
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional dithiolane monomers for multi-scale, recyclable light-driven additive manufacturing†
Benjamin R.
Nelson
 ab,
Jaxon T.
Cione
a,
Bruce E.
Kirkpatrick
abc,
Kendra M.
Kreienbrink
be,
Abhishek P.
Dhand
d,
Jason A.
Burdick
abde,
C. Wyatt
Shields IV
aef,
Kristi S.
Anseth
abe and
Christopher N.
Bowman
*ae
ab,
Jaxon T.
Cione
a,
Bruce E.
Kirkpatrick
abc,
Kendra M.
Kreienbrink
be,
Abhishek P.
Dhand
d,
Jason A.
Burdick
abde,
C. Wyatt
Shields IV
aef,
Kristi S.
Anseth
abe and
Christopher N.
Bowman
*ae
aDepartment of Chemical and Biological Engineering, University of Colorado Boulder, Boulder CO 80303, USA. E-mail: christopher.bowman@colorado.edu
bBioFrontiers Institute, University of Colorado Boulder, Boulder, CO 80303, USA
cMedical Scientist Training Program, University of Colorado Anschutz Medical Campus, Aurora CO 80045, USA
dDepartment of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA
eMaterials Science & Engineering Program, University of Colorado Boulder, Boulder, CO 80303, USA
fBiomedical Engineering Program, University of Colorado, Boulder, Boulder, CO 80303, USA
First published on 2nd April 2025
Abstract
In this work, we develop a tetrafunctional monomer incorporating 1,2-dithiolanes as the reactive group, lipoic acid pentaerythritol ethoxylate, which is capable of photopolymerization and is suitable for light-based additive manufacturing with high spatial resolution across various length scales. This monomer polymerizes in either the presence or absence of exogenous photoinitiator. Using dynamic light processing and two photon lithography techniques, parts were printed on size scales ranging from multiple cm to μm, with resolution as small as 1 μm. As a result of the dithiolane polymerization, linear disulfides are formed, forming covalent adaptable networks directly from the polymerization reaction. Furthermore, through heating and dilution in solvent, the network was recycled back to the lipoic acid functional monomer with approximately 95% monomer recovery, which was subsequently repolymerized to achieve nearly identical modulus evolution as a function of exposure time. This work represents an advance in the development of multifunctional dithiolane monomers, as well as recyclable resins for additive manufacturing that are capable of polymerization with or without exogenous photoinitiators.
Introduction
Compared to linear thermoplastics, thermoset polymers are attractive for high performance applications owing to their shape stability and resistance to physical deformation.1 Furthermore, light-based crosslinking affords spatiotemporal resolution in the production of thermosets, enabling precise manipulation of shape and stiffness.2 Developments in photopolymerization have led to a range of new technologies, including UV and visible light-curable coatings,3 photolithography-based 3D printing,4 and biofabrication techniques.5 Recent advances in vat photopolymerization methods for 3D printing have improved upon the flexibility, speed, and resolution of printing.6 In particular, digital light processing (DLP) has emerged as a versatile light-driven additive manufacturing method that uses planar light projection to crosslink a liquid photo-reactive resin into a 3D object in a layer-by-layer manner. Although DLP enables rapid fabrication, object resolution is often limited by the pixel size (typically about 25 to 50 μm). As a complementary technique, two-photon lithography (2PL) uses a pulsed laser to scan and raster a photoreactive resin in a point or line-by-line manner. As a result, features with sub-micron resolution are readily fabricated with 2PL, and 2PL has continued to increase the resolution achieved within printed parts and features,7 but the process is less scalable. However, compared to traditional thermoplastic extrusion printing methods, DLP and 2PL technologies achieve much smaller feature sizes and more complex geometries, while also facilitating faster printing.8 Despite the promise of these 3D printing modalities for the fabrication of multifunctional materials, their use in the processing of dynamic materials has been largely limited.As printing technologies improve, these systems become more attractive for applications including rapid prototyping, high throughput and automatable manufacturing, and fabrication of non-moldable parts.6a However, the permanent crosslinks that give rise to thermoset materials and their desirable properties also restrict their recycling and reprocessing. Covalent adaptable networks (CANs) have been proposed as a potential solution to overcome these limitations, through either remolding parts to a new physical shape or degrading them to soluble oligomers or small molecules/monomers that can be subsequently reused.9 Specifically, complete chemical recycling to monomer or partial degradation to functional oligomers is promising for sustainable 3D printing, as these products are then capable of subsequent reutilization in a variety of applications rather than only being subject to mechanical reprocessing. However, such systems often require the addition of virgin monomer to the recycled resin for reprinting which diminishes the ultimate fraction of material that is recyclable.10 While recent studies have identified systems capable of polymerization and subsequent recycling back to a functional monomer, many of these resins are not amenable to subsequent photopolymerization.11 A fundamental limitation remains that CANs are significantly more amenable to reprocessing rather than true recycling, which greatly limits their applicability for light-based additive manufacturing.
Disulfide bonds, one of the oldest forms of dynamic chemistries in polymers,12 are attractive for the chemical recycling of thermosets back to a repolymerizable monomer. 1,2-Dithiolanes, a class of ring strained cyclic disulfides, have long been known to undergo a light-driven ring-opening polymerization reaction,13 and recently are undergoing a renaissance in the production of polymers. Notably, this ring-opening reaction directly results in the formation of linear disulfides, integrating a dynamic chemistry into the polymer backbone. With a reported ceiling temperature of approximately 140 °C, dithiolanes have been investigated for depolymerization back to the ring-closed monomer.14 Recent studies have prepared both linear and crosslinked polymers from dithiolanes through a variety of polymerization reactions, including direct photolysis,15 thermally driven reactions,16 and radical,17 cationic,18 and anionic19 polymerizations. Furthermore, radical copolymerizations of dithiolanes and various alkenes have been reported, enabling systematic control over the dynamic content of these polymers.20
To date, there has been limited exploration of multifunctional dithiolanes in crosslinked thermosets. Most network-forming strategies thus far have relied on modification of the carboxylic acid present on many dithiolanes to add supramolecular interactions or an additional crosslinking moiety, or the side-chain functionalization of a linear polymer with dithiolanes to enable crosslinking.21 However, for use in recyclable additive manufacturing applications, the development of multifunctional monomers utilizing dithiolanes as the reactive species would have significant advantages. Recently, such monomers have been prepared as the interest in both dithiolanes as polymerizable moieties and additive manufacturing grows.22 Dithiolanes are an attractive chemistry for the development of CANs through 3D printing, as the polymerization directly results in the formation of dynamic bonds, unlike many other dynamic chemistries where the exchangeable bond must be included within the monomer. Furthermore, as a ceiling temperature polymer, which is able to revert to the ring closed form, dithiolanes have the potential to depolymerize back to a repolymerizable monomer through simple heating and dilution. Thus, using a simple disulfidation reaction, it is possible to modulate the dynamic content of the material post-polymerizaiton. In this work, we synthesize a novel tetrafunctional dithiolane monomer, lipoic acid pentaerythritol ethoxylate (LAPEO) using Fischer esterification, which is an easily scalable and inexpensive synthesis strategy. LAPEO is capable of photopolymerization into crosslinked thermosets both with and without photoinitiator. When heat is applied to the resultant network, thermal stress relaxation driven by disulfide bond rearrangement enables reprocessing of photopolymerized networks. This inherent dynamic behavior is tempered through a post-polymerization disulfidation reaction with a vinyl ether. Furthermore, in the presence of a suitable excess of solvent, heating above the ceiling temperature causes the polymer to revert to the ring-closed LAPEO monomer, which is then readily recovered and repolymerized, achieving effectively identical mechanical properties in comparison to those of the virgin resin. When used as a photopolymerizable 3D printing resin, LAPEO forms a rubbery network capable of reversion to monomer and subsequent reprinting, enabling a fully circular light-based additive manufacturing resin system amenable to multiple printing methods and length scales. For most 3D printing applications, reactions containing initiator are preferred due to improved reaction rates. However, recycling films produced without initiator eliminates residual initiator and initiator fragments that would otherwise accumulate across recycling generations, which results in increased light attenuation. When dithiolanes are polymerized without initiators, the monomer, polymerization reaction, and final dynamic chemistry allows one to functionally and theoretically infinitely extend the possible number of polymerization cycles. This monomer system advances the state of both dithiolanes as dynamic crosslinked polymers and recyclable 3D printing systems across multiple length scales.
Results and discussion
LAPEO, a tetra-functional dithiolane monomer (Fig. 1a), was synthesized from pentaerythritol ethoxylate 15/4 as a core, and lipoic acid, a commonly available and naturally sourceable dithiolane, as the reactive end group. Briefly, pentaerythritol ethoxylate 15/4 and lipoic acid were refluxed in toluene for 18 hours in the presence of catalytic sulfuric acid (Fig. S1†), reacting the carboxylic acid of the dithiolane with the terminal alcohol of the pentaerythritol ethoxylate to form an ester linkage. The reaction proceeded to high conversion as evidenced by the high dithiolane functionality of the monomers as confirmed by NMR (Fig. S2 and S3,† >95%, additionally characterized by FTIR, Fig. S4†) and high yield of monomer (∼80%). Notably, despite heating of the dithiolanes above 70 °C, i.e., the temperature at which thermal polymerization was observed under other conditions,21a polymer formation was not observed during the synthesis. This behavior is attributed to the ceiling temperature behavior of the dithiolane polymerization and the dilute nature of this reaction (dithiolane concentration of 0.2 M).14 The Fischer esterification was chosen because of the potential for scaleup compared to reactions that involve hazardous or expensive reagents and complex workups unsuitable for industrial production. A viscous yellow liquid monomer, capable of photopolymerization with and without initiator (Fig. 1b), was obtained from this reaction. Using a similar Fischer esterification, a bifunctional control was also synthesized to compare the gelation of a 2 vs. 4 arm system (Fig. S5–S8†). | ||
| Fig. 1 (a) LAPEO monomer structure. (b) Polymerization and depolymerization of dithiolanes to form or to recycle networks, respectively. Inset is a polymerized LAPEO film after solvent removal, illustrating network transparency. (c) Initiator-free polymerization of LAPEO diluted in toluene (monomer at 53 w/w%, 20 mW cm−2 405 nm light). Dashed line indicates beginning of light exposure. (d) LAPEO polymerized with photoinitiator (BAPO, 0.5 w/w%, monomer at 53 w/w%, varied intensities 405 nm light). Dashed lines indicate region of light exposure. (e) Tensile testing of LAPEO films at 0.5% strain per s (monomer polymerized at 53 w/w%, with 0.5 w/w% BAPO added, irradiated with 20 mW cm−2 405 nm light for 5 min per side, 100 μm samples, solvent removed at 60 °C for 18 hours). Three replicate traces are shown. Young's modulus = 3.0 ± 0.1 MPa, ultimate tensile strength = 1.6 ± 0.2 MPa, strain to break = 110 ± 10%. | ||
To limit the optical thickness of the resin (LAPEO ε = 68 M−1 cm−1 at 405 nm, LAPEO ε = 400 M−1 cm−1 at 365 nm in decadic molar extinction, Fig. S9†), monomer was diluted in toluene prior to polymerization (monomer at 53 w/w%). Toluene was chosen as a solvent because of its excellent optical properties. Photopolymerization of LAPEO without initiator proceeded relatively slowly under 405 nm light (20 mW cm−2) with gelation (indicated by a crossover in the storage and loss moduli) observed after approximately 400 s (Fig. 1c). While the initiatorless LAPEO polymerization proceeded much more rapidly when initiated at shorter wavelengths, e.g., 365 nm, significant optical attenuation of the light occurs due to strong absorption at the lower wavelengths. To overcome this speed limitation, the photoinitiator phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide (BAPO, 0.5 w/w%) was added, greatly reducing the time to gelation (Fig. 1d). Again, to limit light attenuation, polymerizations were conducted in solution, with monomer diluted in toluene (Fig. S10†). Both with and without initiator, polymerized films reached in situ storage moduli of approximately 3 MPa. Post polymerization, LAPEO formed an optically transparent elastomer film (Fig. 1b). Of note, the final modulus in the photoinitiator-free system was higher than that of the sample containing BAPO by a factor of approximately 1.5, which may be attributed to both slower initiation (longer kinetic chains) and solvent loss over time. To account for solvent loss after polymerization, all subsequent mechanical measurements were performed on dried samples. Solvent was removed in an oven at 60 °C for 18 hours prior to mechanical testing, resulting in approximately 20% linear shrinkage. Dynamic mechanical analysis revealed a Tg of −30 °C with a full width half max of 15 °C, indicating a relatively homogenous network structure (Fig. S11†). Furthermore, differential scanning calorimetry indicates a higher Tg of −11 °C (Fig. S12†).
In tensile testing, these materials exhibit a high strain to break of 110 ± 10%, reaching an ultimate tensile strength of 1.6 ± 0.2 MPa. Furthermore, given the sulfur content of these networks, these materials also have excellent optical properties as far as clarity and refractive index. The refractive index of the monomer is 1.512 at 589.3 nm, rising to 1.529 after polymerization for a Δn of 0.017. Additionally, these materials exhibit high transmission through the visible region (Fig. S13†).
Since the crosslinks present in this material are disulfide bonds, the network remains dynamic after polymerization. By raising the temperature, stress relaxation increases proportional to the rate of disulfide metathesis. To characterize this behavior, samples (100 μm thick) were polymerized using 0.5 wt% BAPO and exposed to 20 mW cm−2 405 nm light for 5 minutes per side, followed by solvent removal at 60 °C for 18 hours. Stress relaxation was performed at various temperatures, holding the strain at 5%. At ambient temperatures, stress relaxation is limited; however, at elevated temperatures the samples relieve significant amounts of stress. At 150 °C, the stress is completely relaxed within 8 hours (Fig. 2a). An activation energy of 148 ± 2 kJ mol−1 was calculated from the Arrhenius behavior of the stress relaxation, which is similar to values reported for other disulfide-crosslinked CANs.23
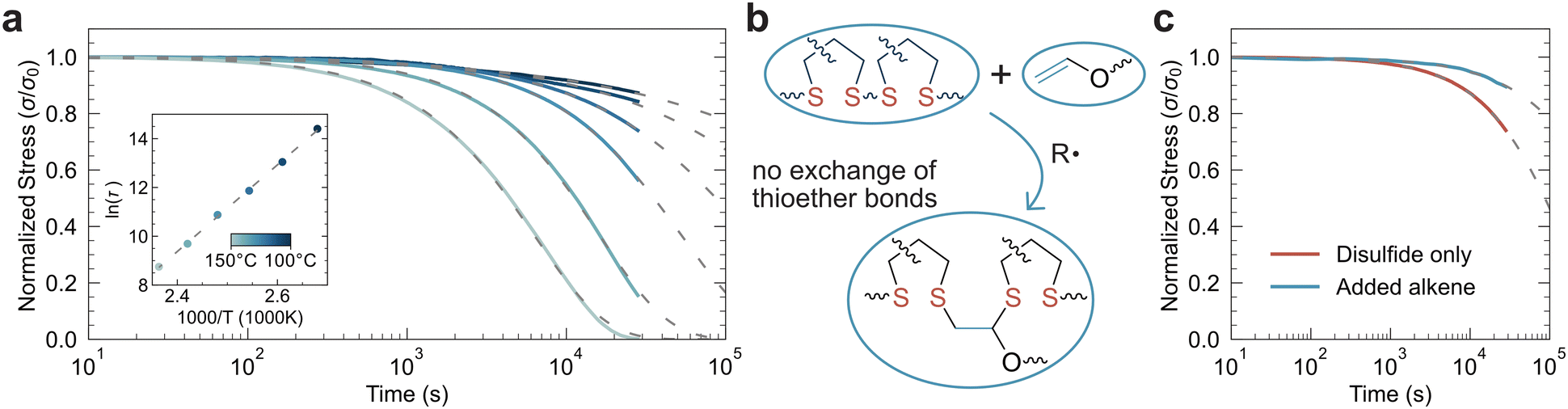 | ||
| Fig. 2 Dynamic behavior of polymerized films. (a) Stress relaxation at temperatures varied from 100–150 °C (darkest blue = 100 °C, lightest blue = 150 °C). Dashed lines indicate the stretched exponential fit. Inset: Arrhenius plot of tau as a function of temperature to calculate the activation energy for stress relaxation. From the fit, Ea = 148 ± 2 kJ mol−1. (b) Scheme for the disulfidation reaction with a vinyl ether which subsequently limits dynamic behavior. (c) Stress relaxation with and without disulfidation post-processing at 120 °C. The relaxation times were found to be, with disulfidation τ = 8.1 × 105 s and without disulfidation, τ = 1.4 × 105 s. | ||
While dynamic bond rearrangement is a valuable tool for altering material structures and recycling, it is not always a desirable feature in the final material produced, particularly when the dynamic chemistry is thermally triggered. To remove disulfides from the network without overall modification to the network architecture, the disulfidation reaction was employed. Using a monofunctional alkene monomer, the disulfide bond was replaced by two thioethers, effectively inserting a carbon–carbon single bond (from the alkene) flanked by thioether linkages in place of the previous disulfide (Fig. 2b). Previous studies have shown that vinyl ethers are particularly favorable for this reaction, so ethylene glycol vinyl ether (EGVE) was selected.24 60 w/w% EGVE and 0.5 w/w% BAPO were dissolved in acetone, and the LAPEO film was swelled overnight in this solution. Since there is no effective change in the network connectivity, and only a minor increase in  (since EDVE has a molecular weight of 88.11, a single addition of EGVE across a disulfide theoretically increases
(since EDVE has a molecular weight of 88.11, a single addition of EGVE across a disulfide theoretically increases  by 11%) there should be a limited effect on the modulus of the material, which was confirmed with in situ rheology (Fig. S14†). Comparing the stress relaxation of films at 120 °C, there was a 5-fold increase in the characteristic time for stress relaxation after disulfidation (Fig. 2c). Notably, these materials are more brittle after the disulfidation, reaching a strain to break of 25 ± 3% at a similar ultimate tensile strength of 1.7 ± 0.3 MPa. We attributed this behavior to decreased dynamic content of the network, which reduces the bond exchange rate and correspondingly its extensibility (Fig. S15†). However, upon disulfidation, the refractive index of the material remains constant at 1.529, but the attenuation in the UV region is shifted to shorter wavelengths, likely through the conversion of both linear and cyclic disulfides to thioethers (Fig. S16†).
by 11%) there should be a limited effect on the modulus of the material, which was confirmed with in situ rheology (Fig. S14†). Comparing the stress relaxation of films at 120 °C, there was a 5-fold increase in the characteristic time for stress relaxation after disulfidation (Fig. 2c). Notably, these materials are more brittle after the disulfidation, reaching a strain to break of 25 ± 3% at a similar ultimate tensile strength of 1.7 ± 0.3 MPa. We attributed this behavior to decreased dynamic content of the network, which reduces the bond exchange rate and correspondingly its extensibility (Fig. S15†). However, upon disulfidation, the refractive index of the material remains constant at 1.529, but the attenuation in the UV region is shifted to shorter wavelengths, likely through the conversion of both linear and cyclic disulfides to thioethers (Fig. S16†).
While many CANs are capable of reprocessing by harnessing their ability to rearrange the network structure, dithiolanes are uniquely suited for both reprocessing and recycling because of their ceiling temperature behavior, which enables complete reversion to reactive monomers. To revert dithiolanes back to their ring-closed state, polymers were heated in the presence of a solvent. Due to the ceiling temperature behavior, a true sink condition (i.e. effective zero concentration of monomer present in the solvent at all times) is not needed, and rather, the concentration only needs to be sufficiently low to favor the equilibrium shift. This behavior is an advantage that ceiling temperature polymers have compared to other CANs, as all exchangeable networks may be depolymerized in a true sink condition; however, the volume of solvent required is much lower for a ceiling temperature polymer. Kim and coworkers recently reported on the apparent impact of the chosen solvent, indicating that N,N-dimethylformamide (DMF) is important to the degradation process, building on previous work by Feringa and coworkers that also relied on DMF.25 DMF is highly hygroscopic and contains trace water, leading to the equilibrium formation of dimethylamine and formic acid through hydrolysis.26 Under these conditions, dimethylamine is present at approximately 0.1% under ambient conditions and likely catalyzes depolymerization of poly(lipoic acid) crosslinks.27 Considering these results and the requirement of a relatively high boiling point, DMF was selected for the recycling process for our networks. Heating polymerized LAPEO samples (∼100 mg, 53 w/w% in toluene, polymerized using 20 mW cm−2 for 5 min per side with 0.5 w/w% BAPO, solvent removed at 60 °C for 18 hours) to 120 °C in 10 mL of DMF for 3 hours resulted in complete degradation of the polymer network. Removal of the depolymerization solvent in vacuo resulted in 95 ± 4% of the monomer being recovered by mass over three separate trials. NMR spectroscopy indicated full reversion to monomer, with no remaining polymer peaks (Fig. 3b). Additionally, GPC results confirmed near complete reversion to monomer with a small fraction of remaining oligomeric content (Fig. S17†). However, any residual oligomer did not greatly affect the polymerization of the recycled monomer, as the reaction proceeded similarly to the virgin monomer when photoinitiator was added (Fig. 3c). This conversion back to monomer is a yield similar to many other depolymerizable polymers28 and did not require the addition of any catalysts or other small molecules beyond solvent. Furthermore, the temperatures necessary for this reversion are relatively mild compared to many thermally reversible polymerizations, which may be beneficial or disadvantageous depending on the desired operating temperature range for the device.29 A feature of this approach with respect to this reversibility is that the temperature sensitivity of reversion and exchange are tunable through changes in the monomer and polymer structures used here,30 as well as modulation of the disulfidation reaction. Films that have undergone the disulfidation reaction are no longer recyclable, revealing disulfidation as an effective tool for modulating this key property.
 | ||
| Fig. 3 Recycling of LAPEO polymerized films. (a) Scheme for the ring-closing monomer reversion reaction. 95 ± 4% monomer was recovered on three recycling experiments. (b) NMR of virgin and depolymerized monomer indicating full reversion to monomer. (c) In situ photorheology of virgin monomer compared to recycled monomer photopolymerization reactions. | ||
To assess the ability to integrate the LAPEO into photolithographic processes, photopatterned polymerizations were conducted at varied length scales with multiple printing techniques. To accommodate versatile methods of printing, it is worthwhile to note that LAPEO is capable of being printed both with and without initiator, a feature partly enabled by the tetrafunctional geometry of the monomer, which allows for network formation at lower conversions than in the bifunctional case. In comparison to a matched bifunctional monomer (PEG core, matched dithiolane concentration and arm length), the photoinitiated times to gelation are comparable, with the tetrafunctional LAPEO having a slightly shorter gelation time. However, in the non-initiated case, LAPEO gels much more rapidly, enabling initiator free printing at considerably lower light doses (Fig. S18†).
First, to investigate the mesoscale printing capabilities, a commercial DLP printer with a 405 nm light source was used to fabricate centimeter-scale 2.5D structures (e.g., University of Colorado buffalo logo) with high fidelity. Since photocrosslinking of LAPEO without photoinitiator proceeds slowly with 405 nm light at intensities commonly used in projection (Fig. 1c), initiator (BAPO, 0.5 w/w%) was added to the resin to speed up the maskless DLP-based light projection, resulting in centimeter-scale patterns on the order of seconds. In contrast, when the length scale of the features required is microns, no initiator was used for 2PL, as the local light dose used is much higher using the confocal lasers. Specifically, using a confocal microscope equipped with a two-photon laser, 2PL at 730 nm was used to print a rendering of the Great Wave Off Kanagawa without the need for exogenous photoinitiator (Fig. 4b). To quantify the resolution achieved with 2PL, positive and negative features of varying sizes were printed and quantified (Fig. 4c). In both positive and negative features, resolutions of 1 μm were achieved in the xy plane, and there is linearity in feature size when printing across size scales (Fig. 4d). Expanding on the scale and complexity, 3D objects were printed using DLP for larger objects (Fig. S19†) and a Nanoscribe 2PL system for smaller objects (Fig. 4e). As with the 2D 2PL, no exogenous initiator was used in the printing of the micron scale parts. 2PL enables rapid production of arrays of microscale printed parts (Fig. S20†) with high fidelity and resolution, showcasing this resin as a tool for rapid prototyping.
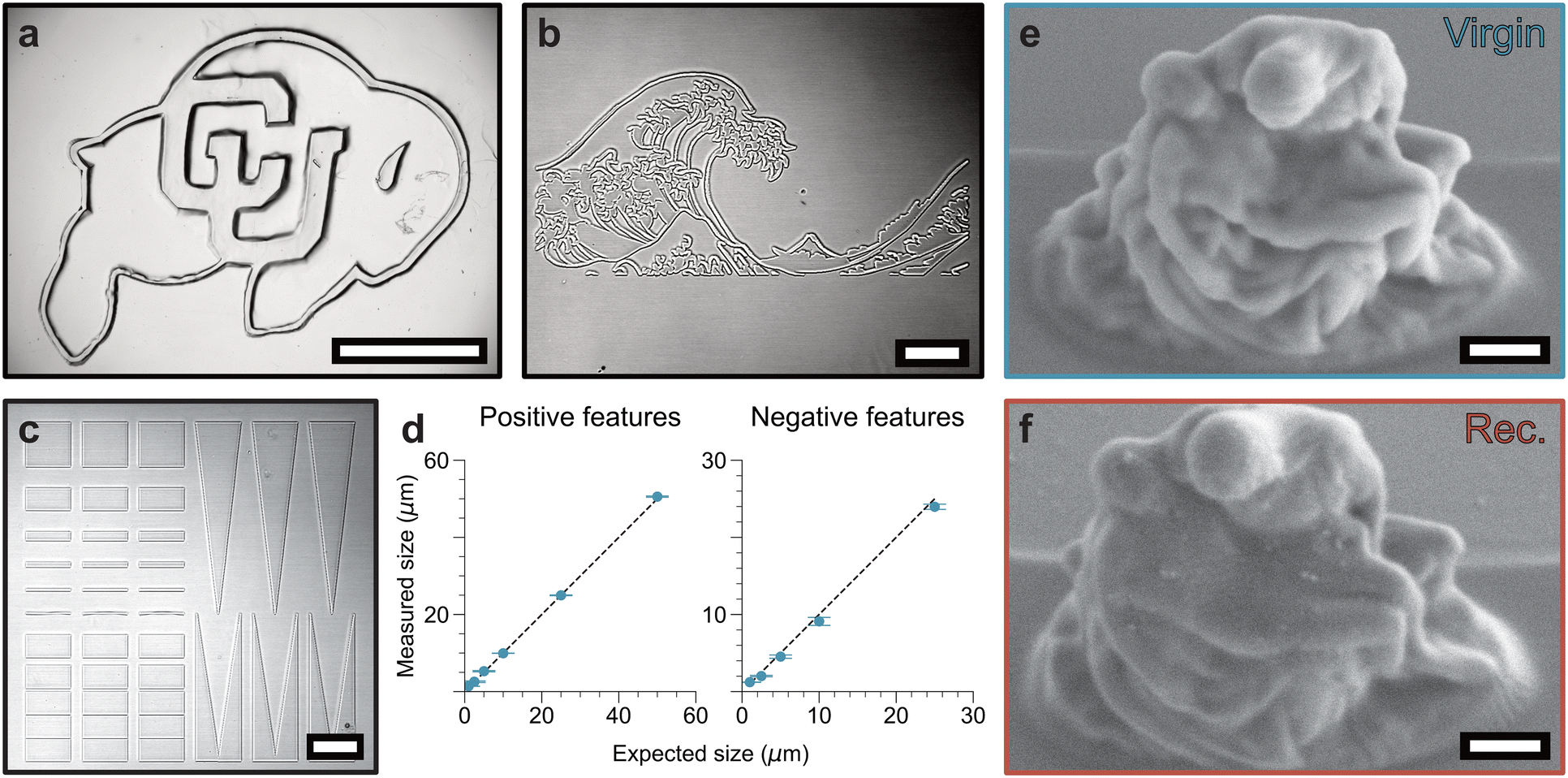 | ||
| Fig. 4 Lithography of LAPEO across length scales. (a) University of Colorado buffalo logo printed in LAPEO using a DLP projector with photoinitiator (405 nm, 0.5 w/w% BAPO, scale bar = 5 mm). (b) Great Wave Off Kanagawa printed via 2PL in LAPEO without initiator (730 nm, scale bar = 50 μm). (c) Resolution test of LAPEO using 2PL (730 nm, scale bar = 50 μm). (d) Quantification of printed features compared to intended feature size. (e) La Pietà printed with Nanoscribe 2PL without initiator (790 nm, scale bar = 10 μm). (f) La Pietà printed from recycled LAPEO with Nanoscribe 2PL with initiator (790 nm, 0.5 wt% BAPO, scale bar = 10 μm). | ||
Finally, printed parts were recycled using the same recycling techniques as applied to bulk polymerized LAPEO to yield repolymerizable monomer. The recovered monomer was subsequently reprinted using any of the techniques explored here. As a proof-of-concept demonstration, a part printed by DLP was recycled to resin and diluted in toluene with initiator. This new resin was then printed using 2PL to achieve a part with similar feature sizes and resolution as the virgin resin (Fig. 4f). This recycling and reprinting capability represents a key component of a circular economy of 3D printing uniquely suited for rapid prototyping.
Conclusion
Results herein demonstrate advances in dithiolane crosslinked thermosets and formulations as recyclable additive manufacturing resins. Specifically, LAPEO was developed as a tetrafunctional dithiolane monomer via a scalable Fischer esterification synthesis. This monomer is capable of photopolymerization with and without initiator to form rubbery elastomers. Harnessing the ceiling temperature behavior of dithiolanes, films were recycled back to a reactive monomer, which was subsequently repolymerized. Finally, LAPEO is well suited to photolithography, with high resolution and pattern fidelity across multiple length scales. Taken together, the recycling capabilities of this resin, the flexibility of the initiator-free crosslinking, and the printing at multiple length scales make this system uniquely capable of rapid prototyping with additive manufacturing.Methods
Materials
All chemicals were purchased from Sigma and used as received unless otherwise noted. 405 nm light was supplied via a Thorlabs 405 nm LED collimated light source.LAPEO synthesis
In a 1 L two-necked round bottom flask (RBF), lipoic acid (4.14 g, 20.075 mmol) was combined with pentaerythritol ethoxylate (15 repeats ethylene glycol/4 arms, 2 g, 2.509 mmol) and a magnetic stir bar. The round bottom flask was then placed into an oil bath and connected to a Dean–Stark apparatus and a condenser. 100 mL toluene was added, and the mixture was purged with argon and heated to 130 °C while stirring. Once fully refluxing, 3 drops of sulfuric acid diluted in 1 mL of toluene was added dropwise. The RBF was then covered in aluminum foil and refluxed while stirring for 18 hours. After the toluene was removed in vacuo, the monomer was refluxed in ethyl acetate with activated charcoal to remove carbonized impurities for 3 hours. This mixture was filtered through sand and Celite to remove the charcoal, and the filtrate was washed with saturated sodium bicarbonate (2×) and brine (1×). The organic phase was dried over magnesium sulfate and concentrated in vacuo. Monomer was further purified via flash chromatography with a gradient from 0–20% MeOH in DCM. A viscous, optically transparent yellow substance remained after concentration in vacuo with a yield of approximately 80%. Products were analyzed with 1H NMR (Fig. S2†).1H NMR (400 MHz, CDCL3, δ): 4.28–4.09 (m, 2H), 3.75–3.42 (m, 16H), 3.24–3.08 (m, 2H), 2.54–2.43 (m, 1H), 2.41–2.31 (m, 2H), 1.98–1.88 (m, 1H), 1.76–1.40 (m, 1H).
2 arm dithiolane monomer synthesis
In a 1 L two-necked round bottom flask (RBF), lipoic acid (5.5 g, 26.67 mmol) was combined with hexaethylene glycol (6 repeats ethylene glycol, 2 g, 6.667 mmol) and a magnetic stir bar. The round bottom flask was then placed into an oil bath and connected to a Dean–Stark apparatus and a condenser. 100 mL toluene was added, and the mixture was purged with argon and heated to 130 °C while stirring. Once fully refluxing, 3 drops of sulfuric acid diluted in 1 mL of toluene was added dropwise. The RBF was then covered in aluminum foil and refluxed while stirring for 18 hours. After the toluene was removed in vacuo, the monomer was refluxed in ethyl acetate with activated charcoal to remove carbonized impurities for 3 hours. This mixture was filtered through sand and Celite to remove the charcoal, and the filtrate was washed with saturated sodium bicarbonate (2×) and brine (1×). The organic phase was dried over magnesium sulfate and concentrated in vacuo. Monomer was further purified via flash chromatography with a gradient from 0–20% MeOH in DCM. A viscous, optically transparent yellow substance remained after concentration in vacuo with a yield of approximately 80%. Products were analyzed with 1H NMR (Fig. S6†).1H NMR (400 MHz, CDCL3, δ): 4.28–4.09 (m, 2H), 3.75–3.42 (m, 11H), 3.24–3.08 (m, 2H), 2.54–2.43 (m, 1H), 2.41–2.31 (m, 2H), 1.98–1.88 (m, 1H), 1.76–1.40 (m, 1H).
Absorbance measurements
LAPEO extinction coefficients were measured using UV/Vis spectroscopy (Thermo Fisher Scientific Evolution 300) at wavelengths from 300–500 nm with data intervals of 1 nm through quartz cuvettes with a path length of 1 cm. Molar extinction coefficients were calculated using Beer's Law. Film % transmittance was measured using the same instrument through a 200 μm thick film over a range from 200–1100 nm.Attenuation measurements
The light penetration depth (Dp) at a given wavelength, i.e., the depth at which 1/e fraction of the light is absorbed, was calculated using eqn (1),31 where A is the absorbance of the liquid resin at a given wavelength as measured on a nanodrop (Thermo Scientific) and b is the pathlength through which the absorbance was measured. LAPEO monomer plus 0.5 wt% BAPO was diluted in toluene to the various concentrations measured. | (1) |
Rheology
Dynamic mechanical analysis
Films were prepared as described above for the thermal stress relaxation measurements. DMA was performed using a TA instruments RSA-2. A cyclic strain of 0.03% was applied at a frequency of 1 Hz, and the temperature was ramped from −80 to 40 °C at a rate of 3 °C min−1.Tensile testing
Tensile testing was conducted using a TA instruments RSA-2 on rectangles ∼5 mm on length, 3.5 mm in width, and 100 μm thick. Samples were subjected to a strain rate of 0.5%/s until failure.Differential scanning calorimetry
Films were prepared for DSC as described above for DMA. DSC was performed on samples with a heat cool heat cycle at 5 °C min−1 from −80 to 180 °C. First cooling and second heating steps are shown.Refractive index measurements
Refractive index was measured with an Anton Paar Abbemat refractometer at a wavelength of 589.3 nm.LAPEO recycling
100 μm thick films with masses of ∼100 mg were prepared by irradiating 53 w/w% LAPEO in toluene with 0.5 w/w% BAPO through RainX coated glass slides with 20 mW cm−2 405 nm light. Polymerized films were added to 10 mL of DMF in 20 mL scintillation vials. The samples were heated to 120 °C for 3 hours, and the DMF was removed in vacuo. The mass of the recovered sample was compared to the original mass of the sample, and 1H NMR (400 MHz, CDCL3) and GPC (in chloroform) were used to compare the initial and recovered monomers.Photopatterning
Photopatterning was performed using a Zeiss LSM 710 laser scanning confocal microscope. LAPEO was photopatterned neat without initiator on an acrylated cover slip. Laser power used was approximately 1 mW, with a pixel size of 2.37 μm and a pixel dwell time of 6.3 μsec.DLP printing
Maskless meso-scale patterning and 3D printing were performed using a Lumen Alpha DLP printer (Volumetric Inc., USA) with 405 nm light source and 35 μm pixel size as described previously.32 Briefly, LAPEO resins were dispended within the vat and each layer (50 μm) was subjected to light exposure (12 seconds, 20 mW cm−2). Resin was composed of 53 w/w% LAPEO in toluene with 0.5 w/w% BAPO initiator. To prevent adhesion of resin and subsequent delamination of the print, a thin layer of fluorinated oil (Krytox, 500 μL) was added to the vat before dispensing the resin. Acrylated cover slips were adhered to the build platform to facilitate attachment of the base layers during printing. The vat was filled with excess LAPEO to prevent drying or evaporation of the object to be printed during the entire process. Post-printing, the objects were removed from the build platform, rinsed with toluene, and subsequently imaged.Nanoscribe printing
Three-dimensional microstructures were printed using the Nanoscribe Photonic Professional GT2. STL designs were created in Blender and converted to print files using DeScribe (Nanoscribe). Prints were completed using Nanowrite. Prints were completed using the oil-immersion (ZEISS – Immersol 518F) configuration with the 63× objective on a borosilicate substrate coated with ∼0.5 nm of Pt to improve finding the interface. Reflectivity of the Pt film caused bubbling due to overexposure at the surface, so the first 10 base layers (∼0.3 μm each) were printed at a lower laser power than the bulk structure. Standard LAPEO prints were completed with CoreLaserPower (CLP) = 70% (where 100% = 50 mW), CoreScanSpeed (CSS) = 8000 μm s−1, BaseLaserPower (BLP) = 30%, and BaseScanSpeed (BSS) = 9000 μm s−1. Recycled La Pietà was printed with CLP = 55%, CSS = 8000 μm s−1, BLP = 32.5%, and BSS = 9000 μm s−1. Prints were developed in toluene for 10 minutes.Secondary electron microscopy (SEM)
SEM images were taken using the Hitachi TM-4000 Plus II with EDS tabletop SEM. Due to the non-continuous nature of the metal coatings over the entire stencil and/or particle surface, the low vacuum charge reduction mode was used at 5–10 kV. Images were taken with either the secondary electron (SE) mode or a mix of SE and backscattering modes.PVD coating
Sputter coating was used for all PVD in this paper using a Cressington 108 auto sputter coater at ∼0.5 Å s−1. ∼0.5 nm were deposited on borosilicate substrates prior to printing. ∼2 nm were deposited onto printed structures before imaging with SEM.Data availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Raw data are available from the authors upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge funding from the Industry University Cooperative Research Center for Fundamentals and Applications of Photopolymerizations (C. N. B. and B. R. N), DARPA W911NF-19-20024 (C. N. B. and K. S. A.), and the National Institutes of Health R01s DE016523 and DK120921 (K. S. A.), Graduate Assistantship in Areas of National Need (B. R. N). C. W. S. is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trusts. C. W. S. would like to thank the Packard Foundation for their support of this project. The authors thank Dr Benjamin Fairbanks, Dr Marianela Lemon, and Dr Monica Ohnsorg for helpful discussions, and Gabriel Seymour for assistance with preliminary experiments.References
- (a) R. Kienle, P. V. D. Meulen and F. Petke, J. Am. Chem. Soc., 1939, 61, 2258 CAS; (b) H. Staudinger and E. Husemann, Ber. Dtsch. Chem. Ges. (A and B Series), 1935, 68, 1618 Search PubMed.
- S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187 CAS.
- M. Sangermano, I. Roppolo and M. Messori, in Photocured Materials, ed. A. Tiwari and A. Polykarpov, The Royal Society of Chemistry, 2014 Search PubMed.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mulhaupt, Chem. Rev., 2017, 117, 10212 CAS.
- L. Moroni, J. A. Burdick, C. Highley, S. J. Lee, Y. Morimoto, S. Takeuchi and J. J. Yoo, Nat. Rev. Mater., 2018, 3, 21 CAS.
- (a) R. Chaudhary, P. Fabbri, E. Leoni, F. Mazzanti, R. Akbari and C. Antonini, Prog. Addit. Manuf., 2022, 8, 331 Search PubMed; (b) J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland, A. Ermoshkin, E. T. Samulski and J. M. DeSimone, Science, 2015, 347, 1349 CAS; (c) C. C. Cook, E. J. Fong, J. J. Schwartz, D. H. Porcincula, A. C. Kaczmarek, J. S. Oakdale, B. D. Moran, K. M. Champley, C. M. Rackson, A. Muralidharan, R. R. McLeod and M. Shusteff, Adv. Mater., 2020, 32, e2003376 Search PubMed.
- A. Mauri, P. Kiefer, P. Neidinger, T. Messer, N. M. Bojanowski, L. Yang, S. Walden, A. N. Unterreiner, C. Barner-Kowollik, M. Wegener, W. Wenzel and M. Kozlowska, Chem. Sci., 2024, 15, 12695–12709 CAS.
- H. H. Hwang, W. Zhu, G. Victorine, N. Lawrence and S. Chen, Small Methods, 2018, 2, 1700277 Search PubMed.
- M. J. Webber and M. W. Tibbitt, Nat. Rev. Mater., 2022, 7, 541 Search PubMed.
- (a) S. Pruksawan, Y. T. Chong, W. Zen, T. J. E. Loh and F. Wang, Chem. – Asian J., 2024, e202400183 CAS; (b) E. M. Maines, M. A. Polley, G. Haugstad, B. Zhao, T. M. Reineke and C. J. Ellison, ACS Applied Polymer Materials, 2024 Search PubMed.
- C. Shi, R. W. Clarke, M. L. McGraw and E. Y.-X. Chen, J. Am. Chem. Soc., 2022, 144(5), 2264–2275 CrossRef CAS PubMed.
- A. V. Tobolsky, W. J. MacKnight and M. Takahashi, J. Phys. Chem., 1964, 68(8), 787–790 CrossRef CAS.
- J. A. Barltrop, P. M. Hayes and M. Calvin, J. Am. Chem. Soc., 1954, 76(17), 4348–4367 CrossRef CAS.
- M. Raeisi and N. V. Tsarevsky, J. Polym. Sci., 2021, 59, 675–684 CrossRef CAS.
- C. Y. Shi, Q. Zhang, B. S. Wang, M. Chen and D. H. Qu, ACS Appl. Mater. Interfaces, 2021, 13(37), 44860–44867 CrossRef CAS PubMed.
- A. Kisanuki, Y. Kimpara, Y. Oikado, N. Kado, M. Matsumoto and K. Endo, J. Polym. Sci., Part A:Polym. Chem., 2010, 48, 5247–5253 CrossRef CAS.
- F. Sanda and T. Endo, J. Polym. Sci., Part A:Polym. Chem., 2001, 39, 265–276 CrossRef CAS.
- B. S. Wang, Q. Zhang, Z. Q. Wang, C. Y. Shi, X. Q. Gong, H. Tian and D. H. Qu, Angew. Chem., Int. Ed. Engl., 2023, 62, e202215329 Search PubMed.
- (a) X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 3822–3833 Search PubMed; (b) Y. Liu, Y. Jia, Q. Wu and J. S. Moore, J. Am. Chem. Soc., 2019, 141, 17075 CrossRef CAS PubMed.
- (a) H. Tang and N. V. Tsarevsky, Polym. Chem., 2015, 6, 6936–6945 RSC; (b) C. Tong, J. A. J. Wondergem, D. Heinrich and R. E. Kieltyka, ACS Macro Lett., 2020, 9, 882–888 CAS.
- (a) Q. Zhang, C. Y. Shi, D. H. Qu, Y. T. Long, B. L. Feringa and H. Tian, Sci. Adv., 2018, 4, eaat8192 CAS; (b) Q. Zhang, Y. X. Deng, H. X. Luo, C. Y. Shi, G. M. Geise, B. L. Feringa, H. Tian and D. H. Qu, J. Am. Chem. Soc., 2019, 141, 12804–12814 CAS; (c) B. Sieredzinska, Q. Zhang, K. J. V. D. Berg, J. Flapper and B. L. Feringa, Chem. Commun., 2021, 57, 9838–9841 Search PubMed.
- (a) T. O. Machado, C. J. Stubbs, V. Chiaradia, M. A. Alraddadi, A. Brandolese, J. C. Worch and A. P. Dove, Nature, 2024, 629, 1069–1074 Search PubMed; (b) M. Chen, R. Yang, H. Wu, Q. Wang, C. Shi, S.-W. Zhou, D. Yang, F.-Y. Liu, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2024, 63, e202409200 Search PubMed.
- C. A. Lindberg, E. Ghimire, C. Chen, S. Lee, N. D. Dolinski, J. M. Dennis, S. Wang, J. J. de Pablo and S. J. Rowan, J. Polym. Sci., 2023, 62(5), 905–915 Search PubMed.
- J. T. Kamps, S. M. Soars, N. J. Bongiardina, B. D. Fairbanks and C. N. Bowman, Tetrahedron, 2022, 109, 132683 CAS.
- (a) J. Lyu, G. Song, H. Jung, Y. I. Park, S. H. Lee, J. E. Jeong and J. C. Kim, ACS Appl. Mater. Interfaces, 2024, 16, 1511 CAS; (b) Y. Deng, Q. Zhang, D. H. Qu, H. Tian and B. L. Feringa, Angew. Chem., Int. Ed. Engl., 2022, e202209100 CAS.
- M. E. O'Kane, J. A. Smith, T. I. Alanazi, E. J. Cassella, O. Game, S. van Meurs and D. G. Lidzey, ChemSusChem, 2021, 14, 2537 Search PubMed.
- W. K. Kwok, W. G. Lee and S. I. Miller, J. Am. Chem. Soc., 1969, 91, 468 CAS.
- (a) C. Shi, W. T. Diment and E. Y. Chen, Angew. Chem., Int. Ed., 2024, 63, e202405083 CAS; (b) H. Wakefield IV, N. J. Fromel, J. Jiang, I. Kevlishvili, Y. Yao, S. L. Craig, H. J. Kulik and R. S. Klausen, Polym. Chem., 2024, 15, 5016 Search PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501 CAS.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Biomacromolecules, 2006, 7, 2827 CAS.
- T. J. Kolibaba, J. P. Killgore, B. W. Caplins, C. I. Higgins, U. Arp, C. C. Miller, D. L. Poster, Y. Zong, S. Broce, T. Wang, V. Talačka, J. Andersson, A. Davenport, M. A. Panzer, J. R. Tumbleston, J. M. Gonzalez, J. Huffstetler, B. R. Lund, K. Billerbeck, A. M. Clay, M. R. Fratarcangeli, H. J. Qi, D. H. Porcincula, L. B. Bezek, K. Kikuta, M. N. Pearlson, D. A. Walker, C. J. Long, E. Hasa, A. Aguirre-Soto, A. Celis-Guzman, D. E. Backman, R. L. Sridhar, K. A. Cavicchi, R. J. Viereckl, E. Tong, C. J. Hansen, D. M. Shah, C. Kinane, A. Pena-Francesch, C. Antonini, R. Chaudhary, G. Muraca, Y. Bensouda, Y. Zhang and X. Zhao, Addit. Manuf., 2024, 84, 104082 CAS.
- A. P. Dhand, M. D. Davidson, H. M. Zlotnick, T. J. Kolibaba, J. P. Killgore and J. A. Burdick, Science, 2024, 385, 566 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00199d |
| This journal is © The Royal Society of Chemistry 2025 |