. Hermens, N. Elders, K. J. van den Berg and B. L. Feringa, J. Am. Chem. Soc., 2023, 145, 17211–17219 CrossRef CAS PubMed.
- L.-L. Shen, H.-S. Mun and J.-H. Jeong, Eur. J. Org. Chem., 2010, 6895–6899 CrossRef CAS.
- V. E. Meyer and G. G. Lowry, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 2843–2851 CrossRef CAS.
- N. A. Lynd, R. C. Ferrier Jr and B. S. Beckingham, Macromolecules, 2019, 52, 2277–2285 CrossRef CAS.
- H.-D. Scharf and J. Janus, Chem. Ber., 1978, 111, 2741–2744 CrossRef CAS.
- M. Mimura, A. Kanazawa and S. Aoshima, Macromolecules, 2019, 52, 7572–7583 CrossRef CAS.
- A. T. N. Belarmino, S. Froehner, D. Zanette, J. P. S. Farah, C. A. Bunton and L. S. Romsted, J. Org. Chem., 2003, 68, 706–717 CrossRef CAS PubMed.
- J. Azuma, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 6110–6119 CrossRef CAS.
- K. Kamigaki, S. Aoshima and A. Kanazawa, ACS Macro Lett., 2024, 13, 754–760 CrossRef CAS PubMed.
- B. H. Northtop and R. N. Coffey, J. Am. Chem. Soc., 2012, 134, 13804–13817 CrossRef PubMed.
- Y. Yu, M. Kim, G. S. Lee, H. W. Lee, J. G. Kim and B.-S. Kim, Macromolecules, 2021, 54, 10903–10913 CrossRef CAS.
- C. Walling and W. Helmreich, J. Am. Chem. Soc., 1959, 81, 1144–1148 CrossRef CAS.
-
R. J. Andrews and E. A. Grulke, in Polymer Handbook, Fourth Edition, ed. J. Brandrup, E. H. Immergut and E. A. Grulke, Wiley-Interscience, 1999, ch. VI, p. 193 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, polymerization data, and DSC data. See DOI: https://doi.org/10.1039/d5py00255a |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Arihiro
Kanazawa
and
Arihiro
Kanazawa