
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRelease performance and crystallization of racemic and enantiopure praziquantel amorphous solid dispersion in various media†
Benedito Roberto
de Alvarenga Junior
 and
Lynne S.
Taylor
*
and
Lynne S.
Taylor
*
Department of Industrial and Molecular Pharmaceutics, College of Pharmacy, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: lstaylor@purdue.edu; Fax: +765-494-6545; Tel: +765-496-6614
First published on 8th July 2025
Abstract
Praziquantel (PZQ) is the first-line treatment for schistosomiasis, but its low aqueous solubility and extensive first-pass metabolism limit PZQ's bioavailability. Furthermore, the commercial formulation of PZQ includes the inactive (S)-PZQ enantiomer, which causes unwanted side effects and a bitter taste. This work aimed to evaluate the impact of chirality on PZQ's performance in amorphous solid dispersion (ASD) formulations prepared from both racemic and the active (R)-PZQ enantiomer, with additional studies on polymer type and processing method. ASDs of (R,S)-PZQ and (R)-PZQ at 30% drug loading were prepared with HPMCAS MF and HPMC E5 via solvent evaporation (SE) and hot-melt extrusion (HME). Release testing was conducted in aqueous media with different pH values and in biorelevant media simulating fasted- and fed-state conditions. Results demonstrated that ASDs significantly enhanced PZQ concentrations, with the amorphous solubility being up to 8-fold higher than that of the corresponding crystalline form. HPMCAS-based ASDs showed pH-dependent release, with poor release at gastric pH but achieving near-complete release with crystallization inhibition at intestinal pH conditions, while HPMC-based ASDs exhibited faster gastric release but reduced stability due to crystallization, which was confirmed by polarized light microscopy (PLM) and powder X-ray diffraction (PXRD). (R)-PZQ-HPMCAS ASDs outperformed (R,S)-PZQ-HPMCAS ASDs in simple media at pH 6.5 at high target concentration, which was attributed to a slightly higher amorphous solubility. However, both ASDs exhibited comparable release in fasted-state media due to bile salt-enhanced solubility. PZQ-ASDs showed crystallization when evaluated in FeSSIF-V2 and did not release well. Different processing methods minimally affected release profiles, highlighting HME's potential as a scalable, solvent-free method. These findings suggest that (R)-PZQ-HPMCAS is a promising alternative to commercial racemic PZQ formulations, potentially reducing side effects and improving patient compliance through allowing for a reduced pill burden.
1. Introduction
Schistosomiasis is a tropical disease caused by trematode worms of the genus Schistosoma that occurs in regions where water is contaminated or sanitation conditions are inadequate.1,2 According to the World Health Organization (WHO), it is estimated that more than 250 million people worldwide required preventive treatment in 2021.3 Currently, praziquantel (PZQ) is an anthelmintic drug recommended as the first-line treatment for all types of schistosomiasis and is also included in the WHO Model List of Essential Medicines for the treatment of both adults and children.3,4PZQ belongs to Biopharmaceutics Classification System (BCS) class II and is characterized by high permeability and low solubility (0.4 mg mL−1 in water at 25 °C), which results in a slow dissolution rate in the gastrointestinal (GI) tract and consequently low bioavailability.5–7 Furthermore, PZQ undergoes an extensive first-pass effect metabolism, converting into inactive metabolites rapidly.6,8–10 In this context, it is necessary to administer a high drug dosage, usually 20 mg kg−1 three times a day at intervals of 4 to 6 hours or as a single dose of 40 mg kg−1 for preventive chemotherapy.11,12 Therefore, investigations are warranted to obtain formulations with enhanced solubility and bioavailability to decrease the PZQ therapeutic dose and, thereby, the tablet size, which is difficult to swallow, in particular for pediatric treatment.
PZQ [2-(cyclohexylcarbonyl)-1,2,3,6,7,11b-hexahydro-4H-pyrazino[2,1a]isoquinolin-4-one] is commercially available in tablet form as the innovator product Biltricide®, as well as various generic versions, and is dosed as a crystalline racemic mixture at a proportion of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.12,13 The (R)-PZQ enantiomer is responsible for the pharmacological activity, while the (S)-PZQ enantiomer has little or no antischistosomal action and provides a bitter taste along with side effects such as abdominal pain, fever, nausea, and vomiting.8,14–16 Given the drawbacks associated with PZQ, a possible way of overcoming its low bioavailability is to increase PZQ solubility through amorphous solid dispersion (ASD).
:1.12,13 The (R)-PZQ enantiomer is responsible for the pharmacological activity, while the (S)-PZQ enantiomer has little or no antischistosomal action and provides a bitter taste along with side effects such as abdominal pain, fever, nausea, and vomiting.8,14–16 Given the drawbacks associated with PZQ, a possible way of overcoming its low bioavailability is to increase PZQ solubility through amorphous solid dispersion (ASD).
ASDs consist of a homogeneous dispersion of the active pharmaceutical ingredient (API) in an inert excipient carrier in the amorphous state.17,18 Since the API is in the amorphous state within an ASD formulation, no energy is necessary to break the crystal lattice, which increases the apparent water solubility, dissolution rate, and bioavailability.19–21 Moreover, improvements in release and wettability due to the presence of a hydrophilic polymer are also factors that may contribute to enhanced drug release.22,23
Several studies have reported release studies of PZQ-ASD formulations prepared by solvent evaporation (SE),24–28 the fusion method,29 co-precipitation,29,30 co-grinding,31 spray drying,32 and hot-melt extrusion (HME).33,34 Most of these studies performed dissolution testing in simple aqueous media. It is also valuable to evaluate the dissolution profile in media that simulate fasted and fed states, in particular for poorly soluble compounds.35 While the impact of biorelevant media has been studied for a formulation containing crystalline PZQ,36 to the best of our knowledge, no studies have explored the influence of biorelevant media on the release rate of PZQ-ASD formulations. In addition, there are limited studies on the release properties of ASDs prepared with the pharmacologically active (R)-PZQ enantiomer.
Therefore, this work aims to evaluate the impact chirality on the PZQ release behavior from ASDs in aqueous media at different pH conditions and in fasted/fed-state simulated GI fluids (FaSSGF, FaSSIF, and FeSSIF-V2), while also varying polymer type and processing method. ASDs of (R,S)-PZQ and (R)-PZQ at 30% drug loading (DL) were prepared with HPMCAS MF and HPMC E5 LV via solvent evaporation (SE) and hot-melt extrusion (HME). Crystallization during release studies was analyzed using polarized light microscopy (PLM) and powder X-ray diffraction (PXRD). Fig. 1 shows the chemical structure of (R)-PZQ, (S)-PZQ, and polymers.
 | ||
| Fig. 1 Chemical structures of (R)-PZQ (a), (S)-PZQ (b), HPMCAS (c), and HPMC (d). | ||
2. Experimental section
2.1. Materials
(R)-PZQ (purity 99.9%) was sourced from Tongli Biomedical Co. (Zhangjiagang, China).(R,S)-PZQ (purity 95%+) was sourced from ChemShuttle (Burlingame, CA). Hydroxypropyl methylcellulose acetate succinate MF grade (HPMCAS-MF; AQOAT AS-MF, Shin-Etsu, Tokyo, Japan) and hydroxypropyl methylcellulose E5 LV grade (HPMC; Methocel E5 Premium LV, The Dow Chemical Company, Midland, MI) were the polymers used in this work. Dimethylsulfoxide (DMSO), dichloromethane (DCM), methanol (MeOH), sodium chloride (NaCl), sodium hydroxide (NaOH), hydrochloric acid (HCl), and glacial acetic acid (CH3COOH) were purchased from Fisher Chemical (Fair Lawn, NJ). Sodium phosphate monobasic anhydrous (NaH2PO4) and maleic acid (C4H4O4) were purchased from Acros Organics (Geel, Belgium). ortho-Phosphoric acid 85% (H3POH4, Switzerland) was obtained from Merck, while sodium acetate anhydrous (CH3COONa) was purchased from Macron Fine Chemicals (Center Valley, PA). Biorelevant simulated gastric and intestinal fluids, including FaSSIF/FaSSGF, and FeSSIF-V2 were purchased from Biorelevant (London, UK).
2.2. Methods
For the fasted-state simulated media preparations, 0.06 g of Biorelevant 3F powder was added in 900 mL of the prepared HCl solution at pH 1.6, while 2.24 g was dissolved in 900 mL of PBS at pH 6.5 to produce FaSSGF and FaSSIF, respectively. The fed-state simulated medium was prepared by adding 9.60 g of Biorelevant FeSSIF-V2 powder to 900 mL of maleate buffer at pH 5.8. The SIF powder was solubilized, and the volume was made up to 1 L with the appropriate buffer. For two-stage experiments in biorelevant media, Biorelevant 3F powder was added to a concentrated PBS at pH 7.3 to obtain FaSSIF at a concentration 10 times higher than required by the Biorelevant media protocol. Biorelevant media were equilibrated for 2 hours before use and were used within 48 h. Table 1 shows the composition of fasted- and fed-state simulated GI media.
| Composition | Fasted state | Fed state | |
|---|---|---|---|
| FaSSGF | FaSSIF | FeSSIF-V2 | |
| SIF powder (g) | 0.06 | 2.24 | 9.76 |
| Sodium taurocholate (mmol L−1) | 0.08 | 3.00 | 10 |
| Phospholipids (mmol L−1) | 0.02 | 0.75 | — |
| Lecithin (mmol L−1) | — | — | 2 |
| Glycerol monooleate (mmol L−1) | — | — | 5 |
| Sodium oleate (mmol L−1) | — | — | 0.8 |
| HCl solution pH 1.6 (L) | 1 | — | — |
| PBS pH 6.5 (L) | — | 1 | — |
| Maleate buffer (L) | — | — | 1 |
| Osmolarity (mOsm kg−1) | 120 | 270 | 387 |
| Buffer capacity (mM L−1 pH−1) | Unbuffered | 10 | 25 |
:1 v/v) to prepare ASDs with DL of 30% for (R,S)-PZQ and (R)-PZQ. The ASDs were subjected to further drying overnight in a vacuum oven at room temperature to remove residual solvents. A 6750 Freezer/Mill cryogenic impact mill (SPEX SamplePrep, Metuchen, NJ) was used to pulverize ASDs, and the powders were passed through a 60 mesh (250 μm) sieve to obtain a uniform particle size.
000 rpm (37 °C, 15 minutes) using an Optima L-100 XP ultracentrifuge (SW 41Ti rotor) (Beckman Coulter, Inc., Brea, CA). The supernatant was diluted in acetonitrile and water (60:40 v/v) and 10 μL was injected into an Agilent high-performance liquid chromatography (HPLC) 1260 Infinity Series (Agilent Technologies, Santa Clara, CA), equipped with a G4225A HiP degasser, quaternary pump G1311B 1260 Quat Pump, an automatic sampler G1329B 1260 ALS, column compartment G1316A 1260 TCC, and UV-Vis detector G1314F 1260 VWD. An Eclipse plus C18 column (4.6 × 150 mm, 5 μm) was used with a mobile phase comprising acetonitrile and water (60:40 v/v) at a flow rate of 1 mL min−1, an injection volume of 10 μL, and an ultraviolet (UV) detector set at 220 nm. Data were collected and processed using OpenLab CDS ChemStation Edition, Rev. C.01.05. A stock solution of (R,S)-PZQ at 20 mg mL−1 in the mobile phase was prepared, and PZQ solubility was obtained from a standard curve in the range of 1–300 μg mL−1.
For single-stage experiments, ASD powders of (R,S)-PZQ and (R)-PZQ were added to 50 mL of PBS at pH 6.5, FaSSIF, or FeSSIF-V2. The two-stage release experiments were performed under gastric conditions at pH 1.6, 3.0, or 5.0, for 1 h, then shifted to pH 6.5 for an additional hour. The drug release was conducted in 45 mL of gastric fluid at pH 1.6, followed by the addition of 5 mL of concentrated PBS pH 7.3 to adjust the pH of the solution to pH 6.5. Release studies in media at pH 3.0 and pH 5.0 were conducted in 47 mL and 48 mL, respectively, and after 1 h, volumes were made up to 50 mL with concentrated PBS at pH 7.3. The fasted-state two-stage experiments were performed in 45 mL of FaSSGF (pH 1.6), followed by the addition of 5 mL of 10× concentrated FaSSIF solution (0.57 mol L PBS, pH 7.3) to achieve 50 mL of FaSSIF pH 6.5. The fed-state condition was performed in 45 mL of acetate buffer pH 4.5, followed by the addition of 5 mL of 10× concentrated FeSSIF-V2 (pH 12) to achieve 50 mL FeSSIF-V2 pH 5.8.
A 5 mm fiber optic dip probe was used to monitor drug concentration in aqueous and fasted-state media, and for fed-state media, a 2 mm fiber optic dip probe was required. Second derivative analysis was applied to correct the spectral baseline, and a calibration curve of the area under the curve (AUC) for the range 278–285 nm was used to calculate the drug concentration in all media, except for FeSSIV-V2, for which a second derivative analysis at 280 nm was employed. Calibration curves in solutions at pH 1.6, 4.5, 6.5, FaSSGF, FaSSIF, and FeSSIF-V2 were built over the concentration range of 10–1500 μg mL−1, while those at pH 3.0 and 5.0 ranged from 10–1400 μg mL−1.
3. Results
3.1. Amorphous and crystalline solubility of PZQ
Table 2 presents the amorphous and crystalline solubilities of PZQ in aqueous media with different pH values and in fasted- and fed-state simulated media. The solubility of crystalline PZQ in different media at 37 °C ranged from 0.17 to 0.31 mg mL−1 for (R,S)-PZQ, and from 0.23 to 0.43 mg mL−1 for (R)-PZQ. The corresponding amorphous solubility for (R,S)-PZQ ranged from 1.17 to 1.54 mg mL−1, while that of (R)-PZQ ranged from 1.20 to 1.67 mg mL−1. The PZQ solubilities are in line with the previously reported values for racemic and enantiopure PZQ in aqueous media.28,40,41 Solubility was found to be independent of pH, consistent with the molecular structure. Furthermore, solubilization by micellar species present in biorelevant media was low. Finally, little difference was noted between the amorphous solubility values for (R,S)-PZQ versus (R)-PZQ, while the crystalline form of (R)-PZQ showed a slightly higher solubility than (R,S)-PZQ.| Medium | Amorphous solubility (mg mL−1) | Crystalline solubility (mg mL−1) | (R,S)-PZQ A/Ca | (R)-PZQ A/Ca | ||
|---|---|---|---|---|---|---|
| (R,S)-PZQ | (R)-PZQ | (R,S)-PZQ | (R)-PZQ | |||
| a Ratio between amorphous and crystalline PZQ solubility. | ||||||
| pH 1.6 | 1.41 ± 0.02 | 1.40 ± 0.02 | 0.19 ± 0.01 | 0.27 ± 0.00 | 7.4 | 5.2 |
| pH 3.0 | 1.26 ± 0.02 | 1.26 ± 0.01 | 0.17 ± 0.00 | 0.23 ± 0.02 | 7.6 | 5.5 |
| pH 4.5 | 1.43 ± 0.01 | 1.35 ± 0.04 | 0.21 ± 0.01 | 0.29 ± 0.01 | 6.8 | 4.7 |
| pH 5.0 | 1.23 ± 0.02 | 1.20 ± 0.01 | 0.16 ± 0.01 | 0.24 ± 0.01 | 7.7 | 5.0 |
| pH 5.8 | 1.30 ± 0.09 | 1.33 ± 0.01 | 0.18 ± 0.00 | 0.27 ± 0.00 | 7.2 | 4.9 |
| pH 6.5 | 1.17 ± 0.05 | 1.39 ± 0.05 | 0.17 ± 0.01 | 0.27 ± 0.01 | 6.9 | 5.1 |
| FaSSGF | 1.55 ± 0.02 | 1.63 ± 0.04 | 0.22 ± 0.01 | 0.28 ± 0.00 | 7.0 | 5.8 |
| FaSSIF | 1.49 ± 0.11 | 1.67 ± 0.04 | 0.22 ± 0.01 | 0.32 ± 0.01 | 6.8 | 5.2 |
| FeSSIF-V2 | 1.74 ± 0.09 | 1.70 ± 0.10 | 0.31 ± 0.01 | 0.43 ± 0.00 | 5.6 | 4.0 |
3.2. Powder release studies of PZQ-ASDs
The influence of pH and fasted/fed-state simulated media on drug release from ASD powders manufactured by SE or HME, containing enantiopure versus racemic PZQ and formulated with HPMCAS MF or HPMC E5 LV, was evaluated. These polymers have demonstrated promising drug release and stability at a DL of 30% in previous studies,42 and hence were evaluated further. | ||
| Fig. 2 Single- and two-stage release experiments with PZQ ASD samples prepared by SE and HME in aqueous media. PZQ target concentrations: 0.667 mg mL−1 for A and B; 1.5 mg mL−1 for C and D. | ||
At a PZQ target concentration of 1.5 mg mL−1, (R,S)-PZQ-HPMCAS prepared by different processing methods showed a similar release profile in PBS pH 6.5 (single-stage), reaching 78% and 71% for the HME and SE methods, respectively (Fig. 2C). Additionally, (R)-PZQ-HPMCAS SE showed a higher dissolution rate than (R)-PZQ-HPMCAS HME, but by the end of the experimental time frame, both samples achieved a drug release of approximately 88%. As observed at the low target concentration, the HPMCAS-based ASD released poorly in pH 1.6 medium (Fig. 2D). After shifting to pH 6.5, the drug release of (R,S)-PZQ-HPMCAS peaked at 73% and 69% for samples obtained by SE and HME methods, respectively, while the (R)-PZQ-HPMCAS SE drug release achieved 91%.
For the ASD formulations containing HPMC, the (R,S)-PZQ-HPMC SE sample reached a drug release of 82% at the end of the single-stage experiments at a concentration of 667 μg mL−1 (Fig. 2A). The same formulation showed a drug release of 96% at 68 minutes and the concentration level remained stable at intestinal pH (Fig. 2B). The ASD composed of (R)-PZQ with HPMC E5 LV presented a maximum drug release of 86% (∼25 min) and 99% (∼49 min) in the single-stage and two-stage experiments, respectively. Subsequently, these samples experienced a slight drop of 1% in drug concentration after 120 minutes, likely due to drug crystallization.
The (R,S)-PZQ-HPMC SE sample released 55% at 39 min and increased slowly to 65% in the single-stage experiment at a target concentration of 1.5 mg mL−1. In contrast, (R)-PZQ-HPMC SE showed a drug release of 65% at the same time, and afterwards, dropped to 60% over the experimental time frame period (Fig. 2C). In the two-stage experiments (Fig. 2D), HPMC-based ASDs exhibited similar drug release extent, with the formulation containing the PZQ racemate releasing slower (71% at 60 minutes) than the enantiopure form (71% at 26 minutes). After reaching maximum release, (R,S)-PZQ-HPMC SE remained almost constant over the dissolution experiment, whereas (R)-PZQ-HPMC SE concentration decreased.
Two-stage experiments were also carried out with media at pH 3.0 or pH 5.0, followed by a shift to pH 6.5. The HPMCAS-based ASDs did not exceed a drug release of 25% in the first dissolution state (gastric pH), but the release increased after shifting to intestinal pH. The samples prepared with HPMCAS presented similar drug release profiles and ranged from 62% to 77% in dissolution experiments at low target concentration (Fig. S1A and S1B†), and from 43% to 58% at high target concentration (Fig. S1C and S1D†). At low PZQ target concentration, HPMC-based ASDs showed very similar drug release profiles, with a drug release higher than 85% up to 35 min, which remained steady over the intestinal pH (Fig. S1A and S1B†). On the other hand, at high target concentration, HPMC-based ASDs released slightly faster in pH 5.0 than in pH 3.0. For (R)-PZQ-HPMC SE, a maximum drug release of 72% was observed at pH 5.0 and 68% at pH 3.0, and subsequently, the drug concentration decreased in both experiments (Fig. S1C and S1D†) due to PZQ crystallization.
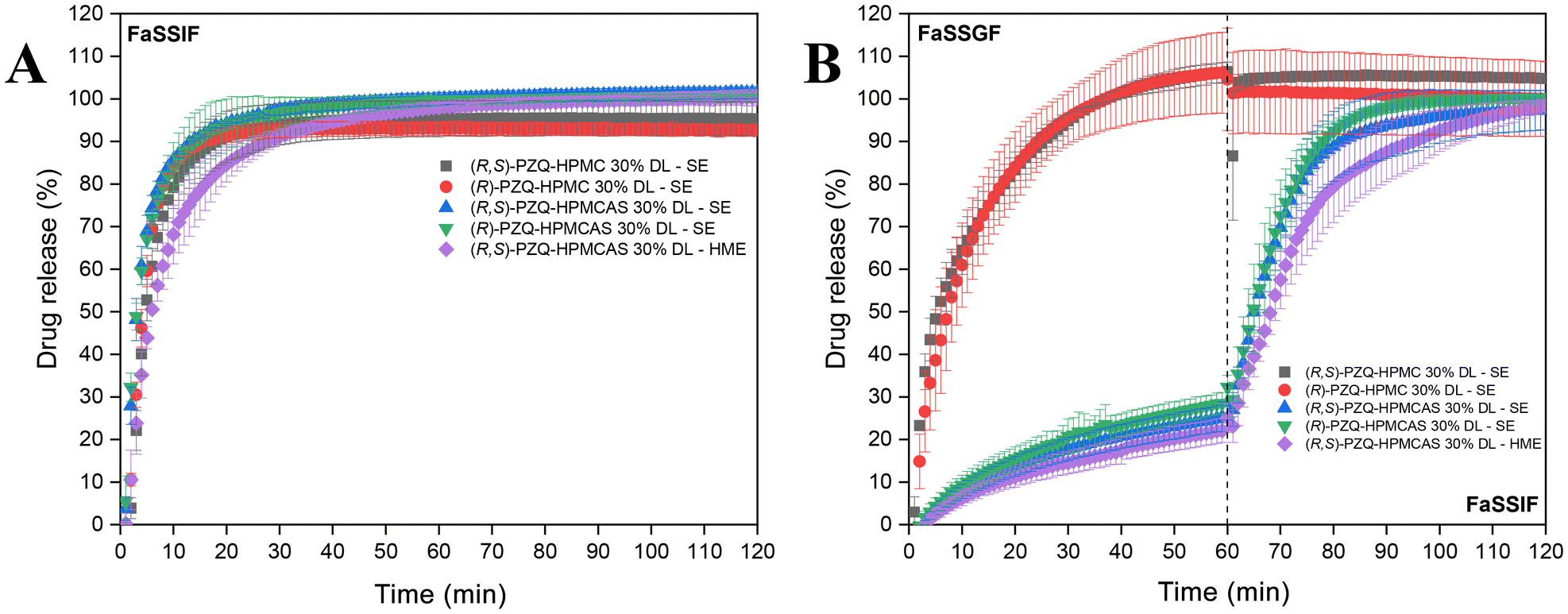 | ||
| Fig. 3 Single- and two-stage release experiments in fasted-state simulated media with PZQ ASD samples prepared by SE and HME. PZQ target concentrations: 0.667 mg mL−1 for A and B; 1.5 mg mL−1 for C and D. | ||
HPMC-based ASDs achieved 93% (∼25 min) in FaSSIF and complete drug release in simulated gastric media at low target concentration (Fig. 3A and B). (R,S)-PZQ-HPMC and (R)-PZQ-HPMC reached respectively 82% (∼40 minutes) and 75% (∼19 minutes) at a high target concentration in the simulated intestinal fluid, and then exhibited a 30% drop in release (Fig. 3C). A decrease in the release was also observed after reaching ∼75% release for formulations with HPMC in the two-stage experiments (Fig. 3D). The drop in release reflects a concentration decreased resulting from PZQ crystallization. HPMC has been demonstrated to be less effective in drug stabilization against crystallization than either HPMCAS MF or LF grades.42 The ASD samples had an increase in the drug release in FaSSIF medium compared to in PBS pH 6.5 medium at high target concentration, which was more evident with samples prepared with the racemic form of PZQ and HPMCAS. This increase in drug release is likely due to the slightly enhanced solubility of the drug in the FaSSIF medium (Table 2). Bile salts present in the medium can improve drug solubility through micellar incorporation, improving the dissolution rate.45–47 However, this effect is relatively minor for PZQ.
 | ||
| Fig. 4 Single- and two-stage release experiments in fed-state simulated media with PZQ ASD samples prepared by SE and HME. PZQ target concentrations: 0.667 mg mL−1 for A and B; 1.5 mg mL−1 for C and D. | ||
HPMC-based ASDs presented complete release (∼35 minutes) at pH 4.5 and 70% release at low and high target concentration, respectively. Upon shifting to FeSSIF-V2 medium, a decrease in drug release was observed for HPMC-based ASDs, which may be related to drug crystallization.
3.3. PLM analysis
PLM images were collected during release studies at 5, 60, and 120 minutes to evaluate the presence of crystals at a PZQ target concentration of 1.5 mg mL−1. PLM images showed particles with no evidence of birefringence for HPMCAS-based ASDs in all instances (Fig. S2–S7†). Even in experiments performed in fed-state simulated media using a two-stage dissolution approach, where drug release was less than 32%, no evidence of PZQ crystallization in HPMCAS-based ASDs was observed using PLM. Thus, the particles appear to consist of undissolved ASD.Birefringence was clearly observed in both the racemic and enantiopure forms of PZQ with HPMC E5 LV after 60 min (Fig. 5, S8, and S9†). Birefringent particles with needle-like and rectangular morphologies were observed for (R)-PZQ-HPMC SE and (R,S)-PZQ-HPMC SE samples, respectively. Since HPMC-based ASDs already presented evidence of crystallization in both single-stage and two-stage experiments in aqueous media, PLM images were not collected from these samples in FaSSGF or FaSSIF.
 | ||
| Fig. 5 PLM images of (R)-PZQ-HPMC ASD obtained during the release studies in aqueous media. | ||
In fed-state simulated media, HPMC-based ASDs showed no presence of crystals in acetate buffer pH 4.5, but crystallization was observed at the beginning of dissolution in the single-stage experiments and after shifting to FeSSIF-V2 medium (Fig. 6 and S10†).
 | ||
| Fig. 6 PLM images of neat HPMC E5 LV, (R,S)-PZQ-HPMC SE, and (R)-PZQ-HPMC SE obtained during the release studies in fed-state simulated media. | ||
3.4. PXRD analysis
 | ||
| Fig. 7 Diffractograms of neat PZQ and PZQ ASDs prepared by solvent evaporation after release studies in single and two-stages experiments. (A) (R,S)-PZQ-HPMC and (B) (R)-PZQ-HPMC in aqueous media, (C) PZQ-ASDs formulated with HPMC, and (D) PZQ-ASDs with HPMCAS in fed-state simulated media. | ||
The (R,S)-PZQ-HPMC ASD sample exhibited small peaks at 7.8°, 15.0°, 19.8°, and 25.2° 2θ, characteristic of crystalline (R,S)-PZQ (Fig. 7A). Samples from release studies with a pH switch from 5.0 to 6.5 presented a small peak at 31.5°, attributed to NaOH. The (R)-PZQ-HPMC SE ASD presented characteristic peaks of the neat crystalline (R)-PZQ at 8.3°, 14.9°, 17.4°, 19.6°, 21.8°, 24.3°, and 27.3° 2θ (Fig. 7B). Some peaks were slightly shifted due to sample non-uniformity in the aluminum cavity holder. Since PXRD was utilized solely for qualitative analysis, the variations in signal intensities of (R)-PZQ-HPMC and (R,S)-PZQ-HPMC SE should not be attributed only to crystalline concentration in the sample. Additionally, factors such as crystal size and degree of crystalline ordering, as well as sample mass also affect signal intensity.
Formulations prepared with racemic and enantiopure forms of PZQ presented crystalline peaks with either HPMC or HPMCAS in fed-state simulated media in both single and two-stage experiments (Fig. 7C, D, and S12†). The peaks were more intense for HPMC ASDs, while they were much smaller for HPMCAS-based ASDs. ASD formulations containing HPMCAS with (R,S)-PZQ or (R)-PZQ in aqueous media or fasted-state simulated media were not analyzed by PXRD, since these samples showed no evidence of crystallinity in PLM images or a decrease in drug release during the dissolution experiments.
4. Discussion
4.1. Crystalline and amorphous solubility of PZQ
The solubility of PZQ was determined in aqueous media across various pH levels, as well as in fasted- and fed-state simulated GI media. The amorphous solubility of (R,S)-PZQ and (R)-PZQ was approximately 8- and 6-fold greater than in the corresponding crystalline state. According to the t-test, there is no statistically significant difference in amorphous solubility at the 95% confidence level between the racemic and enantiopure forms of praziquantel (PZQ) across different media, except in PBS pH 6.5, where a significant difference was observed in the amorphous solubility.The amorphous and crystalline solubilities of PZQ exhibited minimal variation in aqueous media. This result is consistent with the calculated pKa of 1.14 (Fig. S13†),48 which suggests limited ionization-dependent solubility changes across the studied pH range. The solubility of PZQ in both amorphous and crystalline forms exhibited higher values in FeSSIF-V2, probably due to the higher concentration of bile salts (10 mmol L−1 sodium taurocholate) present in this medium. However, the solubility enhancement observed in FeSSIF-V2 was modest which suggests that PZQ has limited interaction with micellar components. Interestingly, it has been reported that both high fat and high carbohydrate diets increase the bioavailability of praziquantel, with a high carbohydrate diet resulting in a greater increase.49 Thus, the positive food effect could depend on factors other than an increase in drug solubility which is quite moderate in FeSSIF-V2.
4.2. Impact of polymer type on drug release
ASD formulations can enhance drug solubility leading to supersaturated solutions upon dissolution, resulting in higher membrane flux and improved absorption and bioavailability. The polymer used to form the ASD, serving an important role in maintaining drug supersaturation by preventing both nucleation and crystal growth – processes involved in crystallization.50,51 HPMCAS and HPMC have been widely employed as drug crystallization inhibitors.52,53 Furthermore, a previous study has demonstrated that they are able to delay crystallization of PZQ,42 motivating the studies performed herein.The influence of the polymer on PZQ release was evaluated in ASD samples composed of HPMCAS MF or HPMC E5 LV with (R,S)-PZQ and (R)-PZQ prepared via SE. ASD samples prepared with HPMC E5 LV showed a greater extent of drug release in the first dissolution stage than those prepared with HPMCAS MF in experiments initiated at pH 1.6, 3.0, 4.5, 5.0, fasted- and fed-state simulated media (two-stage) (Fig. 2–4, and S1†). (R)-PZQ-HPMC SE showed a decrease in drug release with time in all dissolution media studied at a high PZQ dose concentration, and (R,S)-PZQ-HPMC SE had a decrease in drug release with time only in the fasted- and fed-state simulated media (Fig. 3 and 4). These observations indicate that (R)-PZQ has a higher tendency to crystallize from highly supersaturated solutions than the racemic form. This observation is consistent with a previous study that found that (R)-PZQ had a shorter nucleation induction time than the racemic form, from supersaturated solutions.42 The release results further demonstrated that HPMC E5 LV was not sufficiently effective in preventing PZQ crystallization during dissolution, as confirmed by PLM and PXRD (Fig. 5 and S9†). ASD samples with HPMC E5 LV (approximately 70%) showed little difference in the extent of release for different pH conditions. On the other hand, formulations containing HPMCAS MF did not show a decrease in drug concentration with time, except in fed-state simulated media, consistent with HPMCAS MF being a better nucleation inhibitor.47,54,55 XRD analysis revealed that formulations containing HPMCAS exhibited drug crystallization in FeSSIF-V2. Thus, the higher concentration of bile salts and lecithin present in FeSSIF-V2 promoted crystallization, which could not be as effectively inhibited by HPMCAS when compared to buffer. A previous study with the poorly soluble compounds, posaconazole and atazanavir, found that induction times were much longer in buffer than in FaSSIF when HPMCAS was present in both instances.56 In other words, the crystallization inhibitory power of a polymer was diminished when bile salts and lecithin were present. Therefore, the increased crystallization tendency following (R,S)-PZQ and (R)-PZQ ASD dissolution in FeSSIF-V2 is in broad agreement with these previous observations.
HPMCAS-based ASDs released poorly at low pH, but had similar or greater release than HPMC E5 LV ASDs after 2 h in PBS pH 6.5 (single stage), pH-shift experiment from pH 1.6 to pH 6.5 fasted state media, and in FeSSIF-V2 (single stage). HPMCAS has pH-dependent solubility and is insoluble at gastric pH due to the minimal ionization of succinoyl groups, explaining why ASD samples with this polymer showed limited dissolution at low pH, with drug release below 30%. This aligns with its resistance to gastric dissolution and its role as an enteric polymer, solubilizing in media with pH ∼ 5.5–6.
No decrease in drug release with time was observed with HPMCAS-based ASDs, suggesting that HPMCAS MF better inhibits crystallization from supersaturated PZQ solutions than HPMC E5 LV. It is worth noting that neither polymers could prevent PZQ crystallization in FeSSIF-V2, as observed through PLM and PXRD analyses, although the extent of crystallization was much smaller for HPMCAS formulations (Fig. 7). Samples formulated with HPMC E5 LV showed nearly immediate PZQ crystallization in FeSSIF-V2 (Fig. 6 and S10†), which could be related to the rapid decrease in drug release extent during the transition to FeSSIF-V2 in the two-stage experiments. Further studies are needed to understand the stability of supersaturated PZQ solutions in fed-state simulated media.
4.3. Impact of chirality on drug release
The therapeutic efficacy of PZQ is attributed mostly to (R)-PZQ while the (S) enantiomer and its metabolites do not contribute to the therapeutic effect, but does lead to side effects and a bitter taste.57 A formulation produced with only (R)-PZQ is expected to eliminate most of the drawbacks of the racemic form, and would further allow the tablet size/overall dose to be reduced by half. Given this, the exclusion of (S)-PZQ is crucial to improve dosing of PZQ. In this work, (R)-PZQ-ASDs were evaluated to determine whether they could provide better performance and serve as an alternative to the commercially available racemic crystalline PZQ mixture. If bioavailability enhancement could be achieved, as has been observed in preclinical models with racemic PZQ-ASDs,24,27,58 the dose could be further reduced, mitigating the increased production costs of isolating the (R) enantiomer.The enantiomer (R)-PZQ formulated with HPMC showed a similar or better dissolution rate and extent of drug release than the corresponding ASD with (R,S)-PZQ in aqueous media at low or high target concentrations. After achieving the maximum extent of drug release, the (R)-PZQ-HPMC SE ASD exhibited a faster decrease in drug concentration at the high target concentration than (R,S)-PZQ-HPMC SE, particularly in pH-shift experiments from pH 3.0 or 5.0 to 6.5, in PBS pH 6.5 (single-stage) and FaSSIF (single stage). Crystals of both the enantiopure and racemic forms of PZQ formulated with HPMC were clearly observed during PLM monitoring, which was also confirmed by PXRD analysis after release studies. Notably, (R,S)-PZQ-HPMC and (R)-PZQ-HPMC showed a greater drug release in FaSSIF as compared to in PBS pH 6.5 at high concentration, but the presence of bile salts may have also contributed to the faster crystallization of (R)-PZQ. No significant difference was observed between the enantiopure and racemic mixture of PZQ formulated with either HPMCAS or HPMC in FeSSIF-V2. PZQ crystallization tendency is theoretically associated with the crystal lattice energy and solvation of components that vary for each drug solid state form and affect the crystallization kinetics.59 Since the (R,S)-PZQ anhydrous (Form A) and (R)-PZQ hemihydrate were formed herein, polymers can interact differently with the drug based on its conformational structure, thereby impacting crystal growth, and the thermodynamic driving force for crystallization likely differs between racemate and enantiomer. In the enantiopure (R)-PZQ, all molecules have the same stereochemistry, which can lead to more efficient molecular packing and stronger intermolecular interactions. This can promote a higher nucleation rate compared to the racemic compound, where the presence of both R- and S-enantiomers leads to less favorable packing and where the counter-enantiomer can act as an “impurity”.60 The greater crystallization tendency of an enantiomer over a racemate in the amorphous form has been noted previously.60
The enantiopure form and racemic PZQ mixture, when formulated with HPMCAS, released drug similarly in all release conditions studied, except in PBS pH 6.5 and pH shift experiment from pH 1.6 to 6.5, where (R)-PZQ-HPMCAS (SE) had a better release rate and extent than (R,S)-PZQ-HPMCAS (SE). Based on the calculated pKa of 1.14, PZQ remained predominantly as a neutral form under the conditions studied, suggesting minimal pH-dependent solubility and little-to-no ionization in low-pH conditions. Indeed, the amorphous and crystalline solubilities of (R,S)-PZQ and (R)-PZQ showed no substantial differences across the pH range investigated. The improved release observed for (R)-PZQ-HPMCAS compared to (R,S)-PZQ-HPMCAS is likely attributable to the slightly higher amorphous solubility of (R)-PZQ in PBS pH 6.5. This is supported by the maximum concentrations achieved during dissolution, which were approximately 1.4 mg mL−1 for (R)-PZQ-HPMCAS SE and 1.1 mg mL−1 for (R,S)-PZQ-HPMCAS SE, in agreement with the amorphous solubility data presented in Table 1.
4.4. Impact of processing method type on drug release
(R,S)-PZQ has good thermal stability upon reaching its melting point (Tm) of 136–142 °C,61 but undergoes significant thermal decomposition above 200 °C.62 (R)-PZQ possesses a slightly lower Tm 113–115 °C,63 and, unless influenced by chirality-specific interactions, this enantiomer should likely follow a similar decomposition temperature to that of the racemic PZQ mixture. The glass transition temperature (Tg) of (R,S)-PZQ has been reported to be around 38 °C,14 in good agreement with the value obtained herein (around 37 °C, Fig. S14†). The Tg of (R)-PZQ was observed at around 34 °C, consistent with its lower Tm. The thermal stability of PZQ makes it a promising candidate for preparation via HME, where the typical processing temperature exceeds the drug's melting point and is 15–60 °C higher than the polymer's glass transition temperature64 (around 154 °C for HPMC65 and 120 °C for HPMCAS66), although this latter criterion can be reduced if plasticization by the drug occurs.67–69 Based on the Tg values of (R,S)-PZQ and (R)-PZQ, plasticization of both polymers would occur. Consequently, X-ray amorphous extrudates of both systems could be prepared at reasonable temperatures. Additionally, HME samples benefit from the inherent advantages of the technique, such as continuous manufacturing, solvent-free processing, potential for automation, and the possibility of real-time monitoring.70To evaluate the impact of the processing method on PZQ release, (R,S)-PZQ and (R)-PZQ were formulated with HPMCAS MF via SE and HME. HPMC-based ASDs via SE had a higher tendency to crystallize compared to those prepared with HPMCAS under most dissolution conditions studied and, therefore, formulations with HPMC were not prepared by HME. The racemic form of PZQ prepared by both SE and HME exhibited a very similar dissolution profile, achieving an equivalent dissolution rate and extent of release in most media. Thus, the (R,S)-PZQ-HPMCAS sample was studied under all conditions, whereas (R)-PZQ, where material was limited, was evaluated only using single-stage experiments in PBS pH 6.5 and FeSSIF-V2 at a high target concentration.
Despite presenting a slightly slower dissolution rate in PBS pH 6.5, the (R)-PZQ-HPMCAS HME ASD presented the same extent of release as the formulation prepared by SE at the end of the experiment. HPMCAS-based ASDs prepared by HME released slightly less than those prepared by SE in FeSSIF-V2, which can be attributed to different extents of drug crystallization as confirmed by PXRD analysis. These results demonstrated that preparation approach for both the enantiopure and racemic forms of PZQ ASDs had no discernable impact on PZQ dissolution.
5. Conclusions
This work evaluated the impact of polymer type, processing method, and chirality on PZQ release in ASD formulations of racemic and enantiopure PZQ at a DL of 30% in simple aqueous media and under fasted/fed-state media conditions. The medium pH had no influence on PZQ drug release when formulated with HPMC E5 LV, but these formulations exhibited lower stability to crystallization than HPMCAS-based ASDs during release studies. PZQ crystallization was observed through PLM images and PXRD analyses in HPMC-based ASDs under all release conditions. Although formulations containing HPMCAS MF released poorly at gastric pH due to low solubility of HPMCAS MF at low pH, (R,S)-PZQ-HPMCAS and (R)-PZQ-HPMCAS demonstrated good release in PBS pH 6.5 and fasted-state simulated media. HPMCAS-based ASDs showed no evidence of crystallization, except in fed-state simulated media, and HPMCAS was more effective as a crystallization inhibitor compared to HPMC E5 LV.The enantiopure (R)-PZQ formulated with HPMCAS MF released more than (R,S)-PZQ-HPMCAS in PBS pH 6.5 at high target concentration, but both formulations had a similar performance in fasted-state simulated media due to enhanced solubility of PZQ induced by the presence of bile salts. (R,S)-PZQ-HPMCAS and (R)-PZQ-HPMCAS prepared via HME and SE exhibited a similar drug release profile, demonstrating that HME is a promising technique for producing PZQ-ASDs. (R)-PZQ-HPMCAS appears to be a potential formulation to replace the currently available commercial racemic PZQ mixture, thereby avoiding the side effects and bitter taste associated with the enantiomer (S)-PZQ, as well as lowering the patient pill burden.
Conflicts of interest
The authors declare no conflicts of interest. The authors alone are responsible for the content and writing of the paper.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
The authors would like to acknowledge the Bill and Melinda Gates Foundation (Seattle, WA) for funding this study through award number INV-042627. Under the grant conditions of the Foundation, a Creative Commons Attribution 4.0 Generic License has already been assigned to the Author Accepted Manuscript version that might arise from this submission. Mingxin Qian and Tongli Biomedical Co. are thanked for supplying (R)-PZQ.References
- X. Y. Wang, et al., Prevalence and correlations of schistosomiasis mansoni and schistosomiasis haematobium among humans and intermediate snail hosts: a systematic review and meta-analysis, Infect. Dis. Poverty, 2024, 13(1), 63 CrossRef PubMed.
- K. Mekete, et al., The Geshiyaro Project: a study protocol for developing a scalable model of interventions for moving towards the interruption of the transmission of soil-transmitted helminths and schistosome infections in the Wolaita zone of Ethiopia, Parasites Vectors, 2019, 12(1), 503 CrossRef PubMed.
- World Health Organization, Schistosomiasis, 2025, Available from: https://www.who.int/news-room/fact-sheets/detail/schistosomiasis [2023 February 18th].
- W. H.Organization, WHO model list of essential medicines: 23rd list, 2023, World Health Organization, 2023 Search PubMed.
- A. Gaggero, et al., Co-grinding with surfactants as a new approach to enhance in vitro dissolution of praziquantel, J. Pharm. Biomed. Anal., 2020, 189, 113494 CrossRef CAS PubMed.
- A. Borrego-Sánchez, et al., Molecular and crystal structure of praziquantel. Spectroscopic properties and crystal polymorphism, Eur. J. Pharm. Sci., 2016, 92, 266–275 CrossRef PubMed.
- E. D. Costa, et al., Unexpected solvent impact in the crystallinity of praziquantel/poly(vinylpyrrolidone) formulations. A solubility, DSC and solid-state NMR study, Int. J. Pharm., 2016, 511(2), 983–993 CrossRef CAS PubMed.
- D. N. O. Kuevi, et al., Challenges and Proven Recommendations of Praziquantel Formulation, J. Clin. Pharm. Ther., 2023, 2023(1), 3976392 Search PubMed.
- F. C. Lombardo, B. Perissutti and J. Keiser, Activity and pharmacokinetics of a praziquantel crystalline polymorph in the Schistosoma mansoni mouse model, Eur. J. Pharm. Biopharm., 2019, 142, 240–246 CrossRef CAS PubMed.
- A. Borrego-Sánchez, et al., Praziquantel–Clays as Accelerated Release Systems to Enhance the Low Solubility of the Drug, Pharmaceutics, 2020, 12(10), 914 CrossRef PubMed.
- T. K. Špehar, et al., Investigation of Praziquantel/Cyclodextrin Inclusion Complexation by NMR and LC-HRMS/MS: Mechanism, Solubility, Chemical Stability, and Degradation Products, Mol. Pharm., 2021, 18(11), 4210–4223 CrossRef PubMed.
- P. L. Bonate, et al., Extrapolation of praziquantel pharmacokinetics to a pediatric population: a cautionary tale, J. Pharmacokinet. Pharmacodyn., 2018, 45(5), 747–762 CrossRef PubMed.
- P. Olliaro, P. Delgado-Romero and J. Keiser, The little we know about the pharmacokinetics and pharmacodynamics of praziquantel (racemate and R-enantiomer), J. Antimicrob. Chemother., 2014, 69(4), 863–870 CrossRef CAS PubMed.
- I. D'Abbrunzo, G. Procida and B. Perissutti, Praziquantel Fifty Years on: A Comprehensive Overview of Its Solid State, Pharmaceutics, 2024, 16(1), 27 CrossRef PubMed.
- T. Meyer, et al., Taste, A New Incentive to Switch to (R)-Praziquantel in Schistosomiasis Treatment, PLoS Neglected Trop. Dis., 2009, 3(1), e357 CrossRef PubMed.
- S.-K. Park, et al., Mechanism of praziquantel action at a parasitic flatworm ion channel, Sci. Transl. Med., 2021, 13(625), eabj5832 CrossRef CAS PubMed.
- R. Iyer, et al., Amorphous Solid Dispersions (ASDs): The Influence of Material Properties, Manufacturing Processes and Analytical Technologies in Drug Product Development, Pharmaceutics, 2021, 13(10), 1682 CrossRef CAS PubMed.
- J. A. Baird and L. S. Taylor, Evaluation of amorphous solid dispersion properties using thermal analysis techniques, Adv. Drug Delivery Rev., 2012, 64(5), 396–421 CrossRef CAS PubMed.
- A. K. P. Mann, et al., Producing Amorphous Solid Dispersions via Co-Precipitation and Spray Drying: Impact to Physicochemical and Biopharmaceutical Properties, J. Pharm. Sci., 2018, 107(1), 183–191 CrossRef CAS PubMed.
- S. Mohapatra, et al., Effect of Polymer Molecular Weight on the Crystallization Behavior of Indomethacin Amorphous Solid Dispersions, Cryst. Growth Des., 2017, 17(6), 3142–3150 CrossRef CAS.
- A. Budiman, et al., Effect of Drug–Polymer Interaction in Amorphous Solid Dispersion on the Physical Stability and Dissolution of Drugs: The Case of Alpha-Mangostin, Polymers, 2023, 15(14), 3034 CrossRef CAS PubMed.
- C. S. Ferreira Marques, et al., Solid dispersion of praziquantel enhanced solubility and improve the efficacy of the schistosomiasis treatment, J. Drug Delivery Sci. Technol., 2018, 45, 124–134 CrossRef CAS.
- K. Zheng, et al., Effect of Particle Size and Polymer Loading on Dissolution Behavior of Amorphous Griseofulvin Powder, J. Pharm. Sci., 2019, 108(1), 234–242 CrossRef CAS PubMed.
- Y. Liu, et al., Dissolution and oral bioavailability enhancement of praziquantel by solid dispersions, Drug Delivery Transl. Res., 2018, 8(3), 580–590 CrossRef CAS PubMed.
- P. Torre, S. Torrado and S. Torrado, Preparation, Dissolution and Characterization of Praziquantel Solid Dispersions, Chem. Pharm. Bull., 1999, 47, 1629–1633 CrossRef.
- S. Orlandi, et al., Structural Elucidation of Poloxamer 237 and Poloxamer 237/Praziquantel Solid Dispersions: Impact of Poly(Vinylpyrrolidone) over Drug Recrystallization and Dissolution, AAPS PharmSciTech, 2018, 19(3), 1274–1286 CrossRef CAS PubMed.
- N. El-Lakkany, S. H. S. el-Din and L. Heikal, Bioavailability and in vivo efficacy of a praziquantel–polyvinylpyrrolidone solid dispersion in Schistosoma mansoni-infected mice, Eur. J. Drug Metab. Pharmacokinet., 2012, 37(4), 289–299 CrossRef CAS PubMed.
- P. R. Dametto, et al., Development and physicochemical characterization of solid dispersions containing praziquantel for the treatment of schistosomiasis, J. Therm. Anal. Calorim., 2017, 127(2), 1693–1706 CrossRef CAS.
- M. V. Chaud, et al., Solid dispersions with hydrogenated castor oil increase solubility, dissolution rate and intestinal absorption of praziquantel, Braz. J. Pharm. Sci., 2010, 46, 473–481 CrossRef CAS.
- M. V. Chaud, et al., Development and Evaluation of Praziquantel Solid Dispersions in Sodium Starch Glycolate, Trop. J. Pharm. Res., 2013, 12, 163–168 Search PubMed.
- M. Cugovčan, et al., Biopharmaceutical characterization of praziquantel cocrystals and cyclodextrin complexes prepared by grinding, J. Pharm. Biomed. Anal., 2017, 137, 42–53 CrossRef PubMed.
- M. Münster, et al., Multiparticulate system combining taste masking and immediate release properties for the aversive compound praziquantel, Eur. J. Pharm. Sci., 2017, 109, 446–454 CrossRef PubMed.
- J. Boniatti, et al., Direct Powder Extrusion 3D Printing of Praziquantel to Overcome Neglected Disease Formulation Challenges in Paediatric Populations, Pharmaceutics, 2021, 13(8), 1114 CrossRef CAS PubMed.
- U. Bhatt, U. S. Murty and S. Banerjee, Theoretical and experimental validation of praziquantel with different polymers for selection of an appropriate matrix for hot-melt extrusion, Int. J. Pharm., 2021, 607, 120964 CrossRef CAS PubMed.
- S. Klein, The use of biorelevant dissolution media to forecast the in vivo performance of a drug, AAPS J., 2010, 12(3), 397–406 CrossRef CAS PubMed.
- T. Eason, et al., Revisiting the Dissolution of Praziquantel in Biorelevant Media and the Impact of Digestion of Milk on Drug Dissolution, Pharmaceutics, 2022, 14(10), 2228 CrossRef CAS PubMed.
- European Pharmacopoeia Commission, Recommendations on Methods for Dosage Forms Testing, in European Pharmacopoeia, European Directorate for the Quality of Medicines (EDQM), Strasbourg, 2014 Search PubMed.
- Biorelevant, Which Ingredients Are in FeSSIF-V2 Powder? n.d. February 26, 2025; available from: https://biorelevant.com/learning_center/Which-ingredients-are-fessif-v2-powder/.
- G. A. Ilevbare and L. S. Taylor, Liquid–Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations, Cryst. Growth Des., 2013, 13(4), 1497–1509 CrossRef CAS.
- M. Qian, et al.Crystal form of (R)-Praziquantel and preparation method and application thereof, U.S. Patent No. 9657017, U.S. Patent and Trademark Office, 2017.
- S. K. El-Arini, D. Giron and H. Leuenberger, Solubility properties of racemic praziquantel and its enantiomers, Pharm. Dev. Technol., 1998, 3(4), 557–564 CrossRef CAS PubMed.
- H. Polyzois, et al., Amorphous Solid Dispersion Formation for Enhanced Release Performance of Racemic and Enantiopure Praziquantel, Mol. Pharm., 2024, 21(10), 5285–5296 CrossRef CAS PubMed.
- M. Choudhari, et al., Emerging Applications of Hydroxypropyl Methylcellulose Acetate Succinate: Different Aspects in Drug Delivery and Its Commercial Potential, AAPS PharmSciTech, 2023, 24(7), 188 CrossRef CAS PubMed.
- J. Zhang, et al., Advances in the development of amorphous solid dispersions: The role of polymeric carriers, Asian J. Pharm. Sci., 2023, 18(4), 100834 Search PubMed.
- R. Holm, A. Müllertz and H. Mu, Bile salts and their importance for drug absorption, Int. J. Pharm., 2013, 453(1), 44–55 CrossRef CAS PubMed.
- J.-n. Yu, et al., Enhancement of oral bioavailability of the poorly water-soluble drug silybin by sodium cholate/phospholipid-mixed micelles, Acta Pharmacol. Sin., 2010, 31(6), 759–764 CrossRef CAS PubMed.
- C. Pigliacelli, et al., Interaction of polymers with bile salts – Impact on solubilisation and absorption of poorly water-soluble drugs, Colloids Surf., B, 2023, 222, 113044 CrossRef CAS PubMed.
- Chemaxon, Playground, Chemaxon Ltd., Budapest, Hungary, 2025. Available from: https://playground.calculators.cxn.io/ Search PubMed.
- N. Castro, et al., Bioavailability of praziquantel increases with concomitant administration of food, Antimicrob. Agents Chemother., 2000, 44(10), 2903–2904 CrossRef CAS PubMed.
- R. B. Chavan, et al., Evaluation of the inhibitory potential of HPMC, PVP and HPC polymers on nucleation and crystal growth, RSC Adv., 2016, 6(81), 77569–77576 RSC.
- J. M. Miller, et al., A Win–Win Solution in Oral Delivery of Lipophilic Drugs: Supersaturation via Amorphous Solid Dispersions Increases Apparent Solubility without Sacrifice of Intestinal Membrane Permeability, Mol. Pharm., 2012, 9(7), 2009–2016 CrossRef CAS PubMed.
- A. Butreddy, Hydroxypropyl methylcellulose acetate succinate as an exceptional polymer for amorphous solid dispersion formulations: A review from bench to clinic, Eur. J. Pharm. Biopharm., 2022, 177, 289–307 CrossRef CAS PubMed.
- D. D. Shah and L. S. Taylor, Chemistry and ionization of HPMCAS influences the dissolution and solution-mediated crystallization of posaconazole amorphous solid dispersions, J. Pharm. Sci., 2025, 114(1), 223–233 CrossRef CAS PubMed.
- D. T. Friesen, et al., Hydroxypropyl Methylcellulose Acetate Succinate-Based Spray-Dried Dispersions: An Overview, Mol. Pharm., 2008, 5(6), 1003–1019 CrossRef CAS PubMed.
- W. Curatolo, J. A. Nightingale and S. M. Herbig, Utility of hydroxypropylmethylcellulose acetate succinate (HPMCAS) for initiation and maintenance of drug supersaturation in the GI milieu, Pharm. Res., 2009, 26(6), 1419–1431 CrossRef CAS PubMed.
- A. Elkhabaz, et al., Crystallization Kinetics in Fasted-State Simulated and Aspirated Human Intestinal Fluids, Cryst. Growth Des., 2021, 21(5), 2807–2820 CrossRef CAS.
- I. Meister, et al., Activity of Praziquantel Enantiomers and Main Metabolites against Schistosoma mansoni, Antimicrob. Agents Chemother., 2014, 58(9), 5466–5472 CrossRef PubMed.
- X. Wen, et al., Preparation and In Vitro/In Vivo Evaluation of Orally Disintegrating/Modified-Release Praziquantel Tablets, Pharmaceutics, 2021, 13(10), 1567 CrossRef CAS PubMed.
- C. Rodríguez-Ruiz, et al., Structural, Physicochemical, and Biopharmaceutical Properties of Cocrystals with RS- and R-Praziquantel–Generation and Prolongation of the Supersaturation State in the Presence of Cellulosic Polymers, Cryst. Growth Des., 2022, 22(10), 6023–6038 CrossRef.
- B. Atawa, et al., Impact of chirality on the Glass Forming Ability and the crystallization from the amorphous state of 5-ethyl-5-methylhydantoin, a chiral poor glass former, Int. J. Pharm., 2018, 540(1), 11–21 CrossRef CAS PubMed.
- Sigma-Aldrich, Praziquantel. n.d. March 9, 2025; available from: https://www.sigmaaldrich.com/US/en/product/sigma/p4668.
- R. M. Mainardes, M. P. D. Gremião and R. C. Evangelista, Thermoanalytical study of praziquantel-loaded PLGA nanoparticles, Rev. Bras. Cienc. Farm., 2006, 42, 523–530 CrossRef CAS.
- M. Qian, R-Praziquantel Preparation Method, Suzhou Tongli Biomedical Co., Ltd, United States, 2015 Search PubMed.
- J. M. C. de Assis, et al., Hot-melt extrudability of amorphous solid dispersions of flubendazole-copovidone: An exploratory study of the effect of drug loading and the balance of adjuvants on extrudability and dissolution, Int. J. Pharm., 2022, 614, 121456 CrossRef CAS PubMed.
- N. Nyamweya and S. W. Hoag, Assessment of polymer-polymer interactions in blends of HPMC and film forming polymers by modulated temperature differential scanning calorimetry, Pharm. Res., 2000, 17(5), 625–631 CrossRef CAS PubMed.
- A. L. Sarode, et al., Stability assessment of hypromellose acetate succinate (HPMCAS) NF for application in hot melt extrusion (HME), Carbohydr. Polym., 2014, 101, 146–153 CrossRef CAS PubMed.
- Y. Li, et al., Interactions between drugs and polymers influencing hot melt extrusion, J. Pharm. Pharmacol., 2014, 66(2), 148–166 CrossRef CAS PubMed.
- G. Verreck, The Influence of Plasticizers in Hot-Melt Extrusion, in Hot–Melt Extrusion, Pharm. Appl., 2012, 93–112 CAS.
- D. K. Tan, et al., Innovations in Thermal Processing: Hot-Melt Extrusion and KinetiSol® Dispersing, AAPS PharmSciTech, 2020, 21(8), 312 CrossRef CAS PubMed.
- S. Tambe, et al., Hot-melt extrusion: Highlighting recent advances in pharmaceutical applications, J. Drug Delivery Sci. Technol., 2021, 63, 102452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5pm00117j |
| This journal is © The Royal Society of Chemistry 2025 |