
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking the cellular delivery bottleneck: recent developments in direct cytosolic delivery of biologics
Harini
Nagaraj
a,
Victor
Lehot
a,
Nourina
Nasim
a,
Yagiz Anil
Cicek
a,
Ritabrita
Goswami
a,
Taewon
Jeon
ab and
Vincent M.
Rotello
 *a
*a
aDepartment of Chemistry, University of Massachusetts Amherst, 710 North Pleasant Street, Amherst, Massachusetts 01003, USA. E-mail: rotello@chem.umass.edu
bDepartment of Biochemistry and Molecular Biology, University of Massachustts Amherst, 710 North Pleasant Street, Amherst, MA 01003, USA
First published on 2nd July 2025
Abstract
Proteins and nucleic acid therapeutics represent a significant and growing share of the pharmaceutical landscape. The majority of biological and therapeutic applications of these biomolecules require access to the cytosol. Delivery of biologics directly to the cytosol is made difficult by the impermeability of the cell membrane. As a result, most delivery strategies have utilized endocytic uptake pathways to deliver biologics into the cell. However, endosomally entrapped cargo often faces limited escape efficiency and is prone to degradation within endo/lysosomal compartments. The emergence of delivery vehicles capable of bypassing endocytosis and directly traversing the cell membrane offers a promising approach to improve the cytosolic delivery efficiency of biomolecules. Here, we highlight recent developments in endocytosis-independent delivery systems for biologics and ways to accurately assess cytosolic delivery of biologics. Strategies employing covalent and non-covalent modification of biomolecules will be reviewed, along with strategies incorporating both covalent and supramolecular processes.
1. Introduction
Proteins and nucleic acid-based therapeutics are promising strategies for treatment due to their specificity and precision in treating ‘undruggable’ disease targets.1–3 Nucleic acid-based therapeutics are a broad class and include messenger RNA (mRNA) and plasmid DNA (pDNA) that promote the expression of proteins in the cell, as well as RNA interference (RNAi) technologies, such as micro-RNA (miRNA) and small interfering RNA (siRNA) that decrease (knockdown) protein expression.4 Nucleic acids are highly charged species, making them impermeable to the cell membrane.5 Due to their susceptibility to nuclease degradation, nucleic acids require encapsulation within nanocarriers for protection and effective delivery into cells and then into the cytosol to perform their function.6Proteins present a second major family of biomacromolecular therapeutics.7 Native proteins play dynamic and diverse roles in all aspects of cell function, making them promising therapeutics.8,9 Current protein therapeutics on the market are encompassed by monoclonal antibodies,10 enzyme replacement therapies,11 interferons,12 hormones, and fusion proteins.13,14 Protein therapeutics provide numerous advantages over traditional small-molecule drugs, including high target specificity, fewer side effects, and enhanced effectiveness in treating various genetic mutations.15 Despite revolutionizing the medical field, protein therapeutics still face challenges pertaining to therapeutic efficacy.16 These challenges arise from the inability of proteins to cross the cell membrane effectively to access the intracellular locations required for their activity.5
Several approaches have been utilized to transport biomacromolecules into cells and cytosol, including mechanical techniques such as microinjection and electroporation.17 While mechanical methods provide direct access to the cytosol, they require specialized instruments and cannot be readily translated to 3D tissues and organs.18–20 Chemical strategies for delivery have also been developed, including cell-penetrating peptides (CPPs)21 and ligands.22–25 However, at low concentrations, CPPs are often internalized by the cells via endocytic pathways with limited escape efficiency, resulting in degradation in the endo/lysosomal compartments.26,27 This limitation can be circumvented by increasing CPP concentration, but higher dosages often result in cytotoxicity and unwanted side effects in a systemic environment.27–29 While CPPs can independently deliver biologics, they are increasingly employed to modify nanocarriers, to enhance intracellular uptake efficiency and are often attached to the surface via electrostatic interactions or via covalent coupling strategies.30 For nanocarriers that utilize this strategy, endosomal escape efficiency is observed to be predominantly dependent on the surface charge, which should be a critical consideration for future nanocarrier design.31–33
Nanocarriers, such as inorganic nanoparticles,34,35 lipids,36,37 and polymers38,39 have been utilized widely for intracellular delivery of proteins and nucleic acids due to their broad design space.40 Endocytosis is the preferred mode of uptake for most nanocarrier-based delivery systems.41,42 Most nanocarriers encapsulating protein and nucleic acid-based therapeutics enter the cells through endocytic mechanisms such as clathrin- and caveolae-mediated endocytosis, macropinocytosis and phagocytosis.43–46 The cargo then often becomes entrapped within endosomes, and is eventually degraded in the endolysosomal compartments, without gaining access to the cytosol, thereby limiting the efficacy of the delivery system for intracellular targets.47,48
Cytosolic access of biologics is crucial to achieving optimal therapeutic efficacy for most biologics, either for direct activity in the cytosol, or for reaching the nucleus, and other intracellular organelles.49,50 Substantial progress has been made to address endosomal entrapment by designing strategies to escape these compartments, however the reported endosomal escape efficiency using these strategies is low, generally <10%.51–53
Direct delivery of biologics to the cytosol circumvents the challenges associated with endosomal escape, thereby increasing the therapeutic potential.54,55 In this review, we will focus on recent advancements in the development of non-endocytic, direct cytosolic delivery systems for proteins and nucleic acids. Methods for assessing direct cytosolic delivery will be discussed, and key modifications and encapsulation strategies that enhance direct cytosolic delivery of biologics will be highlighted.
2. Assessment of cytosolic localization and direct delivery mechanism
Accurate evaluation of subcellular cargo localization and internalization mechanisms are both crucial for developing effective delivery systems. This section will highlight common challenges in validating cytosolic access and direct cytosolic delivery mechanisms while discussing effective strategies to evaluate the delivery processes.Unambiguous assessment of the subcellular localization of the biomolecule cargo can be challenging and subject to confirmation bias.56 Perhaps the most common pitfall is the distinction between diffuse and punctate fluorescent signals, qualitative indicators of cytosolic access, and endosomal entrapment (Fig. 1).57 Inaccurate evaluation or experimental conditions, such as excessive particle concentrations, oversaturation, out-of-frame fluorescence, or improper qualitative analysis, can lead to punctate signals being mistakenly identified as diffuse.58–60 The use of dye-labeled biomolecules can introduce another source of uncertainty: dyes can be cleaved chemically and enzymatically.61,62 The optical properties of dyes can vary with pH, a classic example being fluorescein, which is non-fluorescent at endosomal pH. Additionally, dye labeling can significantly alter the physico-chemical properties and intracellular trafficking of the labeled cargo, potentially leading to incorrect conclusions about the delivery mechanism.63,64 Cell fixation can lead to intracellular redistribution of monitored cargo, so it should be avoided or used cautiously when studying the cytosolic localization of biomolecules.65,66
 | ||
| Fig. 1 Delivery of biologics using nanocarriers through two different pathways: endocytosis (left) or direct cytosolic entry (right). Endosomal entrapment presents initially as punctate fluorescence, while diffuse fluorescence indicates cytosolic distribution. | ||
Cytosolic access can be validated through the activity of functional proteins or oligonucleotides within the cytosol or through extensive cytometry experiments.67 Small intrinsically fluorescent proteins (<60 kDa), such as GFP, or nucleic acids diffuse passively from the cytosol to the nucleus through the nuclear pores in the membrane.54,68 Fluorescence in the nucleus thus provides the most straightforward and unambiguous observation to validate the cytosolic access of delivered protein cargo.
However, while cytosolic localization can result from direct delivery or endosomal escape, experimental evidence alone is insufficient to determine the specific delivery mechanism. Caution should be taken when working with inhibitors of endocytic pathways, as most of these inhibitors were shown to be non-specific.69 In particular, the use of cholesterol-depleting agents such as methyl-β-cyclodextrin inhibits clathrin-independent processes such as caveolae-mediated endocytosis as well as lipid raft formation, or cholesterol-enriched microdomains that can facilitate direct cytosolic delivery.70 Performing delivery experiments at 4 °C is a commonly used strategy to inhibit energy-dependent pathways such as endocytosis, but it also reduces membrane fluidity, interfering with non-endocytic pathways.71
Organelle-specific markers are other common tools used to study the intracellular trafficking of delivered cargo.72 A common challenge in interpreting these studies is treating the absence of co-localization between the cargo and endo–lysosomal markers (e.g., Lysotracker series) as evidence that the cargo was delivered via a non-endocytic pathway. These pH-responsive fluorescent markers can be quenched as the endo/lysosomal pH changes and so, the absence of colocalization with the delivered cargo does not conclusively prove direct cytosolic delivery.73–75
The most reliable method for establishing delivery of biomolecules to cells is live-cell video microscopy.76 This technique allows real-time visualization of the delivery process, providing clear insights into the dynamics of cargo internalization and subcellular localization.77 Unlike static imaging methods, live-cell microscopy eliminates the potential ambiguity caused by oversaturation or artifacts, ensuring a more accurate assessment of the delivery mechanism.
The articles discussed in this review have provided experimental results supporting cytosolic localization (usually, diffuse fluorescence signal) and a direct cytosolic delivery mechanism.
3. Covalent modification of biologics for cytosolic delivery
3.1 Cell-penetrating peptide conjugates
Cell-penetrating peptides (CPPs) have significant potential for delivering a wide variety of biomolecules, including therapeutic proteins and nucleic acids (Fig. 2a). CPPs are composed of 5–30 amino acids and are typically positively charged at physiological pH due to the presence of arginine/lysine residues.78 The uptake mechanism of CPPs have been extensively studied over the years, and several hypotheses have been proposed. However, no singular uptake pathway applies to all CPPs, as the internalization route is influenced by several factors, including structure and concentration of CPP, interaction of CPP with the cell membrane, and nanoparticles carried along by the CPPs.79 Direct membrane penetration and endosomal uptake via one of the endocytic pathways have been suggested as the two main uptake mechanisms.80 Direct membrane translocation has been observed at high concentrations of CPP when they are attached to relatively smaller cargos such as fluorophores.27 For larger molecules such as proteins, endocytosis is the most common route of uptake, necessitating endosomal escape for these proteins to reach the cytosol and other subcellular organelles.81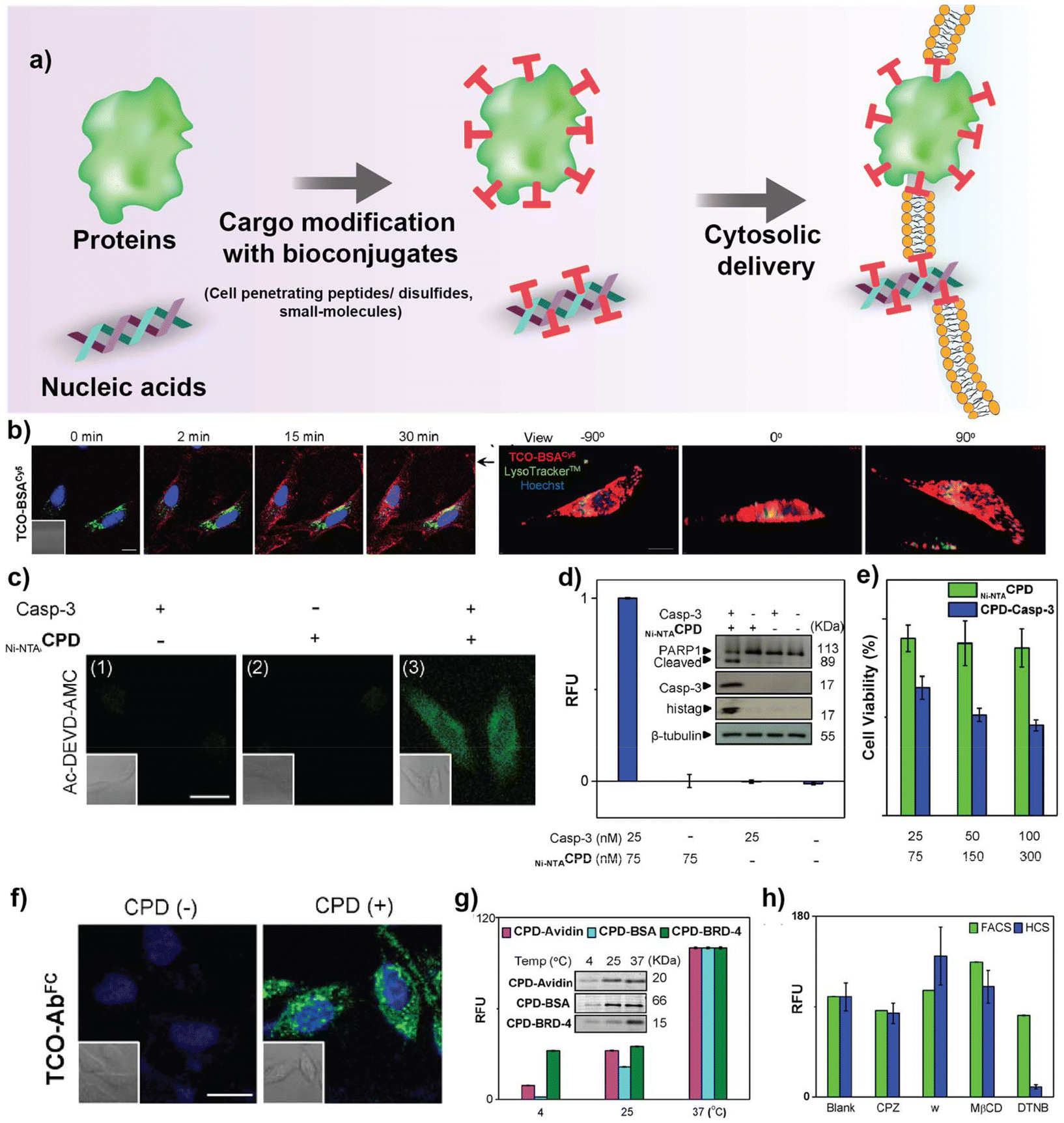 | ||
| Fig. 2 (a) Schematic representation of covalently modified biologics for cytosolic access. (b) Real-time confocal images showing cytosolic delivery of HeLa cells treated with CPD-BSA, Lysotracker and Hoechst dye at different time points. Z stack images on the right showing CPD-BSA delivery in HeLa cells after 2 h of treatment. (c) Representative confocal images showing cytosolic delivery of CPD-Casp-3 in HeLa cells after treatment with Ac-DEVD-AMC (fluorogenic substrate for casp-3). (d) Relative fluorescence units (RFU) of enzymatic Casp-3 assay of HeLa cell lysates. Western blot image demonstrating PARP1 cleavage in HeLa lysates upon treatment with caspase-3. (e) Viablity of HeLa cells treated with CPD-Casp-3 quantified by XTT assay. (f) Confocal images depicting AF-488-CPD-Ab delivery to HeLa cells treated with Hoechst dye. (g) Temperature-dependent protein uptake in HeLa cells, quantified by flow cytometry. (h) Quantification of protein uptake in HeLa cells pre-treated with different inhibitors, chlorpromazine (CPZ), wortmannin (w), methyl-β-cyclodextrin (MβCD), and 5,5′-dithiobis-2-nitrobenzoic acid (DTNB). Reproduced from ref. 97 with permission from American Chemical Society, copyright 2015. | ||
Recently, cyclic CPPs82,83 have been reported to promote direct cytosolic delivery of proteins and nucleic acids, suggesting that chemical modifications such as attaching specific conjugates could further enhance delivery capacity.84 Salehi et al. synthesized a series of cyclic peptides and their linear counterparts containing positively charged arginine (R) residues and hydrophobic tryptophan (W) or diphenylalanine (Dip) residues for effective siRNA delivery.85 [WR]5 cyclic peptide improves molecular transportation capabilities in conjugation with gold, silver, and selenium nanoparticles.86,87 Salehi et al. hypothesized that by replacing W residues with more hydrophobic Dip residues in [WR]5, cellular uptake efficiency of siRNA can be improved further. Preliminary cytotoxicity and cell proliferation studies demonstrated effective growth inhibition of SK-OV-3 ovarian cancer cells (76%) with [DipR]5 at 10 μM, while HEK 293 epithelial cells (6%) remained largely unaffected. Delivery capabilities of fluorescently labeled siRNA was demonstrated with [DipR]5 and was able to achieve effective cellular uptake of siRNA at N/P 10 (30-fold) and 20 (50-fold) compared to siRNA alone in MDA-MB-231 cells. Further, mechanism of uptake was evaluated with endocytic inhibitors using flow cytometry and confocal microscopy. The cargo seemed to be largely unaffected in the presence of endocytic inhibitors, although a combination of uptake mechanisms could not be completely ruled out.
The Hackenberger group reported bioorthogonal conjugation of cyclic azido-TAT peptide with a mutant GFP that carries a homopropargyl glycine at the N terminus.88 The cyclic CPP was designed with alternating L and D amino acids for maximal uptake efficiency and a flexible spacer region containing the reactive azido group to facilitate effective conjugation. cTAT-GFP was uptaken directly to the cytosol at all the concentrations tested (50–150 μM), while the linear TAT-GFP showed very low intracellular GFP signal even at the highest concentrations (150 μM). This methodology was further applied for the cytosolic delivery of nanobodies and fluorescent proteins.89 Despite the overall success in achieving direct cytosolic access, high concentrations of the cargo protein were used (over 100 μM), generally unrealistic levels for in vivo use.
Delivery of CPP–protein and CPP–antibody conjugates is challenging, and often requires laborious protein engineering. Through the recent work from the Hackenberger group, cytosolic delivery of various cargoes (TAMRA, mCherry, Cre recombinase, and IgG) were shown with both synthetic and recombinant CPPs using thiol-reactive cyclic R10 peptides while still preserving their function.90 The thio-nitro-benzoic-acid-activated R10 peptide (TNB-R10) was reported to show quick cellular uptake due to the assistance from the thiol-reactive head groups to form nucleation zones to cross the membrane at both 37 °C and 4 °C, consistent with direct membrane transduction in an energy-independent manner.
Mandal et al. recently reported the use of a hydrophobic derivative, 4-(4-dimethylaminophenylazo) benzoic acid (DABCYL), to modify cR10 peptides for improved cell uptake efficiency of synthetic ubiquitin cargos via non-endocytic routes.91 Ubiquitin analogues were synthesized with a PEG linker containing a cysteine residue to mediate the formation of disulfide bridges with the CPP, and the N-terminus was labeled with TAMRA. The CPP-TAMRA ubiquitin conjugates were delivered to U2OS cells, and the confocal microscopy analysis showed cytosolic delivery and nuclear localization. To investigate the role of DABCYL in cytosolic delivery, Cy5-labeled cR10 and Cy5-labeled DABCYL-cR10 were synthesized and studied under time-lapse microscopy in U2OS cells. Nucleation zones were observed after 30 seconds of incubation of both peptides, with more nucleation zones in cells treated with 500 nM of Cy5-DABCYL-cR10 at 37 °C. Nucleation zones are the points of non-endocytic entry through the cell membrane. It was then observed that these zones disappeared following gradual diffusion of fluorescence throughout the cytosol.
Recently, the Cheng group investigated the use of a family of cationic helical polypeptides – PVBLGn-8 to induce pore formation in cell membranes to facilitate efficient siRNA delivery.92 Unlike CPPs such as penetratin and oligoarginine, PVBLGn-8 was able to retain its membrane disruptive properties regardless of the pH environment. The helical conformation of PVBLGn-8 was essential for direct cytosolic delivery, as demonstrated by experiments showing that its disrupted analogue, PVBDLG100-8, failed to mediate siRNA delivery, highlighting the critical role of membrane permeation in this process. siRNA uptake was further examined by incubating HeLa-Luc cells with PVBLG100-8 before treatment with siRNA. siRNA was able to enter HeLa-Luc cells pretreated with PVBLG100-8, likely through membrane pores formed by the polypeptide. Delivery of TAMRA-siRNA was further evaluated at 4 °C as well as with endocytic inhibitors and the data suggests that siRNA uptake with PVBLGn-8 occurs via diffusion through membrane pores created by the helical peptide.
3.2 Cell-penetrating disulfide conjugates
Cell-penetrating disulfides (CPDs) have been rapidly emerging as an alternative to synthetic CPPs due to their endocytosis-independent cellular uptake mechanism.93,94 They are synthetic mimics of polyarginine-rich CPPs, except the polypeptide backbones are replaced with poly(disulfide)s and undergo rapid degradation upon cell uptake, catalyzed by endogenous glutathione (GSH), resulting in spontaneous cargo release. Moreover, CPDs employ a thiol-mediated cell uptake mechanism, which is not affected by endocytosis inhibition, resulting in efficient cytosolic delivery.Recently, Guo et al. developed a series of degradable poly(disulfide)s containing phenylboronic acids (PBA) and guanidine groups for efficient binding of PBA-CPDs to proteins via multiple non-covalent interactions.95 These CPDs demonstrated efficient cytosolic delivery of a wide range of native proteins (phycoerythrin, ovalbumin, BSA, β-galactosidase, GFP, horseradish peroxidase, ribonuclease A, IgG, saporin, cytochrome C, trypsin, Cre recombinase) or small peptides without any chemical modification. Cellular uptake studies at 4 °C, or in the presence of inhibitors indicated a predominant thiol-mediated uptake, bypassing the endocytosis-dependent pathways. Both β-galactosidase and Cre recombinase retained their functional capabilities upon delivery to cells and were able to retain about 80% enzymatic activity and 70% gene editing efficiency respectively. Further, hyaluronic acid (HA)-coated PBA-CPD was explored for the delivery of saporin in vivo for the treatment of HeLa-xenografted tumor model and demonstrated efficient tumor accumulation and tumor repression as compared to the control groups without HA.
The Qian group developed pH-responsive cell-penetrating poly(disulfide)s (CPDIMD) for targeted protein delivery to cancer cells.96 The CPDIMDs utilizes imidazole groups instead of the conventional cationic arginine residues. Under normal physiological conditions, the imidazole groups remain neutral, which reduces nonspecific interactions during blood circulation. However, in the acidic tumor microenvironment (TME), these groups become protonated (pKa ≈ 6.8), turning the polymer positively charged. This charge reversal allowed for enhanced cellular uptake by combining thiol-mediated and counterion-mediated uptake with no endosomal entrapment, as demonstrated through the delivery of fluorescently labeled IgG. A 2-fold increase in uptake of IgG was observed at pH 6.5 compared to pH 7.5, demonstrating the pH sensitivity of CPDIMD. Endocytic inhibitors were used to study the entry mechanism further, and IgG uptake remained largely unaffected, consistent with direct cytosolic delivery. In vivo biodistribution studies demonstrated more tumor accumulation and less non-specific uptake with CPDIMD as compared to CPDArg, confirming the cancer-selective delivery capabilities of CPDIMD.
Shao Q. Yao's group reported a cyclic polydisulfide CPD-mediated method for the intracellular delivery of proteins, therapeutic antibodies, and small-molecule drugs.97 Proteins were conjugated with CPDs, either covalently (via biorthogonal chemistry) or non-covalently (via affinity interaction), which enabled the proteins to be delivered into the cytosol of cells without being entrapped by endocytic vesicles (Fig. 2b–h). Three thiol-containing initiators—biotin, nitrilotriacetic acid (NTA), and tetrazine (Tz)—were used to synthesize BiotinCPD, Ni-NTACPD, and TzCPD for efficient cargo attachment with Histidine tagged BSA, avidin and BRD4 respectively. BSA, avidin, and BRD-4 proteins conjugated with the respective CPDs demonstrated direct cytosolic delivery without endosomal entrapment, proven by using endocytic inhibitors (chlorpromazine, wortmannin, methyl-b-cyclodextrin). To further explore the therapeutic potential of the CPDs, delivery of active caspase-3 was tested in HeLa cells. Caspase-3 retained 20% of the enzymatic activity and induced apoptosis. Further, Alexa fluor-tagged IgG was used as a model antibody to demonstrate successful delivery to HeLa cells.
3.3 Other small molecule bioconjugates for cytosolic delivery of biologics
In addition to CPPs and CPDs, other small molecules have been used to modify biomolecule cargos, facilitating their interaction with cell membranes, and enabling their direct cytosolic delivery. The Raines group developed a strategy to enhance protein cellular uptake by esterifying carboxyl groups on the protein surface, reducing anionic character, and increasing hydrophobicity.98 The esterified proteins showed improved cellular internalization, with uptake correlating to the number of ester groups on the protein. Confocal microscopy confirmed that esterified GFP demonstrated direct cytosolic delivery, while unmodified GFP did not enter cells, and a “supercharged” variant entered cells via endocytosis, showing a punctate fluorescence pattern. Further, esterified GFP with a nuclear localization signal (nlsGFP) demonstrated diffuse fluorescence not only in the cytosol but also co-localized with the nuclear stain, exhibiting successful transport to the nucleus (Fig. 3a and b). This approach leverages the masking of anionic charges to promote protein uptake and overcome cellular barriers. Additionally, the esterification was reversible, as incubation with mammalian cell extract led to the complete removal of the ester labels, mimicking the behavior of prodrugs. | ||
| Fig. 3 Confocal images depicting (a) cellular internalization of GFP esterified variant containing diazo compound (GFP-1) (15 μM) in CHO-K1 cells at 37 °C or 4 °C, stained with Hoechst 33342 and wheat germ agglutinin (WGA)-Alexa Fluor 647 (b) nuclear internalization of GFP and esterified GFP (GFP-1) in CHO-K1 cells treated with nuclear localization signal GFP (nlsGFP) or nuclear localization signal GFP-1 (nlsGFP-1). Reproduced from ref. 98 with permission from American Chemical Society, copyright 2017. | ||
Zhou et al. recently demonstrated efficient cytosolic delivery of miRNA and siRNA using guanidinium-containing disulfide (GDS) molecule via a thiol-mediated cellular uptake mechanism, bypassing endo/lysosomal entrapment.99 Complexation of GDS with the RNA resulted in the formation of disulfide-based nanospheres (DBNP), which upon cellular internalization underwent reductive depolymerization by endogenous glutathione, releasing the cargo in the process. Cellular uptake mechanism was confirmed by pre-treating cells with endocytic inhibitors and thiol-blocking reagents, prior to treatment with siRNA/miRNA, and results indicated a thiol-mediated cellular uptake, demonstrated via flow cytometry, confocal microscopy. KRAS siRNA delivered to PANC-1 pancreatic cancer cells effectively silenced gene expression and reduced downstream protein expression levels, as confirmed by western blot analysis. miRNA-34a targeting Notch-1 and CD44 was delivered to MDA-MB-231 cells to evaluate the gene silencing effects. Flow cytometry results revealed that DBNP-miR34a induced apoptosis (24.9%) more efficiently in comparison to RNAiMAX (18.9%). Therapeutic effect of DBNP-siKRAS (1 mg kg−1) was further evaluated in PANC-1 xenograft tumor-bearing nude mice and results revealed a 2-fold reduction in tumor growth compared to the control groups.
Recently, a simple small-molecule tag was developed by conjugating Coomassie blue (CB) with cholesterol.100 CB dye facilitated non-covalent binding to proteins via hydrophobic interactions, while cholesterol was used to decorate the protein-tag complex to promote interaction with the cellular membrane. The tag, linked by a bifunctional linker to enhance solubility and mask negative charges, facilitates the insertion of proteins between lipid bilayers, promoting their eventual release into the cytosol through membrane dissociation. The cholesterol-tagged protein delivery system was tested with proteins ranging from 6.5 to 150 kDa to assess its size selectivity. Fluorescently labeled proteins demonstrated distinct intracellular fluorescence patterns, with smaller proteins (e.g., aprotinin, 6.5 kDa) showing homogeneous distribution via direct membrane permeation, while larger proteins (e.g., BSA (66 kDa) and IgG (150 kDa)) exhibited punctate fluorescence, indicating endocytic uptake. To exclude the possibility of endocytic uptake followed by efficient endosomal escape, the authors conducted confocal microscopy experiments at 4 °C, which inhibits endocytosis. At low tag/protein ratios, aprotinin was mostly confined to the cell membrane, while at the optimal tagging ratio, homogeneous intracellular fluorescence was observed, confirming direct membrane permeation. This approach was however limited to the delivery of small proteins, as cargos larger than 30 kDa yielded punctate fluorescent signals.
Recently, the Yin lab developed a ‘pro-protein’ system that utilizes LAT1-mediated transport for selective delivery to cancer cells, bypassing endocytosis.101,102 Native proteins were engineered with an H2O2-responsive domain (4-nitrophenyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl carbonate) and a LAT1 substrate (3,4-dihydroxy-L-phenylalanine) to facilitate efficient intracellular delivery. The LAT1-mediated delivery system was shown to selectively target cancer cells, with pro-protein uptake ranging from 35–55% in tumor cell lines (e.g., MDA-MB-231, HeLa) and minimal uptake (<3.5%) in normal cells. Cytosolic delivery was inhibited by incubation at 4 °C and by LAT-1 selective inhibitor, but not by endocytosis inhibitors, suggesting a LAT-1-mediated direct cytosolic delivery mechanism. The efficacy of the system was further demonstrated by the successful cytosolic delivery of pro-enzymes, including β-galactosidase (β-gal) and horseradish peroxidase (HRP) to cancer cells. β-Gal restored enzymatic activity (∼80%), and HRP exhibited substantial activity in the presence of H2O2, confirming the functional restoration of the delivered enzymes. Further, the gene editing capabilities of Cas9 ribonucleoprotein (RNP) targeting polo-like kinase 1 (PLK1) was evaluated in HeLa cells and were able to demonstrate significant DNA cleavage (21.1% indels), downregulated PLK1 mRNA and protein levels by 54.1% and 58.5%, respectively, and triggered apoptosis.
4. Polyplex and nanoplex delivery approaches
4.1. Direct encapsulation
The covalent modification strategies described above present challenges in terms of retaining activity relative to the native biomacromolecules. The cargo must be sufficiently modified to enable efficient delivery, while minimizing excessive alterations that could negatively impact the structure and function. Furthermore, direct modification of biomolecules leads to smaller size systems which are subject to renal clearance and are vulnerable to enzymatic degradation during circulation. On the other hand, strategies based on cargo encapsulation can lead to nano-sized particles with more favorable pharmacokinetic profiles (Fig. 4a). | ||
| Fig. 4 (a) Scheme depicting direct cytosolic delivery of biologics using nanocarriers (inorganic, polymeric, liposomes). (b) Confocal microscopy images showing FITC-BSA delivery using gemini amphiphiles (GA) in HeLa cells pre-treated with endocytic inhibitors. (c) Relative mean fluorescence intensity (MFI) changes recorded with various endocytic inhibitors and delivery conditions. (d) T7E1 assay after intracellular delivery of ribonucleoprotein/gemini amphiphiles (RNP/GA) complexes in 293T cells targeting EGFP loci. (e) Fluorescence images of 293T-EGFP cells treated with RNP/GA complexes. (f) Flow cytometry analysis in 293T-EGFP cells depicting relative MFI changes between experimental and control groups. Reproduced from ref. 105 with permission from American Chemical Society, copyright 2023. | ||
Tang et al. demonstrated a rapid and efficient protein delivery strategy using nanoparticle-based supramolecular nanocapsules (NPSCs).103 The protein–NPSC complexes were formed by mixing proteins with HKRK gold nanoparticles (AuNPs) to provide supramolecular stabilization. Template emulsions were created by combining AuNPs in phosphate buffer and oil (linoleic acid), which were then mixed with the protein–AuNP solution to form the final protein–NPSCs. This approach enabled direct delivery of functional proteins in their native forms, overcoming challenges associated with protein stability and cellular uptake. GFP was used as a model protein to study intracellular distribution as well as the delivery capabilities of NPSCs. An even distribution throughout the cytosol and nucleus was observed using confocal microscopy and live-cell video imaging, supporting membrane fusion as the primary mechanism of uptake. Notably, the NPSCs were not taken up as intact entities by the cells, reinforcing the idea that the protein release occurred through fusion with the cell membrane rather than endocytotic uptake. To demonstrate the therapeutic potential of the system, NPSCs were utilized to deliver caspase 3, owing to its crucial role in apoptosis. CASP3-NPSCs were incubated in HeLa cells and demonstrated 72 ± 6% apoptosis, confirmed by double staining with Yopro-1 (a dye to detect apoptotic nuclei) and 7-AAD (used to detect membrane disruption).
The Miserez group recently developed a short pH-responsive peptide (HBpep) coacervate platform conjugated with disulfide bond-containing moieties that disassemble in thiol-rich intracellular environments.104 These coacervate microdroplets deliver a wide range of biomacromolecules, from short therapeutic peptides (726 Da) to large proteins (430 kDa), and mRNA, with results consistent with direct delivery into the cytosol. Dye-labeled payloads showed diffuse fluorescent signals, consistent with cytosolic localization. The mechanism of uptake was further verified by pretreating the cells with cholesterol/endocytic inhibitors and incubation at 4 °C, and the results confirm a non-endocytic mode of uptake. Luciferase-encoding mRNA transfection using HBpep was studied in HepG2, HEK 293 cell lines and compared with the conventional transfection reagents (PEI, Lipofectamine 2000 and 3000). At the optimal peptide concentration, the transfection efficiencies of coacervates were higher than those of PEI and Lipofectamine 3000, although slightly lower than Lipofectamine 2000 in HepG2 cells. The coacervates exhibited transfection efficiency comparable to that of Lipofectamine 2000 in HEK293 cells. Further, the transfection efficiency of EGFP-encoding mRNA was quantified using FACS and demonstrated 72% and 81.6% EGFP expression in HepG2 and HEK293 cells respectively.
Le et al. recently developed a family of gemini amphiphiles (GAs) for direct cytosolic delivery of proteins.105 Different amines were chosen to vary GA charge. A series of α-substituted aldehydes and carboxylic acids (C12–C20) with a range of alkyl tail lengths were employed to tune hydrophobicity. GAs containing C18 and C19 proved to be the most efficient in delivering FITC-BSA to HeLa cells, with delivery efficiencies of 94% and 89% respectively, while the shorter alkyl chain tail (C14 and C15) provided cell viability less <50% (Fig. 4b and c). To demonstrate therapeutic applicability, delivery of Cas9 ribonucleoprotein (Cas9 RNP) was assessed in 293T-EGFP cells and were able to demonstrate a 45% decrease in the GFP expression. Furthermore, T7E1 assay results from targeting the KRAS gene in SW-480 cells, AAVS1, and HBB (hemoglobin subunit beta) in 293T cells collectively depict efficient genome editing at the target loci. KRAS gene targeting was further tested in vivo. Tumor tissues were harvested and tested for the insertion and deletion frequencies using T7E1 assay. An indel frequency of 17.9% to 22.1% was detected, which was further verified using Sanger sequencing (Fig. 4d–f).
Jana et al. reported a size-dependent mechanism for cellular uptake using arginine-terminated gold nanoparticles (AuNPs), highlighting a critical threshold of <10 nm diameter.106 Specifically, AuNPs smaller than 10 nm with 100 ± 30 average number of arginine molecules per particle entered cells via energy-independent direct membrane penetration, enabling immediate access to the cytosol. In contrast, larger nanoparticles (∼16, 40 nm) were taken up by energy-dependent endocytosis, emphasizing how nanoparticle size influences cellular internalization pathways. As delivery vectors, these sub-10 nm AuNPs exhibited the ability to rapidly transport electrostatically bound fluorescent labeled bovine serum albumin directly into the cytosol, bypassing endocytosis. These findings highlighted the potential of arginine-functionalized, ultra-small AuNPs as a versatile platform for intracellular protein delivery.
Recently, the Rotello group developed a series of guanidinium-functionalized poly(oxanorborneneimide) polymers (PONI) with varying side chains (C3, C5, C7) and molecular weights (17, 30, 65 kDa) to study the role of amphiphilicity on siRNA complexation and delivery efficiency.107 PONI-Guan homopolymers formed discrete polyplexes with siRNA via electrostatic interactions, demonstrating over 95% encapsulation efficiency and preventing siRNA from enzymatic degradation when exposed to RNase A. Cellular uptake efficiency of AF488-siRNA was evaluated in LPS-induced M1 macrophages and results showed maximum uptake efficiency was achieved with the 65 kDa polymers of all chain lengths. The uptake mechanism was further investigated by pre-treating cells with small-molecule inhibitors and results revealed cholesterol dependence on uptake. Furthermore, the ability of polyplex-mediated siRNA delivery to regulate TNF-α levels for immune modulation was investigated in vitro and results revealed 70% gene silencing when treated with 65 kDa C3 and C5-Guan polyplexes. In vivo efficacy of Cn-Guan/si_TNF-α polyplexes against lung inflammation was evaluated in LPS-challenged BALB/c mice model. Highly efficient TNF-α knockdown was achieved with polyplexes of all three chain lengths at a low 0.14 mg kg−1 siRNA dosage, highlighting the therapeutic potential of Cn-Guan polyplexes (Fig. 5a–e).
 | ||
| Fig. 5 (a) Flow cytometry of AF488-siRNA uptake in M1-polarized RAW 264.7 cells after treatment with Cn-Guan/AF488-siRNA polyplexes with different molecular weights and G/P ratios. (b) Quantification of eGFP knockdown in M1-polarized RAW 264.7:eGFP cells after treatment with Cn-Guan/si_eGFP polyplexes. (c) Confocal laser scanning microscopy images depicting eGFP knockdown in M1-polarized RAW 264.7:eGFP cells. (d) qRT-PCR analysis of TNF-α mRNA levels from pulmonary tissue sections relative to β-actin levels. (e) Relative bronchial wall thickness analysis from H&E stained lung tissue sections. Reproduced from ref. 107 with permission from Elsevier, copyright 2024. | ||
The Csiszár group used fusogenic liposomes composed of DOPE and DOTAP (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine and 1,2-dioleoyl-3-trimethylammonium-propane chloride, respectively) to encapsulate water-soluble proteins, allowing their direct delivery to the cytosol.108 The formation of these proteoliposomes relied on electrostatic interactions between the cationic liposomes and the encapsulated cargo, limiting the application of this system to negatively charged proteins. Upon short incubation times (10–15 min), this system allowed direct cytosolic delivery of fluorescent proteins (EGFP, Dendra2, and R-phycoerythrin) in CHO cells, yielding diffuse fluorescence signals, and binding peptides in both CHO cells and primary rat cardiac myofibroblasts, where the binding capabilities were retained.
The Csiszár group later carried out a systematic study to identify key features in the compositions of their fusogenic liposomes which allowed fusion-driven cell entry.109 Using a BODIPY FL-DHPE tracer with concentration dependent spectral changes to assess the cell entry mechanisms at play, the study distinguished between fusion and endocytic uptake. Fusion events resulted in a green monomer signal due to dye dilution, while endocytosis caused a red-shifted dimer signal due to dye aggregation in endosomes. The results confirmed that fusion efficiency increased with the zeta potential of liposomes, with saturation occurring at a DOTAP concentration of around 50 mol%, reaching approximately 90% fusion efficiency. Of the six cationic lipids (DMTAP, DOTAP, DOTMA, DOEPC, MVL5, DC-cholesterol) tested for their fusion capabilities, DOTAP and DOTMA liposomes achieved over 90% fusion, DMTAP and DOEPC had ∼30% efficiency, and MVL5 and DC-cholesterol showed minimal or no fusion, highlighting the significant impact of lipid composition on fusogenicity. Further, the role of aromatic components and neutral lipids were examined and found that aromatic components (above 5 mol% of the composition) were necessary and sufficient to allow efficient cell entry through fusion-like mechanism. Neutral helper lipids were found to provide liposomes with increased stabilization and reduced cytotoxicity but could also strongly impact the cell entry mechanism and efficiency.
Sarkar et al. generated micelles using a guanidinium-terminated polyaspartic acid and used these for delivery of proteins and pDNA.110 Cells pre-treated with endocytic inhibitors prior to incubation with rhodamine B dye-conjugated polymer suggested guanidinium-phosphate salt bridging occurred between the micelle with the cell membrane. Transient deformation of the membrane then led to direct cytosolic delivery. To test the cell delivery performance of polyaspartic micelles, FITC-conjugated BSA was used as a model protein and delivered to KB cells. The results showed cytosolic access within the first 2 h and that the direct cytosolic delivery is strictly dependent on the size and colloidal properties of the nano-assembly. To test the use of polyaspartic acid as a gene transfection agent, GFP pDNA was used as a model cargo. Various cell lines (KB, SHSY5Y, astrocytes, and progenitors) were utilized to study transfection after incubating polymer-pDNA or lipofectamine-pDNA nano-assemblies. Immunoblot assay was utilized to quantitatively determine the relative GFP expression in cells and was able to obtain higher GFP expression with the polymeric carrier in comparison to lipofectamine. Furthermore, the tentative number of protein/DNA bound to the micelles was found to be about 3–4 proteins/DNA per micelle and about 20–50% of the micelle surface is free, consistent with guanidinium groups at the micelle surface driving translocation.
The Hoffmann group investigated the use of neutralization reagents to enhance the transfer efficiency of nucleic acids (NAs) via fusogenic liposomes (FLs).111 While FLs alone were inefficient at transferring NAs, pre-incubation with positively charged molecules, like protamine, significantly improved transfer efficiency. Further, fusogenic liposomes (FLs) containing DOPE/DOTAP/DiR (1/1/0.1) were tested for their mRNA transfer capabilities in mammalian cells and were shown to achieve 80% transfer efficiency, while formulations lacking DiR were significantly less effective (<20%). Replacing DOTAP with the multivalent lipid MVL-5 showed reduced fusion efficiency despite similar surface charge densities, and high MVL-5 concentrations inhibited liposomal fusogenicity. Replacing DOPE with DOPC prevented fusion and greatly reduced transfer efficiency (4%). mRNA transfer efficiency was further quantified, demonstrating 1700 fusion events per cell in 10 minutes and successful mRNA delivery into primary cortical neurons, highlighting the versatility and potential of FLs for efficient gene transfer in primary tissues.
4.2. Cargo co-engineering
Cargo co-engineering is an integrative approach involving the rational modification of therapeutic payloads to enable efficient encapsulation with the delivery vehicles (Fig. 6a). This coordinated design enhances stability, promotes efficient cellular uptake, and enables controlled biodistribution in systemic environments.112 | ||
| Fig. 6 (a) Schematic representation of cargo modification and encapsulation for cytosolic delivery. (b) Scheme depicting nanocomposite preparation via streptavidin conjugation with biotinylated components followed by encapsulation with PONI-Guan polymers. (c) Confocal microscopy images of b-GFP delivery at varying charge ratios. (d) Quantification of delivered b-GFP in HEK 293T cells using imaging flow cytometry. (e) Viability of HEK 293T cells after treatment with b-GrA nanocomposites and in the presence of small-molecule inhibitors: chlorpromazine (CPM) and methyl-β-cyclodextrin (MβCD). Reproduced from ref. 116 with permission from American Chemical Society, copyright 2022. | ||
Recently, the Futaki group developed a branched CPP FcB(L17E)3 containing three copies of an attenuated cationic amphiphilic lytic peptide L17E (a mutant of M-lycotoxin), with an Fc-binding peptide for efficient complexation with IgG.113 Interestingly, FcB(L17E)3 when mixed with Alexa488-IgG formed coacervates, facilitating efficient translocation of IgG to the cytosol. The negative charges imparted by Alexa488 on IgG was found to be critically important in the liquid droplet/coacervate formation. Transient permeabilization of cell membrane followed by actin polymerization and membrane ruffling was proposed as the mechanism of action induced by the coacervates from inhibitor experiments. Delivery of other proteins modified with negative charged molecules was demonstrated using Alexa Fluor 594-labeled anti-pore complex antibody and an anti-mCherry nanobody tagged with supercharged GFP.
Rotello and co-workers reported a protein–particle co-engineering strategy for direct cytosolic protein delivery. In this method, proteins were genetically engineered with a peptide chain comprised of different lengths of oligo-glutamic acid tags (E-tag) at their N or C terminus.114 This allowed for self-assembly of the protein cargo with the arginine-functionalized gold nanoparticles (ArgNPs) via guanidine-carboxylate electrostatic interactions. These assemblies provided direct cytosolic delivery in a membrane-fusion like process. Confocal microscopy of E-tagged GFP in mammalian cells showing diffuse fluorescence throughout the cytosol including the nucleus indicated direct cytosolic delivery bypassing endocytosis. Live cell imaging videos by video confocal microscopy further confirmed the membrane-fusion like mechanism where these supramolecular assemblies were shown to fuse with the cell membrane, through interaction of ArgNPs with the cell membrane, releasing the E-tagged GFP into the cytosol within seconds after attaching to the membrane. Moreover, delivery after pre-treatment with endocytosis inhibitors did not significantly alter uptake, all consistent with a direct delivery pathway. Besides GFP, other proteins were also delivered including functional Cre recombinase and gene editing ribonucleoprotein CRISPR Cas9 systems.38
Inspired by these results, the co-engineering strategy was then adapted to deliver E-tagged proteins with polymeric delivery vehicles to increase the design space and to provide a more scalable platform for direct cytosolic protein delivery.115 Briefly, Poly(oxanorborneneimide) polymers (PONI) featuring cationic guanidinium moieties (PONI-Guan) were synthesized via ring opening metathesis polymerization and utilized for protein delivery. The ‘semiarthritic’ (neither too rigid nor too flexible) oxanorbornene backbone and the guanidinium functional group reminiscent of the ArgNPs, play a pivotal role in fusing of the carrier with the membrane.
Recently, Rotello and co-workers reported a modular biotin–streptavidin (STV) assembly-based strategy for cytosolic delivery of proteins.116 In this approach, biotinylated oligo(glutamate) (bE20) and biotinylated proteins were first bound to STV to generate biotin–STV protein bioconjugates (Fig. 6b–e). These conjugates were then self-assembled with the cationic PONI-Guan homopolymer to form discrete supramolecular protein–polymer nanocomposites. This simplified strategy eliminated the need for protein engineering and facilitated efficient cytosolic delivery, as demonstrated through microscopy and nuclear access of biotinylated green fluorescent protein (b-GFP) with imaging flow cytometry in mammalian cells, consistent with their previous reports. This strategy simplifies the process of complexation with the nanovectors, eliminating the need for protein engineering and provides a modular approach for direct cytosolic protein delivery.
A more straightforward approach of protein modification using citraconic anhydride was recently reported to impart an overall negative charge on the protein cargo, enabling them to assemble with positively charged nanovectors.117 Proteins of varying sizes and pI (EGFP, RNase A, ovalbumin, BSA, apo-transferrin, dsRed) were anhydride modified and dye-labeled to assess the delivery efficiency in HEK-293T cells. Diffuse fluorescence was observed in confocal microscopy, suggesting efficient cytosolic access in cells. The mechanism of uptake was assessed using cholesterol depletion agents as well as endocytic inhibitors as previously described and the results indicate a membrane fusion-like mechanism, which was further confirmed using time-lapse imaging. Delivery of active proteins including Cre recombinase and RNase A were studied to establish the functional capabilities of this approach. Modified Cre recombinase was able to excise the dsRed DNA coding sequence between LoxP sites and trigger GFP expression in a Cre reporter HEK-293T cell line. Likewise, anhydride-modified RNase A (an endonuclease) showed 50% apoptosis in HeLa cells, confirming the retention of enzymatic activity.
5. Summary and outlook
Protein and nucleic acid-based therapeutics can be used to treat ‘undruggable’ disorders, opening up new therapeutic pathways. Intracellular delivery using nanocarriers has advanced rapidly. Although endocytic pathways are among the most common modes of cellular uptake, inefficient endosomal escape of the cargo remains a significant challenge. Direct membrane fusion-based approaches offer a strategy for direct delivery to the cytosol delivery, achieving high delivery efficiency by evading endosomal entrapment. While fusion-based approaches are still emerging, they hold the potential to revolutionize protein and nucleic acid-based therapeutic delivery. However, the mechanisms underlying these systems require further exploration and understanding.There remains substantial opportunity for advancing nanocarrier systems for intracellular protein and nucleic acid delivery, particularly to enhance specificity and minimizing toxicity. A key challenge is clinical translation of these promising technologies, would be facilitated if tissue, organ, and cell-type specificity of cargo-loaded nanocarriers are developed. In circulation, nanocarriers often face challenges due to protein corona formation, which prevents the administered therapeutics to reach the site of action. These challenges can be addressed through precise engineering of the physicochemical properties (size, shape, surface charge) of nanocarriers, to influence biodistribution and cell uptake. Further, specificity can be significantly enhanced by functionalizing nanocarriers with ligands/antibodies that specifically recognizes surface markers on cell types of interest.
There is enormous space that can be explored for delivery of biomolecules using nanocarriers. A critical factor driving this momentum is the incorporation of clinical feedback into the development cycle. Insights gained from patient responses and trial outcomes enable researchers to refine formulations, optimize efficacy and safety. A classic example of this evolution is the development of siRNA-based therapeutics Vutrisiran from Onpattro, which illustrates how clinical feedback can improve the delivery profiles and patient outcomes. Given the substantial advancements in delivery strategies for protein and nucleic acid-based therapeutics in recent years, we expect delivery platforms such as those discussed here will make their way into the clinic in the near future.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Acknowledgements
This work is supported by the National Institutes of Health under EB02264 and AI186027.References
- S. Niamsuphap, C. Fercher, S. Kumble, P. Huda, S. M. Mahler and C. B. Howard, Expert Opin. Drug Delivery, 2020, 17, 1189–1211 CrossRef CAS PubMed.
- Y.-W. Lee, D. C. Luther, J. A. Kretzmann, A. Burden, T. Jeon, S. Zhai and V. M. Rotello, Theranostics, 2019, 9, 3280–3292 CrossRef CAS PubMed.
- S. Du, S. S. Liew, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2018, 140, 15986–15996 CrossRef CAS PubMed.
- A. Gupta, J. L. Andresen, R. S. Manan and R. Langer, Adv. Drug Delivery Rev., 2021, 178, 113834 CrossRef CAS PubMed.
- N. J. Yang and M. J. Hinner, Methods Mol. Biol., 2014, 1266, 29–53 CrossRef PubMed.
- Y. Sung and S. Kim, Biomater. Res., 2019, 23, 1–7 CrossRef PubMed.
- X. Liu, F. Wu, Y. Ji and L. Yin, Bioconjugate Chem., 2019, 30, 305–324 CrossRef PubMed.
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- D. M. Ecker, S. D. Jones and H. L. Levine, mAbs, 2014, 7, 9–14 CrossRef PubMed.
- D. Stevens, S. Milani-Nejad and T. Mozaffar, Curr. Treat. Options Neurol., 2022, 24, 573–588 CrossRef PubMed.
- L. S. Castro, G. S. Lobo, P. Pereira, M. G. Freire, M. C. Neves and A. Q. Pedro, Vaccines, 2021, 9, 328 CrossRef CAS PubMed.
- G. J. Weiner, Nat. Rev. Cancer, 2015, 15, 361–370 CrossRef CAS PubMed.
- L. Cheng, Y. Wang, Y. Guo, S. S. Zhang and H. Xiao, Cell Chem. Biol., 2024, 31, 428–445 CrossRef CAS PubMed.
- M. Rahban, F. Ahmad, M. A. Piatyszek, T. Haertlé, L. Saso and A. A. Saboury, RSC Adv., 2023, 13, 35947–35963 RSC.
- J. Wu, J. K. Sahoo, Y. Li, Q. Xu and D. L. Kaplan, J. Controlled Release, 2022, 345, 176–189 CrossRef CAS PubMed.
- D. M. Rad, M. A. Rad, S. R. Bazaz, N. Kashaninejad, D. Jin and M. E. Warkiani, Adv. Mater., 2021, 33, 2005363 CrossRef PubMed.
- A. Fu, R. Tang, J. Hardie, M. E. Farkas and V. M. Rotello, Bioconjugate Chem., 2014, 25, 1602–1608 CrossRef CAS PubMed.
- A. Hamann, A. Nguyen and A. K. Pannier, J. Biol. Eng., 2019, 13, 1–16 CrossRef PubMed.
- Y. An, M. J. Park, J. Lee, J. Ko, S. Kim, D. H. Kang and N. S. Hwang, Adv. Ther., 2020, 3, 1–19 Search PubMed.
- A. F. L. Schneider, A. L. D. Wallabregue, L. Franz and C. P. R. Hackenberger, Bioconjugate Chem., 2019, 30, 400–404 CrossRef CAS PubMed.
- A. R. Stasińska, P. Putaj and M. K. Chmielewski, Bioorg. Chem., 2019, 92, 103223–103223 CrossRef PubMed.
- A. R. Stasińska, P. Putaj and M. K. Chmielewski, Bioorg. Chem., 2019, 95, 103518 CrossRef PubMed.
- J. Winkler, Ther. Delivery, 2013, 4, 791–809 CrossRef CAS PubMed.
- B. B. Mendes, J. Conniot, A. Avital, D. Yao, X. Jiang, X. Zhou, N. Sharf-Pauker, Y. Xiao, O. Adir, H. Liang, J. Shi, A. Schroeder and J. Conde, Nat. Rev. Methods Primers, 2022, 2, 1–21 CrossRef.
- F. Zákány, I. M. Mándity, Z. Varga, G. Panyi, P. Nagy and T. Kovács, Cells, 2023, 12, 1700–1700 CrossRef PubMed.
- A. Gori, G. Lodigiani, S. G. Colombarolli, G. Bergamaschi and A. Vitali, ChemMedChem, 2023, 18, 00236 CrossRef PubMed.
- R. A. Bottens and T. Yamada, Cancers, 2022, 14, 5546 CrossRef CAS PubMed.
- B. R. Liu, C.-W. Chen, Y.-W. Huang and H.-J. Lee, Molecules, 2023, 28, 3367–3367 CrossRef CAS PubMed.
- I. Gessner and I. Neundorf, Int. J. Mol. Sci., 2020, 21, 2536 CrossRef CAS PubMed.
- Y. Behzadipour and S. Hemmati, Biomed. Pharmacother., 2024, 176, 116910–116910 CrossRef CAS PubMed.
- S. M. Ghorai, A. Deep, D. Magoo, C. Gupta and N. Gupta, Pharmaceutics, 2023, 15, 1999–1999 CrossRef CAS PubMed.
- L. Porosk and Ü. Langel, Front. Pharmacol., 2022, 13, 1056467 CrossRef CAS PubMed.
- D. C. Luther, R. Huang, T. Jeon, X. Zhang, Y.-W. Lee, H. Nagaraj and V. M. Rotello, Adv. Drug Delivery Rev., 2020, 156, 188–213 CrossRef CAS PubMed.
- H. Son, S. Lee, J. Lee, S. Kim, M.-H. Kang and J. Park, Surf. Interfaces, 2025, 61, 106096 CrossRef CAS.
- E. Samaridou, J. Heyes and P. Lutwyche, Adv. Drug Delivery Rev., 2020, 154–155, 37–63 CrossRef CAS PubMed.
- H. Mukai, K. Ogawa, N. Kato and S. Kawakami, Drug Metab. Pharmacokinet., 2022, 44, 100450–100450 CrossRef CAS PubMed.
- J. Lv, Q. Fan, H. Wang and Y. Cheng, Biomaterials, 2019, 218, 119358 CrossRef CAS PubMed.
- A. J. Mukalel, R. S. Riley, R. Zhang and M. J. Mitchell, Cancer Lett., 2019, 458, 102–112 CrossRef CAS PubMed.
- M. Cárdenas, R. A. Campbell, M. Y. Arteta, M. J. Lawrence and F. Sebastiani, Curr. Opin. Colloid Interface Sci., 2023, 66, 101705–101705 CrossRef.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2020, 20, 1–24 CrossRef PubMed.
- L. H. Rhym and D. G. Anderson, Med, 2022, 3, 167–187 CrossRef CAS PubMed.
- R. Goswami, T. Jeon, H. Nagaraj, S. Zhai and V. M. Rotello, Trends Pharmacol. Sci., 2020, 41, 743–754 CrossRef CAS PubMed.
- N. Chen, Y. He, M. Zang, Y. Zhang, H. Lu, Q. Zhao, S. Wang and Y. Gao, Biomaterials, 2022, 286, 121567 CrossRef CAS PubMed.
- R. Allen and T. Yokota, Molecules, 2024, 29, 5997 CrossRef CAS PubMed.
- A. M. Butt, N. Abdullah, N. N. I. M. Rani, N. Ahmad and M. C. I. M. Amin, Pharm. Res., 2022, 39, 1047–1064 CrossRef CAS PubMed.
- A. Wittrup, A. Ai, X. Liu, P. Hamar, R. Trifonova, K. Charisse, M. Manoharan, T. Kirchhausen and J. Lieberman, Nat. Biotechnol., 2015, 33, 870–876 CrossRef CAS PubMed.
- E. Xu, W. M. Saltzman and A. S. Piotrowski-Daspit, J. Controlled Release, 2021, 335, 465–480 CrossRef CAS PubMed.
- K. Deprey, L. Becker, J. Kritzer and A. Plückthun, Bioconjugate Chem., 2019, 30, 1006–1027 CrossRef CAS PubMed.
- H. Wang, C. Wang, Y. Zou, J. Hu, Y. Li and Y. Cheng, Giant, 2020, 3, 100022 CrossRef.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2018, 30, 263–272 CrossRef PubMed.
- D. J. Brock, L. Kustigian, M. Jiang, K. Graham, T.-Y. Wang, A. Erazo-Oliveras, K. Najjar, J. Zhang, H. Rye and J.-P. Pellois, Traffic, 2018, 19, 421–435 CrossRef CAS PubMed.
- S. L. Y. Teo, J. J. Rennick, D. Yuen, H. Al-Wassiti, A. P. R. Johnston and C. W. Pouton, Nat. Commun., 2021, 12, 3721 CrossRef CAS PubMed.
- N. Chen, Y. He, M. Zang, Y. Zhang, H. Lu, Q. Zhao, S. Wang and Y. Gao, Biomaterials, 2022, 286, 121567 CrossRef CAS PubMed.
- D. C. Luther, R. Huang, T. Jeon, X. Zhang, Y.-W. Lee, H. Nagaraj and V. M. Rotello, Adv. Drug Delivery Rev., 2020, 156, 188–213 CrossRef CAS PubMed.
- M. Y. M. Corral, D. E. Alvarez and W. Poon, Curr. Opin. Biotechnol., 2024, 85, 103042 CrossRef PubMed.
- D. C. Luther, T. Jeon, R. Goswami, H. Nagaraj, D. Kim, Y.-W. Lee and V. M. Rotello, Bioconjugate Chem., 2021, 32, 891–896 CrossRef CAS PubMed.
- F. Wang, Y. Wang, X. Zhang, W. Zhang, S. Guo and F. Jin, J. Controlled Release, 2014, 174, 126–136 CrossRef CAS PubMed.
- I. Nakase, H. Hirose, G. Tanaka, A. Tadokoro, S. Kobayashi, T. Takeuchi and S. Futaki, Mol. Ther., 2009, 17, 1868–1876 CrossRef CAS PubMed.
- K. Padari, K. Koppel, A. Lorents, M. Hällbrink, M. Mano, M. C. Pedroso and M. Pooga, Bioconjugate Chem., 2010, 21, 774–783 CrossRef CAS PubMed.
- J. Liu and Z. Cui, Fluorescent Labeling of Proteins of Interest in Live Cells: Beyond Fluorescent Proteins, Bioconjugate Chem., 2020, 31, 1587–1595 CrossRef CAS PubMed.
- C. P. Toseland, Fluorescent labeling and modification of proteins, J. Chem. Biol., 2013, 6, 85–95 CrossRef PubMed.
- S. Snipstad, S. Hak, H. Baghirov, E. Sulheim, Ý. Mørch, S. Lélu, E. von Haartman, M. Bäck, K. P. R. Nilsson, A. S. Klymchenko, C. de Lange Davies and A. K. O. Åslund, Cytometry, Part A, 2016, 91, 760–766 CrossRef PubMed.
- T. Thomsen, A. B. Ayoub, D. Psaltis and H.-A. Klok, Biomacromolecules, 2020, 22, 190–200 CrossRef PubMed.
- S. Irgen-Gioro, S. Yoshida, V. Walling and S. Chong, eLife, 2022, 11, e79903 CrossRef CAS PubMed.
- S. Yoshida, B. K. Maity and S. Chong, J. Phys. Chem. B, 2023, 127, 4165–4173 CrossRef CAS PubMed.
- K. Deprey, N. Batistatou, M. F. Debets, J. Godfrey, K. B. VanderWall, R. R. Miles, L. Shehaj, J. Guo, A. Andreucci, P. Kandasamy, G. Lu, M. Shimizu, C. Vargeese and J. A. Kritzer, ACS Chem. Biol., 2022, 17, 348–360 CrossRef CAS PubMed.
- M. Durymanov and J. Reineke, Front. Pharmacol., 2018, 9, 00971 CrossRef PubMed.
- D. Vercauteren, R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders and K. Braeckmans, Mol. Ther., 2010, 18, 561–569 CrossRef CAS PubMed.
- N. Wytinck, D. S. Sullivan, K. T. Biggar, L. Crisostomo, P. Pelka, M. F. Belmonte and S. Whyard, Sci. Rep., 2020, 10, 12773 CrossRef CAS PubMed.
- N. Chen, Y. He, M. Zang, Y. Zhang, H. Lu, Q. Zhao, S. Wang and Y. Gao, Approaches and materials for endocytosis-independent intracellular delivery of proteins, Biomaterials, 2022, 286, 121567 CrossRef CAS PubMed.
- X. Shao, C. Meng, W. Song, T. Zhang and Q. Chen, Adv. Drug Delivery Rev., 2023, 199, 114977 CrossRef CAS PubMed.
- A. H. Ponsford, T. W. Ryan, A. Raimondi, E. Cocucci, S. A. Wycislo, F. Fröhlich, L. E. Swan and M. Stagi, Autophagy, 2020, 17, 1500–1518 CrossRef PubMed.
- Z. Wang, Y. Zhang, Y. Zhu, Y. Fu, X. Zhang, Z. Shen, S. Gong, Z. Meng and S. Wang, Microchem. J., 2022, 180, 107601 CrossRef CAS.
- C. Wang, T. Zhao, Y. Li, G. Huang, M. A. White and J. Gao, Adv. Drug Delivery Rev., 2017, 113, 87–96 CrossRef CAS PubMed.
- D. Jia, M. Cui and X. Ding, Small, 2024, 20, 04482 Search PubMed.
- J. Zalejski, J. Sun and A. Sharma, J. Imaging, 2023, 9, 192 CrossRef PubMed.
- A. Borrelli, A. Tornesello, M. Tornesello and F. Buonaguro, Cell Penetrating Peptides as Molecular Carriers for Anti-Cancer Agents, Molecules, 2018, 23, 295 CrossRef PubMed.
- K. Desale, K. Kuche and S. Jain, Biomater. Sci., 2021, 9, 1153–1188 RSC.
- R. Bajracharya, J. G. Song, B. R. Patil, S. H. Lee, H.-M. Noh, D.-H. Kim, G.-L. Kim, S.-H. Seo, J.-W. Park, S. H. Jeong, C. H. Lee and H.-K. Han, Drug Delivery, 2022, 29, 1959–1970 CrossRef CAS PubMed.
- Y. Kawaguchi and S. Futaki, Curr. Opin. Chem. Biol., 2024, 81, 102482 CrossRef CAS PubMed.
- P. G. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed.
- F. Reichart, M. Horn and I. Neundorf, J. Pept. Sci., 2016, 22, 421–426 CrossRef CAS PubMed.
- H. D. Herce, D. Schumacher, A. Schneider, A. K. Ludwig, F. A. Mann, M. Fillies, M.-A. Kasper, S. Reinke, E. Krause, H. Leonhardt, M. C. Cardoso and C. P. R. Hackenberger, Nat. Chem., 2017, 9, 762–771 CrossRef CAS PubMed.
- D. Salehi, S. Mozaffari, K. Zoghebi, S. Lohan, D. Mandal, R. K. Tiwari and K. Parang, Cells, 2022, 11, 1156 CrossRef CAS PubMed.
- A. N. Shirazi, R. K. Tiwari, D. Oh, B. Sullivan, A. Kumar, Y. A. Beni and K. Parang, Mol. Pharm., 2014, 11, 3631–3641 CrossRef PubMed.
- A. N. Shirazi, D. Mandal, R. K. Tiwari, L. Guo, W. Lu and K. Parang, Mol. Pharm., 2012, 10, 500–511 CrossRef PubMed.
- N. Nischan, H. D. Herce, F. Natale, N. Bohlke, N. Budisa, M. C. Cardoso and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2014, 54, 1950–1953 CrossRef PubMed.
- H. D. Herce, D. Schumacher, A. Schneider, A. K. Ludwig, F. A. Mann, M. Fillies, M.-A. Kasper, S. Reinke, E. Krause, H. Leonhardt, M. C. Cardoso and C. N. Hackenberger, Nat. Chem., 2017, 9, 762–771 CrossRef CAS PubMed.
- A. F. L. Schneider, M. Kithil, M. C. Cardoso, M. Lehmann and C. P. R. Hackenberger, Nat. Chem., 2021, 13, 530–539 CrossRef CAS PubMed.
- S. Mandal, G. Mann, G. Satish and A. Brik, Enhanced Live-Cell, Angew. Chem., Int. Ed., 2021, 60, 7333–7343 CrossRef CAS PubMed.
- N. P. Gabrielson, H. Lu, L. Yin, K. H. Kim and J. Cheng, Mol. Ther., 2016, 20, 1599–1609 CrossRef PubMed.
- E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed.
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- J. Guo, T. Wan, Z. Qi, Y. Zhang, X. Yan, B. Zhang, Q. Pan, B. Li, Z. Li and Y. Ping, Nano Today, 2024, 56, 102283 CrossRef CAS.
- Y. Kong, K. Zeng, Y. Zhang, J. Shao, J. Yan, J.-Y. Liao, W. Wang, X. Dai, Q. Weng, S. Q. Yao, S. Zeng and L. Qian, Chem. Commun., 2022, 58, 1314–1317 RSC.
- J. Fu, C. Yu, L. Li and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed.
- K. A. Mix, J. E. Lomax and R. T. Raines, J. Am. Chem. Soc., 2017, 139, 14396–14398 CrossRef CAS PubMed.
- J. Zhou, J. Zhang, S. Chen, Q. Lin, R. Zhu, L. Wang, X. Chen, J. Li and H. Yang, Acta Biomater., 2023, 170, 401–414 CrossRef CAS PubMed.
- W. Tai, P. Zhao and X. Gao, Sci. Adv., 2020, 6, 0310 Search PubMed.
- Z. Zhao, X. Liu, M. Hou, R. Zhou, F. Wu, J. Yan, W. Li, Y. Zheng, Q. Zhong, Y. Chen and L. Yin, Adv. Mater., 2022, 34, 2110560 CrossRef CAS PubMed.
- R. Yan, X. Zhao, J. Lei and Q. Zhou, Nature, 2019, 568, 127–130 CrossRef CAS PubMed.
- R. Tang, C. S. Kim, D. J. Solfiell, S. Rana, R. Mout, E. M. Velázquez-Delgado, A. Chompoosor, Y. Jeong, B. Yan, Z.-J. Zhu, C. Kim, J. A. Hardy and V. M. Rotello, ACS Nano, 2013, 7, 6667–6673 CrossRef CAS PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- Z. Le, Q. Pan, Z. He, H. Liu, Y. Shi, L. Liu, Z. Liu, Y. Ping and Y. Chen, ACS Cent. Sci., 2023, 9, 1313–1326 CrossRef CAS PubMed.
- P. Panja and N. R. Jana, J. Phys. Chem. Lett., 2020, 11, 2363–2368 CrossRef CAS PubMed.
- R. Goswami, H. Nagaraj, Y. A. Cicek, N. Nasim, S. S. Mirza, M. A. Hassan, R. Mhaske, D. M. Saravanan, C. Noonan, E. Pham, J. Mager and V. M. Rotello, J. Controlled Release, 2025, 378, 1092–1102 CrossRef CAS PubMed.
- S. Kube, N. Hersch, E. Naumovska, T. Gensch, J. Hendriks, A. Franzen, L. Landvogt, J.-P. Siebrasse, U. Kubitscheck, B. Hoffmann, R. Merkel and A. Csiszár, Langmuir, 2017, 33, 1051–1059 CrossRef CAS PubMed.
- R. Kolašinac, C. Kleusch, T. Braun, R. Merkel and A. Csiszár, Int. J. Mol. Sci., 2018, 19, 346 CrossRef PubMed.
- A. K. Sarkar, K. Debnath, H. Arora, P. Seth, N. R. Jana and N. R. Jana, ACS Appl. Mater. Interfaces, 2022, 14, 3199–3206 CrossRef CAS PubMed.
- M. Hoffmann, N. Hersch, S. Gerlach, G. Dreissen, R. Springer, R. Merkel, A. Csiszár and B. Hoffmann, Int. J. Mol. Sci., 2020, 21, 2244 CrossRef CAS PubMed.
- N. Hueppe, F. R. Wurm and K. Landfester, Macromol. Rapid Commun., 2022, 44, 00611 Search PubMed.
- T. Iwata, H. Hirose, K. Sakamoto, Y. Hirai, J. Vincent, M. Akishiba, M. Imanishi and S. Futaki, Angew. Chem., Int. Ed., 2021, 60, 19804–19812 CrossRef CAS PubMed.
- R. Mout, M. Ray, T. Tay, K. Sasaki, G. Y. Tonga and V. M. Rotello, General Strategy for Direct Cytosolic Protein, ACS Nano, 2017, 11, 6416–6421 CrossRef CAS PubMed.
- Y.-W. Lee, D. C. Luther, R. Goswami, T. Jeon, V. Clark, J. Elia, S. Gopalakrishnan and V. M. Rotello, J. Am. Chem. Soc., 2020, 142, 4349–4355 CrossRef CAS PubMed.
- D. C. Luther, Y.-W. Lee, H. Nagaraj, V. Clark, T. Jeon, R. Goswami, S. Gopalakrishnan, S. Fedeli, W. Jerome, J. L. Elia and V. M. Rotello, ACS Nano, 2022, 16, 7323–7330 CrossRef CAS PubMed.
- R. Goswami, V. Lehot, Y. A. Çiçek, H. Nagaraj, T. Jeon, T. Nguyen, S. Fedeli and V. M. Rotello, Pharmaceutics, 2023, 15, 218 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |