
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
BH3·SMe2 addition enables molar mass control via chain stabilization in phosphine–borane dehydropolymerization†
Matthew A.
Wiebe
a,
Jade E. T.
Watson
 a,
Charles
Killeen
a,
J. Scott
McIndoe
a,
Anne
Staubitz
*b and
Ian
Manners‡
a
a,
Charles
Killeen
a,
J. Scott
McIndoe
a,
Anne
Staubitz
*b and
Ian
Manners‡
a
aDepartment of Chemistry, University of Victoria, 3800 Finnerty Rd, V8P 5C2, Victoria, BC, Canada
bOtto-Diels-Institute for Organic Chemistry, University of Kiel, Otto-Hahn-Platz 4, 24118 Kiel, Germany. E-mail: staubitz@oc.uni-kiel.de
First published on 7th February 2025
Abstract
We report the synthesis of high molar mass polyphosphinoboranes using commercially available reagents through thermal dehydropolymerization in the presence of Lewis acids and bases. These dehydropolymerizations produce materials of higher molecular weight compared to the state-of-the-art catalyst, Cp(CO)2FeOTf ([PhPH-BH2]n (2), 5 mol% LiOTf, 2 M in 2-MeTHF, 100 °C, 24 h; Mn = 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Đ = 1.64 cf. 5 mol% Cp(CO)2FeOTf, 2 M in toluene, 100 °C, 24 h, Mn = 40000 g mol−1, Đ = 1.64). We propose a mechanism for the thermal dehydropolymerization of PhPH2·BH3 (1) with additives. Initially, the phosphine–borane adduct dissociates, yielding borane in situ, which acts as a (pre)catalyst for the dehydrogenation of 1. Subsequent addition polymerization occurs as described previously, but the addition of Lewis acids and Lewis bases allows for reversible complexation of both termini. Competition between temporary chain capping and termination events results in fewer termination events over time, leading to high molar mass materials. With this mechanism in mind, we were able to show that added BH3·SMe2 allows for control over the molar mass of the resulting materials. These results show that transition-metal catalysts are not needed in the thermal dehydropolymerization of PhPH2·BH3, and offer a new mechanistic insight that may unlock greater control over the dehydropolymerization of main-group substrates.
000 g mol−1, Đ = 1.64 cf. 5 mol% Cp(CO)2FeOTf, 2 M in toluene, 100 °C, 24 h, Mn = 40000 g mol−1, Đ = 1.64). We propose a mechanism for the thermal dehydropolymerization of PhPH2·BH3 (1) with additives. Initially, the phosphine–borane adduct dissociates, yielding borane in situ, which acts as a (pre)catalyst for the dehydrogenation of 1. Subsequent addition polymerization occurs as described previously, but the addition of Lewis acids and Lewis bases allows for reversible complexation of both termini. Competition between temporary chain capping and termination events results in fewer termination events over time, leading to high molar mass materials. With this mechanism in mind, we were able to show that added BH3·SMe2 allows for control over the molar mass of the resulting materials. These results show that transition-metal catalysts are not needed in the thermal dehydropolymerization of PhPH2·BH3, and offer a new mechanistic insight that may unlock greater control over the dehydropolymerization of main-group substrates.
Introduction
Polymers incorporating main-group elements into their main chain are of interest as inclusion of these elements can lead to desirable properties and useful materials.1–9 Polyphosphinoboranes are polymers with a main chain comprised of alternating phosphorus and boron atoms1,5,10–13 that have been explored as flame retardants,14 materials for soft lithography,15 and solvogels.16 Poly(P-phenyl)phosphinoborane is of particular interest as an inorganic P–B analogue of polystyrene, and was first sought in the 1950s.17,18 However, well-characterized polyphosphinoboranes were not accessed until the turn of the 21st century when the Manners group reported phosphine–borane adduct dehydropolymerization using Rh catalysts.19,20 Several catalysts have since been explored based on Fe,21,22 Ir,23 and Rh (Scheme 1).24–26 More recently, we have worked in collaboration with the Greb group to showcase the ability of a geometrically constrained aluminate species to act as a dehydropolymerization catalyst27 and with the Weller group to characterize the first well-defined block copolymer that is comprised of two distinct polyphosphinoborane blocks.28 | ||
| Scheme 1 Typical conditions for the catalytic dehydropolymerization of phosphine-boranes with catalyst examples. | ||
Polyphosphinoboranes can also be accessed through the direct generation of transient phosphinoborane monomers in situ. Cationic oligomers of phosphanylboranes (e.g. Me3N·BH2PH2BH2·NMe3) were first obtained by the Scheer group via the reaction of Lewis base-stabilized phosphanylboranes with monohalideboranes.29 The polymerisation via the generation of phosphinoboranes in situ was first demonstrated through the synthesis of tBuPH-BH2(NMe3) adducts that lose NMe3 under gentle heating to generate phosphinoborane monomers in solution.30 Later, we reported the synthesis of P-disubstituted polyphosphinoborane polymers ([PhRP-BH2]n; R = Ph, Et) through the cyclic(alkyl)amino carbene (cAAC)-mediated dehydrogenation of phosphine–borane adducts31 or through the deprotonation of phosphine–(triflimido)borane adducts (Scheme 2).32
 | ||
| Scheme 2 Synthesis of polyphosphinoboranes via the targeted generation of phosphine–borane adducts (top) and the work performed in this article (bottom). | ||
The mechanism of phosphine–borane dehydropolymerization is interesting as mechanism-led design of catalysts can result in greater efficiency in accessing materials as well as better control over the polymer microstructure and degree of polymerization.33 However, despite mechanistic studies performed thus far,21,24,25,34,35 the normal protocol for phosphine–borane dehydropolymerization still requires the use of a catalyst that normally takes several steps to synthesize, and forcing reaction conditions (≥16 h, ≥100 °C).13 One report explores the dehydrocoupling of PhPH2·BH3 using B(C6F5)3 (BCF) under milder conditions (3 days, 20 °C), where only a material of low molecular weight (Mw ≤ 3200 g mol−1) was obtained.36 It is therefore desirable not only to find new catalysts but also to investigate new mechanistic principles that are not only concerned with monomer formation, but also with the growth of the polymer chain itself. Some work has been done computationally, where Pomogaeva and Timoshkin have explored the chemistry of PB decamers in silico.37 They found that end-group complexation of phosphine–borane oligomers can prevent backbiting/cyclization of oligomers (and thus terminating the reactive growing chain). End-group capping by either a relatively weak Lewis acid or a relatively weak Lewis base is insufficient to prevent this cyclization event from occurring, but capping at both ends can disfavor backbiting. Computational studies have suggested that BH3 can act as a catalyst for the dehydrogenation of phosphine-boranes.38
Thus, in an effort to better understand the mechanism of dehydropolymerization using the state-of-the-art CpFe(CO)2OTf catalyst, we explored the ability of LiOTf and other Lewis acid–Lewis base pairs to catalyze the dehydropolymerization of PhPH2·BH3, 1.
Results and discussion
Dehydropolymerizations in toluene
First, thermal dehydropolymerization in the absence of any additives was explored as a control reaction and achieved by heating PhPH2·BH3 (1) in toluene at 100 °C for 24 h. The product mixture was analyzed by 31P{1H} and 11B{1H} NMR spectroscopy revealing broad peaks at −48.9 ppm and −34.7 ppm, respectively, indicating that [PhPH-BH2]n (2) had formed.Subsequently, the polymeric material was isolated via repeated precipitations, first in cold iPrOH, and twice more in cold hexanes resulting in a 35% yield of a colorless material. Analysis of the material by gel permeation chromatography (GPC) confirmed that a polymer was present, albeit a polydisperse material of low molar mass (Mn = 12680 g mol−1; Đ = 2.00) (Fig. 1).
 | ||
| Fig. 1 Molar mass values (Mn and Mw, solid orange and pink bars) and dispersity (Đ, striped green bars) of materials obtained from the dehydropolymerization of phosphine-boranes in toluene using no catalyst, Cp(CO)2FeOTf,21 and commercially available additives. *Mn and Mw for the bar chart are taken from the literature.21 Furthermore, at 20 °C, not all salts were fully soluble. | ||
Repeating the above procedure with the addition of 5 mol% LiOTf resulted in the formation of higher molecular weight 2 (Mn = 42200 g mol−1; Đ = 1.69) in 62% yield. These mass values closely match the activity of polymerizations performed with 5 mol% of CpFe(CO)2OTf (Mn = 40000 g mol−1; Đ = 1.70).21 We were surprised by these results as our initial mechanistic studies on Cp(CO)2Fe(OTf) identified Fe-containing oligomers by ESI-MS and established a relationship between [Fe] loading and polymer molar mass, indicating that the iron centre was involved in the dehydrogenation and chain growth steps. However, as the resulting polymer from LiOTf-catalyzed dehydropolymerization has no iron-derived impurities, this is a major advantage as a preceramic polymer. Intrigued by this result, we commenced to explore the ability of other commercially available salts to dehydropolymerize phosphine–borane adducts.
To determine the role of the triflate anion of LiOTf and separate its influence from that of the respective cation, we explored the ability of other triflate and triflimide salts to catalyze the dehydropolymerization of 1. Accordingly, the polymerization described above was performed with 5 mol% Mg(OTf)2 as an additive. This worked well as a catalyst for the dehydropolymerization of 1 to 2 (Mn = 32020 g mol−1; Đ = 1.70, 67% yield). However, using 5 mol% of Sc(OTf)3 resulted merely in a polymer that resembled that obtained from thermal dehydropolymerization (Mn = 12370 g mol−1; Đ = 2.26, 42% yield). Thus, the ability to access high molar mass 2 is not general to all additives. We then explored dehydropolymerization using the closely related salt LiNTf2. Similar to LiOTf and Mg(OTf)2, LiNTf2 addition also resulted in the formation of high, albeit slightly less high, molar mass 2 (Mn = 29260 g mol−1; Đ = 1.73; 58% yield).
As the cation appeared to influence the outcome of the reaction substantially, salts with a weakly coordinating cation in one case and a weakly coordinating anion in the other was used. [Bu4N][OTf] and [Li][B(C6F5)4]·Et2O both resulted in a polymer that resembled that obtained from thermal dehydropolymerization with no additives ([Bu4N][OTf]: Mn = 13110 g mol−1; Đ = 1.68; 30% yield; [Li][B(C6F5)4]·Et2O: Mn = 10980 g mol−1; Đ = 2.29; 36% yield) similar to what was observed for Sc(OTf)3. This result suggests that having a coordinatively unsaturated anion and cation is key to accessing high molar mass 2.
We were curious if it was possible to access a high molar mass material in the thermal dehydropolymerization of 1 using a Lewis acid–Lewis base adduct containing a dative bond, BH3·SMe2. This adduct is comprised of both a weak Lewis acid and a weak Lewis base, and if the components of the adduct (i.e., BH3 or SMe2) were to coordinate with the respective P- and B-termini of the growing oligomer, cyclization reactions would be disfavoured according to computational studies by Pomogaeva and Timoshkin.37 Accordingly, performing the polymerization as described above with the addition of 5 mol% of BH3·SMe2 resulted in the formation of a polymeric material as confirmed by GPC (Mn = 27990 g mol−1; Đ = 1.70). These data further support the hypothesis that both a Lewis acid and a Lewis base are needed in situ in order to access a high molar mass material.
Dehydropolymerizations in 2-MeTHF
As the presence of Lewis acids and Lewis bases in situ appeared to assist in the formation of high molar mass 2, we targeted the thermal dehydropolymerization of 1 in a coordinating solvent rather than in a weakly coordinating arene solvent. Not only would this precaution remove any ambiguity over the solubility of the additives and the actual loading, but it would also assist in realizing the more complete dissociation of the additives. The resulting greater stabilizing effect on the growing chain would produce higher molar mass materials. Furthermore, in the dehydropolymerizations of closely related amine–borane adducts, performing reactions in coordinating solvents can result in the formation of higher molar mass materials as chain transfer reactions are disfavoured.39 Accordingly, we performed the dehydropolymerization of 1 in 2-MeTHF. It has a higher boiling point than THF and is derived from sustainable sources,40 but its polarity is comparable with a dielectric constant of 6.97 for 2-MeTHF and 7.5 for THF.41Performing the dehydropolymerization of 1 as a 2 M solution in 2-MeTHF without any other additives also resulted in the formation of 2 after 24 h at 100 °C as determined by 31P and 11B NMR spectroscopy. Subsequent isolation was performed in the same manner as described above, resulting in a good yield of 70%. Analysis of the polymeric material by GPC revealed that it had a very high molar mass (Mn = 80680 g mol−1; Đ = 1.66; Fig. 2). Notably, this mass is double that obtained from thermal dehydropolymerizations performed under comparable reaction conditions using a 5 mol% loading of CpFe(CO)2OTf in toluene. Moreover, in our initial study on the dehydropolymerization of PhPH2·BH3 using CpFe(CO)2OTf, we found that performing the dehydropolymerization in 1,4-dioxane resulted in the formation of a higher molar mass material (Mn = 67000 g mol−1; Đ = 1.35).21 Furthermore, we assessed the ability of salts to catalyze the formation of high molar mass materials and found similar results to those obtained in toluene, where LiOTf, Mg(OTf)2, LiNTf2, and BH3·SMe2 would produce high molar mass materials and Sc(OTf)3, [Bu4N][OTf], and [Li][B(C6F5)4]·Et2O produced materials of lower molecular weights (Fig. 2). Notably, polymerizations performed with [Bu4N][OTf] produced materials of significantly lower molecular weights.
 | ||
| Fig. 2 Molar mass values (Mn and Mw, solid orange and pink bars) and dispersity (Đ, striped green bars) of materials obtained from the dehydropolymerization of phosphine-boranes in 2-MeTHF using no catalyst, in dioxane using Cp(CO)2FeOTf*,21 and in 2-MeTHF using commercially available additives. | ||
We were surprised by the performance of the thermal dehydropolymerization of 1 without any additives in 2-MeTHF as the studies in toluene suggest that both a Lewis acid and a Lewis base must be present in situ for a high molar mass material to be accessed. We suggest that under the reaction conditions, a small fraction of 1 can undergo adduct dissociation into phenylphosphine (PhPH2) and borane (BH3). Gas-phase computational studies into the bonding of phosphine–borane adducts estimate that bond dissociation is unfavourable and only a very little amount of free phosphine and free borane would be present in solution (ΔG° = 13.8 kcal mol−1; K = 8.26 × 10−9).42 However, this reaction would become more feasible in a donor solvent such as 2-MeTHF, as 2-MeTHF could stabilize borane through a dative interaction. Furthermore, if this dissociation were to occur, both a Lewis base (2-MeTHF, PhPH2) and a Lewis acid (BH3) would be present in situ and would satisfy the conditions established for dehydropolymerizations in toluene.
Mechanistic study
Having identified LiOTf and BH3·SMe2 as good catalysts in the dehydropolymerization of 1, we aimed to monitor the reaction more closely. Dehydropolymerizations of 1 were prepared in 2-MeTHF with LiOTF and BH3·SMe2 as additives and heated to 100 °C as described above. However, reactions were quenched at 0.15, 0.50, 1.0, 2.0, 3.0, 6.0, 16 and 24 hours for analysis by NMR spectroscopy and GPC (Fig. 3). Performing the dehydropolymerization in 2-MeTHF with 5 mol% of LiOTf resulted in a typical reaction progress graph where high conversion (>80%) was only reached at 16 h. Further analysis of each reaction using LiOTf over the 24-hour period by GPC revealed that a significant amount of a high molar mass material was obtained at 6 h (Fig. S3†), meaning that a high molar mass was obtained at low conversions of 1, similar to what was observed for dehydropolymerizations performed with CpFe(CO)2OTf.21 However, dehydropolymerizations performed with 5 mol% BH3·SMe2 in 2-MeTHF appeared to occur at a significantly higher rate compared to LiOTf as a significant conversion (>80%) of 1 had occurred within 6 h. Analysis of the reaction products by GPC revealed that a significant amount of a high molar mass material was obtained within 3 h (Fig. S4†). Comparing the conversion of 1 to 2 in both dehydropolymerizations with the Mn of the isolated materials reveals that high molar mass materials (Mn > 20000 g mol−1) are obtained at low conversions (<50%), but GPC traces obtained at higher conversions (>50%) reveal the formation of materials with even higher molar mass values (Mn > 40000 g mol−1).
 | ||
| Fig. 3 Conversion vs. Mn plot using LiOTf and BH3·SMe2 additives. Dehydropolymerizations were performed as 2 M solutions of 1 in 2-MeTHF and heated to 100 °C for 0.15 to 24 h. | ||
To identify if the Lewis acidic and Lewis basic components of the additives could be observed as end-groups in samples of 2, we conducted end-group analysis using electrospray ionization mass spectrometry (ESI-MS). Samples of 2 were prepared in DCM, and both positive and negative mode spectra were recorded. For polymerizations performed with both LiOTf and BH3·SMe2, a series of peaks separated by m/z 122, the mass of the [PhPH-BH2] monomer, were observed and signals corresponding to oligomers with up to 23 repeat units could be identified (Fig. S10†). Closer inspection of the positive mode spectra with peaks in the m/z 1 250–1500 range (Fig. S11†) indicated that polymerizations carried out with 5 mol% of LiOTf predominantly featured decamers and undecamers with H and PhPH2 end-groups ([H{PhPH-BH2}n·PhPH2]+; e.g., n = 10, m/z 1330.53). In contrast, for polymerizations performed with 5 mol% of BH3·SMe2, positive mode spectra within the same range revealed signals corresponding to oligomers with BH2(SMe2) end-groups ([(BH2(SMe2))·{PhPH-BH2}n·PhPH-BH2]+; e.g., n = 10, m/z 1416.45) in addition to those with H and PhPH2 end-groups. In the negative mode, ESI-MS spectra for polymerizations performed with either LiOTf or BH3·SMe2 consistently showed a series of peaks corresponding to P-phenylphosphinoborane decamers with BH3 and H end-groups ([H3B·{PhPH-BH2}nH]−; n = 10, m/z 1234.75). Thus, for polymerizations performed in LiOTf, end-groups incorporating either Li+ or [OTf]− could not be observed by ESI-MS. However, as both components of LiOTf are weakly coordinating, it is possible that these potential end-groups are displaced in the work-up, sample preparation, or in the ESI-MS instrument itself. In contrast, for polymerizations performed with BH3·SMe2, both BH3 and SMe2 were observed as chain caps. The source of BH3 end-groups could either be from 1 or BH3·SMe2, but the presence of the BH2(SMe2) chain-ends implicates that BH3·SMe2 has a role in chain propagation of the growing polymer.
We then explored the ability to regenerate reactive chain ends through continued late-stage step-growth polymerization of isolated polymers. Accordingly, an isolated sample of 2 of moderate molecular weight (Mn = 57740 g mol−1, Đ = 1.65, from Table S1 entry 24†) was dissolved in a minimal amount of 2-MeTHF and the resulting solution was transferred to a J. Young's NMR tube. This tube was then sealed and heated to 100 °C for 24 hours. Afterwards, the resulting polymeric material was isolated as described above and analysed by 31P NMR spectroscopy and GPC. Analysis of the 31P and 31P{1H} NMR spectra revealed that only the peak for linear polymeric 2 was present, and no peaks indicative of branching points were observed (Fig. S8†). Then, examination of the GPC chromatogram revealed that the polymerization did continue, resulting in the formation of a material of a higher molar mass, but with a higher dispersity (Mn = 149420 g mol−1, Đ = 2.40) (Fig. S9†). Furthermore, the GPC trace revealed what appears to be a bimodal material, indicating that polymers with different end-groups undergo continued growth at different rates.
Based on our findings from the dehydropolymerizations of 1 in toluene and 2-MeTHF, along with time-dependent studies and end-group analyses of 2, we propose the mechanism illustrated in Scheme 3. At elevated temperatures, a small amount of free borane (BH3) is generated in situ through phosphine–borane cleavage (Scheme 3a). This free BH3 acts as the dehydrogenation (pre)catalyst, facilitating the formation of phosphinoborane monomers (M). It is possible that borane-mediated dehydrogenation occurs via an initial hydride abstraction, followed by the formation of M with concomitant loss of H2. Monomers can self-initiate (rate constant ki) to form the active species P, or react with residual Lewis acid LA or Lewis base LB (Scheme 3b). Evidence for this mechanism includes the significantly faster polymerization rates observed when BH3 (from BH3·SMe2) is added: >80% conversion of 1 is reached within 6 hours, and high molar mass, low-dispersity materials are formed within 3 hours. In contrast, polymerizations without added BH3 (e.g., those using LiOTf alone) achieve >80% conversion only after 6 hours, with high molar mass materials appearing at similar times. The active species (P) can undergo termination reactions (kt) to produce 2.
 | ||
| Scheme 3 Proposed mechanism for the dehydropolymerization of phosphine–borane adducts at elevated temperatures with three distinct steps a, b, and c, as discussed in the main text. | ||
Thus, a potential role of the additive is in the stabilization of the growing chain P. From the polymerizations performed in toluene and 2-MeTHF, additives that had both Lewis acidic and basic components were required to access high molar mass materials (Fig. 1 and 2). Furthermore, ESI-MS of polymeric materials obtained using added BH3·SMe2 revealed the presence of oligomers with end groups corresponding to BH2(SMe2) (Fig. 4). Further, polymerizations performed in coordinating solvents accessed materials of higher molar mass than those performed in non-coordinating solvents (Fig. 2). Moreover, performing polymerizations in the presence of a strong Lewis acid (Sc(OTf)3)43 consistently resulted in materials of relatively low molecular weights (Fig. 1 and 2). Therefore, we propose that the stabilization may be occurring through the reversible coordination of weak Lewis acids and weak Lewis bases to chain termini (Scheme 3c). This would allow for the attenuation of the reactivity of the growing oligomer chain, through accessing a chain-capped dormant species, LB-P-LA, allowing for reactions that compete with chain termination including cyclization reactions.37 The dormant species, LB-P-LA, could release either LB or LA to access P-LA or LB-P. Either dissociation would result in the formation of an active species which can continue growth via addition polymerization through reactions with M. Furthermore, either LB-P or P-LA can readily recombine (krecom) with either LA or LB to access the dormant species. However, reactions between LB-P and P-LA would also allow for the reformation of the dormant species coupled with an increase in the chain length. Moreover, the ability to favor the formation of the dormant species either through the choice of Lewis acid and Lewis base, or the loading of the chosen Lewis acid and Lewis base may allow for greater control over the polymerization of phosphinoboranes produced in situ. Similarly, the dissociation of borane has been shown to be relevant in the thermolytic and catalytic dehydropolymerization of linear aminoborane dimers44 and in the spontaneous dehydrogenation of N-aryl substituted amine-boranes (ArNH2·BH3).45 It should be noted that strong Lewis bases have been shown to depolymerize polyphosphinoboranes.46 Thus, while the presence of Lewis bases may assist in chain growth in early stages of the polymerization, if they are too nucleophilic, they may result in the depolymerization of the resulting material. This mechanism is reminiscent of those postulated for quasi-living radical polymerizations.47–50
 | ||
| Fig. 4 ESI-MS positive mode spectrum of 1 prepared in 2-MeTHF in the presence of 5 mol% LiOTf (top) and BH3·SMe2 (bottom) over the range of 1300–1500 m/z. Orange boxes highlight the peak for [H[PhPH-BH2]10,11·PhPH2]+ and pink boxes highlight the peaks for [(BH2(SMe2))·[PhPH-BH2]9,10·PhPH-BH2]+. | ||
Molecular weight control using added BH3·SMe2
If the above mechanism were in operation, it may be possible to control the degree of polymerization as a greater concentration of Lewis base and Lewis acid in situ would favor dormant species and result in a material of lower molecular weight. Accordingly, 5 different loadings were explored (1 mol%, 2.5 mol%, 5 mol%, 7.5 mol%, and 10 mol%) of either LiOTf or BH3·SMe2, while maintaining a concentration of 2 M of 1 in 2-MeTHF. The dehydropolymerization reactions were performed at 100 °C over the course of 24 hours. Dehydrocoupling was determined to have occurred in each case and subsequent isolation of the materials resulted in moderate to good yields (50% to 74%). Analysis of the polymers produced with 1–10 mol% of LiOTf added revealed that while low loadings of LiOTf resulted in a material of higher molar mass, no real correlation between LiOTf loading and the obtained polymer molar mass could be observed at loadings at or greater than 5 mol%. However, polymerizations performed with added BH3·SMe2 revealed a clear relationship between loading and degree of polymerization across the conditions attempted (1–10 mol% of BH3·SMe2), obtaining materials over a wide mass range with similar dispersity (Mn = 83120–57740 g·mol−1; Đ ca. 1.66; Fig. 5). This control over the resulting material further suggests that Lewis base and Lewis acid additives favor the formation of a dormant species, as proposed in Scheme 3.
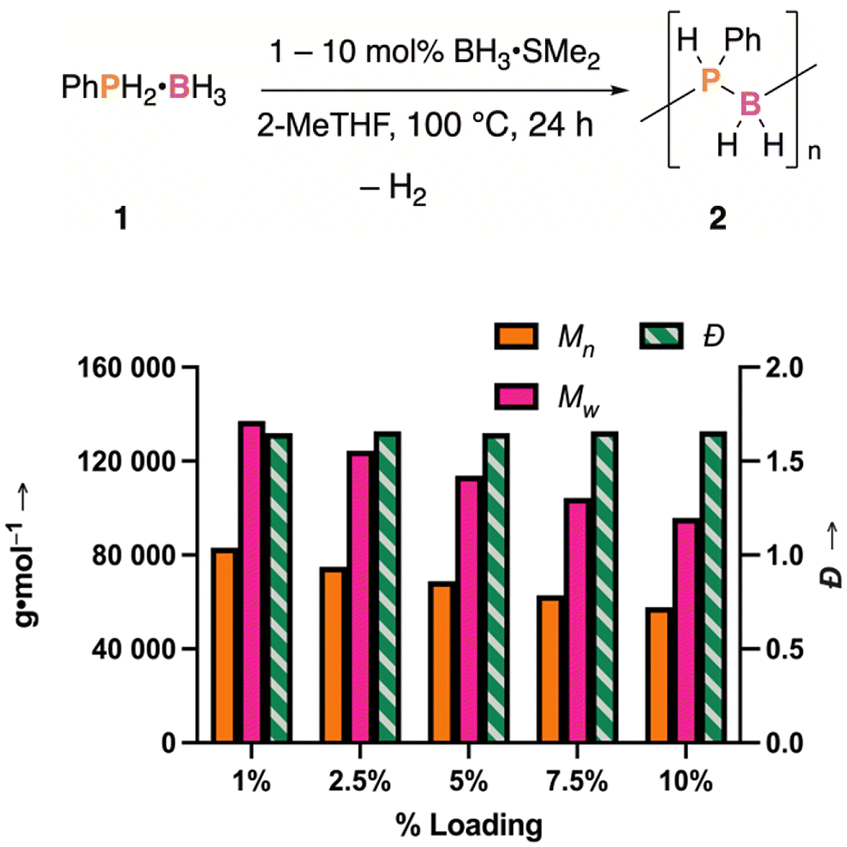 | ||
| Fig. 5 Molar mass values (Mn and Mw, solid orange and pink bars) and dispersity (Đ, striped green bars) of isolated materials from the dehydropolymerization of 1 in 2-MeTHF with added BH3·SMe2. | ||
Conclusions
We report the synthesis of high molar mass polyphosphinoboranes using commercially available reagents. We propose that thermal dehydropolymerization occurs in three major steps: adduct dissociation to give free borane, allowing for the second step where free borane acts as a (pre)catalyst for the dehydrogenation of 1. The third phase is the polymerization step where reactive phosphinoboranes in situ catenate to yield linear chains that can then undergo termination events to yield polymeric materials. However, in the presence of additives, such as LiOTf or BH3·SMe2, phosphinoboranes and their chains can participate in Lewis acid base chemistry at either terminus, competing with termination events and allowing for further chain growth to access polymers. This approach allowed for the synthesis of high molar mass 2 from the dehydropolymerization of 1 in 2-MeTHF, where the presence of additives can afford control over the resulting materials, with BH3·SMe2 showing a clear relationship to Mn of the resulting material over a wide mass range (Mn = 57740–83120 g mol−1). Overall, this work has implications for the field of phosphine–borane dehydropolymerization in that high molar mass materials can be accessed using readily available reagents. While the polymerizations reported here still operate under conditions typical of transition-metal catalyzed dehydropolymerization of phosphine-boranes, this work shows that transition metals are unnecessary in accessing high molar mass polyphosphinoboranes. Moreover, this work provides evidence for an alternative mechanism that may allow for greater control over the addition polymerization of phosphinoborane monomers produced in situ.
Author contributions
Conceptualization: M.A.W., A.S., and I.M. contributed equally. Data curation: M.A.W. (lead), J.E.T.W. (supporting), and C.K. (supporting). Formal analysis: M.A.W. (lead), J.E.T.W. (supporting), and C.K. (supporting). Funding acquisition: I.M. (lead). Investigation: M.A.W. (lead), J.E.T.W. (supporting), and C.K. (supporting). Methodology: M.A.W. (lead), J.E.T.W. (supporting), and C.K. (supporting). Project administration: M.A.W. and A.S. contributed equally. Resources: I.M. (lead) and J.S.M. (supporting). Supervision: A.S. (lead), I.M. (supporting), and J.S.M. (supporting). Validation: M.A.W. (lead). Visualization: M.A.W. (lead). Writing – original draft: M.A.W. (lead). Writing – review and editing: A.S. (lead), all other authors contributed equally.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was largely performed on the traditional territories of the Ləkwəŋən (Songhees and Esquimalt) Peoples. M. A. W. would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for a PGS-D fellowship, the University of Victoria for the Dean's Award for Indigenous Graduate Students and a President's Research Award, the Government of British Columbia for British Columbia Graduate Scholarships, and the Irving K. Barber Scholarship Society and the Manitoba Métis Federation for awards. The authors further acknowledge the funding that made this work possible, primarily through an NSERC DG and a C150 grant awarded to Ian Manners.References
- T. Baumgartner and F. Jaekle, Main Group Strategies Towards Functional Hybrid Materials, John Wiley & Sons, Incorporated, Newark, 2018 Search PubMed.
- F. Jaekle, Chem. Rev., 2010, 110, 3985–4022 Search PubMed.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- R. De Jaeger and M. Gleria, Prog. Polym. Sci., 1998, 23, 179–276 CrossRef CAS.
- A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2016, 45, 922–953 RSC.
- T. Chivers and I. Manners, Inorganic Rings and Polymers of the p-Block Elements, RSC Publishing, 2009 Search PubMed.
- A.-M. Caminade, E. Hey-Hawkins and I. Manners, Chem. Soc. Rev., 2016, 45, 5144–5146 RSC.
- J. E. Mark, Acc. Chem. Res., 2004, 37, 946–953 Search PubMed.
- A. L. Colebatch and A. S. Weller, Chem. – Eur. J., 2019, 25, 1379–1390 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 Search PubMed.
- E. M. Leitao, T. Jurca and I. Manners, Nat. Chem., 2013, 5, 817–829 CrossRef CAS PubMed.
- F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 Search PubMed.
- D. Han, F. Anke, M. Trose and T. Beweries, Coord. Chem. Rev., 2019, 380, 260–286 Search PubMed.
- A. W. Knights, M. A. Nascimento and I. Manners, Polymer, 2022, 247, 124795 CrossRef CAS.
- T. J. Clark, J. M. Rodezno, S. B. Clendenning, S. Aouba, P. M. Brodersen, A. J. Lough, H. E. Ruda and I. Manners, Chem. – Eur. J., 2005, 11, 4526–4534 CrossRef CAS PubMed.
- A. W. Knights, S. S. Chitnis and I. Manners, Chem. Sci., 2019, 10, 7281–7289 RSC.
- A. B. Burg and R. I. Wagner, J. Am. Chem. Soc., 1953, 75, 3872–3877 CrossRef.
- A. B. Burg, J. Inorg. Nucl. Chem., 1959, 11, 258 CrossRef CAS.
- H. Dorn, R. A. Singh, J. A. Massey, A. J. Lough and I. Manners, Angew. Chem., Int. Ed., 1999, 38, 3321–3323 CrossRef CAS PubMed.
- H. Dorn, R. A. Singh, J. A. Massey, J. M. Nelson, C. A. Jaska, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2000, 122, 6669–6678 CrossRef CAS.
- A. Schäfer, T. Jurca, J. Turner, J. R. Vance, K. Lee, V. A. Du, M. F. Haddow, G. R. Whittell and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 4836–4841 CrossRef PubMed.
- N. T. Coles, M. F. Mahon and R. L. Webster, Organometallics, 2017, 36, 2262–2268 CrossRef CAS.
- U. S. D. Paul, H. Braunschweig and U. Radius, Chem. Commun., 2016, 52, 8573–8576 Search PubMed.
- M. A. Huertos and A. S. Weller, Chem. Sci., 2013, 4, 1881–1888 RSC.
- T. N. Hooper, A. S. Weller, N. A. Beattie and S. A. Macgregor, Chem. Sci., 2016, 7, 2414–2426 RSC.
- T. N. Hooper, M. A. Huertos, T. Jurca, S. D. Pike, A. S. Weller and I. Manners, Inorg. Chem., 2014, 53, 3716–3729 Search PubMed.
- F. Schön, L. M. Sigmund, F. Schneider, D. Hartmann, M. A. Wiebe, I. Manners and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202202176 CrossRef PubMed.
- J. J. Race, A. Heyam, M. A. Wiebe, J. Diego Garcia-Hernandez, C. E. Ellis, S. Lei, I. Manners and A. S. Weller, Angew. Chem., Int. Ed., 2023, 62, e202216106 Search PubMed.
- C. Marquardt, C. Thoms, A. Stauber, G. Balazs, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 3727–3730 CrossRef CAS PubMed.
- C. Marquardt, T. Jurca, K. C. Schwan, A. Stauber, A. V. Virovets, G. R. Whittell, I. Manners and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13782–13786 CrossRef CAS PubMed.
- N. L. Oldroyd, S. S. Chitnis, V. T. Annibale, M. I. Arz, H. A. Sparkes and I. Manners, Nat. Commun., 2019, 10, 1–9 Search PubMed.
- M. A. Wiebe, S. Kundu, E. A. LaPierre, B. O. Patrick and I. Manners, Chem. – Eur. J., 2023, 29, e202202897 Search PubMed.
- P. S. Chum and K. W. Swogger, Prog. Polym. Sci., 2008, 33, 797–819 Search PubMed.
- C. A. Jaska and I. Manners, J. Am. Chem. Soc., 2004, 126, 9776–9785 Search PubMed.
- T. N. Hooper, M. A. Huertos, T. Jurca, S. D. Pike, A. S. Weller and I. Manners, Inorg. Chem., 2014, 53, 3716–3729 Search PubMed.
- J.-M. Denis, H. Forintos, H. Szelke, L. Toupet, T.-N. Pham, P.-J. Madec and A.-C. Gaumont, Chem. Commun., 2003, 54–55 RSC.
- A. V. Pomogaeva and A. Y. Timoshkin, J. Phys. Chem. A, 2021, 125, 3415–3424 Search PubMed.
- L. Wang, T. Zhang, H. He and J. Zhang, RSC Adv., 2013, 3, 21949–21958 Search PubMed.
- H. C. Johnson, E. M. Leitao, G. R. Whittell, I. Manners, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2014, 136, 9078–9093 CrossRef CAS PubMed.
- C. S. Slater, M. J. Savelski, D. Hitchcock and E. J. Cavanagh, J. Environ. Sci. Health, A, 2016, 51, 487–494 CrossRef CAS PubMed.
- D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156–159 CrossRef CAS.
- A. Adamson and P. Burk, Comput. Theor. Chem., 2014, 1032, 12–19 Search PubMed.
- J. N. Bentley, S. A. Elgadi, J. R. Gaffen, P. Demay-Drouhard, T. Baumgartner and C. B. Caputo, Organometallics, 2020, 39, 3645–3655 CrossRef CAS.
- A. P. M. Robertson, E. M. Leitao and I. Manners, J. Am. Chem. Soc., 2011, 133, 19322–19325 Search PubMed.
- H. Helten, A. P. M. Robertson, A. Staubitz, J. R. Vance, M. F. Haddow and I. Manners, Chem. – Eur. J., 2012, 18, 4665–4680 Search PubMed.
- C. Marquardt, O. Hegen, A. Vogel, A. Stauber, M. Bodensteiner, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2018, 24, 360–363 Search PubMed.
- D. Mardare and K. Matyjaszewski, Macromolecules, 1994, 27, 645–649 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 Search PubMed.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 Search PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The synthesis and characterization of polymers, NMR spectra, mass spectra, and GPC traces. See DOI: https://doi.org/10.1039/d4py01362j |
| ‡ Deceased Dec 3, 2023. |
| This journal is © The Royal Society of Chemistry 2025 |