
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ-Conjugated polymers consisting of heavier group 13 elements
Shunichiro
Ito
 ab and
Kazuo
Tanaka
*ab
ab and
Kazuo
Tanaka
*ab
aDepartment of Polymer Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: tanaka@poly.synchem.kyoto-u.ac.jp
bDepartment of Technology and Ecology, Graduate School of Global Environmental Studies, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan
First published on 17th April 2025
Abstract
Boron, aluminum, gallium, indium, and thallium are group 13 elements that can induce various electronic properties and unique functions when incorporated into main-chain conjugation through polymers. As vacant p-orbitals in these elements interact with Lewis bases, stimuli responsiveness can be induced. Additionally, the chemical and thermal stability can be enhanced by connecting with extra Lewis bases as supporting ligands. Moreover, superior optoelectronic properties, such as light absorption, emission, and carrier mobility, are often observed from group 13 element-containing π-conjugated systems. The introduction of boron into conjugated systems has been widely applied not only for improving material properties but also for providing new functionalities for conventional polymers. In contrast, there are limited examples of polymers possessing the heavier group 13 elements in their repeating units. According to recent studies, it has been shown that the chemical, physical, and material properties of π-conjugated compounds can be unexpectedly modulated by these heavier group 13 elements. In this review, we mainly explain the synthesis and fundamental photophysical properties of conjugated polymers consisting of the heavier group 13 elements in their main-chains.
 Shunichiro Ito | Shunichiro Ito received his Ph.D. in Polymer Chemistry from Kyoto University in 2020. During his Ph.D. studies, he worked as a Research Fellow of the Japan Society for the Promotion of Science. He also spent the summer of 2017 as a visiting researcher in the group of Prof. John R. Reynolds at the Georgia Institute of Technology. He has been an Assistant Professor at the Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University since 2020. His research is currently directed toward developing novel photo- and electro-functional materials based on metal complexes. |
 Kazuo Tanaka | Kazuo Tanaka received his Ph.D. degree in 2004 from Kyoto University, and worked at Stanford University, USA, Kyoto University, and RIKEN as a postdoctoral fellow. In 2007, he moved to the Department of Polymer Chemistry, Graduate School of Engineering, Kyoto University, and in 2018, he was promoted to Professor. His research projects focus on the design of new functional materials for optics and nanotechnology based on heteroatom-containing conjugated polymers and organic–inorganic polymer hybrids. |
Introduction
Group 13 elements, boron, aluminum, gallium, indium, and thallium have a general ground-state electronic configuration ns2np1, where n denotes the principal quantum number. These elements commonly form a +3 formal oxidation states to satisfy the octet rule, whereas heavier elements often exhibit lower oxidation states, such as +1 and +2, due to the so-called ‘inert pair effect’.1 For example, the most common oxidation state of thallium is known to be +1. Furthermore, the ionization energy of these elements is not a monotonic function of the principal quantum number (Table 1).2 As a consequence, the structures, stability, reactivity, and chemical and physical properties of these compounds with the heavier group 13 elements, from Al to Tl, are expected to be different from those of the corresponding boron-containing compounds.3 One of the most representative examples of their differences is the available coordination number. Typical coordination numbers of boron(III) are three or four, and the other group 13 elements often form five- and six-coordinate structures as well. The large ionic sizes and plumper valence orbitals of the heavier elements should play a significant role in the multi-coordinated structures.| B | Al | Ga | In | Tl | |
|---|---|---|---|---|---|
| a Data taken from ref. 2. | |||||
| Ground-state electron configuration | [He]2s22p1 | [Ne]3s23p1 | [Ar]4s24p1 | [Kr]5s25p1 | [Xe]6s26p1 |
| Ionization energy/kJ mol −1 | |||||
| E → E+ | 800.637 | 577.539 | 578.844 | 558.299 | 589.351 |
| E+ → E2+ | 2427.07 | 1816.68 | 1979.41 | 1820.71 | 1971.03 |
| E2+ → E3+ | 3659.75 | 2744.78 | 2963 | 2704 | 2878 |
| E3+ → E4+ | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 025.9 025.9 |
11577.46 |
6175 | 5210 | (4900) |
Owing to their wide versatility—including semiconductivity, light absorption and emission, non-linear optical properties, and Lewis acidity,—much effort has been devoted to studying the electronic structures of π-conjugated polymers involving boron. By modulating structures, various advantageous chemical and physical properties derived from their polymeric structures, such as excellent processability, elasticity, high local concentrations, environmental sensitivity, and extension of π-conjugation, can be obtained. Thus far, many reviews describing such boron-containing polymeric materials have been published.4–15 In particular, the incorporation of boron-coordinating structures into π-conjugated systems has been revisited because they allowed access to n-type semiconducting molecules and polymers.16 However, the number of polymers containing the heavier congeners of B, Al, Ga, In, and Tl remains limited.17,18 The heavier elements often play a critical role in drastically improving small-molecular material properties, which originate from the chemical structure of a molecule, intermolecular interactions, molecular motions, and other unpredictable factors.
As a representative example, the efficiency of preliminary organic light-emitting diodes (OLEDs) consisting of 8-hydroxylquinolinate complexes is usually higher with aluminum complexes as compared to those with boron complexes.19 Moreover, a device composed of gallium tris(8-hydroxylquinoline) showed higher efficiency as compared to those of aluminum and indium complexes probably because of the superior charge-transporting property of the gallium complex.20 Such striking differences in chemical and physical properties among group 13 elements result from differences in atomic and ionic size, orbital energies, nature of bonds, interactions with neighboring atoms, and intermolecular interactions.
These observations have encouraged the development of a new series of materials composed of these elements and comparison of their chemical and physical properties. Notably, the elemental dependency of optoelectronic properties, especially in the solid state, cannot generally be inferred from boron chemistry. Therefore, it is necessary to discover the appropriate elements that maximize the material properties for each practical application. Furthermore, the adoption of heavier homologues into polymer structures may accentuate the differences between boron and other elements and lead to advanced functionalities, such as room-temperature phosphorescence, stimuli-responsive luminescence, and higher electrical conductivity, as demonstrated in small molecule systems.
There has been a focus on the synthesis of inorganic and organometallic molecular compounds composed of the heavier group 13 elements to clarify their molecular structures, chemical reactivity, and consistency between experimental and theoretical data.21–23 However, the incorporation of those species into π-conjugated polymer backbones has been prevented because it is likely that most compounds consisting of the heavier group 13 elements decompose under common polymerization conditions, such as metal-catalyzed cross-coupling reactions. Thus, alternative synthetic approaches are required to construct π-conjugated polymers composed of these classes of complexes. One successful approach is the post-polymerization functionalization method, where element-containing structures are constructed after polymerization reactions. This method facilitates the development of polymers that incorporate relatively unstable moieties and allows easy control of the functionalization ratio by simply adjusting the reaction stoichiometry.24 Another approach is the kinetic and thermodynamic stabilization of monomers by employing elaborate supporting ligands, which enables us to assess the material properties of these classes of compounds.25,26
This review mainly focuses on recent synthetic advances that can be used to obtain fully characterized π-conjugated polymers and oligomers involving aluminum and gallium. In addition, an indium-containing homocatenate oligomer is also described for stimulating further development in this field. The chemical and physical properties of these materials will be discussed.
Aluminum-containing π-conjugated polymers
Aluminum-containing inorganic and organometallic complexes have been applied in the broad field of chemistry involving organic reagents,27 catalysts,28–31 optoelectronic materials,32 photodynamic therapies,33 and energy-storage materials,34 likely because of their high reactivity coming from the strong Lewis acidity, oxophilicity, and photoactivity. However, their high reactivity has hampered the growth of versatile polymers consisting of aluminum-based compounds in the main-chains. Therefore, this section focuses on the most actively studied systems based on aluminum tris(8-quinolinolate), Alq3, and its derivatives (Fig. 1 and Table 2). Nevertheless, more recently, aluminum complexes have garnered attention because of their prominently intriguing photophysical and photochemical functions, such as, efficient fluorescence,35 phosphorescence,36 thermally activated delayed fluorescence,37 circularly polarized luminescence,38 stimuli-responsive luminescence,39,40 and photosensitizing ability.41 Dramatically improved chemical stability has been observed in some cases,36,38 motivating the community to develop polymeric materials involving those aluminum-based compounds. | ||
| Fig. 1 (a) Chemical structure of Alq3. Conjugated polymers composed of Alq3-type complexes with (b) poly(8-quinolinol) and (c) poly(aryleneethynylene) scaffolds. | ||
| λ abs/nm | λ PL/nm | PLQY | Condition | Ref. | |
|---|---|---|---|---|---|
| Alq3 | 384 | 521 | 0.13 | Chloroform solution | 45 |
| Alq′2q | 375 | 507 | 0.07 | Chloroform solution | 45 |
| 1 | 359 | 489 | 0.23 | Chloroform solution | 54 |
| 564 | Film | ||||
| 2pre-q(6/4-OEH) | 423 | 472 | 0.34 | Chloroform solution | 55 |
| 429 | 491, 536 | Film | |||
| 2pre-r(6/4-OEH) | 413 | 471 | 0.49 | Chloroform solution | |
| 417 | 486, 522 | Film | |||
| 2pre-q(8/2-OEH) | 426 | 475 | 0.32 | Chloroform solution | |
| 428 | 493, 550 | Film | |||
| 2pre-r(8/2-OEH) | 427 | 476 | 0.34 | Chloroform solution | |
| 437 | 490, 526 | Film | |||
| 2-q(6/4-OEH) | 419 | 472 | 0.22 | Chloroform solution | |
| 425 | 473, 507 | Film | |||
| 2-r(6/4-OEH) | 413 | 471 | 0.54 | Chloroform solution | |
| 445 | 474, 504 | Film | |||
| 2-q(8/2-OEH) | 435 | 474 | 0.25 | Chloroform solution | |
| 439 | 570 | Film | |||
| 2-r(8/2-OEH) | 433 | 476 | 0.37 | Chloroform solution | |
| 442 | 493, 529 | Film |
Aluminum tris(8-quinolinolate), Alq3 (Fig. 1a), and its derivatives are the most important family of aluminum complexes in the field of optoelectronics because Alq3 possesses a superb ability for electron transport accompanied by satisfactory luminescence in solid states. As Alq3 was applied as the emitting and electron-transporting layer in the first organic light-emitting diode,32 tremendous research efforts have been devoted to developing functionalized Alq3-based materials for improved luminescent and electronic properties, processability, and chemical stability. In particular, immobilization by covalent bonds within polymer backbones enhances film formability and device stability. Some examples of Alq3-decorated polymers with non-conjugated backbones are poly(bisphenol-A sulfone),42 polynorbornene,43,44 phenol resin,45 poly(methyl methacrylate),46–52 and polystyrene.53
These studies successfully demonstrated that polymeric complexes can be applied both to solution-processed device fabrication and in preparing luminescent micelles.49 Most polymers in these studies were prepared with post-polymerization complexation to prevent the chemical decomposition of complex moieties. It was also reported that Alq3 is labile and that a ligand exchange reaction often takes place to generate cross-linking, which will result in insoluble polymers, followed by the generation of low-molecular weight polymers with low yields.42 The solubility of polymers can be improved by introducing alkyl chains46 and by directly polymerizing Alq3-decorated monomers using methods, such as ring-opening metathesis43 and radical polymerizations.47
Incorporating Alq3 into the main-chain of π-conjugated polymers has also been attempted for modulating and improving its optoelectronic properties. Post-polymerization complexation of 8-quinolinol-based conjugated polymers was applied to obtain fully characterized Alq3-based polymers in pioneering works.54,55 A soluble poly(8-quinolinol), 1pre, was synthesized by enzyme-catalyzed polymerization and subjected to subsequent complexation with bis(2-methyl-8-quinolinolato)ethylaluminum (Alq′2(Et)) to give 1 (Fig. 1b).54 Horseradish peroxidase (HRP) was used for the polymerization of 8-quinolinol in the presence of hydrogen peroxide as an oxidizing reagent. The number- and weight-average molecular weights (Mn and Mw) were determined to be 2500 and 3400, respectively, by GPC analysis. Importantly, the 1H and 13C NMR spectra of 1pre showed that the polymerization predominantly proceeded regioselectively at the 4 and 7 positions of the 8-quinolinol ring. The subsequent reaction of 1pre with Alq′2(Et) resulted in the complete conversion of the quinolinol repeating units into the aluminum complex of 1. It was reported that 1 exhibits photoluminescence in solution and in the solid state, while 1pre shows little emission. Interestingly, the relative photoluminescence quantum yield (PLQY) of 1 (0.27) is higher than that of Alq′2q (0.07). The longest-wavelength absorption maximum (λabs) of Alq3 and 1 was determined to be 375 and 359 nm, respectively. The hypsochromic shift of λabs might originate from the electron-withdrawing property of the poly(8-quinolinol-4,7-diyl) backbone.
As metal-catalyzed cross-coupling reactions offer more reliable control of regioselectivity than oxidative coupling polymerizations, the palladium-catalyzed Sonogashira–Hagihara reaction was used for preparing poly(aryleneethynylene)-type conjugated polymers consisting of 8-quinolinol units in their main-chain (Fig. 1c).55 The polymerization reactions were carried out using three types of silyl-protected 8-quinolinol monomers with different substitution positions (q–r) and diethynyl- and dibromo–dialkoxyphenylene co-monomers. The subsequent deprotection of the silyl groups using tetrabutylammonium fluoride (TBAF) afforded the precursor polymers 2pre. The Mn values of the silyl-protected polymers ranged from 8500 to 13000, which corresponds to approximately eight monomeric units of monomer q in the polymer chain for the case of 2pre-q(6/4-OEH).
Although treatment of 2pre with Alq′2(Et) was attempted to give the corresponding Alq3-incorporated conjugated polymers, the dodecyloxy-based polymers (the –OC12 series) yielded insoluble products probably due to cross-linking caused by a ligand exchange reaction of the Alq3-type scaffold. However, 2-ethylhexyloxy-based polymers resulted in desired soluble polymers 2 because the branched alkoxy groups retarded the intermolecular ligand exchange. FT-IR spectroscopy and elemental analysis indicated that the conversion of the phenolic hydroxyl group to the aluminum complex was approximately 30% in the case of 2-q(6/4-OEH), suggesting that its single polymer chain could statistically contain approximately two aluminum complex units. Photoluminescence was emitted by both 2pre and 2 in solution and in solid-state films. The photoluminescence spectra of 2pre-q(6/4-OEH) and 2-q(6/4-OEH) were almost the same shape and peaked at 472 nm, which was assignable to the aromatic polymer chain rather than the aluminum complex moiety. In the solid-state thin films, there were additional emission bands attributed to an excimer-like adduct for 2pre-q(6/4-OEH) (536 nm) and an aluminum complex unit for 2-q(6/4-OEH) (507 nm). Hence, it was implied that the intermolecular energy transfer from the aryleneethynylene main-chains to the aluminum complexes could occur in the film of 2-q(6/4-OEH).
Aluminum porphyrins and salen complexes are one of the most widely investigated families of aluminum-based π-conjugated molecules because of their promising catalytic activities. Taking advantage of their microporous structures, it has been revealed in recent years that conjugated microporous polymers (CMPs) consisting of aluminum complexes exhibit outstanding catalytic properties.56–58 Representative aluminum complex-based CMPs 3,564,57 and 558 were synthesized by post-polymerization complexation, one-pot polymerization and complexation, and direct polymerization, respectively (Fig. 2).
 | ||
| Fig. 2 Conjugated microporous polymers composed of (a) aluminum salen with an alkylene linker, (b) aluminum porphyrin with a phenylene linker, and (c) aluminum porphyrin with a tetraphenylmethane linker. | ||
High-resolution transmission electron microscopy images and gas adsorption analysis of CMP 3 revealed nanometer-sized pores (approximately 0.5 nm) and 798 m2 g−1 of BET surface area. Its CO2 uptake at 298 K and 760 mmHg reached 76.5 mg g−1, which is comparable with that of representative metal–organic frameworks. As a result, cobalt porphyrin CMP 3 exhibited exceptionally high catalytic activity in converting propylene oxide (PO) and CO2 to propylene carbonate (PC) in the presence of tetrabutylammonium bromide as a co-catalyst, achieving 78.2% conversion at atmospheric pressure and room temperature.
Similar highly efficient catalytic activity was also reported for CMP 4 with turnover frequencies (TOF) of up to 443.6 h−1 at room temperature and atmospheric pressure. Furthermore, due to the cationic units in CMP 5, co-catalyst was not required, and remarkably high TOF of up to 2200 h−1 resulted for the conversion of epichlorohydrin and CO2 to the corresponding cyclic carbonate. Further synthetic efforts toward group 13 complex-based polymers involving gallium59 could open a novel avenue for achieving highly efficient and chemically stable catalytic systems.
Gallium-containing π-conjugated polymers
Gallium complexes have been developed for achieving not only optoelectronic materials, as mentioned in the introduction, but also antitumor compounds60 and radiopharmaceuticals.61–63 Therefore, it can be envisioned that the incorporation of those compounds into polymer scaffolds could lead to remarkable improvements in their electronic and optical properties and cooperative functions, such as a theranostic ability.64 However, because their chemical structures are usually labile, examples of gallium-containing π-conjugated polymers are limited (Fig. 3 and 4 and Table 3). This section focuses mainly on the synthetic protocols and the photophysical properties of these polymers. | ||
| Fig. 3 Conjugated polymers composed of Mamx-stabilized (a) gallafluorene and (b) gallium-bridged phenylene. | ||
 | ||
| Fig. 4 Conjugated polymers composed of β-diketiminate boron and gallium complexes. | ||
| λ abs/nm | λ PL/nm | PLQY | Condition | Ref. | |
|---|---|---|---|---|---|
| 6-C | 326 | 383 | 0.37 | Chloroform solution | 73 |
| 326 | 385 | 0.05 | Film | ||
| 6-O | 366 | 411 | 0.34 | Chloroform solution | |
| 365 | 420 | 0.04 | Film | ||
| 6-Flu | 383 | 416, 438 | 0.69 | Chloroform solution | |
| 383 | 430, 454 | 0.06 | Film | ||
| 6-BTH | 404 | 486 | 0.20 | Chloroform solution | |
| 407 | 515 | 0.07 | Film | ||
| 6-BTz | 442 | 479 | 0.53 | Chloroform solution | |
| 441, 469 | 479, 531 | 0.09 | Film | ||
| 6-CDT | 481 | 531, 569 | 0.32 | Chloroform solution | |
| 478 | 572 | 0.004 | Film | ||
| 6-BTA | 374, 477 | 643 | 0.43 | Chloroform solution | |
| 377, 485 | 641 | 0.08 | Film | ||
| 7-Ph | 275 | 378 | 0.32 | Chloroform solution | 74 |
| 7-PhO | 277, 325 | 392 | 0.23 | ||
| Biphenyl | 250 | 314 | <0.01 | ||
| 7-Model-terphenyl | 266, 318 | 384 | 0.16 | ||
| 7-Model1 | 263 | 319 | 0.01 | ||
| 7-Model2 | 269 | 322 | 0.04 | ||
| 8-B-FL | 399 | 545 | <0.01 | Chloroform solution | 98 |
| 399 | 545 | 0.07 | Film | ||
| 8-B-CBZ | 397 | 545 | <0.01 | Chloroform solution | |
| 404 | 552 | 0.07 | Film | ||
| 8-B-BT | 404 | 581 | <0.01 | Chloroform solution | |
| 417 | 575 | 0.07 | Film | ||
| 8-Ga-FL | 411 | 571 | <0.01 | Chloroform solution | |
| 415 | 575 | 0.05 | Film | ||
| 8-Ga-CBZ | 410 | 576 | <0.01 | Chloroform solution | |
| 414 | 573 | 0.05 | Film | ||
| 8-Ga-BT | 420 | 610 | <0.01 | Chloroform solution | |
| 424 | 601 | 0.03 | Film |
The 2,4-di-t-butyl-6-(N,N-dimethylaminomethyl)phenyl group, known as the Mamx ligand (the structure is shown in Fig. 3a),65 thermodynamically and kinetically stabilizes group 13 organoelement compounds, leading to the isolation of gallium-containing polymers such as poly(galla[1]ferrocenophane).66 Thus, the Mamx ligand has opened an avenue for the synthesis and evaluation of π-conjugated compounds consisting of group 13 elements, such as 9-heterofluorenes,67–70 dithienoheteroles,71 and dibenzoheteropins.72 These compounds are stable under atmospheric conditions and can be purified with typical silica-gel column chromatography. It is important to note that the gallafluorene and indafluorene derivatives exhibited fluorescence and phosphorescence in room-temperature solutions, while the borafluorene and alumafluorene derivatives showed only fluorescence. Furthermore, the addition of tris(pentafluorophenyl)borane (B(C6F5)3) to solutions of bora-, aluma-, and gallafluorenes induced phosphorescence from their triplet exciplex of the heterofluorenes and B(C6F5)3.67,68
Mamx-stabilized gallafluorene is sufficiently stable to be directly subjected to the palladium-catalyzed Suzuki–Miyaura, Sonogashira–Hagihara, and Migita–Kosugi–Stille coupling reactions to give a variety of conjugated polymers 6 (Fig. 3a).73 Their absorption and photoluminescence spectra highly relied on the co-monomers, and their emission colors covered the visible region. Notably, the fluorescence lifetime measurements indicated that all their emission bands were assigned to fluorescence, based on their nanosecond-order lifetimes. No apparent phosphorescence was observed even at 77 K. Compared to the spectra in solution states, the redshifted emission spectra of 6-O, 6-Flu, 6-BTH, and 6-BTz suggested intermolecular interactions.
In particular, the film of 6-BTz exhibited two distinct bands in the absorption (441 and 469 nm) and emission (479 and 531 nm) spectra. The lower-energy bands were absent in its dilute solution. The intensity ratio of these two peaks depended on the solution concentration and film-preparation method. Therefore, it was suggested that the intermolecular interactions in concentrated states resulted in those additional absorption and emission bands. Furthermore, the results of cyclic voltammetry showed that the gallafluorene moiety should function as a stronger electron donor than 9,9-dialkylfluorene because the synthesized gallafluorene polymers have higher highest occupied molecular orbital (HOMO) levels than the corresponding fluorene-based polymers.
The effects of gallium atoms on π-conjugation of polymers were evaluated by incorporating the Mamx-stabilized triarylgallium unit into the poly(p-phenylene) scaffolds (Fig. 3c).74 Homopolymerization with nickel-mediated Yamamoto coupling and copolymerization with palladium-catalyzed Suzuki–Miyaura cross-coupling successfully yielded 7-Ph and 7-PhO, respectively. Importantly, compared to the corresponding model compounds, the apparent red shifts of the absorption and emission spectra by polymerization were observed. The dimeric model, 7-model2, exhibited absorption and emission bands between 7-Ph and 7-model1. These observations strongly indicate that main-chain π-conjugation should extend through the gallium atoms. In addition, using theoretical calculations employing periodic boundary conditions for 7-Ph, the bandwidths of the highest occupied and lowest unoccupied crystal orbitals (HOCOs and LUCOs) were calculated to be 0.112 and 0.347 eV, respectively. These values certainly indicate that the electronic conjugation in the LUCO apparently extends over the main-chain involving the gallium atoms.
In the context of material science, a major limitation of typical organic luminophores is that their planar π-conjugated structures, which efficiently emit in dilute solutions but experience luminescence loss in aggregated or crystalline states due to concentration quenching. On the contrary, AIE- and CIE-active materials exhibit efficient luminescence in aggregate and crystalline states, in particular, respectively. Their solutions show only lower efficiency emission.75–78 Therefore, those classes of luminophores are utilized in a wide range of practical applications, such as organic light-emitting diodes, bioimaging,79 and chemosensors.80
Recently, β-diketiminate ligands, which have been utilized for isolating various reactive species,81,82 have enabled us to develop luminescent complexes and demonstrate their aggregation- and crystallization-induced emission (AIE and CIE) properties.36,39,83–92 Furthermore, the incorporation of these complexes into π-conjugated polymer backbones results in emissive film materials with productive responses to external stimuli.84,90,93–97 The mechanisms behind the AIE and CIE properties of β-diketiminate complexes have been proposed: the sterically hindered substituents, e.g., aromatic rings, in the ligands hampered detrimental intermolecular interactions, which caused concentration quenching in concentrated states. The quite weak luminescence in dilute solutions could stem from rapid nonradiative quenching processes through vibronic couplings88 and conical intersections.90
In this context, the post-polymerization complexation method was applied to achieve π-conjugated polymers containing gallium β-diketiminate complexes (Fig. 4).98 The ligand polymers 8pre-FL, 8pre-CBZ, and 8pre-BT were synthesized using the palladium-catalyzed Suzuki–Miyaura coupling reaction. The subsequent treatment of these polymeric ligands with trichlorogallium in the presence of triethylamine afforded the corresponding gallium polymers 8-Ga-FL, 8-Ga-CBZ, and 8-Ga-BT. NMR, FT-IR, and XRF analyses revealed that complexation reactions were completely accomplished at all the β-diketiminate coordination sites. The Suzuki–Miyaura coupling polycondensation employing the diiodo-β-diketiminate boron complex directly yielded the corresponding boron polymers 8-B-FL, 8-B-CBZ, and 8-B-BT. Notably, the direct Suzuki–Miyaura polymerizations of the corresponding gallium-containing monomer likely failed because of a more labile Ga–Cl bond as compared to B–F. Therefore, the post-polymerization method was required for achieving gallium-containing polymers. The enhancement of the PLQY of the film state compared to the solution state clearly indicates their AIE properties derived from the β-diketiminate scaffold. Remarkably, the gallium-based polymers exhibited bathochromic shifts in the absorption and emission spectra compared to the corresponding boron-based polymers.
Cyclic voltammetry measurements and theoretical calculations suggested that the LUMO level was significantly stabilized by replacing the central element boron with gallium, while the HOMO level was not drastically affected. As a result, the HOMO–LUMO gap of the gallium polymers should be lower than that of the boron polymers, leading to the observed bathochromic shifts. Notably, the HOMO–LUMO gap of the small molecules of the gallium and boron complexes is comparable, although the HOMO and LUMO of the gallium complexes are lower than those of the boron complexes. Consequently, the effects of the central atom were likely manifested by incorporating the complexes into the π-conjugated polymers because the relatively electron-rich co-monomers should limit the HOMO level of the polymers.
Except for the series of polymerizations with metal catalysts, electrochemical polymerization has also been applicable for preparing homogenous thin films of conjugated polymers directly onto electrodes. As the obtained polymer films are usually insoluble in any solvent, electropolymerized films can be utilized for further applications, such as transparent electrodes, thin-film sensors, and precursors for preparing inorganic nanoparticles.99–101 As thiophene and its derivatives are easily polymerized on anodes, thiophene-terminated monomers are often applied for this approach.
In 2010, a typical example of electrochemical polymerization with a gallium-containing monomer was reported (Fig. 5).102 To fabricate conducting polymer films as a precursor of wide-bandgap semiconducting Ga2S3 nanoparticles, the monomer was designed. It was composed of a gallium Schiff-base complex decorated with bithiophene, allowing for anode polymerization. First, a five-coordinate neutral monomer possessing a chloride ligand on the gallium atom was used in the preparation. As expected, electrochemical polymerization successfully occurred to form polymeric precursor 9-neutral on various working electrodes, including carbon-coated gold TEM grids, indium tin oxide, stainless steel, and platinum buttons. For the preparation of Ga2S3 nanoparticles, the obtained thin films were repeatedly treated with the following cycle: dichloromethane solution of H2S, methanol solution of Ga(NO3)2, and then the dichloromethane solution of H2S again. This process was similar to that used for the preparation for cadmium sulfide nanoparticles.99 However, the desired Ga2S3 nanoparticles were not obtained due to the low reactivity of 5-coordinated gallium.
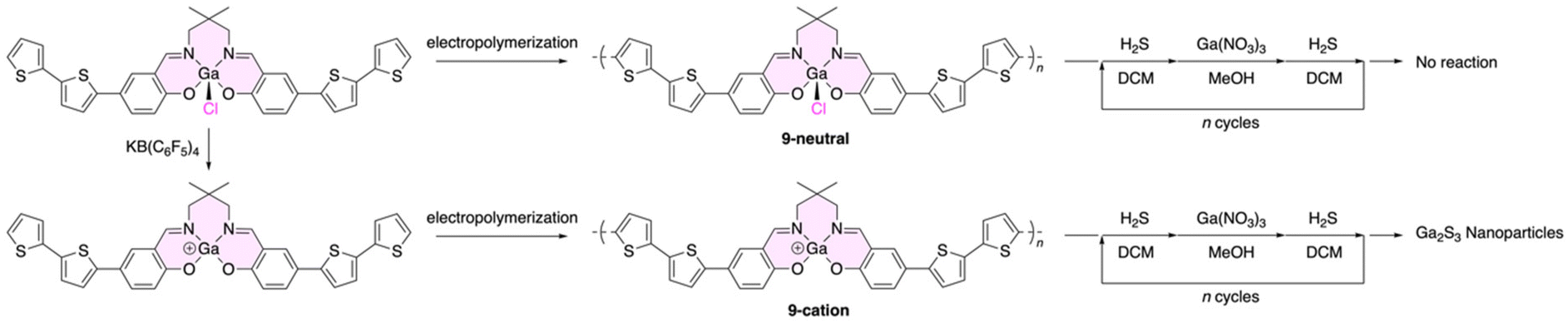 | ||
| Fig. 5 Electrochemical polymerization of gallium complexes of a Schiff base and subsequent preparation of gallium sulfide nanoparticles. | ||
The addition of KB(C6F5)4 induced the release of the apical chloride ligands, followed by the generation of cationic species, and the reactivity of gallium was enhanced. This cationic 2-coordinated monomer was also polymerized in a similar manner to give polymer 9-cation, which was further subjected to the nanoparticle preparation process. TEM observations supported the generation of Ga2S3 nanoparticles on polymer films. Moreover, the size of the nanoparticles was tunable and controllable by changing the number of preparation cycles. The average sizes of nanoparticles were 2.99 and 3.41 nm for two and four cycles, respectively.
Indium- and thallium-containing π-conjugated molecules
Thus far, π-conjugated polymers involving indium and thallium even in solutions have not been isolated for clear characterization of their structures. However, several unusual polymeric or oligomeric structures and their intriguing photophysical properties have been observed in crystalline states. One of the most striking examples, a homocatenated oligomeric structure of indium β-diketiminate complex (7), was reported.103 In the crystalline state,103 homocatenated structures are composed of direct covalent bonds between the same elements as carbon–carbon bonds. Low-valent group 14, 15, and 16 elements often exhibit homocatenation in solutions and solid states,17,18,104 whereas the group 13 elements rarely form homocatenates because of their chemical and thermal instability.104β-Diketiminate ligands have been widely applied for separating such unstable species because of the facile tunability of their steric and electronic demands. Careful screening of the steric hindrance of β-diketiminate ligands for indium revealed that the 3,5-dimethylphenyl groups were suitable for obtaining a homocatenated hexamer, 10, in the solid state (Fig. 6). The formal oxidation state of the internal four indium atoms was +1, while the terminal two indium atoms were divalent because of the iodine substituents. Such low oxidation states of group 13 atoms should be more easily accessible for the heavier atoms, such as indium and thallium, because of the inert pair effect.
 | ||
| Fig. 6 Homocatenated indium oligomer observed in a crystalline state. | ||
Indeed, a similar trimeric structure was observed in the crystal of the thallium β-diketiminate complex.105 These homocatenated polymers/oligomers can be applied as synthetic precursors of metal nanoparticles. The single-crystal X-ray analysis revealed that the In–In bond lengths increased as the distance from the iodine terminals increased, and they were within the range previously reported for In(II) complexes. In addition, the UV–Vis absorption spectrum of this complex in a hexane solution showed an absorption maximum at 349 nm, which could be attributed to the σ–σ* transition and implied the existence of such oligomeric structures even in solution.
Conclusions and perspectives
Various studies concerning the incorporation of the heavier group 13 elements into polymeric structures have recently been conducted. We explained polymerization methods, such as post-polymerization functionalization, direct polymerization of element-containing monomers, solubilization with sidechains and copolymerization, metal-catalyzed polymerization, electrochemical polymerization, and stabilization of reactive monomers and yielded polymers and oligomers. Such progress in synthetic methodology enabled us to functionalize parent small conjugated molecules through polymerization, for instance, improving solution processability, modulating luminescent properties, regulating frontier orbital energies, and enhancing thermal stability. Such control of the optoelectronic properties is crucial to fabricate practical devices, such as OLEDs and organic photovoltaics. The substitution of light elements for the heavier congeners is an orthogonal functionalization strategy with the conventional introduction of substituents. Furthermore, we illustrated that polymerization and oligomerization broaden the application range of group 13 elements as precursors for inorganic nanoparticles.As veiled structure–property relationships remain in the field of group 13 element-containing polymers that have not been experimentally accessible, new synthetic strategies must be developed. Moreover, the unique chemical and physical functionalities, such as large coordination numbers, low-valent species, and redox activity, must be exploited. For instance, it was theoretically predicted that polyborole could possess metal-like properties due to its quite small bandgap.106 If a heavier element is incorporated into the polyheterole backbone instead of boron, systematic change of the electronic properties ranging from metallic to semiconductor-like can occur. Although the chemical instability of group 13 heteroles prevents the isolation of polyborole and its heavier congeners, it might be achieved by insulation of polymer backbones.107 In addition, we witnessed the emergence and development of iminoborane (B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)-containing conjugated polymers, which show the extension of π-conjugation through the BN bonds.108,109 The heavier congeners of BN, involving GaP and InP, might provide novel electronic states for conjugated polymers, such as weakly aromatic Ga2P and In2P rings,110 possibly resulting in higher hyperpolarizability.111 Furthermore, the inert pair effect in the heavier atoms could allow us to utilize high and low oxidation states, expanding the possibilities for redox applications, such as transition metal-like catalysts.112 In addition, the heavier congeners often improve the material properties involving charge carrier mobility,113 phosphorescence,114 and stimuli-responsiveness.115
N)-containing conjugated polymers, which show the extension of π-conjugation through the BN bonds.108,109 The heavier congeners of BN, involving GaP and InP, might provide novel electronic states for conjugated polymers, such as weakly aromatic Ga2P and In2P rings,110 possibly resulting in higher hyperpolarizability.111 Furthermore, the inert pair effect in the heavier atoms could allow us to utilize high and low oxidation states, expanding the possibilities for redox applications, such as transition metal-like catalysts.112 In addition, the heavier congeners often improve the material properties involving charge carrier mobility,113 phosphorescence,114 and stimuli-responsiveness.115
Further improvements in the synthetic approach for isolation of many unstable structures and polymerization methods applicable to a wider range of monomers will expand the frontiers of the heavier group 13 elements by involving thallium chemistry, which is not able to be extrapolated from the chemistry of boron. In this context, the incorporation of these species into polymeric scaffolds could provide enhanced chemical stability because the hydrophobic polymeric structures could expel polar-reactive species, such as water. In addition, we witnessed that photochemical and electrochemical reactions and their cooperative usages have opened new horizons in the field of organic chemistry. The application of these powerful synthetic methodologies in inorganic and organometallic chemistries can play pivotal roles for accessing novel chemical structures.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was partially supported by a Grant-in-Aid for Early-Career Scientists (for S. I., JSPS KAKENHI Grant Number 23K13793) and for Scientific Research (B) (for K. T., JSPS KAKENHI Grant Number 24K01570).References
- A. Das, U. Das and A. K. Das, Coord. Chem. Rev., 2023, 479, 215000 CrossRef CAS.
- The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, 2011 Search PubMed.
- J. Lewiński, J. Zachara, I. Justyniak and M. Dranka, Coord. Chem. Rev., 2005, 249, 1185–1199 CrossRef.
- C. D. Entwistle and T. B. Marder, Angew. Chem., Int. Ed., 2002, 41, 2927–2931 CrossRef CAS PubMed.
- C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS.
- F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- N. Matsumi and Y. Chujo, Polym. J., 2008, 40, 77–89 CrossRef CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef PubMed.
- K. Tanaka and Y. Chujo, Macromol. Rapid Commun., 2012, 33, 1235–1255 CrossRef CAS PubMed.
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 633–643 CrossRef CAS.
- Y. Ren and F. Jäkle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- M. Gon, K. Tanaka and Y. Chujo, Bull. Chem. Soc. Jpn., 2017, 90, 463–474 CrossRef CAS.
- H. Helten, Chem. – Asian J., 2019, 14, 919–935 CrossRef CAS PubMed.
- S. Ito, M. Gon, K. Tanaka and Y. Chujo, Polym. Chem., 2021, 12, 6372–6380 RSC.
- J. Huang, X. Wang, Y. Xiang, L. Guo and G. Chen, Adv. Energy Sustainability Res., 2021, 2, 2100016 CrossRef CAS.
- J. Miao, Y. Wang, J. Liu and L. Wang, Chem. Soc. Rev., 2021, 51, 153–187 RSC.
- A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2015, 45, 922–953 RSC.
- F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS PubMed.
- S. Anderson, M. S. Weaver and A. J. Hudson, Synth. Met., 2000, 111, 459–463 CrossRef.
- P. E. Burrows, L. S. Sapochak, D. M. McCarty, S. R. Forrest and M. E. Thompson, Appl. Phys. Lett., 1994, 64, 2718–2720 CrossRef CAS.
- C. J. Broan, J. P. L. Cox, A. S. Craig, R. Kataky, D. Parker, A. Harrison, A. M. Randall and G. Ferguson, J. Chem. Soc., Perkin Trans. 2, 1991, 87–99 RSC.
- J. S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- D. A. Atwood and M. J. Harvey, Chem. Rev., 2001, 101, 37–52 CrossRef CAS PubMed.
- Y. Kawano, Y. Ito, S. Ito, K. Tanaka and Y. Chujo, Macromolecules, 2021, 54, 1934–1942 CrossRef CAS.
- C. Cui, H. W. Roesky, H. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- H. W. Roesky and S. S. Kumar, Chem. Commun., 2005, 4027–4038 RSC.
- M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Chem. Rev., 2021, 121, 12784–12965 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2009, 39, 486–494 RSC.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- L. Luo, J. M. Younker and A. V. Zabula, Science, 2024, 384, 1424–1428 CrossRef CAS PubMed.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- K. Lang, J. Mosinger and D. M. Wagnerová, Coord. Chem. Rev., 2004, 248, 321–350 CrossRef CAS.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- F. L. Portwich, Y. Carstensen, A. Dasgupta, S. Kupfer, R. Wyrwa, H. Görls, C. Eggeling, B. Dietzek, S. Gräfe, M. Wächtler and R. Kretschmer, Angew. Chem., Int. Ed., 2022, 61, e202117499 CrossRef CAS PubMed.
- S. Ito, T. Hosokai, K. Tanaka and Y. Chujo, Commun. Chem., 2024, 7, 202 CrossRef CAS PubMed.
- K. Hoshi, H. Sasabe, Y. Chiba, N. Yoshida, T. Nakamura, K. Nagasawa, Y. Sayama, H. Katagiri and J. Kido, Adv. Opt. Mater., 2024, 12, 2303303 CrossRef CAS.
- T. Ono, K. Ishihama, A. Taema, T. Harada, K. Furusho, M. Hasegawa, Y. Nojima, M. Abe and Y. Hisaeda, Angew. Chem., Int. Ed., 2021, 60, 2614–2618 CrossRef CAS PubMed.
- Y. Aoyama, Y. Sakai, S. Ito and K. Tanaka, Chem. – Eur. J., 2023, 29, e202300654 CrossRef CAS PubMed.
- F. Kobayashi, A. Yoshida, M. Gemba, Y. Takatsu and M. Tadokoro, Dalton Trans., 2024, 53, 11689–11696 RSC.
- J.-W. Wang, F. Ma, T. Jin, P. He, Z.-M. Luo, S. Kupfer, M. Karnahl, F. Zhao, Z. Xu, T. Jin, T. Lian, Y.-L. Huang, L. Jiang, L.-Z. Fu, G. Ouyang and X.-Y. Yi, J. Am. Chem. Soc., 2023, 145, 676–688 CrossRef CAS PubMed.
- J. Lu, A. R. Hlil, Y. Meng, A. S. Hay, Y. Tao, M. D'Iorio, T. Maindron and J. Dodelet, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2887–2892 CrossRef CAS.
- A. Meyers and M. Weck, Macromolecules, 2003, 36, 1766–1768 CrossRef CAS.
- A. Meyers and M. Weck, Chem. Mater., 2004, 16, 1183–1188 CrossRef CAS.
- T. Iijima and T. Yamamoto, J. Organomet. Chem., 2006, 691, 5016–5023 CrossRef CAS.
- T. Takayama, M. Kitamura, Y. Kobayashi, Y. Arakawa and K. Kudo, Macromol. Rapid Commun., 2004, 25, 1171–1174 CrossRef CAS.
- N. Du, R. Tian, J. Peng and M. Lu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 397–406 CrossRef CAS.
- Q. Mei, N. Du and M. Lu, J. Appl. Polym. Sci., 2006, 99, 1945–1952 CrossRef CAS.
- S. Li, M. Lin, J. Lu and H. Liang, Macromolecules, 2009, 42, 1258–1263 CrossRef CAS.
- C. Wang, W. Zhang, N. Zhou, Y. Qiu, Z. Cheng and X. Zhu, Int. J. Polym. Sci., 2010, 2010, 1–7 Search PubMed.
- J. Luo, C. Yang, L. Liang and M. Lu, J. Polym. Res., 2011, 18, 1197–1206 CrossRef CAS.
- F. Fang, F. Kong, X. Zhang, M. Lin, Y. Zhang and S. Ding, J. Appl. Polym. Sci., 2017, 134, 44573 CrossRef.
- X.-Y. Wang and M. Weck, Macromolecules, 2005, 38, 7219–7224 CrossRef CAS.
- T. Yamamoto and I. Yamaguchi, Polym. Bull., 2003, 50, 55–60 CrossRef CAS.
- T. Iijima, S. Kuroda and T. Yamamoto, Macromolecules, 2008, 41, 1654–1662 CrossRef CAS.
- Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- X. Zhang, H. Zhang, B. Qiu, D. Zhu, S. Zhang, Y. Bian, J. Wang, D. Li, S. Wang, W. Mai, J. Chen and T. Li, Fuel, 2023, 331, 125828 CrossRef CAS.
- M. Chen, X. Liu, Y. Yang, W. Xu, K. Chen and R. Luo, ACS Appl. Mater. Interfaces, 2023, 15, 8263–8274 CrossRef CAS PubMed.
- V. Walter, Y. Gao, N. Grzegorzek, M. Krempe, F. Hampel, N. Jux and R. R. Tykwinski, Angew. Chem., Int. Ed., 2019, 58, 494–498 CrossRef CAS PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2007, 183–194 Search PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- K. A. Morgan, S. E. Rudd, A. Noor and P. S. Donnelly, Chem. Rev., 2023, 123, 12004–12035 CrossRef CAS PubMed.
- Y. Liu, P. Bhattarai, Z. Dai and X. Chen, Chem. Soc. Rev., 2018, 48, 2053–2108 RSC.
- M. Yoshifuji, K. Kamijo and K. Toyota, Tetrahedron Lett., 1994, 35, 3971–3974 CrossRef CAS.
- B. Bagh, J. B. Gilroy, A. Staubitz and J. Müller, J. Am. Chem. Soc., 2010, 132, 1794–1795 CrossRef CAS PubMed.
- T. Matsumoto, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2013, 135, 4211–4214 CrossRef CAS PubMed.
- T. Matsumoto, K. Tanaka, K. Tanaka and Y. Chujo, Dalton Trans., 2015, 44, 8697–8707 RSC.
- T. Matsumoto, H. Takamine, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 2368–2375 RSC.
- T. Matsumoto, S. Ito, K. Tanaka and Y. Chujo, Polym. J., 2018, 50, 197–202 CrossRef CAS.
- T. Matsumoto, K. Tanaka and Y. Chujo, RSC Adv., 2015, 5, 55406–55410 RSC.
- T. Matsumoto, H. Takamine, K. Tanaka and Y. Chujo, Org. Lett., 2015, 17, 1593–1596 CrossRef CAS PubMed.
- T. Matsumoto, K. Tanaka and Y. Chujo, Macromolecules, 2015, 48, 1343–1351 CrossRef CAS.
- T. Matsumoto, Y. Onishi, K. Tanaka, H. Fueno, K. Tanaka and Y. Chujo, Chem. Commun., 2014, 50, 15740–15743 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, e1903530 CrossRef PubMed.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031–3066 CrossRef CAS PubMed.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2014, 20, 8320–8324 CrossRef CAS PubMed.
- R. Yoshii, A. Hirose, K. Tanaka and Y. Chujo, J. Am. Chem. Soc., 2014, 136, 18131–18139 CrossRef CAS PubMed.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 4, 5314–5319 RSC.
- S. Ito, A. Hirose, M. Yamaguchi, K. Tanaka and Y. Chujo, J. Mater. Chem. C, 2016, 4, 5564–5571 RSC.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2017, 1, 1573–1579 RSC.
- S. Ito, K. Tanaka and Y. Chujo, Inorganics, 2019, 7, 100 CrossRef CAS.
- S. Ito, M. Yaegashi, K. Tanaka and Y. Chujo, Chem. – Eur. J., 2021, 27, 9302–9312 CrossRef CAS PubMed.
- S. Ito, M. Hashizume, H. Taka, H. Kita, K. Tanaka and Y. Chujo, Mater. Chem. Front., 2023, 7, 4971–4983 RSC.
- S. Ito, K. Tanaka and Y. Chujo, Dalton Trans., 2024, 53, 14858–14865 RSC.
- K. Singh, I. Siddiqui, V. Sridharan, R. A. K. Yadav, J.-H. Jou and D. Adhikari, Inorg. Chem., 2021, 60, 19128–19135 CrossRef CAS PubMed.
- A. Hirose, K. Tanaka, R. Yoshii and Y. Chujo, Polym. Chem., 2015, 6, 5590–5595 RSC.
- K. Tanaka, T. Yanagida, A. Hirose, H. Yamane, R. Yoshii and Y. Chujo, RSC Adv., 2015, 5, 96653–96659 RSC.
- M. Yamaguchi, S. Ito, A. Hirose, K. Tanaka and Y. Chujo, Polym. Chem., 2018, 9, 1942–1946 RSC.
- S. Ito, M. Fukuyama, K. Tanaka and Y. Chujo, Macromol. Chem. Phys., 2022, 223, 2100504 CrossRef CAS.
- S. Ito, Y. Sakai and K. Tanaka, Macromol. Rapid Commun., 2025, 46, e2400775 CrossRef PubMed.
- S. Ito, A. Hirose, M. Yamaguchi, K. Tanaka and Y. Chujo, Polymers, 2017, 9, 68 CrossRef PubMed.
- M. L. Mejía, K. Agapiou, X. Yang and B. J. Holliday, J. Am. Chem. Soc., 2009, 131, 18196–18197 CrossRef PubMed.
- J. Heinze, B. A. Frontana-Uribe and S. Ludwigs, Chem. Rev., 2010, 110, 4724–4771 CrossRef CAS PubMed.
- C. Friebe, M. D. Hager, A. Winter and U. S. Schubert, Adv. Mater., 2012, 24, 332–345 CrossRef CAS PubMed.
- M. L. Mejía, G. Reeske and B. J. Holliday, Chem. Commun., 2010, 46, 5355–5357 RSC.
- M. S. Hill, P. B. Hitchcock and R. Pongtavornpinyo, Science, 2006, 311, 1904–1907 CrossRef CAS PubMed.
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed.
- M. S. Hill, R. Pongtavornpinyo and P. B. Hitchcock, Chem. Commun., 2006, 3720–3722 RSC.
- U. Salzner, J. B. Lagowski, P. G. Pickup and R. A. Poirier, Synth. Met., 1998, 96, 177–189 CrossRef CAS.
- M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS PubMed.
- T. Lorenz, A. Lik, F. A. Plamper and H. Helten, Angew. Chem., Int. Ed., 2016, 55, 7236–7241 CrossRef CAS PubMed.
- T. Lorenz, M. Crumbach, T. Eckert, A. Lik and H. Helten, Angew. Chem., Int. Ed., 2017, 56, 2780–2784 CrossRef CAS PubMed.
- A. García-Romero, C. Hu, M. Pink and J. M. Goicoechea, J. Am. Chem. Soc., 2025, 147, 1231–1239 CrossRef PubMed.
- D. Jacquemin, J. Phys. Chem. A, 2004, 108, 9260–9266 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- S. Ye, V. Lotocki, H. Xu and D. S. Seferos, Chem. Soc. Rev., 2022, 51, 6442–6474 RSC.
- Z. Cheng, X. Wang, J. Zhao, S. Wang, X. Wu, H. Tong and L. Wang, Macromolecules, 2023, 56, 2972–2979 CrossRef CAS.
- M. Gon, Y. Morisaki, K. Tanimura and K. Tanaka, Dalton Trans., 2024, 53, 11858–11866 RSC.
| This journal is © The Royal Society of Chemistry 2025 |