
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High performance polyurethanes derived from aromatic acetal-containing polyols enabling closed-loop recycling†
Patrick
Schara
a,
Tankut
Türel
 a,
Anna
Cristadoro
b,
Rint P.
Sijbesma
a and
Željko
Tomović
*a
a,
Anna
Cristadoro
b,
Rint P.
Sijbesma
a and
Željko
Tomović
*a
aDepartment of Chemical Engineering and Chemistry, and Institute for Complex Molecular Systems, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: z.tomovic@tue.nl
bBASF Polyurethanes GmbH, Elastogranstraße 60, 49448 Lemförde, Germany
First published on 30th January 2025
Abstract
Polyurethanes (PUs) are widely employed across diverse industries due to their versatility, durability, and mechanical strength. Enhancing their thermal and mechanical performance holds great potential for expanding their applicability and unlocking new market opportunities. This study addresses two key challenges: limited availability of aromatic polyols for high-performance PUs and their recycling issues. Incorporation of aromatic content in polyether polyols has traditionally been difficult using conventional methods. Herein, we developed three novel aromatic acetal-containing polyols through a green and solvent-free protocol via the polycondensation of aldehydes and diols, using acidic heterogeneous catalysts. Resulting polyols, with tailored aromatic content, significantly improved the mechanical strength of PUs, while maintaining low viscosity and easy processability. Besides that, PUs synthesized from these polyols exhibited excellent thermal stability and remarkable water resistance under neutral conditions. Additionally, these materials demonstrated efficient closed-loop recyclability through a novel transacetalization-based depolymerization under mild acidic conditions, yielding high purity monomers in good yields. This work introduces innovative aromatic polyacetal polyols, offering a sustainable approach to high-performance PUs. The approach also leverages the wide availability of diols and aldehydes, enabling the design of PUs with superior properties and closed-loop recycling.
Introduction
Each year, approximately 400 million tons of plastics are produced globally, with polyurethanes (PUs) accounting for approximately 8% of the total due to their exceptional versatility.1 This versatility makes PUs indispensable across a broad spectrum of applications, including rigid and flexible foams, elastomers, adhesives, and thermoplastics, all integral to modern life.2–4 Although numerous grades of PU materials are already available, enhancing their physical properties can expand their applicability and open up new market opportunities.Despite their widespread use, the end-of-life management of PU waste presents a significant and persistent challenge. The majority of PU waste is either landfilled or incinerated, contributing to environmental pollution and the irreversible loss of valuable resources.5–7 Current methods for processing PU waste predominantly rely on mechanical grinding, which involves grinding the waste into small particles and adding them as fillers in new PU product. Unfortunately, this approach produces materials with severely degraded properties, essentially downcycling the PU waste into low-value applications, such as carpet underlay and mats.8,9
To address these limitations, substantial research has been dedicated towards chemical recycling as a more sustainable alternative to mechanical recycling for PU materials. Common chemical recycling methods such as glycolysis,10–14 hydrolysis,15–17 aminolysis,18–21 and acidolysis,22–24 offer the potential to break down PUs into smaller components. However, these methods often require harsh reaction conditions, including high temperatures, pressures or excessive amounts of solvents and reactants. Additionally, they typically yield complex mixtures of polyols and oligomers that are cumbersome to isolate and purify to the same quality as virgin polyols, limiting their reuse in producing new PUs. These challenges have impeded the widespread commercial adoption of chemical recycling, leaving incineration and landfilling as dominant waste management strategies for PU materials.
To enhance the recyclability of PU materials, significant research efforts have focused on incorporating cleavable bonds into PU structures, particularly within the polyol component. Acetal functionalities have emerged as a promising candidate due to their stability under neutral or basic conditions and selective hydrolysis under acidic environments.25–29 In our recent works, we developed acetal polyols via solvent-free synthesis using heterogeneous catalysts. These polyols were then utilized for the preparation of PU materials amenable to closed loop recycling.30
Beyond improving recyclability, another key area of PU research is the incorporation of aromatic groups into the polyol structures, which can significantly enhance mechanical strength, thermal stability, and fire retardance.31–37 Typically, commercial polyether polyols are synthesized through the ring-opening of either propylene oxide or ethylene oxide, which does not allow for the incorporation of significant aromatic content. Additionally, the use of aromatic polyols presents its own challenge: increasing the aromatic content often raises the melting points and viscosities of polyols, complicating the bulk synthesis of PUs, which significantly limits their industrial applicability. For example, polyols used for the synthesis of PU cast elastomers are frequently processed at temperatures ranging from 45 to 75 °C, while for thermoplastic PU typically at processing temperature of ∼80 °C.4 Therefore, ensuring that polyols are processable within this temperature range is crucial for their successful application in polyurethane production. Consequently, polyether polyols with high aromatic content remain scarce in the literature, with most examples featuring only limited aromatic incorporation.
In this work, improvements in both the mechanical performance and recyclability of PU materials are addressed. Specifically, we developed a green protocol for the synthesis of an array of acetal-containing polyols with aromatic motifs via the polycondensation of diols and aldehydes, utilizing either aromatic aldehydes or aromatic diols (Fig. 1A). This strategy enabled the design of highly aromatic polyols with a broad range of properties. The resulting PU materials not only exhibited superior mechanical properties to the conventional PUs but also demonstrated efficient closed-loop recyclability (Fig. 1B). The hydrolysis of these acetal-containing PUs allowed the recovery of the original monomers with high yields and purity. Furthermore, monomer recovery process was enhanced through transacetalization, utilizing environmentally benign alcohols and acids. Notably, this work marks the first use of transacetalization method for the depolymerization of acetal containing polymer materials. This approach enables efficient recycling under milder conditions, avoiding the harsh conditions and long reaction times typically required by conventional hydrolysis methods. Furthermore, this work highlights the potential of acetal chemistry to advance the development of sustainable, recyclable polyurethanes with superior mechanical performance, addressing key challenges in both recyclability and material properties.
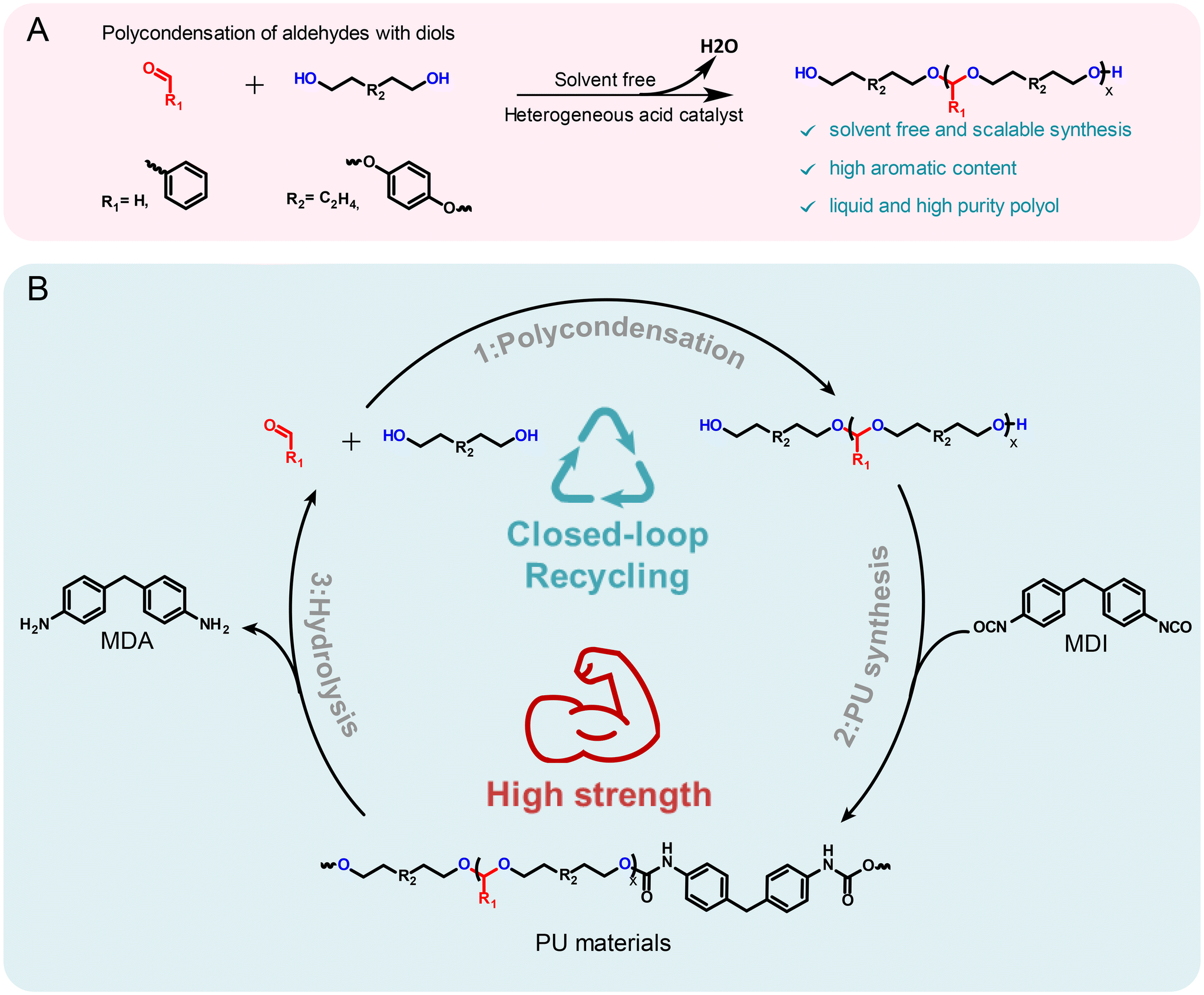 | ||
| Fig. 1 Synthesis and utilization of aromatic acetal-containing polyol in high performance closed-loop recyclable polyurethanes. (A) Synthetic route for aromatic acetal-containing polyols via the condensation of diols with aldehydes, facilitated by a heterogeneous acid catalyst. (B) Proposed closed-loop recycling scheme of acetal-containing high-performance PU materials back into original monomers. | ||
Results and discussion
Synthesis and characterization of aromatic acetal-containing polyols
Advancing previous synthetic methods for aliphatic acetal polyols (APs) described in our previous work,30 herein, we developed a green protocol for the synthesis of novel aromatic APs. Using this new approach, three novel aromatic polyols, AP1–AP3, were prepared, with aromatic contents ranging from 11 to 35 wt%. AP1 and AP2 were synthesized via the polycondensation of paraformaldehyde with 1,6-hexanediol (H16) and 1,4-bis(2-hydroxyethoxy)benzene (HEB) in varying ratios, utilizing a commercial silica-alumina heterogeneous acid catalyst, Siral 70 (see ESI for more details, Scheme S1–S3†). Specifically, AP1 was produced with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2, while AP2 utilized a ratio of 1:0.8. AP2 was optimized to maximize HEB content without increasing the melting point and viscosity of the polyol beyond practical limits for polyurethane applications. An alternative method for incorporating aromatic units into APs as a side group involved the condensation of aromatic aldehydes with diols utilizing a commercial copper-based metal organic framework heterogeneous acid catalyst, C300. AP3, synthesized through the condensation of benzaldehyde and H16 using the same heterogeneous acid catalyst, achieved the highest aromatic content among the polyols at 35 wt%, as confirmed by 1H-NMR spectra (Fig. S1–S3†). After filtration of the reaction mixture to remove the heterogeneous acid catalyst, the resulting polyols were transparent, colorless, and had an acid value below the detection limit (0.08 mg KOH g−1). Table 1 provides a comprehensive summary of the prepared APs, including their structures and properties. The purity and structure of AP1–3 were confirmed with 1H-NMR and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (Fig. 2 and Fig. S1–S3†). The 1H-NMR spectra (Fig. 2) displayed characteristic acetal proton signals for each polyol. AP1 and AP2 showed three distinct acetal signals at 4.66, 4.75, and 4.85 ppm, corresponding to the different acetal groups within the copolymer structure of H16 and HEB repeating units (Fig. S4†). For both AP1 and AP2 the monomer ratio matches the feed ratio closely. Additionally, the relative intensities of the acetal signals indicate that HEB incorporation is statistical, with acetals bridging two HEB units (4.85 ppm) being only a minor product in both AP1 and AP2. These findings confirm the statistical incorporation of HEB throughout the polyols in proportions consistent with the monomer feed ratios. As expected, AP3 displayed a single acetal proton signal, with no detectable impurities. MALDI-TOF-MS analysis was employed to further validate the polymer structures. The mass spectra for AP1 and AP2 showed a spacing of 130.10 and 210.08 Da between the peaks, corresponding precisely to the mass values of the H16 and HEB repeating units, respectively. The absolute peak values also matched the theoretical molar mass of the polymer chains with varying numbers of repeating units, with an additional sodium cation (e.g., [M + Na]+ for AP1, the peak at 1131.75 Da corresponds to a polymer chain with seven H16 units and one HEB unit). For AP2, the mass spectra showed similar peaks as AP1, but with different relative intensities due to the different monomer ratio. The [M + Na]+ peak at 1161.63 Da represents a polymer chain with four H16 units and three HEB units. In contrast, AP3 exhibited a simpler mass spectrum with 206.13 Da spacing between the peaks, consistent with the mass of H16-benzaldehyde repeating units. The absolute peak values for AP3 also aligned with the expected polymer structure (e.g., [M + Na]+ at 1171.72 Da, corresponds to a polymer chain with six H16 repeating units).
:0.2, while AP2 utilized a ratio of 1:0.8. AP2 was optimized to maximize HEB content without increasing the melting point and viscosity of the polyol beyond practical limits for polyurethane applications. An alternative method for incorporating aromatic units into APs as a side group involved the condensation of aromatic aldehydes with diols utilizing a commercial copper-based metal organic framework heterogeneous acid catalyst, C300. AP3, synthesized through the condensation of benzaldehyde and H16 using the same heterogeneous acid catalyst, achieved the highest aromatic content among the polyols at 35 wt%, as confirmed by 1H-NMR spectra (Fig. S1–S3†). After filtration of the reaction mixture to remove the heterogeneous acid catalyst, the resulting polyols were transparent, colorless, and had an acid value below the detection limit (0.08 mg KOH g−1). Table 1 provides a comprehensive summary of the prepared APs, including their structures and properties. The purity and structure of AP1–3 were confirmed with 1H-NMR and matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (Fig. 2 and Fig. S1–S3†). The 1H-NMR spectra (Fig. 2) displayed characteristic acetal proton signals for each polyol. AP1 and AP2 showed three distinct acetal signals at 4.66, 4.75, and 4.85 ppm, corresponding to the different acetal groups within the copolymer structure of H16 and HEB repeating units (Fig. S4†). For both AP1 and AP2 the monomer ratio matches the feed ratio closely. Additionally, the relative intensities of the acetal signals indicate that HEB incorporation is statistical, with acetals bridging two HEB units (4.85 ppm) being only a minor product in both AP1 and AP2. These findings confirm the statistical incorporation of HEB throughout the polyols in proportions consistent with the monomer feed ratios. As expected, AP3 displayed a single acetal proton signal, with no detectable impurities. MALDI-TOF-MS analysis was employed to further validate the polymer structures. The mass spectra for AP1 and AP2 showed a spacing of 130.10 and 210.08 Da between the peaks, corresponding precisely to the mass values of the H16 and HEB repeating units, respectively. The absolute peak values also matched the theoretical molar mass of the polymer chains with varying numbers of repeating units, with an additional sodium cation (e.g., [M + Na]+ for AP1, the peak at 1131.75 Da corresponds to a polymer chain with seven H16 units and one HEB unit). For AP2, the mass spectra showed similar peaks as AP1, but with different relative intensities due to the different monomer ratio. The [M + Na]+ peak at 1161.63 Da represents a polymer chain with four H16 units and three HEB units. In contrast, AP3 exhibited a simpler mass spectrum with 206.13 Da spacing between the peaks, consistent with the mass of H16-benzaldehyde repeating units. The absolute peak values for AP3 also aligned with the expected polymer structure (e.g., [M + Na]+ at 1171.72 Da, corresponds to a polymer chain with six H16 repeating units).
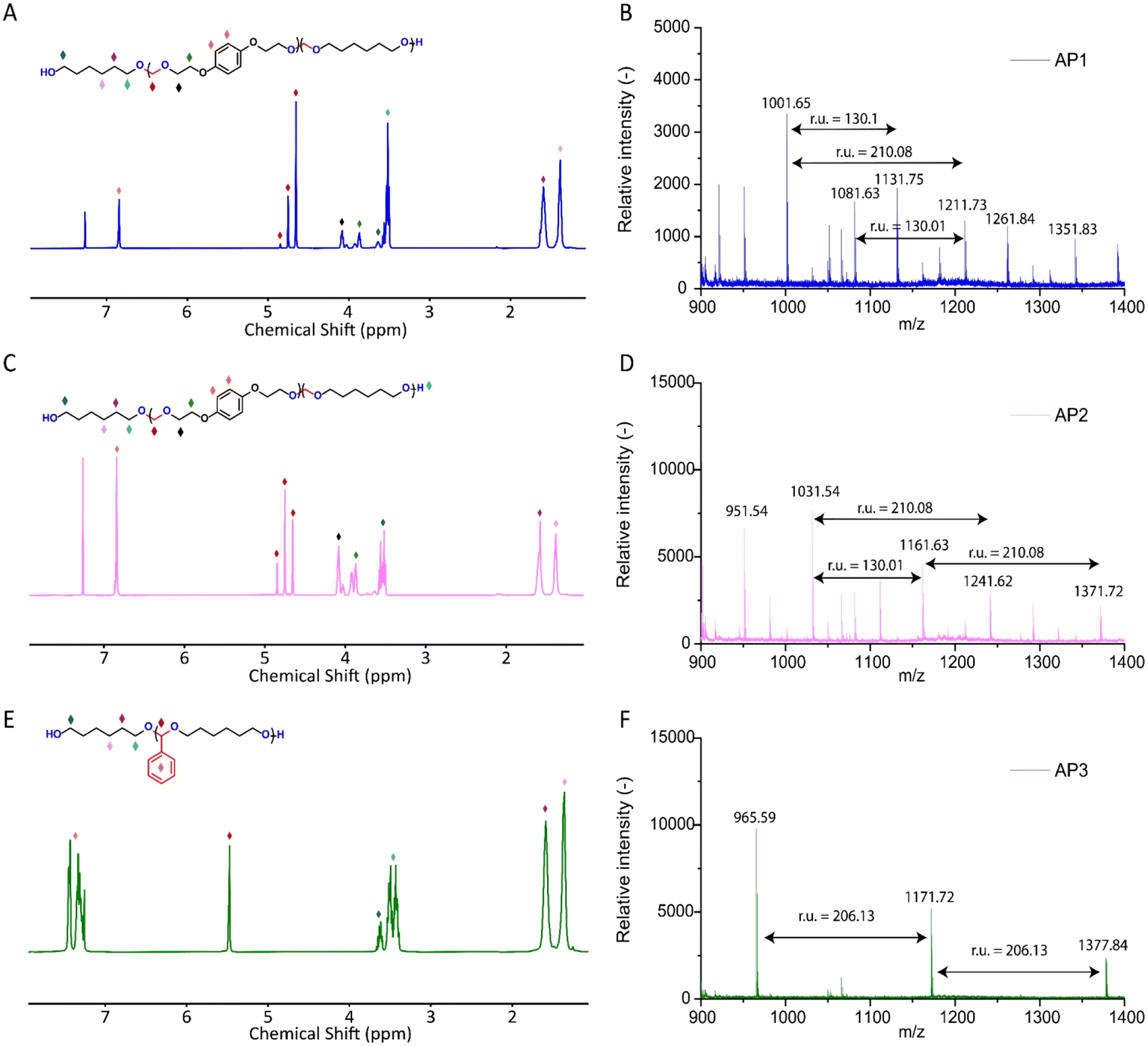 | ||
| Fig. 2 1H-NMR (400 MHz, CDCl3) and MALDI-TOF mass spectra of AP1 (A and B), AP2 (C and D) and AP3 (E and F). | ||
| Polyol name | Polyol structure | Aromatic content (wt%) | Viscosity at 65 °C (mPa s) | M n based on 1H-NMR (g mol−1) | OH# (mg KOH g−1) |
|---|---|---|---|---|---|
| AP 1 |

|
11 | 97 | 1370 | 77 |
| AP 2 |
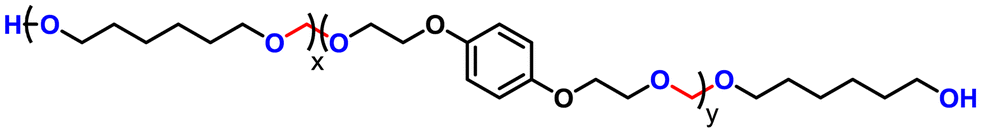
|
23 | 250 | 1400 | 81 |
| AP 3 |
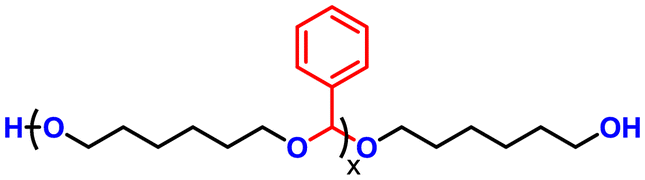
|
35 | 330 | 1430 | 84 |
The viscosities of the polyols were measured at 65 °C across a range of shear rates (Fig. S5†). AP1 and AP2 exhibited shear-thickening behavior, while AP3 demonstrated shear-thinning behavior. The viscosity plateau values for AP1, AP2, and AP3 were 97, 250, and 330 mPa s, respectively. These relatively low viscosity values make these polyols suitable for utilization in PU preparation under bulk processing conditions. Low-viscosity polyols are particularly advantageous in industrial applications as they facilitate efficient mixing, handling, and processing, ultimately contributing to cost-effectiveness and scalability in PU production.
Gel permeation chromatography (GPC) was performed to determine the molar mass distribution of the polyols (Fig. S6†). The GPC traces for AP1–3 revealed nearly identical distribution patterns, with no detectable monomer residues and a dispersity of approximately 1.6. The smooth and symmetrical GPC curves for all APs indicated uniformity in the polymer chains, suggesting a well-controlled polymerization process.
Differential scanning calorimetry (DSC) was employed to investigate the thermal properties of the polyols (Fig. S7†). Both AP1 and AP2 exhibited distinct crystallization and melting behavior. AP1 showed a melting point around 10 °C, while AP2 displayed a broad melting peak between 30 and 70 °C, indicative of its higher aromatic content as compared with AP1. Additionally, the glass transition temperatures (Tg) of AP1, AP2, and AP3 were found to be −70 °C, −47 °C, and −43 °C, respectively, reflecting the increasing rigidity of the polyols with higher aromatic content.
The hydrolytic stability of AP1–AP3 was investigated in an acidic water/THF mixture at 50 °C using p-toluenesulfonic acid as an acid catalyst utilizing in situ1H-NMR spectroscopy (Fig. S8–S10†). The obtained 1H-NMR spectra were used to calculate the percentage of remaining acetal bonds over time (Fig. 3). AP1 and AP2 demonstrated exceptional hydrolytic stability, with no detectable hydrolysis or aldehyde formation after 15 hours, indicating that the acetal bonds in these polyols remain intact under acidic conditions. This result underscores the robust nature of these polyols, particularly AP1 and AP2, in environments where hydrolytic degradation is a concern. In contrast, AP3 underwent rapid hydrolysis under acidic conditions, with complete loss of acetals within 2 hours, accompanied by the formation of the benzaldehyde. Notably, the acetal concentration in AP3 approached zero, rather than reaching an equilibrium with the free aldehyde present in the solution, which is in contrast to the previous observations in studies involving acetals derived from acetaldehyde.30 The significant difference between the hydrolytic susceptibility of the benzaldehyde based polyol AP3 compared to the formaldehyde based AP1 and AP2 are consistent with previous literature. Accordingly, the hydrolytic stability of acetals under acidic conditions is influenced by the substituents on the acetal bridge. Acetals with a methylene bridge (i.e., AP1 and AP2) exhibit significantly higher stability compared to those with additional substituents such as AP3. This is due to the stabilizing effect of the side groups on the cationic hydrolysis intermediate.38
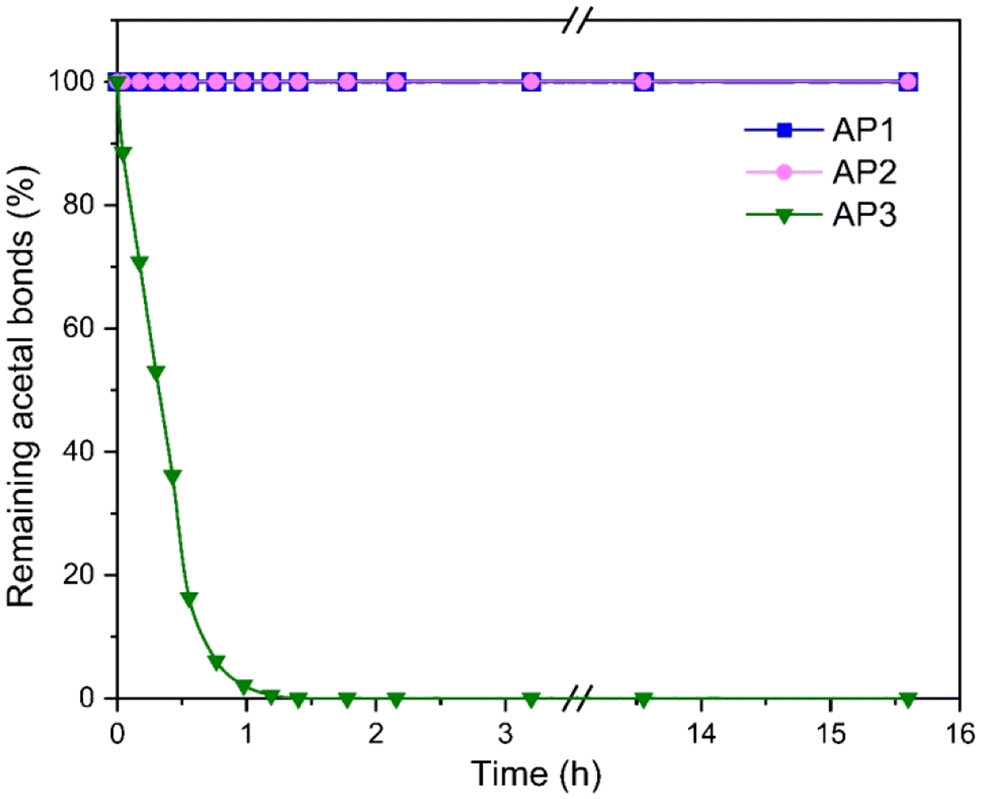 | ||
| Fig. 3 Degree of hydrolysis of AP1–AP3 under acidic conditions measured by 1H NMR over a period of 16 h (200 mg polyol in 1 ml THF-d8 with 0.2 g of a 20 mM p-toluenesulfonic acid aqueous solution). | ||
Synthesis and characterization of acetal polyol containing PU materials
Acetal containing PU materials were synthesized by reacting AP1–3 with 4,4′-methylene diphenyl diisocyanate (4,4′-MDI) and H16, maintaining an NCO-to-hydroxyl ratio of 1.01:1 in all formulations. The successful formation of urethane linkages was confirmed by FTIR, as indicated by the appearance of a carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretch at 1700 cm−1 and the disappearance of the isocyanate (NCO) stretching band at 2260 cm−1 (Fig. S11†). The carbonyl region around 1700 cm−1 consists of two distinct stretching peaks: the non-hydrogen bonded CO at 1730 cm−1 and the hydrogen-bonded CO at 1700 cm−1, which is typical for phase-separated polyurethanes.39,40 Since hydrogen bonding predominantly occurs in the hard phase, the degree of phase separation between soft and hard phases can be inferred by comparing the intensity of these two carbonyl signals. The ratio of the hydrogen-bonded to non-hydrogen-bonded carbonyl groups was calculated by comparing the areas under the peaks (Fig. S12†). Among the PUs, PU AP3 exhibited the highest ratio of 2.6, suggesting the greatest degree of phase separation. This was followed by PU AP1 (2.1), and PU AP2 (1.4). These results suggest that the presence of HEB increases interphase mixing, leading to reduced hydrogen bonding compared to PU AP3. GPC measurements were performed to determine the molar mass of the PUs, which showed weight-average molar mass (Mw) exceeding 100 kg mol−1 for all samples (Fig. S13†). PU AP1 and PU AP3 showed similar dispersities, with values of 2.8 and 2.6, respectively. PU AP2 possessing a higher molecular weight, exceeded the exclusion limit of the GPC column, leading to an high apparent molar mass shoulder at the exclusion limit. Thermal properties of the acetal-containing PU materials were evaluated using thermogravimetric analysis (TGA), and DSC. TGA demonstrated good thermal stability for all three PU materials, with initial degradation temperatures (Td5%) around 310 °C, which could be correlated with the degradation of the urethane bonds (Fig. 4A).39 At higher temperatures, PU AP3 degraded almost completely by 400 °C, leaving less than 10 wt% of material, while PU AP1 and PU AP2 retained about 60 wt% at this temperature. PU AP1 and PU AP2 showed two distinct temperatures of maximum degradation rate (Td,max) at approximately 340 °C and 430 °C, with the latter attributed to the degradation of the polyol soft phase (Fig. 4A).39 The differences in thermal characteristics can be attributed to the lower thermal stability of the aromatic acetal groups present in PU AP3 compared to the formaldehyde-based acetal groups in PU AP1 and PU AP2. DSC analysis revealed a soft phase Tg of −36 °C, 1 °C, and −18 °C for PU AP1, PU AP2, and PU AP3, respectively (Fig. 4B). Notably, PU AP2 exhibited the highest Tg, despite its constituent polyol (AP2) having a glass transition temperature comparable to that of AP3 (∼−45 °C). The substantial increase in the Tg of PU AP2 compared to the more modest increase observed in the Tg of PU AP3 relative to their respective acetals, could be explained by the mixing of the hard phase in the soft phase.41 The degree of phase separation, could be predicted based on the differences in Tg of the PUs with respect to their constituent acetals polyols. Accordingly, the degree of phase separation was lowest in PU AP2, followed by PU AP1 and highest in PU AP3, aligning with the findings from the FTIR analysis.
O) stretch at 1700 cm−1 and the disappearance of the isocyanate (NCO) stretching band at 2260 cm−1 (Fig. S11†). The carbonyl region around 1700 cm−1 consists of two distinct stretching peaks: the non-hydrogen bonded CO at 1730 cm−1 and the hydrogen-bonded CO at 1700 cm−1, which is typical for phase-separated polyurethanes.39,40 Since hydrogen bonding predominantly occurs in the hard phase, the degree of phase separation between soft and hard phases can be inferred by comparing the intensity of these two carbonyl signals. The ratio of the hydrogen-bonded to non-hydrogen-bonded carbonyl groups was calculated by comparing the areas under the peaks (Fig. S12†). Among the PUs, PU AP3 exhibited the highest ratio of 2.6, suggesting the greatest degree of phase separation. This was followed by PU AP1 (2.1), and PU AP2 (1.4). These results suggest that the presence of HEB increases interphase mixing, leading to reduced hydrogen bonding compared to PU AP3. GPC measurements were performed to determine the molar mass of the PUs, which showed weight-average molar mass (Mw) exceeding 100 kg mol−1 for all samples (Fig. S13†). PU AP1 and PU AP3 showed similar dispersities, with values of 2.8 and 2.6, respectively. PU AP2 possessing a higher molecular weight, exceeded the exclusion limit of the GPC column, leading to an high apparent molar mass shoulder at the exclusion limit. Thermal properties of the acetal-containing PU materials were evaluated using thermogravimetric analysis (TGA), and DSC. TGA demonstrated good thermal stability for all three PU materials, with initial degradation temperatures (Td5%) around 310 °C, which could be correlated with the degradation of the urethane bonds (Fig. 4A).39 At higher temperatures, PU AP3 degraded almost completely by 400 °C, leaving less than 10 wt% of material, while PU AP1 and PU AP2 retained about 60 wt% at this temperature. PU AP1 and PU AP2 showed two distinct temperatures of maximum degradation rate (Td,max) at approximately 340 °C and 430 °C, with the latter attributed to the degradation of the polyol soft phase (Fig. 4A).39 The differences in thermal characteristics can be attributed to the lower thermal stability of the aromatic acetal groups present in PU AP3 compared to the formaldehyde-based acetal groups in PU AP1 and PU AP2. DSC analysis revealed a soft phase Tg of −36 °C, 1 °C, and −18 °C for PU AP1, PU AP2, and PU AP3, respectively (Fig. 4B). Notably, PU AP2 exhibited the highest Tg, despite its constituent polyol (AP2) having a glass transition temperature comparable to that of AP3 (∼−45 °C). The substantial increase in the Tg of PU AP2 compared to the more modest increase observed in the Tg of PU AP3 relative to their respective acetals, could be explained by the mixing of the hard phase in the soft phase.41 The degree of phase separation, could be predicted based on the differences in Tg of the PUs with respect to their constituent acetals polyols. Accordingly, the degree of phase separation was lowest in PU AP2, followed by PU AP1 and highest in PU AP3, aligning with the findings from the FTIR analysis.
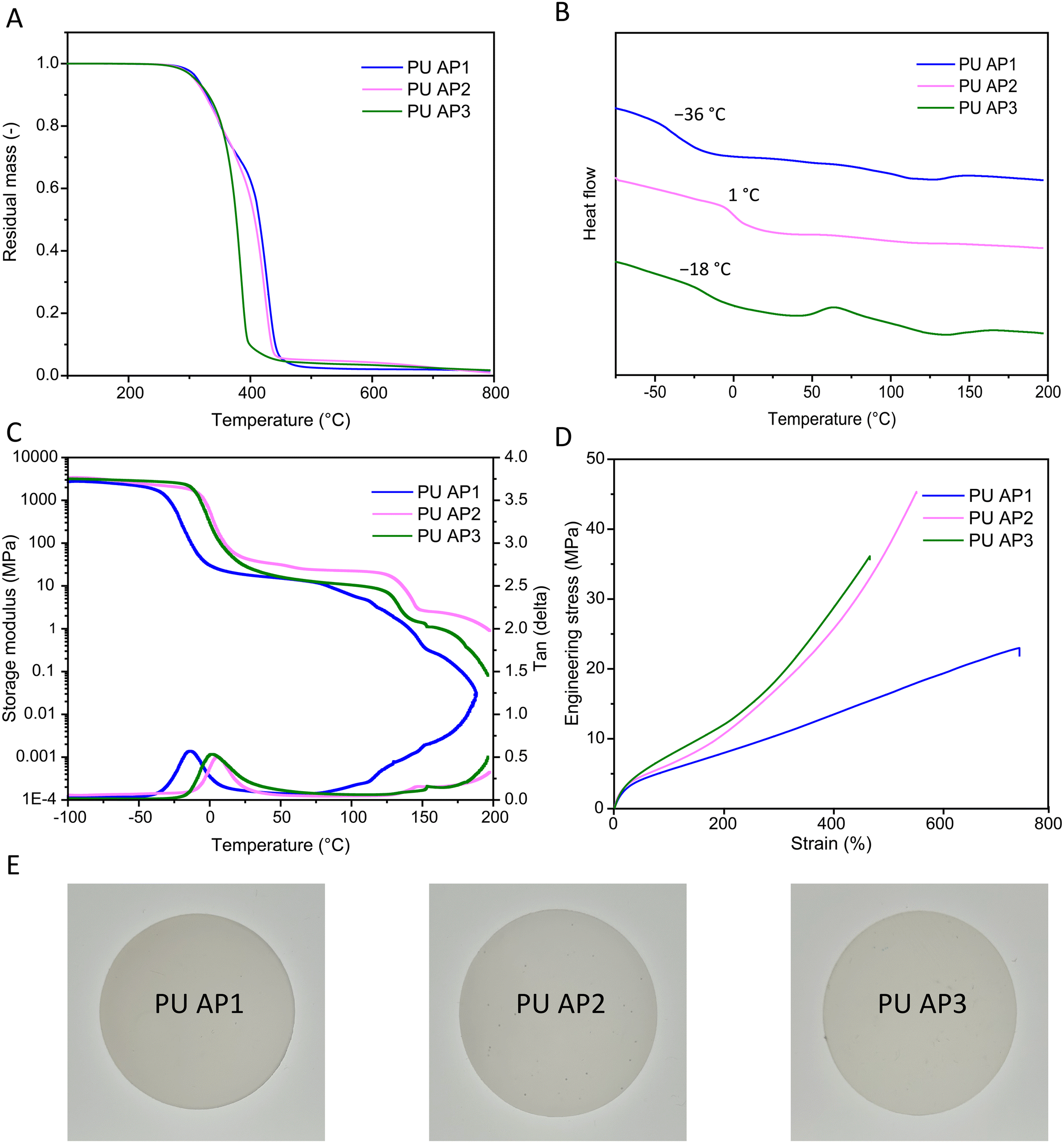 | ||
| Fig. 4 Thermo-mechanical properties of the acetal-containing PUs. (A) TGA (B) DSC, (C) DMA curves, (D) stress–strain, (E) photographs of the bulk cast material. | ||
Dynamic mechanical analysis (DMA) provided further insights into the thermomechanical behavior. Tg values, derived from the maxima of tan(δ) curves, were −14 °C, 2 °C, and 7 °C for PU AP1, PU AP2, and PU AP3, respectively, which followed a similar trend to those observed in DSC. Melting points were indicated by a drop in storage modulus around 130 °C. Notably, the modulus did not immediately drop to zero, indicating solid-like behavior persisted up to 200 °C, highlighting the mechanical robustness of the materials at elevated temperatures (Fig. 4C).
Tensile testing revealed excellent mechanical properties across all PU samples. PU AP1 showed the highest elongation of 663 ± 22% and the lowest stress at break of 23 ± 1 MPa, making it the most flexible material among the samples. In contrast, PU AP2 and PU AP3, having higher aromatic content, exhibited lower flexibility. PU AP2 stood out for its exceptional mechanical performance, including high elongation at break (560 ± 38%), the highest stress at break (47 ± 8 MPa), and Young's modulus (37 ± 2 MPa), resulting in a remarkable toughness exceeding 100 MJ m−3, equal to or even surpassing high-performance polyurethanes (Table 2 and Fig. 4D).32,42,43
| PU materials | T d5% (°C) | T g (DSC), (°C) | T g (DMA), (°C) | Young's modulus (MPa) | Stress at break (MPa) | Elongation (%) | Toughness (MJ m−3) |
|---|---|---|---|---|---|---|---|
| PU AP1 | 313 | −36 | −14 | 24 ± 2 | 23 ± 1 | 663 ± 22 | 96 ± 7 |
| PU AP2 | 308 | 1 | 6 | 37 ± 2 | 47 ± 8 | 560 ± 38 | 105 ± 21 |
| PU AP3 | 310 | −18 | 2 | 28 ± 3 | 37 ± 1 | 470 ± 4 | 75 ± 3 |
To assess the hydrolytic stability of the PU material, tensile test specimens of PU AP2 were fully submerged in distilled water at 70 °C for 48 hours. After drying under vacuum at 70 °C for 5 hours, the mechanical properties of the samples remained nearly identical to those of the original, non-exposed specimens. Additionally, the exposed samples showed no visible changes compared to the original films, and the GPC curve of the exposed sample was nearly identical to that of the original, further confirming excellent hydrolytic stability (Fig. 5 and Table 3)
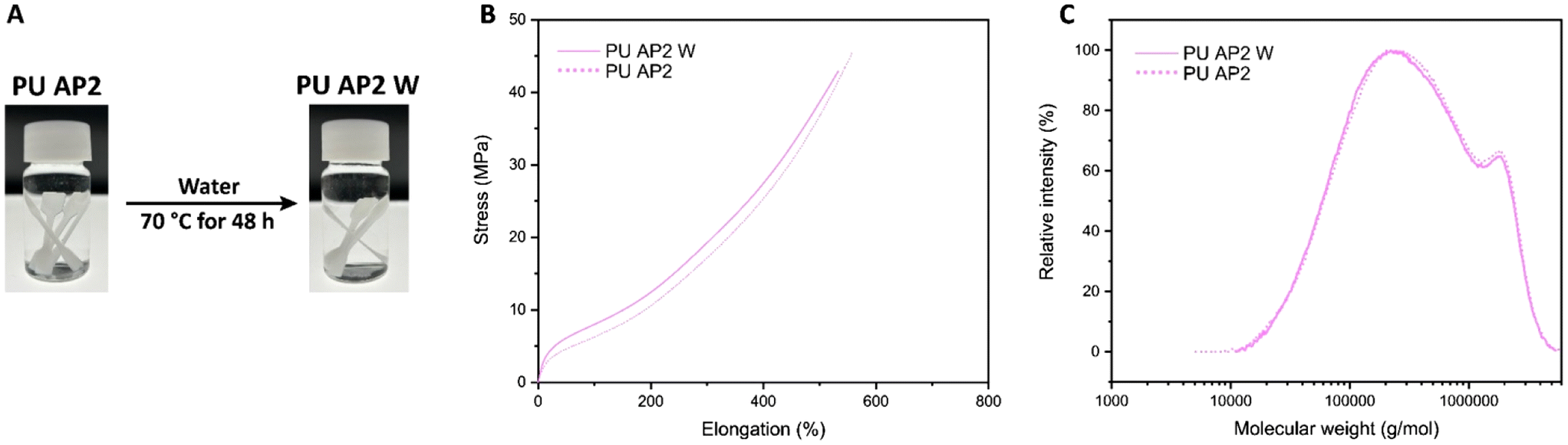 | ||
| Fig. 5 (A) Photographs of PU AP2 before and after water exposure, (B) Stress–strain curves, and (C) GPC traces of the specimens. | ||
| PU materials | Elongation (%) | Stress at break (MPa) | Young's modulus (MPa) | Toughness (MJ m−3) | M n based on GPC (Kg mol−1) |
|---|---|---|---|---|---|
| PU AP2 | 560 ± 38 | 47 ± 8 | 37 ± 2 | 105 ± 21 | 147 |
| PU AP2 W | 529 ± 7 | 45 ± 1 | 45 ± 2 | 103 ± 3 | 147 |
Closed-loop recycling of PU materials
To explore the potential for closed-loop recycling of PU AP1–3, we investigated their hydrolysis under acidic conditions. This process allows the acetal-containing PUs to be chemically recycled back into their original components (i.e., diols, aldehydes), and a urethane-rich hard phase, which can be easily separated through filtration and distillation. The isolated hard phase was further depolymerized into diols and 4,4′-Methylenedianiline (MDA), the precursor of 4,4′-MDI (Fig. 6A).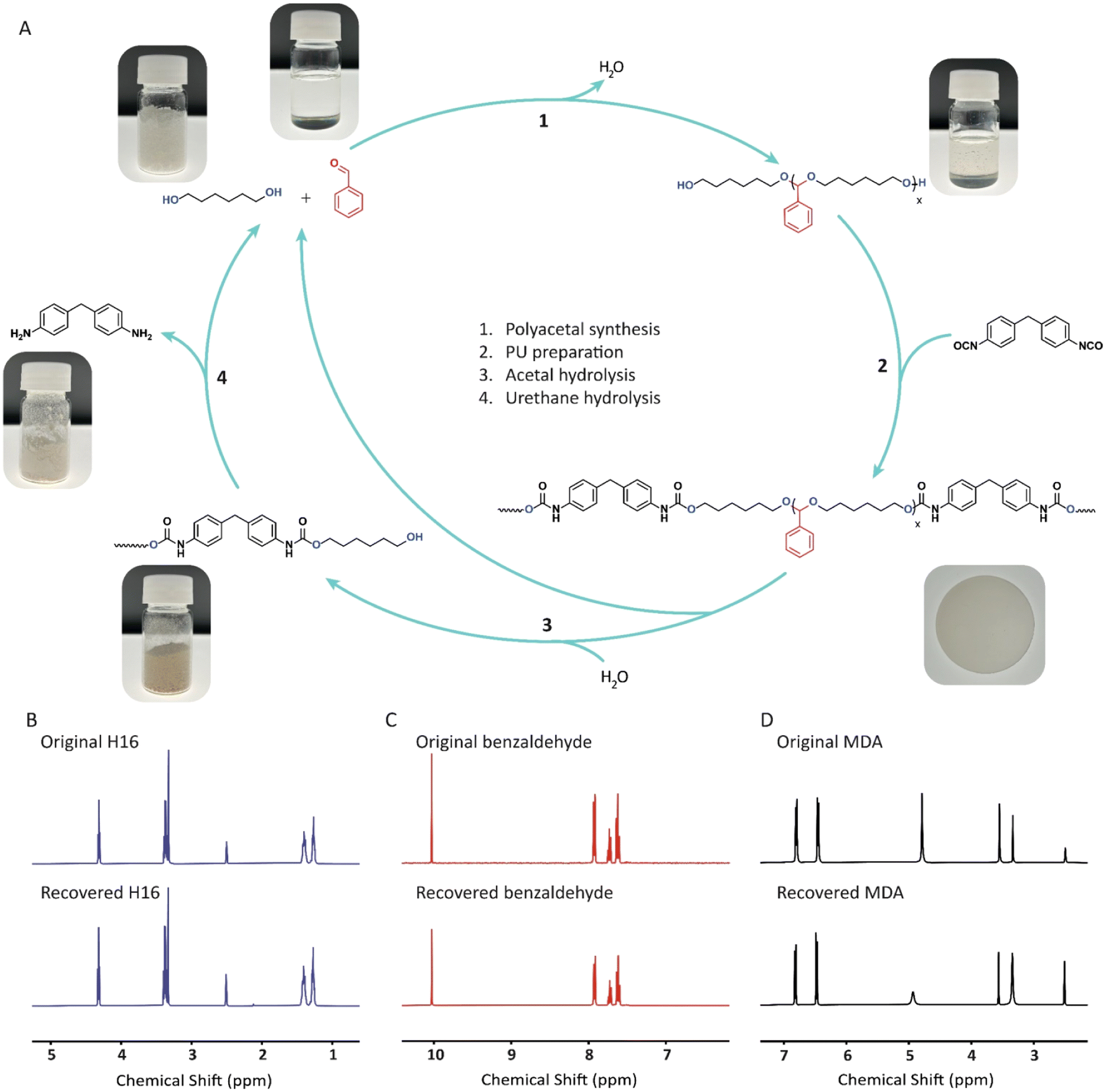 | ||
| Fig. 6 (A) Synthesis and chemical recycling of PU AP3. (B, C and D) 1H-NMR spectra (400 MHz, DMSO-d6) of the original (top) and recovered (bottom) monomers, H16 (B), Benzaldehyde (C), and MDA (D). | ||
Significant differences in hydrolytic stability under acidic conditions were observed among the PU samples, particularly between PU AP1–2 and PU AP3, in accordance with the results shown in Fig. 3. PU AP3 was quickly hydrolyzed under relatively mild conditions, whereas PU AP1 and PU AP2 exhibited much greater hydrolytic stability under acidic conditions. Both 0.3 M HCl and 1 M H3PO4 were effective in hydrolyzing the acetal bonds fully in PU AP3, producing free benzaldehyde within 24 hours, as confirmed by 1H-NMR spectra (Fig. S14 and S15†). In contrast, attempts to recycle PU AP2 were hampered by its higher acetal stability and low swelling in water, resulting in significantly slower hydrolysis rates. Even treatment with 2 M and 4 M HCl solutions yielded partial hydrolysis, leaving a considerable amount of acetals intact.
To accelerate hydrolysis and reduce the required acidity, we investigated aqueous acidic solutions in the presence of several green organic solvents such as 2-methyltetrahydrofuran (Me-THF), acetone, and ethanol. Among these, ethanol demonstrated the highest efficacy, as confirmed by 1H-NMR analysis (Fig. S16†). The effectiveness of ethanol is attributed to its role in the trans-acetalization reaction, wherein it inserts into the acetal linkages of the polyol chain, producing soluble oligomers which are more readily hydrolyzed (Scheme S4†). This results in a synergistic depolymerization process that simultaneously utilizes hydrolysis and trans-acetalization pathways. The depolymerization of acetal containing polymers using this strategy has never been demonstrated. This process generates highly volatile aldehydes or acetals, which can be easily recovered via condensation from the gas stream.44,45 Such a method would allow for the efficient recovery of monomers in industrial processes.
In the depolymerization studies, both PU AP2 and PU AP3 were treated with an ethanol-2 M H3PO4(aq) (9:1) mixture at reflux temperature. For instance, PU AP3 (44 g in centimeter-sized chunks) was fully depolymerized within 5 hours. Upon completion of the hydrolysis, water was added to induce the precipitation of the hard phase, which was then removed via filtration. The resulting filtrate, containing H16 and benzaldehyde, was extracted with ethyl acetate, to isolate both building blocks, followed by vacuum distillation of each component. Accordingly, benzaldehyde was isolated with a yield of 57%. On the other hand, the precipitated hard phase was further subjected to base-catalyzed hydrolysis with 8 wt% TBD and 16 wt% water at 120 °C. After 36 hours, H16 was recovered from the water-soluble phase by vacuum distillation and combined with the portion obtained from the first hydrolysis step, giving a total recovery yield of 90%. Meanwhile, MDA was recovered from the water-insoluble residue through vacuum distillation and further purified via recrystallization from water, achieving a final yield of 68%. All recovered monomers exhibited high purity, as illustrated in Fig. 6 and Fig. S17–S22.†PU AP2 (20 g) was also depolymerized under the same conditions to achieve monomer recovery yields of 69%, 79% and 74% for HEB, H16 and MDA, respectively. The corresponding 1H-NMR spectra of the recovered monomers are shown in Fig. S23–S27.† The successful depolymerization of both PU AP2 and PU AP3via ethanol-assisted trans-acetalization demonstrates that even hydrolytically stable acetal-containing PUs can be effectively closed-loop recycled using green acids and solvents under mild conditions.
Conclusion
Herein, we successfully developed three aromatic acetal-containing polyols through the polycondensation of aldehydes and diols using acidic heterogeneous catalysts. The aromatic content of these polyols could be modulated as high as 35 wt% while still maintaining an easily processable, low viscosity liquid polyol. Furthermore, these polyols exhibited a wide range of properties, including varying degrees of hydrolytic stability under acidic conditions. When utilized to produce PU materials, the resulting materials demonstrated exceptional mechanical and thermal properties, highlighting their potential in high-performance applications. For example, PU AP2 exhibited a unique combination of mechanical properties, with a high elongation at break (560 ± 38%), the highest stress at break (47 ± 8 MPa), and a high Young's modulus (37 ± 2 MPa). Such characteristics contributed to a toughness of over 100 MJ m−3, surpassing the performance of many conventional polyurethanes. These results underscore the versatility of polycondensation of aldehydes and diols as a method for designing acetal-containing polyols with tailored properties, such as high mechanical performance. A key novel advancement of this work is the efficient depolymerization of the synthesized PUs using ethanol, which facilitates transacetalization of the acetal units utilizing phosphoric acid, thus enabling closed-loop recycling under milder conditions compared to conventional hydrolysis methods. The urethane-rich hard phase remaining after this process was also successfully depolymerized into well-defined monomers. Specifically, PU AP3 was depolymerized into 1,6-hexanediol (H16) and benzaldehyde, while the hard phase yielded 4,4′-methylenedianiline (MDA) and H16, with a final yield of 90% for H16, 57% for benzaldehyde, and 68% for MDA. Similarly, PU AP2 was hydrolyzed into 1,6-hexanediol, 1,4-bis(2-hydroxyethoxy)benzene, and MDA, achieving yields of 69%, 79%, and 74%, respectively. Given the broad availability and diversity of diols and aldehydes, this synthetic approach opens the door for the design of acetal-containing polyols with customized features such as varying polarity, crystallinity, aromatic content, or biobased content. Consequently, this work paves the way for the development of sustainable polyurethane materials with novel properties, advancing the concept of closed-loop recycling in the field of high-performance plastics.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† The data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Xianwen Lou (TU Eindhoven) for MALDI-TOF-MS measurements, and SASOL (Germany) for providing the Siral catalysts. The authors acknowledge financial support from BASF Polyurethanes GmbH (Germany) and the Dutch Ministry of Economic Affairs and Climate Policy (TKI project CHEMIE.PGT.2020.022).References
- Plastics Europe, Plastics-the Facts 2021, (accessed August 9, 2023).
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- B. Eling, Ž. Tomović and V. Schädler, Macromol. Chem. Phys., 2020, 221, 2000114 CrossRef CAS.
- D. Randall and S. Lee, The polyurethanes book, Wiley, 2003 Search PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, V. Vlcek, P. Christopher and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2023, 11, 6114–6128 CrossRef CAS.
- C. Liang, U. R. Gracida-Alvarez, E. T. Gallant, P. A. Gillis, Y. A. Marques, G. P. Abramo, T. R. Hawkins and J. B. Dunn, Environ. Sci. Technol., 2021, 55, 14215–14224 CrossRef CAS PubMed.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Waste Manage., 2018, 76, 147–171 CrossRef PubMed.
- J. Banik, D. Chakraborty, M. Rizwan, A. H. Shaik and M. R. Chandan, Waste Manage. Res., 2023, 41, 1063–1080 CrossRef CAS PubMed.
- T. Vanbergen, I. Verlent, J. De Geeter, B. Haelterman, L. Claes and D. De Vos, ChemSusChem, 2020, 13, 3835–3843 CrossRef CAS PubMed.
- P. Jutrzenka Trzebiatowska, H. Beneš and J. Datta, React. Funct. Polym., 2019, 139, 25–33 CrossRef CAS.
- D. Simón, A. M. Borreguero, A. de Lucas and J. F. Rodríguez, Polym. Degrad. Stab., 2015, 116, 23–35 CrossRef.
- R. Donadini, C. Boaretti, A. Lorenzetti, M. Roso, D. Penzo, E. Dal Lago and M. Modesti, ACS Omega, 2023, 8, 4655–4666 CrossRef CAS PubMed.
- M. B. Johansen, B. S. Donslund, E. Larsen, M. B. Olsen, J. A. L. Pedersen, M. Boye, J. K. C. Smedsgård, R. Heck, S. K. Kristensen and T. Skrydstrup, ACS Sustainable Chem. Eng., 2023, 11, 10737–10745 CrossRef CAS.
- S. Zamani, J.-P. Lange, S. R. A. Kersten and M. P. Ruiz, Polymers, 2022, 14, 4869 CrossRef CAS PubMed.
- Y. Branson, S. Söltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jäger and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2023, 62, e202216220 CrossRef CAS PubMed.
- S. Motokucho, Y. Nakayama, H. Morikawa and H. Nakatani, J. Appl. Polym. Sci., 2018, 135, 45897 CrossRef.
- I. Olazabal, A. González, S. Vallejos, I. Rivilla, C. Jehanno and H. Sardon, ACS Sustainable Chem. Eng., 2023, 11, 332–342 CrossRef CAS PubMed.
- M. Grdadolnik, B. Zdovc, A. Drinčić, O. C. Onder, P. Utroša, S. G. Ramos, E. D. Ramos, D. Pahovnik and E. Žagar, ACS Sustainable Chem. Eng., 2023, 11, 10864–10873 CrossRef CAS PubMed.
- S. Bhandari and P. Gupta, in Recycling of Polyurethane Foams, ed. S. Thomas, A. V. Rane, K. Kanny, A. Vayyaprontavida Kaliyathan and M. G.Thomas, William Andrew Publishing, 2018, pp. 77–87 Search PubMed.
- H. Watando, S. Saya, T. Fukaya, S. Fujieda and M. Yamamoto, Polym. Degrad. Stab., 2006, 91, 3354–3359 CrossRef CAS.
- M. Grdadolnik, A. Drinčić, A. Oreški, O. C. Onder, P. Utroša, D. Pahovnik and E. Žagar, ACS Sustainable Chem. Eng., 2022, 10, 1323–1332 CrossRef CAS PubMed.
- H. He, H. Su, H. Yu, K. Du, F. Yang, Y. Zhu, M. Ma, Y. Shi, X. Zhang, S. Chen and X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 5515–5523 CrossRef CAS.
- Z. Westman, B. Liu, K. Richardson, M. Davis, D. Lim, A. L. Stottlemyer, C. S. Letko, N. Hooshyar, V. Vlcek, P. Christopher and M. M. Abu-Omar, JACS Au, 2024, 4(8), 3194–3204 CrossRef CAS PubMed.
- M. Ionescu, S. Sinharoy and Z. S. Petrović, J. Polym. Environ., 2009, 17, 123 CrossRef CAS.
- A. Iinuma, T. Hashimoto, M. Urushisaki and T. Sakaguchi, J. Appl. Polym. Sci., 2016, 133, 44088 CrossRef.
- P. Schara, A. M. Cristadoro, R. P. Sijbesma and Ž. Tomović, Macromolecules, 2023, 56, 8866–8877 CrossRef CAS.
- T. Türel, Ö. Dağlar, C. Pantazidis and Ž. Tomović, RSC Sustainability, 2024, 2(11), 3311–3319 RSC.
- T. Türel and Ž. Tomović, ACS Sustainable Chem. Eng., 2023, 11, 8308–8316 CrossRef.
- P. Schara, A. Cristadoro, R. P. Sijbesma and Ž. Tomović, ChemSusChem, 2024, e202401595 Search PubMed.
- N. Mahmood, Z. Yuan, J. Schmidt and C. (Charles) Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- L. Liu, P. Hao, R. Zhang, Y. Zhang, Q. Zhou, X. Lu, D. Yan, Y. Li and C. Shi, J. Appl. Polym. Sci., 2023, 140, e54238 CrossRef CAS.
- C. Fu, J. Liu, H. Xia and L. Shen, Prog. Org. Coat., 2015, 83, 19–25 CrossRef CAS.
- F. Yang, H. Yu, Y. Deng and X. Xu, e-Polymers, 2021, 21, 491–499 CrossRef CAS.
- M. Rhein, A. Demharter, B. Felker and M. A. R. Meier, ACS Appl. Polym. Mater., 2022, 4, 6514–6520 CrossRef CAS.
- P. S. Lee and S. M. Jung, J. Appl. Polym. Sci., 2022, 139, 52010 CrossRef CAS.
- A. Duval, D. Vidal, A. Sarbu, W. René and L. Avérous, Mater. Today Chem., 2022, 24, 100793 CrossRef CAS.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306–2317 CrossRef CAS PubMed.
- Y.-S. Jung, S. Lee, J. Park and E.-J. Shin, Polymers, 2022, 14, 4269 CrossRef CAS PubMed.
- B.-X. Cheng, W.-C. Gao, X.-M. Ren, X.-Y. Ouyang, Y. Zhao, H. Zhao, W. Wu, C.-X. Huang, Y. Liu, X.-Y. Liu, H.-N. Li and R. K. Y. Li, Polym. Test., 2022, 107, 107489 CrossRef CAS.
- F. Peng, X. Yang, Y. Zhu and G. Wang, Polymer, 2022, 239, 124429 CrossRef CAS.
- A. Mouren and L. Avérous, Eur. Polym. J., 2023, 197, 112338 CrossRef CAS.
- H. Wang, Y. Li, H. Zhang, X. Lang, X. Wang, L. Cao and C. Zong, J. Polym. Sci., 2025, 63, 121–132 CrossRef CAS.
- H. J. Hagemeyer, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Ltd, 2002 Search PubMed.
- J. Ackermann, P. Radici and P. Erini, United States, US3911046A, 1975 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01428f |
| This journal is © The Royal Society of Chemistry 2025 |