
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glycidyl ethers from acyclic terpenes: a versatile toolbox for multifunctional poly(ethylene glycol)s with modification opportunities†
Sandra
Schüttner‡
,
Gregor M.
Linden‡
,
Elena C.
Hoffmann
 ,
Philipp
Holzmüller
and
Holger
Frey
*
,
Philipp
Holzmüller
and
Holger
Frey
*
Department of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de
First published on 6th December 2024
Abstract
Multifunctional poly(ethylene glycol) copolymers (mfPEGs) are accessible via the anionic copolymerization of functional epoxides with ethylene oxide (EO). Glycidyl ethers are conveniently synthesized from bio-renewable alcohols and epichlorohydrin (ECH). Herein, we present the synthesis of a series of acyclic terpenyl glycidyl ethers (TGEs) and their subsequent copolymerization with ethylene oxide (EO) via anionic ring-opening polymerization (AROP). The resulting library of copolymers with varied side chain length and comonomer composition comprises molar masses in the range of 4800 to 8300 g mol−1 and narrow molar mass distributions (Đ = 1.06–1.13). For the copolymerization of the TGEs with EO, detailed 1H NMR in situ kinetic studies revealed a change from ideally random to slight gradient copolyether microstructures with increasing chain length and hydrophobicity of the respective TGE. The living nature of AROP provides control of molar masses, and optimized reaction conditions, such as low reaction temperatures and a weakly bound cesium counterion, suppress the well-known proton abstraction of monosubstituted epoxides. Since the incorporation of the terpenyl side chains impedes crystallization, thermal properties of the copolyethers can be tailored by the monomer feed ratio. Subsequently, hydrogenation and thiol–ene click reactions at the side chain double bonds were carried out as post-polymerization modifications. The application of potassium azodicarboxylate (PADA) in the diimide reduction of the polymers was demonstrated to possess vast potential for the full hydrogenation of the novel copolymers, offering facile purification options. Overall, the copolymerization of EO and TGEs gives access to biobased, tailormade polyethers with various options for post-functionalization.
Introduction
Poly(ethylene glycol) (PEG) marks a high-value polyether owing to its excellent biocompatibility and extraordinary solubility in aqueous media. Known as the gold standard for pharmaceutical and biomedical purposes, the application range of PEG also includes cosmetics, surface-active materials and polymer electrolytes.1–4 To overcome the limited backbone functionality, copolymerization of ethylene oxide (EO) with functional epoxide building blocks allows for the synthesis of multifunctional PEG (mf-PEG) featuring tailormade material properties.1,5 Particularly glycidyl ethers (GE) represent an underrated monomer class, which is available from a plethora of alcohols and epichlorohydrin (ECH).6,7 Alternatively, the reaction of an alcohol with allyl bromide, followed by epoxidation using meta-chloroperoxybenzoic acid affords the respective GE.8 However, despite the increasing use of renewable resources for polymer synthesis, bio-derived epoxides in general and specifically glycidyl ethers have scarcely been exploited for more sustainable polyethers.9 Simultaneously, several efforts have been reported that aim at more sustainable sources of conventional polyether building blocks, e.g. farnesyl glycidyl ether from farnesol.10 Intriguingly, Sargent and coworkers have recently improved the electrochemical production of ethylene oxide (EO) from CO2 and water.11 Alternatively, “green ethylene” can be transformed into EO utilizing the conventional silver-catalyzed epoxidation.12 Furthermore, “green ECH” is readily available from glycerol, a byproduct in the bio-fuel production from biomass.13,14The use of natural product-based building blocks in glycidyl ether synthesis is a key objective. Fatty acids extracted as value-added chemicals from plant oils have been a long-standing raw material for the chemical industry and for polymer synthesis.15,16 Relevant parameters, which affect the chemical and physical properties, are the length and the degree of unsaturation of the fatty acid chain as well as the stereochemistry of the double bond.17 Relying on fatty acids, the respective alcohols for glycidyl ether synthesis are accessible via reductive transformation into an alcohol and subsequent phase transfer catalysis.16 Verkoyen et al. published an extensive overview of readily available long-chain alkyl epoxides and glycidyl ethers, characterized by their hydrophobicity, including some examples of fatty acid analogs.7,18
As another class of relevant, biomass-derived building blocks, terpenoids and terpenes have been established as attractive monomer precursors.9,15,19–21 Following the “isoprene rule”, acyclic terpenoids vary in their chain length based on their number of linear, head-to-tail condensed (C5H8)n units. Thus, the number of repeat isoprene units allows for the classification into hemi-(C5), mono-(C10) and sesquiterpenoids (C15).22,23 Linear, acyclic terpenoids are abundant in ethereal oils of fruits and plants and their application ranges from pharmaceutical products to flavors, fragrances, and pheromones.24,25 Producing GEs from a range of acyclic terpenoids gives rise to a diverse platform of bio-renewable epoxide monomers (Scheme 1, left). The proposed terpenyl glycidyl ethers (TGEs) are characterized by their unsaturated, hydrophobic side chains. In general, their branched nature impedes crystallization, in stark contrast to their linear fatty acid analogs.8 To date, only a handful of publications on polyethers and polycarbonates have capitalized on these terpene-derived epoxide building blocks, being suitable for anionic and catalytic polymerization techniques.10,26–29 On one hand, the hydrophobic character of farnesyl glycidyl ether in combination with PEG enables the facile synthesis of amphiphilic polyethers.10 On the other hand, the terpenyl side chain flexibility leads to a low glass transition temperature (Tg), thus permitting the generation of flexible, low-Tg polycarbonates.28,29 Moreover, replacing the commonly applied propylene oxide as a hydrophobic building block takes effect on the copolymer microstructure. It is worth emphasizing that the copolymerization of EO with PO entails a strong gradient microstructure, whereas GE and EO are expected to produce a random copolymer.30–32
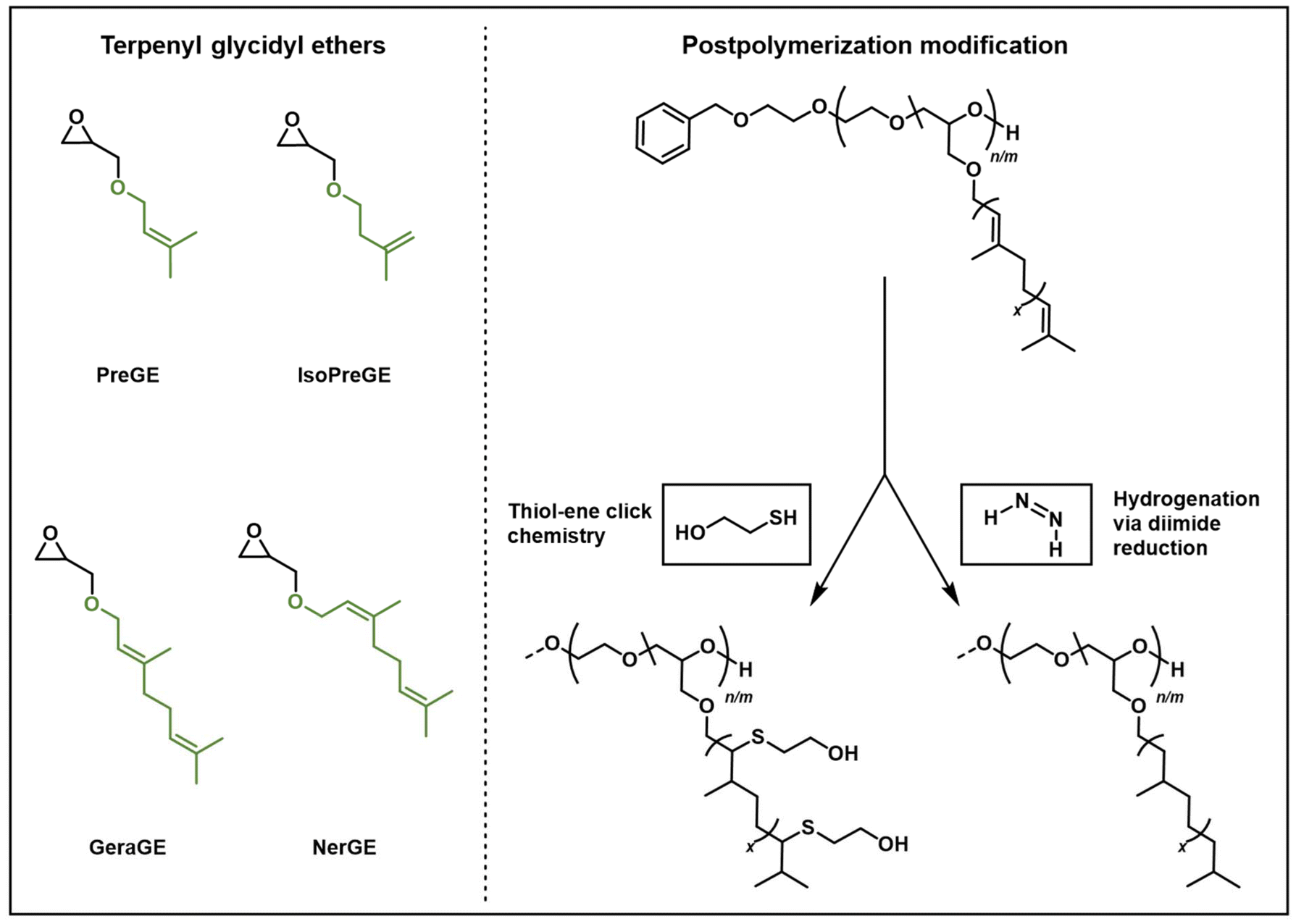 | ||
| Scheme 1 Scope of prepared bio-renewable TGEs for copolyether synthesis (left). The TGE copolymers are amenable to post-polymerization modification e.g. by hydrogenation and thiol–ene click (right). | ||
In addition, the unsaturated side chains of such acyclic TGEs serve as chemoselective handles due to the chemically differing double bonds and, in turn, enable tailor-made materials via post-functionalization.15,19,25,33 For TGEs, cross-metathesis has been demonstrated by Morris and coworkers, who pioneered TGEs for polyether synthesis utilizing prenyl glycidyl ether (PreGE) and isoprenyl glycidyl ether (IsoPreGE).26 In this context, PreGE and IsoPreGE can be considered as a bio-renewable analog of the commonly applied allyl glycidyl ether (AGE).1,26 Moreover, successful click-TAD chemistry on a citronellyl glycidyl ether side chain led to terpene-derived polyether-based organo- and hydrogels.27 Yet, the plentiful modification opportunities have not been investigated extensively. For instance, the thiol–ene click reaction of polymers offers flexible means to create custom materials, as documented in Lowe's comprehensive review.34,35 In addition, hydrogenation is a widely adopted approach that enhances the thermal and oxidative stability of polyisoprene by using gaseous hydrogen and metal catalysts at high temperatures in a reactor.36 Another small-scale modification method is hydrogenation using diimide, which can be generated from various sources and does not require high-pressure equipment.37,38
In this work we present the use of different bio-based TGEs for the synthesis of mf-PEGs. Applying hemi- and monoterpenoids in the synthesis of TGEs (Scheme 1), we expanded the available bio-derived TGE library for polyether design and synthesis based on PreGE, IsoPreGE, geranyl glycidyl ether (GeraGE) and neryl glycidyl ether (NerGE). Statistical polyether copolymers P(EGn-co-TGEm) were prepared via living AROP of EO and the TGEs of varying chain length. The resulting polyethers exhibit the targeted molar masses and low dispersity Đ (≤1.13), while the thermal properties can be fine-tuned by the applied TGE content. 1H NMR in situ kinetics was employed to study the impact of the glycidyl ether side chains on the copolymerization behavior of the respective EO/TGE comonomer pairs and potential gradient formation. Furthermore, the structural variety regarding side chain length and double bond configuration was explored with respect to post-polymerization functionalization (Scheme 1). Aiming at introducing hydroxyl-functionalities, photochemical thiol–ene click chemistry was performed. Further, the double bond was hydrogenated using potassium azodicarboxylate (PADA) diimide reduction, which involves the in situ generation of the diimide via acidic decomposition of PADA. Subsequently, we address some of the limitations associated with the use of para-toluenesulfonyl hydrazide.
Experimental section
Details on reagents used, instrumentation, methodical procedures and synthesis procedures are given in the ESI Sections 1–4.†Results and discussion
Monomer synthesis
The presented TGEs have all been prepared in a phase transfer catalysis, starting from epichlorohydrin and the respective terpenoid.10,29 The products were isolated in high purity after distillation work up, which was performed subject to the monomer boiling temperature Tb (Scheme 2).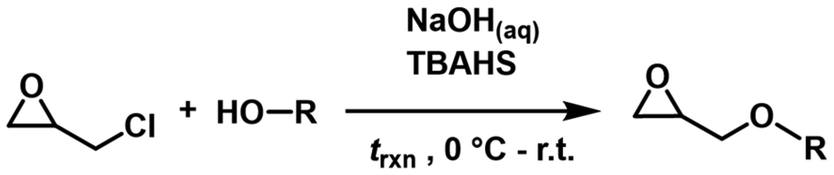 | ||
| Scheme 2 General monomer synthesis via phase transfer catalysis. TBAHS: tetra-n-butylammonium hydrogen sulfate. | ||
During the phase transfer catalysis, elimination and substitution reactions lead to the formation of undesired byproducts, specifically 3-chloro-allyl glycidyl ether and diglycidyl ether.10,29 While the resulting byproducts can be easily separated from the long-chain TGEs (C10 and C15) due to a significant difference in Tbs, the C5-analogs, namely PreGE, IsoPreGE and dihydroprenyl glycidyl ether (DHPreGE), required an altered synthetic protocol.10,29 To suppress byproduct formation, the synthesis approach utilizing the C5-alcohols was performed under prolonged cooling and for shorter reaction times, ultimately resulting in lower overall yields (51–64%) compared to the C10 and C15-analogues GeraGE and NerGE (78–83%). We designed a synthesis protocol that circumvented the purification step via column chromatography and improved the C5-monomer yields compared to literature.26 Detailed NMR characterization for all TGEs (Fig. S1–S14†) as well as an explicit synthetic protocol are provided in the ESI.†
Statistical copolymerization of EO and TGEs
Living statistical copolymerizations of bio-renewable TGEs with EO were performed in a THF-DMSO solvent mixture (VTHF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VDMSO = 5:1) at 40 °C, using conventional AROP conditions, employing cesium 2-(benzyloxy)ethoxide as an initiator salt (Scheme 3).
:VDMSO = 5:1) at 40 °C, using conventional AROP conditions, employing cesium 2-(benzyloxy)ethoxide as an initiator salt (Scheme 3).
 | ||
| Scheme 3 Synthetic strategy for the copolymerization of EO with TGEs of varying terpenyl chain length, resulting in terpenyl-functionalized PEGs. | ||
In order to maintain the remarkable properties of PEG while extending its functionality, we designed a series of copolymers for each TGE by varying the comonomer content between 3.0 and 9.6 mol%. The rationale for this intended low amount of TGE comonomer encompasses significant property changes using small TGE amounts while increasing functionality and maintaining the aqueous solubility of the copolymers. Table 1 summarizes the copolymerization results as well as the thermal characterization data of the statistical P(EGn-co-TGEm) copolymers. Number averaged molar masses (Mn) were calculated via1H NMR end group analysis by referencing to the methylene group of the 2-(benzyloxy)ethanol initiator (Fig. S19, S21, S23, and S25†), in the range of 4800 to 8300 g mol−1.
| Entry | Sample | X TGE,exp /mol% | M n, NMR /g mol−1 | M n,SEC /g mol−1 | Đ | T g /°C | T m /°C | ΔHmc/J g−1 |
|---|---|---|---|---|---|---|---|---|
| a Number averaged molar mass and experimental monomer content determined via1H NMR spectroscopy. b Determined via SEC measurements (RI detector, eluent: DMF, calibration: PEG). c Determined from second heating cycle at a heating rate of 10 °C·min−1. | ||||||||
| 1 | P(EG121-co-PreGE5) | 4.0 | 6190 | 4800 | 1.08 | −59 | 39 | 79 |
| 2 | P(EG113-co-PreGE5) | 4.2 | 5840 | 4600 | 1.06 | −60 | 31 | 74 |
| 3 | P(EG107-co-PreGE10) | 8.5 | 6290 | 4700 | 1.09 | −63 | 21 | 47 |
| 4 | P(EG102-co-PreGE10) | 8.9 | 6070 | 4900 | 1.07 | −62 | 18 | 43 |
| 5 | P(EG96-co-IsoPreGE3) | 3.0 | 4810 | 4700 | 1.07 | −61 | 40 | 76 |
| 6 | P(EG117-co-IsoPreGE7) | 5.6 | 6300 | 5200 | 1.07 | −64 | 29 | 56 |
| 7 | P(EG100-co-IsoPreGE10) | 9.1 | 5980 | 4400 | 1.10 | −66 | 18 | 44 |
| 8 | P(EG126-co-IsoPreGE12) | 8.7 | 7410 | 4800 | 1.11 | −65 | 15 | 44 |
| 9 | P(EG123-co-GeraGE4) | 3.1 | 6250 | 4700 | 1.07 | −61 | 40 | 77 |
| 10 | P(EG117-co-GeraGE6) | 4.9 | 6540 | 4800 | 1.08 | −65 | 26 | 37 |
| 11 | P(EG110-co-GeraGE8) | 6.8 | 6680 | 4200 | 1.07 | −66 | 23 | 40 |
| 12 | P(EG120-co-GeraGE12) | 9.1 | 7960 | 5400 | 1.10 | −63 | 14 | 35 |
| 13 | P(EG109-co-NerGE4) | 3.5 | 5640 | 4400 | 1.07 | −63 | 38 | 70 |
| 14 | P(EG125-co-NerGE8) | 6.0 | 7180 | 5100 | 1.07 | −65 | 32 | 51 |
| 15 | P(EG115-co-NerGE10) | 8.0 | 7160 | 5600 | 1.07 | −67 | 21 | 48 |
| 16 | P(EG123-co-NerGE13) | 9.6 | 7980 | 5500 | 1.08 | — | — | — |
Typical 1H NMR spectra show the anticipated signals of the polyether backbone and the terpenyl side chain, which can be clearly distinguished. Notably, we observed side chain isomerization for all polymers, which is a known phenomenon for allyl glycidyl ether, when applying the general basic conditions of AROP.39–41 We will briefly readdress the isomerization in the kinetics investigation section. Size exclusion chromatography (SEC) indicates well-defined copolymers with monomodal distributions and low dispersity Đ (1.06–1.11) (Fig. S15–S18†). Deviations of the Mn determined by SEC from 1H NMR characterization are ascribed to the structural and polarity difference of the P(EGn-co-TGEm) copolymers compared to the PEG calibration standards for SEC.
In situ 1H NMR copolymerization kinetics of TGEs with EO
Glycidyl ethers can be applied in the copolymerization with EO to introduce functionality along the polyether backbone.5 For a detailed understanding of the resulting materials’ properties, it is pivotal to elucidate the copolymer microstructure.31 Capitalizing on in situ1H NMR kinetic investigations in combination with suitable copolymerization models, it is possible to translate monomer conversion into the respective reactivity ratios of a given comonomer pair.31,42,43 Motivated by the different structure of the terpenyl side chains, we systematically performed in situ1H NMR copolymerization kinetics in a vacuum-sealed NMR tube to compare the copolymerization behavior for all TGE/EO comonomer pairs. It is important to note that we also investigated the fully hydrogenated analogs of the C5, C10 and C15 side chains to evaluate the influence of hydrophobicity on the copolymerization behavior, also aiming at further understanding the role of the allylic double bond. Based on the living AROP copolymerization without chain transfer and termination, we accurately followed monomer conversion, specifically for EO at 2.59 ppm (blue) and PreGE at 3.05 ppm (green, Fig. 1 and Fig. S39, left†). As previously reported, the ideal non-terminal model is known to be valid for the copolymerization of EO and glycidyl ethers. The copolymerizations follow ideal copolymerization behavior, which is expressed by the simple relation: r1 = r2–1.31,44,45 Plotting the individual monomer conversion versus total conversion (Fig. S39, right†), we applied the ideal Jaacks fit (Fig. S27†) to obtain reactivity ratios of rPreGE = 0.94 and rEO = 1.06, revealing an almost ideally random copolymerization for PreGE and EO.46 | ||
| Fig. 1 Selection of 1H NMR spectra (400 MHz, THF-d8:DMSO-d6 = 5:1) of the in situ copolymerization kinetics of PreGE with EO at 40 °C. Relevant proton signals are highlighted in green (PreGE) at 3.05 ppm and blue (EO) at 2.59 ppm. As spectra were collected every 2 min over a period of 51.5 h, only every 30th spectrum is displayed. | ||
Table 2 summarizes the reactivity ratios determined for the statistical copolymerization of EO with the TGEs employed in this work. Detailed in situ kinetic data, characterization as well as the respective SEC traces (Đ ≤ 1.13) are compiled in the ESI, Fig. S27–33 and S39–58.† In general, copolymerization behavior is governed by steric and electronic properties of the epoxide monomers.31 The side chain impact on reactivity ratios is manifest as a collective of three parameters: (i) length, (ii) hydrophobicity, and (iii) flexibility. At first glance, an increase in chain length and hydrophobicity leads to a more pronounced incorporation of EO at the start of the copolymerization. This is consistent with previously reported results for farnesyl glycidyl ether.10 PreGE and IsoPreGE manifest no significantly different copolymerization behavior, although the isoprenyl main chain is more flexible. Both copolymerizations show almost ideally random behavior. On the other hand, the reactivity ratio of EO is higher for all other comonomer pairs, resulting in a (soft) monomer gradient in the copolymer chain. The difference in cis- and trans-stereoconfiguration of NerGE and GeraGE, respectively, appear to be a negligible factor during the copolymerization. In contrast, an increase of side chain hydrophobicity by full hydrogenation appears to be the most influential parameter for the copolymer microstructure, affording a stronger gradient. Altogether, the observed trends are in good agreement with previously reported results, as GEs generally copolymerize in an ideally random manner with EO.31,32 Due to the additional oxygen in the GE side chain, Lynd and coworkers postulated a “crown ether-effect” at the active polymer chain end, enhancing the GE reactivity.32 Following our findings, we hypothesize that the steric demand of the branched terpene structures impedes counterion complexation and, in turn, leads to an inversion of the comonomer reactivity.10 Furthermore, a slight deviation in reactivity ratios could also be due to solvent and counterion effects.31,47
:1) at 40 °C, evaluated by the non-terminal model and the ideal Jaacks fit
| Monomer | Pendant group | r EO | r TGE | R 2 |
|---|---|---|---|---|
| a Errors for reactivity ratios have been rounded up to the last significant digit. b Reactivity ratios of the FarGE/EO comonomer pair have been previously published for identical polymerization conditions.10 | ||||
| Isoprenyl glycidyl ether | C5H9O | 1.03 ± 0.01 | 0.97 ± 0.01 | 0.99 |
| Prenyl glycidyl ether | C5H9O | 1.06 ± 0.01 | 0.94 ± 0.01 | 0.99 |
| Geranyl glycidyl ether | C10H17O | 1.08 ± 0.01 | 0.93 ± 0.01 | 0.99 |
| Neryl glycidyl ether | C10H17O | 1.07 ± 0.01 | 0.93 ± 0.01 | 0.99 |
| Farnesyl glycidyl etherb | C15H25O | 1.18 ± 0.01 | 0.85 ± 0.01 | 0.99 |
| Dihydroprenyl glycidyl ether | C5H11O | 1.18 ± 0.01 | 0.85 ± 0.01 | 0.99 |
| Tetrahydrogeranyl glycidyl ether | C10H21O | 1.21 ± 0.01 | 0.83 ± 0.01 | 0.99 |
| Hexahydrofarnesyl glycidyl ether | C15H31O | 1.25 ± 0.01 | 0.80 ± 0.01 | 0.99 |
In our kinetics study, IsoPreGE and hexahydrofarnesyl glycidyl ether (HHFarGE) show the strongest discrepancy in their copolymerization behavior. To visually demonstrate the difference between the ideally random and gradient structures, we simulated the molar-based composition diagram of a statistical copolymer for EO with IsoPreGE and HHFarGE at an equimolar monomer ratio, respectively. Fig. 2 demonstrates the effect of the reactivity ratio difference on the polymer microstructure. While the isoprenyl side chains are randomly distributed along the polyether chain, the copolymerization with HHFarGE produces a noticeable, but weak gradient copolyether, which determines the nature of the terminal units. HHFarGE units are slightly less incorporated at the onset of the growing polymer chain, whereas the gradient shows a noticeable increase towards the chain terminus. Based on a previous kinetic study by Heatley et al. and for comparison purposes we additionally depict the molar composition diagram for the highly relevant EO/PO comonomer pair (Fig. 2, bottom).30 While statistical P(EGn-co-POm) copolymers feature a significantly stronger gradient composition profile, a nearly random microstructure can be realized by simply substituting PO by a TGE monomer.
 | ||
| Fig. 2 Molar-based composition diagrams of a P(EGn-co-IsoPreGEm) (top, left); a P(EGn-co-HHFarGEm) (top, right) and a P(EGn-co-POm) copolymer (bottom), respectively, modelled at an equimolar monomer ratio, with FX = molar fraction of TGE or PO. Calculations for TGEs are based on the reactivity ratios derived from a linear Jaacks fit46 (top, right), whereas reactivity ratios for the EO/PO comonomer pair are derived from literature (rEO = 2.80; rPO = 0.25).30 | ||
Having revealed the subtle reactivity differences of the available TGEs, we directed our attention to unraveling the unexpected appearance of novel signals during the in situ kinetics of the unsaturated C5-TGEs. Fig. 3 depicts a zoom-in of the time-resolved overlay of 1H NMR spectra between 4.6 and 6.6 ppm, documenting the unexpected appearance of new signals during the copolymerization. Chain transfer to the monomer is a well-known drawback of the alkali metal mediated AROP of substituted epoxides, leading to the formation of new allylic species with signals in a similar down-field region, as evidenced by 1H NMR spectroscopy.48 More importantly, chain transfer would lead to an inaccurate PreGE consumption in our study, falsifying the determined reactivity ratios. Owing to the similarity of the signals’ shifts, we sought to obtain a deeper understanding of our observations regarding (i) signal assignment and (ii) validity of the determined reactivity ratios. Interestingly, our findings indicate the occurrence of base-catalyzed γ-elimination of the prenyl protective group (Scheme S2†) and formation of isoprene during the copolymerization (≤0.5% of overall monomer, Fig. S34†) rather than chain transfer.49–51 A cryotransfer of all volatile compounds following our kinetic study corroborated our hypothesis, as we identified isoprene via GC and NMR analysis (Fig. S35–S37†). Concurrently, a hydroxyl functionality is released, which can act as an additional initiating species due to the degenerative proton transfer in the AROP.1 Yet, as the elimination occurs in trace amounts of less than 1%, a hyperbranched structure is not expected. The aforementioned observations and conclusions are conferrable to the copolymerization of EO and IsoPreGE as a structural isomer of PreGE (Fig. S41†).
 | ||
| Fig. 3 Zoom-in region of stacked 1H NMR spectra (400 MHz, THF-d8:DMSO-d6 = 5:1) of the copolymerization kinetic investigation of PreGE and EO, validating the elimination of isoprene in small quantities. The doublet forming between 4.71 and 4.77 ppm shows the concurrent prenyl-to-isoprenyl isomerization under basic AROP conditions. | ||
We observed that decreasing the base concentration in the NMR tube leads to a decrease in the overall isoprene formation by base-catalyzed elimination. This is in line with the results of the significantly more dilute copolymerization conditions in a flask, for which no isoprene formation has been observed. Additionally, we used dihydroprenyl glycidyl ether (DHPreGE) as a saturated model compound in an in situ kinetic measurement (Fig. S50–S52†), for which, as anticipated, no new signals are witnessed (Fig. S50†). Intriguingly, the elimination reaction was only observed for the unsaturated C5-TGEs, which is presumably favored by a six-membered transition state (Scheme S2†). Conclusively, the chosen copolymerization conditions suppress chain transfer reactions whilst trace elimination does not impact the accurate determination of the reactivity ratios. An additional finding is the partial base-catalyzed prenyl-to-isoprenyl isomerization of the side chain under basic conditions (and vice versa for IsoPreGE), as evidenced in Fig. 3 and Fig. S38.†52 In contrast, the copolymerization EO with neryl and geranyl glycidyl ether (NerGE, GeraGE), respectively, shows little to no isomerization. The different degree of isomerization is further supported by 13C NMR characterization of P(EGn-co-PreGEm) and P(EGn-co-IsoPreGEm) copolymers, which show more pronounced carbon shifts of the isomerized side chain (compare Fig. S20 and S22 vs. Fig. S24 and S26†). Similar in situ isomerization has already been reported for the AROP of EO with AGE, which undergoes a transformation from allyl- to cis-prop-1-enyl ether.39,40 Due to the degree of substitution, PreGE isomerization is sterically promoted towards the isoprenyl structure. Overall, the different copolymerization behavior renders TGE highly promising building blocks to fine-tune the microstructure of mf-PEGs under the established AROP conditions.
Thermal properties of statistical P(EG-co-TGE) copolymers
Differential scanning calorimetry (DSC) of the copolymers not only reveals the thermal properties (Fig. S79–S82†), but also provides indirect support for the copolymer microstructure. PEG is a semicrystalline polyether with a melting temperature (Tm) of 65 °C. Generally, all synthesized copolymers exhibit a Tm that depends on the comonomer content. Accordingly, a gradual TGE increase results in an incremental decrease of Tm and the respective melting enthalpy. In contrast to block-like structures, statistical copolymers with side chain functionalization show significant reduction of the PEG crystallization.5,10 The branched terpenyl side chains lead to regularly distributed steric disturbances along the PEG chain, prohibiting packing in highly ordered crystalline domains. For instance, incorporation of approximately 3 mol% TGE already reduces the Tm by ∼25 °C. Melting points in the range of 30 °C to 40 °C, i.e., in a physiological temperature range are promising for a variety of pharmaceutical applications. Furthermore, branching does also not allow for side chain crystallization for the longer terpenyl side chains (C10), as indicated by the absence of a second Tm.8 As expected for statistical copolymers with a random or soft gradient microstructure, only a single glass transition temperature Tg in the range of −59 °C to −67 °C is observed.53 The prenyl side chain leads to a slightly higher Tg than isoprenyl, which is tentatively ascribed to decreased chain flexibility. The results show that the difference in sidechain length or side chain double bond configuration of the incorporated C5 and C10 TGE chain does not represent a key parameter in tailoring the thermal properties. Overall, the thermal properties of the copolymers observed for a low content of the TGE comonomers are a consequence of the almost ideally random microstructure.Post-polymerization modification
The unsaturated side chains of the P(EG-co-TGE) copolymers are characterized by chemically different double bonds. Therefore, we investigated the different copolymer structures, focusing on their post-polymerization potential capitalizing on thiol–ene click and hydrogenation.Thiol–ene click reaction
Thiol–ene click photo reactions were carried out overnight in DCM using 2,2-dimethoxy-2-phenylacetophenone (DMPA) as a radical photoinitiator and 2-mercaptoethanol as a functional thiol to demonstrate the general applicability. As trisubstituted, internal double bonds represent a particular challenge with respect to modification, P(EG120-co-GeraGE12) was used as the model copolymer (Scheme 4). The ratios of double bonds/DMPA/thiol were varied to obtain fully functionalized copolymers with monomodal molar mass distributions. After the photoreaction, residual 2-mercaptoethanol was removed via aqueous extraction, followed by further purification via dialysis. | ||
| Scheme 4 Thiol–ene modification of P(EG120-co-GeraGE12) employing 2-mercaptoethanol as a functional thiol. The copolymers in entries 4, 8, and 16 were modified in the same manner. For experimental details see Table S1.† | ||
Generally, the reactivity of double bonds towards hydrothiolation decreases from terminal towards internal and subsequently multiple substituted double bonds.54–56 As the formation of the carbon–sulfur bond is reversible and depends on the olefin and thiol structures, it is anticipated that the reverse reaction will be more pronounced for olefins with trisubstituted internal double bonds than less substituted olefins. Terpene-based small molecules with terminal double bonds can be modified with small amounts of excess equivalents of thiol.25 However, the respective modification conditions commonly used for small molecules induce cross-linking of double bond-containing polymers, leading to a multimodal molar mass distribution. This is evidenced in the high Mn of the polymer as determined by SEC (Fig. 4, gray curve). A strategy to overcome crosslinking is the use of large excess thiol equivalents (Fig. 4, compare red, cyan and purple curve) while keeping the required amount of the photoinitiator DMPA at a minimum (Fig. 4, red and green curve). When 200 equivalents of thiol per double bond are applied, a monomodal molar mass distribution is achieved. Comparing the functionalized copolymers to the starting P(EG120-co-GeraGE12) copolymer, a shift of the SEC elugram toward higher molar masses indicates the successful functionalization (dotted blue and purple curve).
 | ||
| Fig. 4 SEC traces of P(EG120-co-GeraGE12) modified with different equivalents of the thiol 2-mercaptoethanol and DMPA with respect to one double bond (Eluent DMF, RI detector, PEG calibration). Purple line represents the SEC curve of the successful modification without crosslinking. | ||
Furthermore, 1H NMR analysis revealed complete conversion of both double bonds in all experiments (Fig. S63–66†). The incisive protons at 2.75 and 2.36 ppm refer to the methine protons from the anti-Markovnikov product of the two double bonds (Fig. S65†). The infrared spectrum illustrates a broad band at approximately 3400 cm−1 due to the added hydroxyl functionalities. The absence of the R–S–H band at 2500 cm−1 indicates complete removal of 2-mercaptoethanol by dialysis (Fig. S69†). MALDI-ToF analysis provides further proof for complete modification, as the peak intervals solely correspond to the EO unit and the double functionalized GeraGE unit with 366 g mol−1 (Fig. S73†). It is noteworthy that unreacted 2-mercaptoethanol was recovered by fractional distillation in yields of 80% (see ESI for details†), reducing the overall thiol equivalents required for functionalization. Equally, we modified P(EG102-co-PreGE10), requiring 600 eq. of thiol per double bond to suppress cross-linking (Fig. S59†). Surprisingly, SEC characterization still indicated a shoulder towards higher molar masses, even when 300 equivalents of thiol were used. P(EG123-co-NerGE13) required similar reaction conditions, despite its similarity to GeraGE. A different reactivity towards hydrothiolation of those species was not expected, however, adjusting the modification conditions enabled us to obtain monomodal distributions for both copolymer types (Fig. S62†).
Hydrogenation via diimide reduction
Typically, the production of diimide involves the thermal decomposition of para-toluenesulfonyl hydrazide (pTSH). Despite its effectiveness, the use of this reagent is accompanied by several limitations. These include the requirement for reflux in solvents with a high boiling point such as ortho-xylene, inefficient byproduct removal, and the occurrence of undesirable reactions towards polyisoprene, such as chain degradation, cyclization, and nucleophilic attack of para-tolylsulfinate anion.57,58 Chain degradation can be reduced by the addition of a tertiary amine and, conclusively, the pTSH route represents a reasonable alternative to catalyzed hydrogenation. While the diimide reduction using pTSH is a well-known hydrogenation method, the use of potassium azodicarboxylate (PADA) as an in situ hydrogenation agent has been scarcely investigated to date.59 Thus, we investigated the post-polymerization hydrogenation of terpenoid structures employing PADA with respect to (i) hydrogenation temperature, (ii) applied PADA equivalents and (iii) the solvent volume required. Detailed experimental procedures and further results (Table S2†) are provided in the ESI.† PADA was utilized to generate the diimide, which upon decomposition produced potassium acetate as a byproduct. The general procedure involved dissolving the dry polymer in dry pyridine, followed by the addition of PADA. A slurry was created, into which dry glacial acetic acid was added dropwise to induce decomposition. It is important to note that the decomposition of PADA is strongly influenced by the concentration of acid (Scheme 5).60 | ||
| Scheme 5 Hydrogenation of P(EG120-co-GeraGE12) employing potassium azodicarboxylate (PADA). Intermediate steps towards diimide are omitted but can be found in the introduction. | ||
The acyclic TGE side chains incorporate two types of double bonds: an in-chain double bond in allyl-position to the oxygen in either cis- or trans-configuration as well as a double bond of varying degree of substitution. In this study, we deliberately selected GeraGE carrying both types of double bonds to highlight the proof-of-principle and robustness of the approach.
First experiments showed that decreasing the temperature from RT to 0 °C only affects the degree of hydrogenation in concentrated solutions, yet the efficiency was reduced to a maximum of 53% (Table S2, entries 1–4†). Next, we increased the volume of the solvent to 3 ml, which increased the degree of hydrogenation from 25% to 53% according to 1H NMR characterization (Table S2, entries 4 and 8†). As the slower decomposition of PADA directly correlates with the slower dosage and reduced concentration of acetic acid in the reaction mixture, improved hydrogenation and reduction of side reactions are observed. A similar effect was observed upon increasing the PADA equivalents from 2.5 to a maximum of 15 per double bond (Table S2, entries 4 and 8†). Fig. 5 illustrates the double bond resonances before and after hydrogenation, respectively, showing a decrease in double bond signals and an increase in the region of aliphatic resonances (full analysis in Fig. S75–S77 of the ESI†).
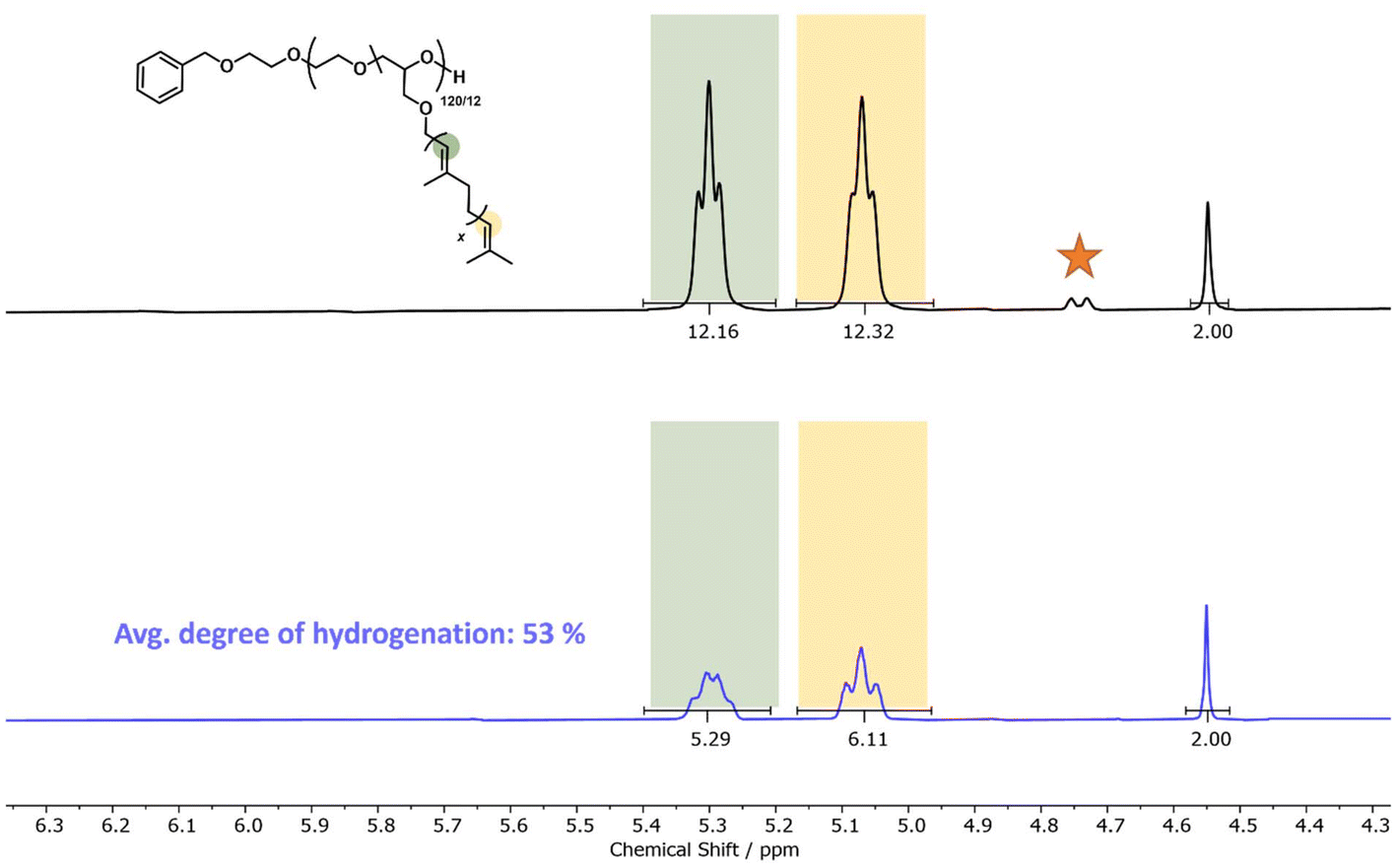 | ||
| Fig. 5 Stacked 1H NMR spectra of P(EG120-co-GeraGE12) before (top) and after (bottom) hydrogenation employing PADA. The double bond with a deficiency of electrons (colored green, resonating at 5.30 ppm) undergoes hydrogenation with a slight preference. The orange star denotes the terminal double bond of the isomerized product and is not considered in the calculations. | ||
This described method of producing diimide using PADA is suitable for post-polymerization hydrogenation, even with partial hydrogenation. We would like to emphasize that the structures presented are difficult to hydrogenate, both due to the trisubstituted double bonds and the attachment to a polymer chain. The reactivity of a double bond is lowered significantly by alkyl substitution.61 For instance, the relative hydrogenation rate constant of 2-methyl-1-pentene, which represents a terminal disubstituted substrate, is 7.3 times higher than that of 2-methyl-2-butene, which is comparable to a terpene unit.37,62 Additionally, upon an increase of acetic acid equivalents (>2 equivalents), we observed a broadening of the molar mass distribution via SEC (Fig. S78†). Similar observations have been reported for the hydrogenation of polyisoprene-based polymers using para-toluenesulfonyl hydrazide, caused by the formation of para-toluene-sulfonic acid as a byproduct.57 Ultimately, the diimide reduction of P(EG-co-TGE) copolymers requires further optimization, but PADA already shows good hydrogenation efficiencies of less substituted double bonds in polymers in model experiments. Nevertheless, it is a particularly interesting method for the hydrogenation of amphiphilic structures, as facile purification is enabled due to the volatile coupling product, i.e., carbon dioxide.
Conclusion
TGEs represent a bio-derived class of epoxide monomers, which have been hardly employed to date. The easy-to-synthesize nature and structural variety of terpenes lead to a versatile monomer platform for polyethers, polycarbonates, and polyester synthesis. We have demonstrated the successful copolymerization of EO with TGEs (PreGE, IsoPreGE, GeraGE, NerGE), introducing branched terpenyl side chains along the polyether backbone. Four systematic series of well-defined statistical copolymers with a TGE content up to 10 mol% have been prepared, maintaining aqueous solubility and crystallizability of PEG, while introducing backbone functionality. A molar mass range between 4800 and 8300 g mol−1 was explored. Generally, the molar mass distribution showed low dispersities Đ in the range of 1.05–1.13. As expected, the random distribution of the branched terpenyl side groups gradually reduced PEG crystallization with increasing TGE content. Thus, thermal properties can be tailored by varying the comonomer ratio. Melting points in the range of 14 to 40 °C were obtained. Only marginal differences in glass transition temperatures were observed, despite the differing side chain length.In situ 1H NMR kinetic investigations revealed a TGE reactivity trend and unraveled the copolymer microstructure. TGE reactivity only slightly decreases with increasing side chain length, flexibility, and hydrophobicity. Ideally random behavior was found for the EO/IsoPreGE comonomer pair, whereas HHFarGE features the strongest, but nevertheless mild gradient in the copolyether series.
The unsaturated double bonds of the terpenyl side chains allow for post-polymerization modification.33 Both modification reactions, thiol–ene click and hydrogenation, addressed the unsaturated double bonds (di- and trisubstituted) of the terpenyl side chains. A representative thiol–ene click reaction was performed using 2-mercaptoethanol on all four types of synthesized copolyethers, comprised of ∼10 mol% of TGE units. We achieved full functionalization for all copolymers, however, the thiol equivalents had to be varied between 200 and 600, depending on the respective TGE side chain to prevent crosslinking. Recycling of the thiol equivalents was possible by up to 80%. Furthermore, the diimide reduction using PADA for side chain hydrogenation has been studied for the first time. Despite the difficulty of the hydrogenation of trisubstituted double bonds, we were able to achieve a hydrogenation of 53%, while the gaseous release of CO2 as the coupling product did not require further purification.
In contrast to commonly reported para-toluenesulfonyl hydrazide for hydrogenation purposes, PADA may have been overlooked as a hydrogenation agent in previous research. However, if side chain double bonds are not desired, the utilization of fully hydrogenated TGEs may prove advantageous, given the low hydrogenation efficiency. While the subtle differences in the reactivity ratios of hydrogenated TGEs compared to their non-hydrogenated counterparts result in a slight gradient microstructure, the resulting copolyethers are likely to exhibit enhanced heat and oxidation resistance while minimizing crosslinking side reactions.
Altogether, we consider the extended TGE library as a versatile platform, allowing for the controlled synthesis of P(EGn-co-TGEm) copolymers as promising PEG derivatives of a partially bio-based nature. The structural variety and potential for postfunctionalization is promising for a diverse scope of applications, ranging from surfactants to polymeric drug carriers and microgels.1,5,63,64
Author contributions
S. S. and G. M. L. contributed equally to the experimental part and writing process. Both contributed to the data evaluation, visualization, and analysis. E. C. H. contributed to the experimental part. S. S. and P. H. collaborated to synthesize terpenoid-derived monomers. H. F. supervised, reviewed, and edited the final work.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the NMR facility at Johannes Gutenberg University for extended measurement periods.References
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef PubMed.
- S. Fusco, A. Borzacchiello and P. A. Netti, J. Bioact. Compat. Polym., 2006, 21, 149–164 CrossRef.
- K. P. Barteau, M. Wolffs, N. A. Lynd, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2013, 46, 8988–8994 CrossRef.
- B. Obermeier, F. Wurm, C. Mangold and H. Frey, Angew. Chem., Int. Ed., 2011, 50, 7988–7997 CrossRef PubMed.
- G. Mouzin, H. Cousse, J.-P. Rieu and A. Duflos, Synthesis, 1983, 117–119 CrossRef.
- P. Verkoyen and H. Frey, Macromol. Rapid Commun., 2020, 41, e2000225 CrossRef PubMed.
- P. Verkoyen, T. Johann, J. Blankenburg, C. Czysch and H. Frey, Polym. Chem., 2018, 9, 5327–5338 RSC.
- F. Della Monica and A. W. Kleij, Polym. Chem., 2020, 11, 5109–5127 RSC.
- S. Schüttner, M. Krappel, M. Koziol, L. Marquart, I. Schneider, T. Sottmann and H. Frey, Macromolecules, 2023, 56, 6928–6940 CrossRef.
- Y. Li, A. Ozden, W. R. Leow, P. Ou, J. E. Huang, Y. Wang, K. Bertens, Y. Xu, Y. Liu, C. Roy, H. Jiang, D. Sinton, C. Li and E. H. Sargent, Nat. Catal., 2022, 5, 185–192 CrossRef.
- T. Pu, H. Tian, M. E. Ford, S. Rangarajan and I. E. Wachs, ACS Catal., 2019, 9, 10727–10750 CrossRef.
- G. Shukla and R. C. Ferrier, J. Polym. Sci., 2021, 14, 243 Search PubMed.
- B. M. Bell, J. R. Briggs, R. M. Campbell, S. M. Chambers, P. D. Gaarenstroom, J. G. Hippler, B. D. Hook, K. Kearns, J. M. Kenney, W. J. Kruper, D. J. Schreck, C. N. Theriault and C. P. Wolfe, Clean:Soil, Air, Water, 2008, 36, 657–661 Search PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef PubMed.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef PubMed.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- M. D. Konieczynska, X. Lin, H. Zhang and M. W. Grinstaff, ACS Macro Lett., 2015, 4, 533–537 CrossRef PubMed.
- C. Wahlen and H. Frey, Macromolecules, 2021, 54, 7323–7336 CrossRef.
- A. W. Kleij, ChemSusChem, 2018, 11, 2842–2844 CrossRef.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- L. Ruzicka, Experientia, 1953, 9, 357–367 CrossRef CAS PubMed.
- S. G. Hillier and R. Lathe, J. Endocrinol., 2019, 242, R9–R22 CAS.
- E. Breitmeier, Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, Wiley-VCH, Weinheim, 2006 Search PubMed.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- S. D. Morrison, R. M. J. Liskamp and J. Prunet, Org. Lett., 2018, 20, 2253–2256 CrossRef CAS PubMed.
- T. Johann, H. A. Houck, T. Dinh, U. Kemmer-Jonas, F. E. Du Prez and H. Frey, Polym. Chem., 2019, 10, 4699–4708 RSC.
- S. Schüttner, C. Gardiner, F. S. Petrov, N. Fotaras, J. Preis, G. Floudas and H. Frey, Macromolecules, 2023, 56, 8247–8259 CrossRef.
- P. Holzmüller, C. Gardiner, J. Preis and H. Frey, Macromolecules, 2024, 57, 5358–5367 CrossRef.
- F. Heatley, G. Yu, C. Booth and T. G. Blease, Eur. Polym. J., 1991, 27, 573–579 CrossRef.
- J. Herzberger, D. Leibig, J. C. Liermann and H. Frey, ACS Macro Lett., 2016, 5, 1206–1211 CrossRef PubMed.
- B. F. Lee, M. Wolffs, K. T. Delaney, J. K. Sprafke, F. A. Leibfarth, C. J. Hawker and N. A. Lynd, Macromolecules, 2012, 45, 3722–3731 CrossRef PubMed.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef PubMed.
- R. K. Iha, K. L. Wooley, A. M. Nyström, D. J. Burke, M. J. Kade and C. J. Hawker, Chem. Rev., 2009, 109, 5620–5686 CrossRef PubMed.
- A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC.
- N. T. McManus and G. L. Rempel, J. Macromol. Sci., Polym. Rev., 1995, 35, 239–285 CrossRef.
- Encyclopedia of reagents for organic synthesis, ed. A. Charette, J. Bode, T. Rovis and R. Shenvi, Wiley, Chichester, 1995 Search PubMed.
- D. J. Pasto and R. T. Taylor, in Organic reactions, Wiley Online Library, Hoboken, N.J., 2004, pp. 91–155 Search PubMed.
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4498–4504 CrossRef PubMed.
- B. Obermeier and H. Frey, Bioconjugate Chem., 2011, 22, 436–444 CrossRef.
- M. Worm, D. Leibig, C. Dingels and H. Frey, ACS Macro Lett., 2016, 5, 1357–1363 CrossRef PubMed.
- A. Natalello, M. Werre, A. Alkan and H. Frey, Macromolecules, 2013, 46, 8467–8471 CrossRef.
- J. Blankenburg, E. Kersten, K. Maciol, M. Wagner, S. Zarbakhsh and H. Frey, Polym. Chem., 2019, 10, 2863–2871 RSC.
- F. T. Wall, J. Am. Chem. Soc., 1941, 63, 1862–1866 CrossRef.
- B. S. Beckingham, G. E. Sanoja and N. A. Lynd, Macromolecules, 2015, 48, 6922–6930 CrossRef.
- V. Jaacks, Makromol. Chem., 1972, 161, 161–172 CrossRef.
- P. Dreier, R. Matthes, R. D. Barent, S. Schüttner, A. H. E. Müller and H. Frey, Macromol. Chem. Phys., 2023, 224, 2200209 CrossRef.
- M. Hans, H. Keul and M. Moeller, Polymer, 2009, 50, 1103–1108 CrossRef.
- F. Guibé, Tetrahedron, 1997, 53, 13509–13556 CrossRef.
- J.-M. Vatèle, Tetrahedron, 2002, 58, 5689–5698 CrossRef.
- P. G. M. Wuts, Greene's protective groups in organic synthesis, Wiley-Interscience, Hoboken, N.J., 4th edn, 2007 Search PubMed.
- T. J. Prosser, J. Am. Chem. Soc., 1961, 83, 1701–1704 CrossRef.
- T. G. Fox, Bull. Am. Phys. Soc., 1956, 1, 123–125 Search PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- A. Matic and H. Schlaad, Polym. Int., 2018, 67, 500–505 CrossRef.
- S. F. Hahn, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 397–408 CrossRef.
- R. Ballini, E. Marcantoni and M. Petrini, Tetrahedron, 1989, 45, 6791–6798 CrossRef.
- C. Schunicht, A. Biffis and G. Wulff, Tetrahedron, 2000, 56, 1693–1699 CrossRef.
- J. T. Groves and K. W. Ma, J. Am. Chem. Soc., 1977, 99, 4076–4082 CrossRef.
- R. A. Back, Res. Chem. Intermed., 1984, 5, 293–323 CrossRef.
- E. W. Garbisch, S. M. Schildcrout, D. B. Patterson and C. M. Sprecher, J. Am. Chem. Soc., 1965, 87, 2932–2944 Search PubMed.
- Y. Liu, W. Hou, H. Sun, C. Cui, L. Zhang, Y. Jiang, Y. Wu, Y. Wang, J. Li, B. S. Sumerlin, Q. Liu and W. Tan, Chem. Sci., 2017, 8, 6182–6187 RSC.
- A. K. Fraser, C. S. Ki and C.-C. Lin, Macromol. Chem. Phys., 2014, 215, 507–515 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI): Detailed information on reagents, instrumentation, experimental and synthetic procedures; NMR characterization of all TGE monomers, additional homo- and copolymer characterization data (NMR, SEC, DSC, MALDI-ToF); evaluation data for in situ1H NMR copolymerization kinetic measurements of TGEs with EO. See DOI: https://doi.org/10.1039/d4py01201a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |