
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfur-containing block polymers from ring-opening copolymerization: coordinative encapsulants for transition metals†
Jenny
Stephan‡
ab,
Merlin R.
Stühler‡
ab,
Christoph
Fornacon-Wood‡
 a,
Mathias
Dimde
b,
Kai
Ludwig
b,
Heinz
Sturm
c,
Jorge L.
Olmedo-Martínez
d,
Alejandro J.
Müller
de and
Alex J.
Plajer
*af
a,
Mathias
Dimde
b,
Kai
Ludwig
b,
Heinz
Sturm
c,
Jorge L.
Olmedo-Martínez
d,
Alejandro J.
Müller
de and
Alex J.
Plajer
*af
aMakromolekulare Chemie, Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: alex.plajer@uni-bayreuth.de
bIntitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34/36, 14195 Berlin, Germany
cBundesanstalt für Materialforschung und -Prüfung (BAM), Unter den Eichen 87, 12205 Berlin, Germany
dPolymat and Department of Polymers and Advanced Materials: Physics, Chemistry and Technology, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018, Donostia-San Sebastián, Spain
eIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, Bilbao 48009, Spain
fBayrisches Polymer Institut (BPI), Universität Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany
First published on 21st January 2025
Abstract
Sulfur-containing polymers can coordinate transition metals via sulfur-centered, chemically soft lone pairs, although this typically occurs in a spatially uncontrolled manner. In this study, we employed the controlled ring-opening copolymerization of oxetane with sulfur-containing comonomers to construct a series of amphiphilic block copolymers featuring thioester and thiocarbonate functionalities. These copolymers self-assemble in aqueous solution into aggregates with a sulfur-rich core capable of coordinating transition metals. This behavior could be resolved by employing cryo-transmission electron tomography and then extended to complexes incorporating functional coligands. Our study demonstrates how selective catalysis can be harnessed to produce functional polymers with tunable metal coordination properties, paving the way for an emerging class of sulfur-containing copolymers.
Introduction
Sulfur-containing polymers can show unique properties and functions their all-oxygen analogues do not exhibit, which are associated with the chemistry of the incorporated sulfur atoms.1–5 For example, due to their chemically soft nature, sulfur-containing copolymers are prime candidates for the binding of transition metals via coordinative L-type interactions with transition metals.6 This phenomenon has been applied in numerous examples in the sequestration of aqueous transition metal impurities, with chemically soft late d-block metals being most strongly bound.7–10 Typically, binding has been utilized in a spatially uncontrolled fashion, meaning that transition metals end up randomly coordinated within a solid polymer matrix. This is perhaps owed to the circumstance that patterned templates to spatially control the coordination, such as block polymers, are hard to access via current polymerization methodologies. Sulfur-containing polymers, such as polythioesters and polythiocarbonates, are often synthesized via polycondensation methodologies which only offer access to a limited number of polymer structures and polymer topology.2,3 In this respect much recent work focused on the ring-opening copolymerisation (ROCOP) of a strained heterocycle with an heteroallene such as carbon disulfide (CS2), carbonyl sulfide (COS), isothiocyanates (RNCS, R = aryl, aryl) or thioanhydrides, which give access to sulfur-containing monomers via a chain-growth rather than a step-growth mechanism from often commercially accessible monomers.11–27 Additionally, for sulfur-containing polymers resulting synthesized through these methodologies, examples of metal binding exist such as recent reports on the sequestration of lead from aqueous solutions.28As part of our recent efforts in sulfur-containing ROCOP, we found that the monomer combination of oxetane with either phenylisothiocyanate (PhNCS), phthalic thioanhydride (PTA) or CS2 yields poly(monothioimidocarbonates), poly(ester-alt-thioester) and poly(dithiocarbonates) respectively (Fig. 1(a)).29–31 Moving from epoxides to oxetane (OX) results in substantially more controlled polymerizations as primary chain-ends are formed for which propagation is thermodynamically more favoured. This led to highly sequence-selective polymerizations that yield high molecular weight semi-crystalline materials with field-leading melting points of up to 181 °C.32 The chemistry of these novel sulfur-containing copolymers as for example their capability to coordinate transition metals remains to be explored. Preliminary experiments in the case of PhNCS suggested the suitability of these methods for block polymer formation via a macro-chain transfer strategy (Fig. 1(b)) that could be employed in self-assembly to form micellar aggregates. However, the extent to which this is transferable to other sulfur-containing ROCOP comonomers remained unresolved. In principle, the formation of these sulfur-containing block copolymers could give access to polymer structures that can spatially confine the coordination of transition metals via combination of the sulfur-containing ROCOP block with a non-coordinating block, which we report in this contribution (Fig. 1(c)).
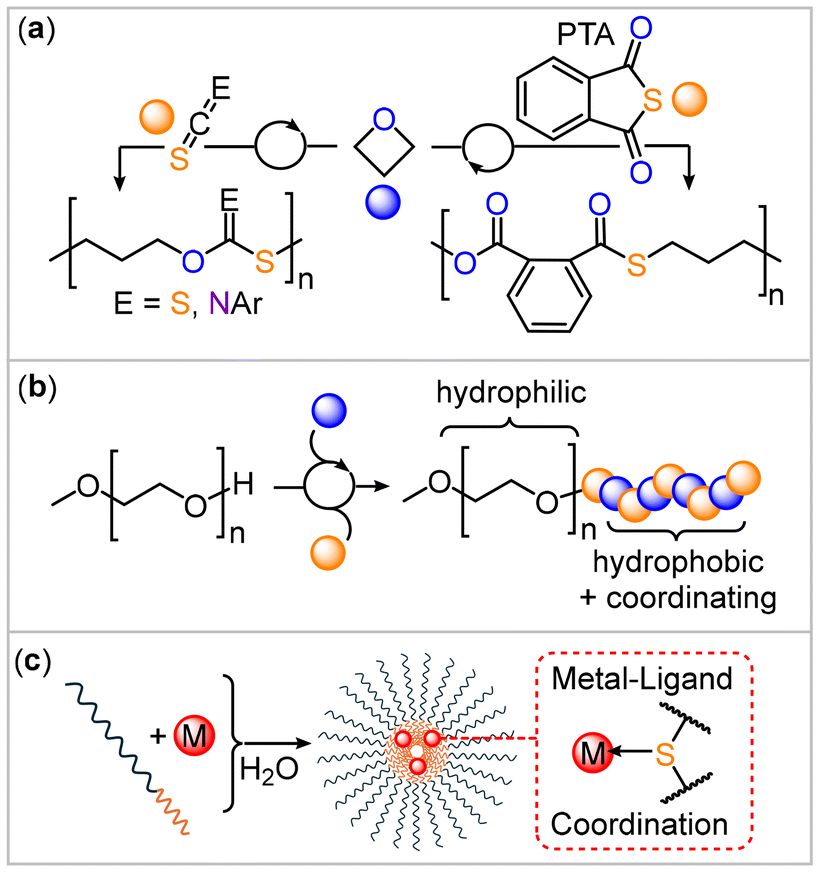 | ||
| Fig. 1 (a) Oxetane copolymerization yielding sulfur-containing copolymers employed in this contribution. (b) Block polymer design yielding (c) micellar aggregates with metal coordinating cores presented in this contribution. | ||
Our study begins by surveying all recently developed oxetane-based ROCOPs for block polymer formation. For this α-Me,ω-OH terminated polyethylene glycol (mPEG-OH, supplied with a number average molecular weight (Mn) of 5 kDa, 20 eq.) was employed as a macro-chain transfer agent to a polymerization containing 1000 eq. of oxetane and 1000 eq. of arylisothiocyanates (ArNCS), PTA or CS2 respectively per equivalent of catalyst at 80 °C. Aryl isothiocyanates with different substituents (C6H5 (PhNCS), 4-methyl-C6H4 (MePhNCS), 4-fluoro-C6H4 (FPhNCS), 3,5-bistrifluoromethyl-C6H4 (CF3PhNCS)) and a chromium(III) based catalyst that has shown quantitative polymerization and >95% linkage selectivity in these types of polymerizations previously were employed.31 In this process, the extension of mPEG-OH by the oxetane copolymers occurs on the basis that the alcohol chain ends are deprotonated in situ by catalyst-bound alkoxide intermediates formed from oxetane ring-opening. Hereby, a mPEG alkoxide is transferred onto the catalyst and thereafter propagates to attach the sulfur-containing block, which we established previously with the combination of oxetane and PhNCS. Conceptually, mPEG-OH functions as a macro-chain transfer agent.33a Following complete oxetane consumption (as established by spectroscopic analysis of aliquots) in the respective ROCOPs outlined in Table 1, the resulting polymer was easily isolated by precipitation from methanol. 1H NMR analysis of the purified polymers indicates successful block polymer synthesis showing a set of resonances between 4.0 and 1.8 ppm corresponding to ring-opened oxetane as well as resonances for aryl hydrogens in addition to the signals corresponding to mPEG at 3.6 ppm. An integrative ratio of 2.4 to 3.4 repeat units of the ROCOP copolymer per repeat unit of the mPEG block is observed. Importantly, the integrative ratio for the two respective blocks remains unchanged after precipitation of a polymer solution in dichloromethane into methanol. The latter represents a good solvent for mPEG but a poor solvent for the stand-alone ROCOP copolymers so that if the respective blocks were not covalently attached, one would precipitate preferentially, thereby changing the ratio of polymers in the mixture. 1H and 13C nuclear magnetic resonance spectroscopy (NMR) reveals that an alternating microstructure of the ROCOP block is obtained for both PTA and all ArNCS, with no signs of ether linkages formation or scrambling from oxygen/sulfur exchange processes.
| Comonomer |
M
n,NMR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kDa) a (kDa) |
M
n,GPCb (kDa) |
T
mc [°C] |
Đ | D [nm] | PDId | CCe [mg L−1] |
|---|---|---|---|---|---|---|---|
| Synthesised at 1 eq. cat.: 1000 eq. OX: 1000 eq. comonomer.a Calculated based on the relative integrals in the 1H NMR spectrum (CDCl3, 400 MHz) assuming a molecular weight of mPEG-OH of 5 kg mol−1 as stated by the supplier.b Determined by gel permeation chromatography (GPC) measurements conducted in THF, using narrow MW polystyrene standards to calibrate the instrument.c Melting point (Tm) determined by differential scanning calorimetry (DSC) from the first heating.d Diameter (D) and polydispersity index (PDI) of self-assembled nano-objects in aqueous solution determined by dynamic light scattering (DLS).e Critical concentration (CC), determined by DLS according to ref. 34.f Other than CC as reported in ref. 31. | |||||||
| PhNCSf | 12.2 | 8.0 | 44, 165 | 1.1 | 126 | 0.27 | 0.01–0.1 |
| FPhNCS | 12.2 | 7.9 | 45, 146 | 1.2 | 146 | 0.22 | 0.5–2 |
| MePhNCS | 11.8 | 9.3 | 45, 142 | 1.1 | 151 | 0.43 | 0.5–2 |
| CF3PhNCS | 16.8 | 11.6 | — | 1.3 | 269 | 0.69 | 1–2 |
| PTA | 15.6 | 6.6 | 49, 83 | 1.3 | 69 | 0.39 | 0.5–2 |
Gel permeation chromatography (GPC) in tetrahydrofuran compared to a narrow polystyrene standard reference shows an increase in the apparent molecular weight relative to the mPEG macroinitiator (see ESI Fig. S36†) and narrow dispersity values of Đ = 1.1–1.3 in all cases qualitatively supporting block polymer formation. Although dispersity values are low, distributions are not perfectly monomodal (see for example Fig. 2(b)) visible as, for example, shoulders at the higher molecular weight part of the GPC trace. Furthermore, some broadening is observed compared to the macroinitiator. This can be tentatively explained by side reactions resembling transesterification as well as chain end-coupling and an exemplary reaction pathway is shown in the ESI (Scheme S6†).33b Hence it is unlikely that the sample exclusively comprises diblock architectures. As summarized in Table 1, the apparent molecular weights by GPC are lower than those determined by NMR presumably due to the differences in chemical structure with respect to the polystyrene standard.
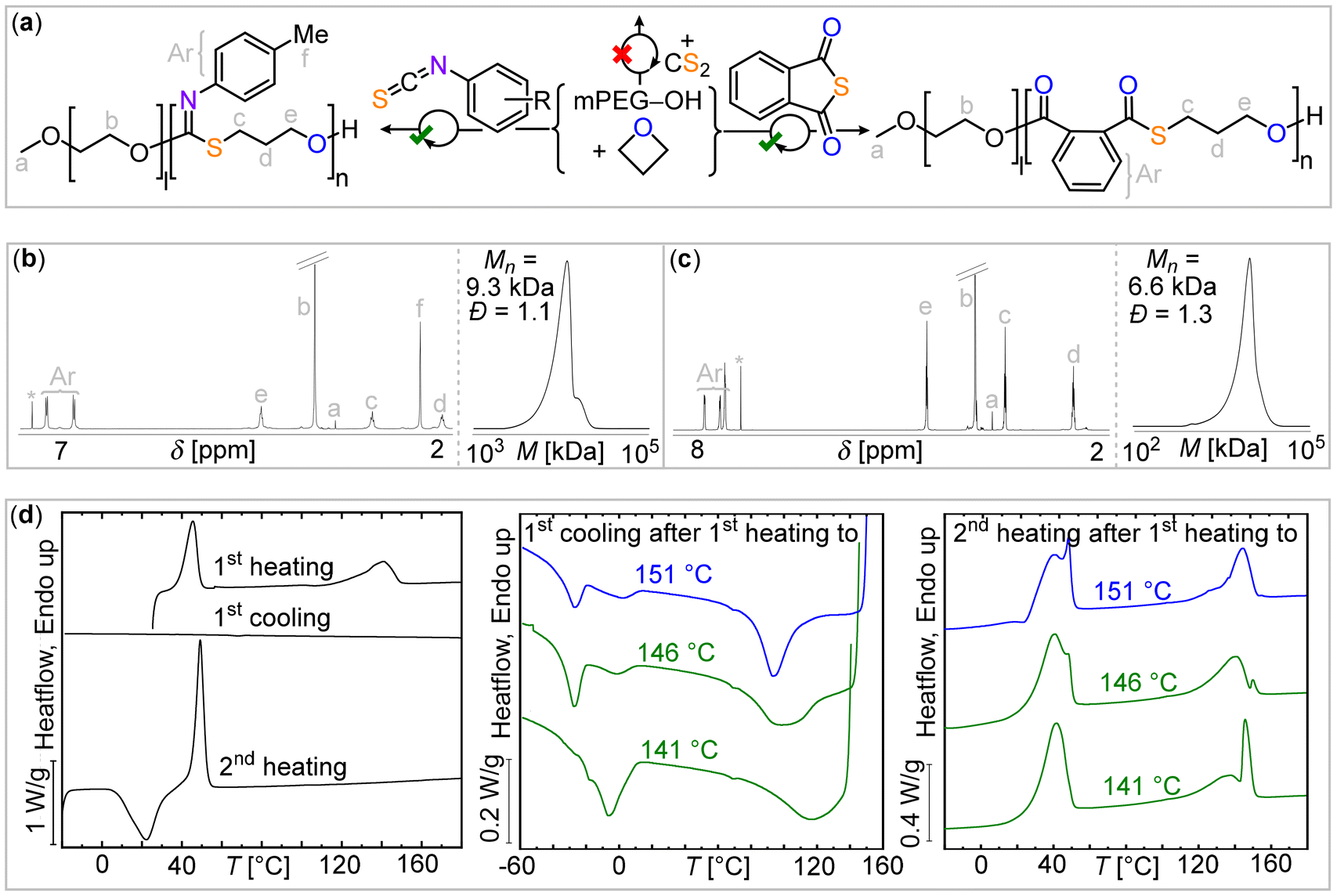 | ||
| Fig. 2 (a) Synthetic scheme of blockpolymerization. 1H NMR spectrum (CDCl3, 400 MHz) and gel permeation chromatography (GPC) traces of purified polymers resulting from (b) MePhNCS and (c) PTA/OX ROCOP in presence of mPEG-OH chaintransfer agent. (d) DSC data of mPEG-b-(MePhNCS-co-OX) block copolymer (left) without and (right) with self-nucleation procedure; for experimental details see ESI† methods section. | ||
Furthermore, the thermal properties of the polymers also support the block formation. Differential scanning calorimetry (DSC) first heating scans of the as-synthesized 4-F-PhNCS (Fig. S8†), 4-Me-PhNCS (Fig. S17†), and PTA-derived block polymers (Fig. S27†) show two distinct melting points for the mPEG and the sulfur-containing blocks. As in the case of the stand-alone ROCOP copolymer, the crystallization from the melt of the sulfur-containing block is very slow. Therefore, the material could not crystallize from the melt, even when employing rates as low as 1 °C min−1. Fig. S8, S17, and S27† show the featureless cooling DSC scans from the melt indicating the absence of crystallization. In the corresponding DSC second heating scans, the temperature region corresponding to the melting of the sulfur-containing block shows a flat baseline, as these blocks could not crystallize during cooling but also do not exhibit cold crystallization. Fig. S17† shows that for the mPEG-b-(OX-co-MePhNCS) copolymer, the mPEG block exhibits cold crystallization during the second heating scan, followed by melting. However, once again no crystallization or melting could be observed for the sulfur-containing blocks.
To accelerate the crystallization of the slow-crystallizing sulfur-containing blocks, we have modified the classical self-nucleation protocol to allow the sample to crystallize.35 In the modified self-nucleation strategy,31 the existing crystals in the as-obtained samples are partially melted (see Fig. S1†). The remaining crystal fragments or self-seeds can efficiently nucleate these sulfur-containing blocks upon cooling from the self-seeding temperature. In this way, crystallization from the self-seeded melt was observed for the MePhNCS derived copolymers.30,31 The DSC cooling scans from three characteristic self-nucleating temperatures for the mPEG-b-(OX-co-FPhNCS) copolymer are presented in Fig. S9† (left) together with the subsequent heating scans (Fig. S9,† right). The appearance of an exotherm corresponding to the sulfur-containing block crystallization can be clearly observed, and the size of the exotherm increases, as expected, with the decrease in the self-nucleation temperature (as a higher number of self-seeds are produced at lower self-nucleation temperatures). In the subsequent heating scans, the cold crystallization of the mPEG block can be observed (for two of the self-nucleation temperatures employed), followed by the melting of these crystals at around 40 °C. At higher temperatures, the melting endotherms corresponding to the sulfur-containing block crystals can be clearly observed at around 150 °C. These results demonstrate the effectiveness of the modified self-nucleation protocol (illustrated in Fig. S1†) in accelerating the overall crystallization of the sulfur-containing blocks. Similar self-nucleation results are presented for the other block copolymers in Fig. S18, S27, and S34.†
Furthermore, a stepwise decomposition profile in the thermogravimetric analysis is observed for the isothiocyanate-derived block copolymers corresponding to the thermal decomposition of the two respective blocks with the ROCOP block decomposing before the mPEG block. However, not all of the recently developed oxetane ROCOPs tolerate chain transfer agents and block polymerization was not as successful with CS2, although without a macro chain-transfer agent, the polymerization is highly controlled, delivering poly(dithiocarbonate).29 Instead, a complex mixture of different (thio)carbonate linkages is formed alongside small-molecule cyclic thiocarbonate byproducts. Furthermore, GPC analysis of the crude mixture does not reveal an increase in the apparent molecular weight compared to the starting mPEG-OH polymer. These observations can be interpreted in light of recent findings showing that CS2 ROCOP is more prone to side reactions originating from alkoxide chain ends than other sulfur-containing ROCOPs. We infer that chain-transfer agents promote the dissociation of alkoxide chain ends from the catalyst through competitive binding to the catalyst, which subsequently leads to linkage scrambling and cyclic thiocarbonate formation via attack on polymer linkages by the dissociated alkoxide.36
With a range of block polymers in hand, we studied their self-assembly behavior in water, as the mPEG block is water-soluble while the oxetane-derived blocks are not. For this purpose, the respective block polymer was dissolved in 1,4-dioxane and then added dropwise into water under vigorous stirring. As previously observed for the mPEG-b-(PhNCS-co-OX) polymer, a cloudy but homogeneous solution was formed by nanoprecipitation for all block polymers.31 Dynamic light scattering (DLS) experiments suggest the presence of nano-objects with sizes between 69 and 269 nm. Although solutions of the different assembled polymers are all stable over time, they show different stabilities with respect to concentration. To investigate this, DLS-supported dilution experiments were conducted to monitor the evolution of the light scattering intensity, steadily decreasing the concentration of the polymers. As the light scattering intensity is proportional to the concentration of a scattering species, this approach efficiently determines the critical concentration at which the aggregates remain stable prior to disassembly. Compared to other techniques, such as fluorescence, DLS has the advantage of allowing a label-free determination of the critical concentration. This not only simplifies determination but also excludes secondary effects between the label (or cargo) and the polymer to influence outcomes, hence not delivering the true stability of the pure aggregates. Although there is some uncertainty due to a gradual transition from a signal decrease to the plateau region in some of the DLS traces of the block polymers, they all exhibit excellent stabilities of below 2 mg L−1 which can rival some of the most performant, highly engineered systems in the literature.34 However, in contrast to those, our copolymers are obtained from simple and in most cases, commercially available or easy-to-synthesize building blocks in a one-pot fashion and are hence attractive candidates for applications. Furthermore, we find that the mPEG-b-(PhNCS-co-OX) exhibits a lower critical concentration and hence higher stability than other aggregates, which can be tentatively attributed to more pronounced π–π interactions within the aggregates as has been previously described for highly stable micelles featuring an aromatic core.34
Accordingly, we thereafter focused our attention on coordination and encapsulation studies employing the assembled mPEG-b-(PhNCS-co-OX) polymer. In order to confirm whether the sulfur-containing block is capable of coordinating transition metals, we first investigated the reaction of the stand-alone PhNCS-co-OX copolymer. Thus, the copolymer was reacted with bright orange bis(benzonitrile)palladium(II)chloride ((PhCN)2PdCl2) featuring easily displaceable benzonitrile coligands and chemically soft PdII for sulfur coordination (Fig. 3(a)). In fact, the addition of a saturated (PhCN)2PdCl2 solution in CH3Cl to a solution of the copolymer results in the immediate precipitation of a dark orange solid, which is insoluble in all common non-coordinating solvents. While the color change already immediately indicates palladium coordination within the precipitate, energy-dispersive X-ray spectroscopy (EDX) in the scanning electron microscope (SEM) was used as a quick test to justify the continuation of the experiment. Since no film could be cast due to a lack of solvent and the sample was only to be pressed at room temperature, the surface quality was sufficient for SEM/EDX (see Fig. S38†). The line intensities differ locally due to the roughness but are qualitatively unambiguous and identify the elements Pd and Cl in addition to the elements of the polymer.
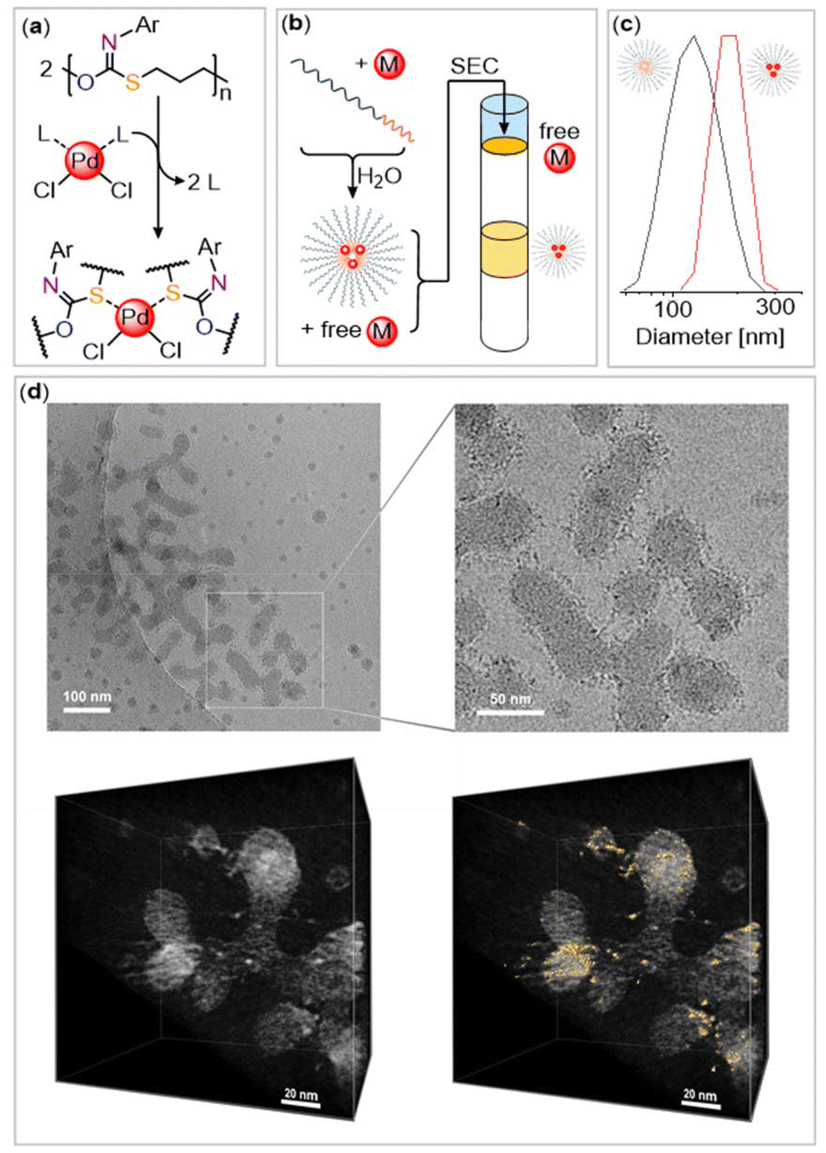 | ||
| Fig. 3 (a) Ligand exchange reaction of polymer with L2PdCl2 (L = PhCN). (b) Purification of PdCl2-loaded aggregates with Sephadex G-25 column. (c) Comparison of the DLS trace before and after PdCl2 encapsulation. (d) Cryo-TEM tomography after palladium encapsulation. Regions of increased electron density visible as dark spots in the 2D image and highlighted in golden in the 3D image correlate to encapsulated PdCl2. | ||
Motivated by this finding, we then turned to coordinating Pd within our aggregates. Therefore, mPEG-b-(PhNCS-co-OX) was dissolved with 20 wt% (PhCN)2PdCl2 with respect to the polymer in 1,4-dioxane and then added dropwise into water under vigorous stirring. Gratifyingly, a stable orange colloidal solution forms, exhibiting a Tyndall effect. DLS reveals a size increase of the aggregates from 126 nm (without PdCl2) to 170 nm, suggesting some morphological adjustment due to loading with the metal salt, thereby supporting encapsulation. Since we suspected that not all PdCl2 was bound during precipitation, we subjected the aggregates to size exclusion chromatography (SEC). As schematically illustrated in Fig. 3(b) we observed the elution of a sharp orange band corresponding to aggregates with PdCl2 while free palladium salt remained on top of the column.
Cryo-transmission electron microscopy (Cryo-TEM) reveals the morphology of the aggregates under native, hydrated conditions rather than dried conditions. Spherical aggregates were observed to merge into elongated worm-like aggregates with an approximate length of 100 to 300 nm and a thickness of 20–40 nm, which were, however, too irregular for statistical evaluation. The elongated worm-like aggregates consist of multiple merging micelles rather than regular cylindrical structures suggesting that the sulfur-containing cores are likely amorphous in nature rather than semi-crystalline. Interestingly, regions of increased electron density in the aggregates are observed, visible as black dots in the images presented in Fig. 3. These likely correspond to the encapsulated PdCl2 as regions rich in palladium, a second-row transition metal, exhibit higher electron density than the surrounding areas comprising lighter elements. Hence, the encapsulated PdCl2 appears to form discrete clusters within the polymer aggregates. We then turned to cryo-electron tomography to spatially resolve where PdCl2 resides. As shown in Fig. 3 areas of increased electron density, highlighted in gold, could be identified in the 3D volume reconstructed from tilt series images (±64°, with 2° increment), and which are highly suspected to be PdCl2. These do not appear to be homogeneously distributed throughout the nanostructure but much rather form a shell around the interior. This could either be due to the coordination of palladium on the surface of a micellar core or within the interior of a vesicular morphology.
Having demonstrated the coordination capability of our block polymers, we proceeded to investigate the coordinative encapsulation of complex fragments featuring functional coligands. Here, the [(bipyridine)2Ru]2+ was chosen, as it contains coligands that induce phosphorescent metal-to-ligand charge transfer (MLCT) transitions. (Bipyridine)2Ru(BArF)2 with hydrophobic tetrakis(3,5-bis(trifluoromethyl)phenyl)borate (BArF) counteranions was employed in encapsulation studies to support encapsulation within the hydrophobic interior of the aggregates with this hydrophobic counter anion.
Initially, the possibility of polymer coordination to the ruthenium complex investigated spectroscopically in a block-non-selective solvent by 1H NMR spectroscopy. Interestingly, the 1H NMR resonances of (bipyridine)2Ru(BArF)2 in CDCl3 exhibit substantial shifting and broadening upon addition of the copolymer with those closest to the ruthenium center being most affected. This clearly indicates a change in coordination environment of the metal and hence coordination of the polymer to ruthenium.
Therefore, mPEG-b-(PhNCS-co-OX) was dissolved with 40 wt% ruthenium complex with respect to polymer in 1,4-dioxane and then added dropwise into water under vigorous stirring, forming a turbid luminescent solution (Fig. 4). Importantly, the complex itself is not water-soluble meaning that the homogeneity of the mixture immediately indicates encapsulation into the aggregates. DLS reveals a size of 122 nm for these, and Cryo-TEM shows a spherical morphology with a homogeneous electron density distribution throughout the aggregate suggesting a uniform distribution of the complex.
 | ||
| Fig. 4 (a) Cryo-TEM image of (bipyridine)2Ru(BArF)2 loaded micelles. (b) Comparative absorption and emission spectra before (grey) and after (black and green) encapsulation. (c) Time-dependent monitoring of the UV/Vis absorption spectra of the 1O2 probe dye 9,10-anthracenediyl-bis(methylene)dimalonic acid in the presence of Ru-loaded micelles in aqueous solution upon irradiation. | ||
Encapsulation is clearly indicated by the optoelectronic properties of the assembly. While (bipyridine)2Ru(BArF)2 in CH3CN solvent shows an emission maximum at approximately 560 nm, it is shifted to 600 nm for the ruthenium complex encapsulated. Having evidenced ruthenium encapsulation, we turned to example applications, such as the generation of singlet oxygen, which is useful in the context of photocatalysis for pollution remediation or organic transformations.37,38 For this, ruthenium-loaded micelles in a buffer solution containing 9,10-anthracenediyl-bis(methylene)dimalonic acid, a dye sensitive to singlet oxygen, were irradiated at 440 nm and ultraviolet–visible (UV-Vis) absorption spectra were recorded every 5 minutes.39 As shown in Fig. 4(c) a continuous intensity decrease over time is observed for the diagnostic absorption bands of the dye between 320 and 420 nm clearly demonstrating the capability of our micellar system to produce singlet oxygen.
In conclusion, we have presented a series of block polymer syntheses in which sulfur-containing oxetane ROCOP polymers were attached to mPEG via a macro-chain transfer strategy. While a variety of isothiocyanates, as well as phthalic thioanhydride, successfully facilitated block polymer formation, side reactions during CS2 ROCOP were exacerbated by the presence of chain-transfer agents. Most block polymers exhibited phase separation in the bulk, resulting in two distinct melting points observed in DSC experiments. Furthermore, the resulting amphiphiles self-assembled into stable nano-objects in aqueous solution featuring a sulfur-containing core that could encapsulate transition metal salts and complex fragments. The latter demonstrated initial activity in singlet oxygen generation, highlighting the potential of our system for applications in photocatalysis and photodynamic therapy.
Data availability
The data supporting this article have been included as part of the electronic ESI.†Conflicts of interest
There are no conflicts of interest.Acknowledgements
We thank the “Verband der Chemischen Industrie” and the “Daimler and Benz Foundation” for financial support (personal scholarships for A. J. P.). We would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG.References
- Y. Sun, C. Zhang and X. Zhang, Chem. – Eur. J., 2024, 30, e202401684 CrossRef CAS PubMed.
- (a) H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed; (b) A. W. Woodhouse, A. Kocaarslan, J. A. Garden and H. Mutlu, Macromol. Rapid Commun., 2024, 45, 2400260 CrossRef CAS PubMed.
- N. M. Bingham, Z. Abousalman-Rezvani, K. Collins and P. J. Roth, Polym. Chem., 2022, 13, 2880–2901 RSC.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- T. Lee, P. T. Dirlam, J. T. Njardarson, R. S. Glass and J. Pyun, J. Am. Chem. Soc., 2022, 144, 5–22 CrossRef CAS PubMed.
- J. R. Dilworth and N. Wheatley, Coord. Chem. Rev., 2000, 199, 89–158 CrossRef CAS.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- X. Deng, R. A. Dop, D. Cai, D. R. Neill and T. Hasell, Adv. Funct. Mater., 2024, 34, 2311647 CrossRef CAS.
- J. M. Scheiger, C. Direksilp, P. Falkenstein, A. Welle, M. Koenig, S. Heissler, J. Matysik, P. A. Levkin and P. Theato, Angew. Chem., Int. Ed., 2020, 59, 18639–18645 CrossRef CAS PubMed.
- W. Cao, F. Dai, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 978–986 CrossRef CAS PubMed.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS PubMed.
- X.-F. Zhu, G.-W. Yang, R. Xie and G.-P. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115189 CrossRef CAS PubMed.
- X.-F. Zhu, R. Xie, G.-W. Yang, X.-Y. Lu and G.-P. Wu, ACS Macro Lett., 2021, 10, 135–140 CrossRef CAS PubMed.
- J.-L. Yang, Y. Wang, X.-H. Cao, C.-J. Zhang, Z. Chen and X.-H. Zhang, Macromol. Rapid Commun., 2021, 42, 2000472 CrossRef CAS PubMed.
- T. M. McGuire and A. Buchard, Polym. Chem., 2021, 12, 4253–4261 RSC.
- D. K. Tran, A. N. Braaksma, A. M. Andras, S. K. Boopathi, D. J. Darensbourg and K. L. Wooley, J. Am. Chem. Soc., 2023, 145, 18560–18567 CrossRef CAS PubMed.
- S. Rupf, P. Pröhm and A. J. Plajer, Chem. Sci., 2022, 13, 6355–6365 RSC.
- C. Gallizioli, D. Battke, H. Schlaad, P. Deglmann and A. J. Plajer, Angew. Chem., Int. Ed., 2024, 63, e202319810 CrossRef CAS PubMed.
- L.-Y. Wang, G.-G. Gu, B.-H. Ren, T.-J. Yue, X.-B. Lu and W.-M. Ren, ACS Catal., 2020, 10, 6635–6644 CrossRef CAS.
- A. J. Plajer and C. K. Williams, Angew. Chem., Int. Ed., 2022, 61, e202104495 CrossRef CAS PubMed.
- D. Silbernagl, H. Sturm and A. J. Plajer, Polym. Chem., 2022, 13, 3981–3985 RSC.
- J. Diebler, H. Komber, L. Häußler, A. Lederer and T. Werner, Macromolecules, 2016, 49, 4723–4731 CrossRef CAS.
- J. Stephan, M. R. Stühler, S. M. Rupf, S. Neale and A. J. Plajer, Cell Rep. Phys. Sci., 2023, 101510 CrossRef CAS.
- T.-J. Yue, M.-C. Zhang, G.-G. Gu, L.-Y. Wang, W.-M. Ren and X.-B. Lu, Angew. Chem., 2019, 131, 628–633 CrossRef.
- S. L. Luyer, B. Quienne, M. Bouzaid, P. Guégan, S. Caillol and N. Illy, Green Chem., 2021, 23, 7743–7750 RSC.
- E. Mongkhoun, P. Guégan and N. Illy, Polym. Chem., 2023, 14, 3729–3738 RSC.
- T.-J. Yue, G. A. Bhat, W.-J. Zhang, W.-M. Ren, X.-B. Lu and D. J. Darensbourg, Angew. Chem., Int. Ed., 2020, 59, 13633–13637 CrossRef CAS PubMed.
- S. Wu, M. Luo, D. J. Darensbourg, D. Zeng, Y. Yao, X. Zuo, X. Hu and D. Tan, ACS Sustainable Chem. Eng., 2020, 8, 5693–5703 CrossRef CAS.
- C. Fornacon-Wood, B. R. Manjunatha, M. R. Stühler, C. Gallizioli, C. Müller, P. Pröhm and A. J. Plajer, Nat. Commun., 2023, 14, 4525 CrossRef CAS PubMed.
- C. Fornacon-Wood, M. R. Stühler, C. Gallizioli, B. R. Manjunatha, V. Wachtendorf, B. Schartel and A. J. Plajer, Chem. Commun., 2023, 59, 11353–11356 RSC.
- J. Stephan, J. L. Olmedo-Martínez, C. Fornacon-Wood, M. Stühler, M. Dimde, D. Braatz, R. Langer, A. J. Müller, H. Schmalz and A. J. Plajer, Angew. Chem., Int. Ed., 2024, 63, e202405047 CrossRef CAS PubMed.
- M. R. Stühler, M. Kreische, C. Fornacon-Wood, S. M. Rupf, R. Langer and A. J. Plajer, Chem. Sci., 2024, 15, 19029–19036 RSC.
- (a) D. J. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC; (b) J. Xu, P. Zhang, Y. Yuan and N. Hadjichristidis, Angew. Chem., Int. Ed., 2023, 62, e202218891 CrossRef CAS PubMed.
- D. Braatz, J. H. Peter, M. Dimde, E. Quaas, K. Ludwig, K. Achazi, M. Schirner, M. Ballauff and R. Haag, J. Mater. Chem. B, 2023, 11, 3797–3807 RSC.
- R. M. Michell, A. Mugica, M. Zubitur and A. J. Müller, in Polymer Crystallization I: From Chain Microstructure to Processing, ed. F. Auriemma, G. C. Alfonso and C. de Rosa, Springer International Publishing, Cham, 2017, pp. 215–256 Search PubMed.
- C. A. L. Lidston, B. A. Abel and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 20161–20169 CrossRef CAS PubMed.
- C. McCullagh, N. Skillen, M. Adams and P. K. J. Robertson, J. Chem. Technol. Biotechnol., 2011, 86, 1002–1017 CrossRef CAS.
- B. König, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef.
- H. Wang, Y. Lai, D. Li, J. Karges, P. Zhang and H. Huang, J. Med. Chem., 2024, 67, 1336–1346 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01415d |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |