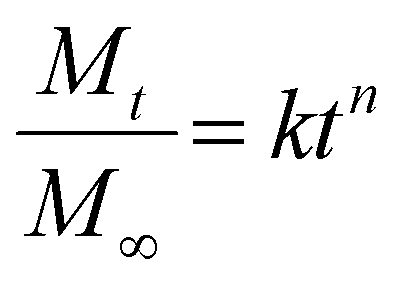DOI:
10.1039/D4PM00296B
(Paper)
RSC Pharm., 2025,
2, 303-317
Comparative analysis of drug release kinetics in polyethylene oxide and xanthan gum matrices with various excipients
Received
17th October 2024
, Accepted 20th December 2024
First published on 23rd December 2024
Abstract
This study aimed to investigate the effect of various pharmaceutical excipients on the drug release kinetics of extended-release formulations composed of polyethylene oxide (PEO) and xanthan gum (XG), using propranolol hydrochloride (PPN) as the model drug. The formulations contained different ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:1, and 3:1 w/w) of PEO or XG to either lactose, dibasic calcium phosphate (DCP), or microcrystalline cellulose (MCC). Compaction analysis revealed that formulations that contain higher excipient content exhibit increased porosity and decreased hardness values. Contact angle measurements indicated that formulations with higher excipient content, particularly with lactose, displayed lower contact angles, which is indicative of increased hydrophilicity. After the in vitro dissolution studies were conducted, the dissolution efficiency (DE), mean dissolution time (MDT), mean dissolution rate (MDR), and similarity factors (f2) were analysed. The findings showed that a higher amount of lactose in both PEO and XG formulations resulted in faster drug release, with the PEO:lactose 1:3 ratio achieving the highest DE (64 ± 8%) and the shortest MDT (77 ± 10 min). Similarly, the XG:lactose 1:3 ratio exhibited the highest DE (61 ± 2%) and fastest MDR (0.20 ± 0.01% min−1), although the effect was less pronounced compared to PEO formulations. The kinetic analysis showed that most PEO formulations followed the Peppas model, indicating non-Fickian transport driven by both diffusion and polymer erosion mechanisms. However, most of the XG formulations followed the Higuchi model. The similarity factors (f2) revealed the influence of excipient type and ratio on the dissolution profiles. Formulations containing a higher amount of MCC displayed higher similarity with the pure polymer profiles. These results give important insights into how excipients can be used to optimise polymeric matrices to regulate drug release in extended-release formulations.
:3, 1:1, and 3:1 w/w) of PEO or XG to either lactose, dibasic calcium phosphate (DCP), or microcrystalline cellulose (MCC). Compaction analysis revealed that formulations that contain higher excipient content exhibit increased porosity and decreased hardness values. Contact angle measurements indicated that formulations with higher excipient content, particularly with lactose, displayed lower contact angles, which is indicative of increased hydrophilicity. After the in vitro dissolution studies were conducted, the dissolution efficiency (DE), mean dissolution time (MDT), mean dissolution rate (MDR), and similarity factors (f2) were analysed. The findings showed that a higher amount of lactose in both PEO and XG formulations resulted in faster drug release, with the PEO:lactose 1:3 ratio achieving the highest DE (64 ± 8%) and the shortest MDT (77 ± 10 min). Similarly, the XG:lactose 1:3 ratio exhibited the highest DE (61 ± 2%) and fastest MDR (0.20 ± 0.01% min−1), although the effect was less pronounced compared to PEO formulations. The kinetic analysis showed that most PEO formulations followed the Peppas model, indicating non-Fickian transport driven by both diffusion and polymer erosion mechanisms. However, most of the XG formulations followed the Higuchi model. The similarity factors (f2) revealed the influence of excipient type and ratio on the dissolution profiles. Formulations containing a higher amount of MCC displayed higher similarity with the pure polymer profiles. These results give important insights into how excipients can be used to optimise polymeric matrices to regulate drug release in extended-release formulations.
1. Introduction
Extended-release (ER) drug delivery systems are designed to release the active pharmaceutical ingredient (API) over a long period of time, maintaining consistent drug concentrations in the bloodstream. By regulating the rate of drug release, ER formulations improve patient compliance, minimise multiple dosing and enhance therapeutic outcomes. These systems are useful for drugs with short half-lives or a narrow therapeutic index.1 The mechanism of drug release from these ER systems involves a combination of diffusion, erosion, and swelling.2 Diffusion occurs as the drug moves out from the polymeric matrix or through pores formed by excipients. Erosion, on the other hand, involves the gradual breakdown of the matrix. Swelling plays an important role in hydrophilic polymers, facilitating the uptake of the dissolution medium and creating a gel layer that controls the release of the drug.3–6 The distance between the diffusion and erosion fronts represents the thickness of the gel layer and is an important factor in drug release kinetics. Factors affecting this release include the molecular weight of the polymer, solubility of the drug and porosity of the matrix.2 This combined mechanism of diffusion, erosion, and swelling plays an important role in the design of controlled release systems, as the interaction between these processes determines the drug release profiles.6 Polyethylene oxide (PEO) and xanthan gum (XG) are two widely used polymers in ER formulations as they can form gel matrices which regulate the release of drugs. PEO is a hydrophilic, synthetic polymer available in different molecular weights and viscosities.7,8 PEO has the capability to regulate the release of both highly water-soluble and poorly soluble drugs from matrix systems.8–11 Additionally, PEO is applied in various ER dosage forms, including controlled-release tablets,7,12,13 pellets,14 hot-melt extrusion (HME) films,15 buccal films,16,17 injection-moulded tablets18 and solid dispersions.19 XG is a high molecular weight extracellular heteropolysaccharide produced through the fermentation of carbohydrates by the bacterium Xanthomonas campestris.20,21 Owing to its hydrophilic nature, XG is commonly employed in the production of hydrophilic matrix systems.22,23 Furthermore, XG has been utilised in a range of ER applications, including sustained-release tablets,24 mini matrices,25 microspheres,26 double-layer tablets,27,28 pellets29 and transdermal films.30
Excipients are an integral component of ER formulations, directly influencing the drug release profile.31,32 Commonly used excipients such as lactose, dicalcium phosphate (DCP), and microcrystalline cellulose (MCC) affect the physical properties of the tablet matrix. Lactose is a water soluble disaccharide that is widely used in pharmaceutical tablets as it enhances wettability and improves flowability.33 DCP is frequently used as a water-insoluble filler in tablet formulations,34 while MCC is particularly used for direct compression (DC) tabletting. Although MCC is insoluble in water, it displays swelling properties when exposed to dissolution media.35,36 To better understand the mechanisms of drug release, this study examines the kinetics of drug release using various mathematical models (zero-order, first-order, Higuchi, and Korsmeyer-Peppas).37–39 Previous studies have extensively explored the use of PEO in ER formulations. For example, controlled-release matrix tablets of verapamil hydrochloride were developed using PEO and various excipients such as lactose, DCP, and MCC. It was noted that higher polymer content decreased the drug release rate, and substituting lactose with DCP or MCC significantly slowed down the release rate.40 Another study examined the influence of polymer molecular weight and excipients on the zero-order release kinetics of verapamil hydrochloride.41 The findings showed that higher molecular weights of PEO and insoluble fillers such as DCP could achieve sustained drug release through swelling and erosion mechanisms. In another investigation, HPMC matrix tablets were formed with diclofenac sodium, incorporating MCC, starch, and lactose. Starch and lactose regulated the release kinetics, achieving zero-order release profiles.42 The influence of excipients on drug release from HPMC matrix systems was also studied, revealing that lactose and MCC enhanced drug diffusion and tablet erosion, while starch resulted in slowing the drug release through synergistic interactions with HPMC.43 The impact of excipient type and level on alprazolam release from HPMC tablets was examined by.44 Similarity factors were used to compare the dissolution profiles, and it was demonstrated that insoluble excipients such as DCP slowed drug release. XG has the ability to extend the drug release of diclofenac sodium, following zero-order kinetics.45 However, swelling-controlled erosional processes have also been observed.46 Factors such as polymer concentration, ionic strength of the dissolution medium, and dissolution apparatus speed can influence the drug release characteristics from XG-based matrices.46 It was found that XG alone can sustain PPN release over 24 h with release kinetics best fitting the Higuchi model. However, its combination with guar gum shifted the release mechanism towards Fickian diffusion.47 Another study discussed the kinetic models for drug dissolution and the importance of selecting appropriate models to understand the release mechanisms from swellable and non-swellable matrices.48 This work investigates the influence of varying excipient ratios (1:3, 1:1, and 3:1) on the drug release profiles and kinetics of two distinct polymers, PEO and XG. The Sessile drop contact angle testing technique is used to determine the impact of the excipients on the hydrophilicity/hydrophobicity of the polymers and its impact on initial swelling. Unlike previous studies which focused on single formulation factors, our work evaluates the influence of different excipient types and their ratios using contact angle measurements and dissolution studies to assess their effect on drug release kinetics. Similarity factors (f2) and dissolution parameters such as dissolution efficiency (DE) and mean dissolution time (MDT) were used to quantitatively assess and compare the dissolution profiles of the formulations. Furthermore, the application of multiple kinetic models gives a deeper insight into the mechanisms governing drug release, thereby contributing to the development of more effective and reliable ER drug delivery systems.
2. Materials and methods
2.1. Materials
Polyethylene oxide (PEO) with a MW of 4000000 (POLYOX™) was generously donated by Colorcon (Dartford, UK). Xanthan Gum (XG) (Xanatural 75™) was kindly provided by CP Kelco, Atlanta, GA, USA. Lactose was supplied by Meggle (Wasserburg, Germany). Dibasic calcium phosphate (DCP) is sourced from Chemische Fabrik Budenheim KG (Budenheim, Germany). Microcrystalline cellulose (MCC) PH102 was obtained from JRS Pharma (Surrey, UK). The active ingredient, propranolol hydrochloride (PPN, pKa 9.45), was purchased from TCI Chemicals (UK). Potassium phosphate monobasic, sodium hydroxide, hydrochloric acid (HCl), and potassium chloride (KCl) for the preparation of the 0.2 M phosphate buffer dissolution medium (pH 6.8) and the 0.1 M hydrochloric acid (HCl) dissolution medium (pH 1.2) were acquired from Fisher (UK).
2.2. Methodology
2.2.1. Compact manufacture.
The formulations were prepared by accurately measuring 312.5 ± 1 mg of a blend containing either PEO or XG, PPN and selected excipients according to the specific ratios outlined in Table 1. The formulations were designed to keep both a consistent drug load and ensure that the polymer excipient ratios (3:1, 1:1, and 1:3) and compact weight remained unchanged. Each blend was thoroughly mixed for 10 min using a Turbula™ mixer (Willy. A Bachofen, Switzerland) before being compressed into 10 mm flat-faced compacts under a force of 10 kN using a Testometric™ hydraulic press (M500-50CT, Testometric Company Ltd, Rochdale, UK). After compression, the resulting compacts were removed from the die and placed in glass vials for subsequent analysis.
Table 1 The quantities of PPN, polymer (PEO/XG), and excipients (lactose, DCP, MCC) in each compact formulation
| Formulation code |
Composition |
PPN (mg) |
Polymer (PEO/XG) (mg) |
Lactose (mg) |
DCP (mg) |
MCC (mg) |
| PPN: propranolol hydrochloride; PEO: polyethylene oxide; XG: xanthan gum; DCP: dicalcium phosphate; MCC: microcrystalline cellulose. |
| P1/X1 |
PEO/XG only |
80 |
232.5 |
— |
— |
— |
| P2/X2 |
PEO/XG:lactose (1:3) |
80 |
58.1 |
174.4 |
— |
— |
| P3/X3 |
PEO/XG:lactose (1:1) |
80 |
116.3 |
116.3 |
— |
— |
| P4/X4 |
PEO/XG:lactose (3:1) |
80 |
174.4 |
58.1 |
— |
— |
| P5/X5 |
PEO/XG:DCP (1:3) |
80 |
58.1 |
— |
174.4 |
— |
| P6/X6 |
PEO/XG:DCP (1:1) |
80 |
116.3 |
— |
116.3 |
— |
| P7/X7 |
PEO/XG:DCP (3:1) |
80 |
174.4 |
— |
58.1 |
— |
| P8/X8 |
PEO/XG:MCC (1:3) |
80 |
58.1 |
— |
— |
174.4 |
| P9/X9 |
PEO/XG:MCC (1:1) |
80 |
116.3 |
— |
— |
116.3 |
| P10/X10 |
PEO/XG:MCC (3:1) |
80 |
174.4 |
— |
— |
58.1 |
2.2.2 Compact hardness and porosity.
To evaluate the compact hardness, compacts were assessed using a Pharmatest Hardness tester (Pharma Test, Hainburg, Germany). The porosity of the compacts was calculated by determining the difference between the apparent density (ρapp) of the compact and the true density of the powder (ρtrue), following eqn (1):| |  | (1) |
2.2.3 Contact angle measurements.
The sessile drop technique was used to measure the contact angle for each formulation using a contact angle goniometer (Ossila, Sheffield, UK). In the contact angle experiments, a volume of 1 μL of deionised water was used. After the liquid was dispensed from the needle, images of the droplet were captured at a rate of 1 frame per second. The average values and standard deviations were measured based on three trials conducted at room temperature. The initial contact angle was obtained using the first image taken 1 s after the droplet was placed.
2.2.4
In vitro release studies.
In vitro dissolution profiles for each compact were obtained using the type 2 USP (Automated-Pharmatest DT 70 Low-Head Dissolution Apparatus) with paddles rotating at 50 ± 1 rpm. To prevent the compact from floating during dissolution, an 8-mesh basket sinker was utilised. The initial 2 h of the test were conducted in 900 mL of 0.1 M hydrochloric acid (HCl) to replicate the gastric environment. Subsequently, the compacts were immediately transferred to another vessel containing 0.2 M phosphate buffer with a pH of 6.8 a further 10 h. Samples were drawn automatically throughout the dissolution process for 12 h at 37 ± 0.5 °C. Absorbance values were measured at 280 nm through a coupled UV spectrophotometer (IDIS-EE software). All experiments were conducted in triplicate.
2.2.5 Dissolution parameters (dissolution efficiency (DE) and mean dissolution time (MDT)).
The mean dissolution time (MDT) represents the average time required for a drug to dissolve under in vitro dissolution settings and is calculated according to eqn (2). It is a model-independent method and is used in dosage forms with variable drug release mechanisms.49–52 Another parameter calculated was the dissolution efficiency (DE) using eqn (3), defined as the area under the dissolution curve up to a specific time, expressed as a percentage of the area of a rectangle 100% dissolution within the same time.53–55| | 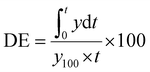 | (2) |
 represents the area under the dissolution curve (AUC) up to time t, where y100 is the maximum dissolution value (usually the percentage of drug released at equilibrium), and t is the total time of the dissolution test.
represents the area under the dissolution curve (AUC) up to time t, where y100 is the maximum dissolution value (usually the percentage of drug released at equilibrium), and t is the total time of the dissolution test.
| |  | (3) |
t
j
represents the midpoint of each dissolution time interval. ΔMj is the amount of drug dissolved during time interval j. n is the total number of dissolution time points.
2.2.6 Similarity factor.
The similarity between the drug release profiles was assessed using the similarity factor (f2), as described in eqn (4).56–58 The f2 value is calculated by the following equation:| | 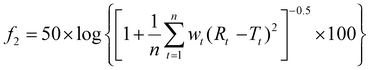 | (4) |
n is the total number of time points considered, Rt refers to the dissolution value of the reference product at a given time t, and Tt represents the dissolution value of the test product at time t and wt is the optional weighting factor applied at each time point.
The similarity factor was calculated using the drug release profile of the pure polymers (PEO or XG) as the reference. An f2 value between 50 and 100 suggests that the two profiles are similar, with values closer to 100 indicating a higher degree of similarity or near-identical release profiles. Values lower than 50 indicate dissimilarity among the two-dissolution profiles.56 The similarity factor was calculated using dissolution data obtained from 3 replicates for each formulation.
2.2.7 Kinetics of drug release.
The drug release kinetics in formulations can be described as either Fickian or non-Fickian diffusion. In Fickian diffusion, the drug release rate is dependent on drug concentration. Zero-order kinetics, on the other hand, implies a constant release of the drug over time. Non-Fickian diffusion drug release is influenced by numerous factors. Based on these variables, the drug release can follow different kinetics models like zero-order, first-order, Higuchi and Korsmeyer-Peppas (power-law) kinetics.
In the zero-order kinetics model, the drug release rate remains constant over time and is independent of the drug concentration.59,60 This model is described by the following equation:
where
Q is the cumulative amount of drug released at time
t,
Q0 is the initial amount of drug (usually zero) and
k0 is the zero-order release constant.
For first-order kinetics, where the drug release rate depends on the concentration of the drug, the release can be represented by eqn (6), where Qt is the amount of drug released at time t, Q0 is the initial amount of drug (usually zero), and k is the first-order rate constant.61
| | | ln(Qt) = lnQ0 + k1 | (6) |
The Higuchi kinetic model represents a drug release model where the drug release rate is proportional to the square root of time, as it occurs when the drug concentration in dosage form exceeds the drug's solubility, this relationship can be described by eqn (7), where Mt is the cumulative amount of drug released at time t, and kH is the Higuchi release constant.62
| | 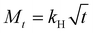 | (7) |
In the Korsmeyer-Peppas (power-law) kinetic model, the rate of drug release is influenced by the polymeric system, encompassing release mechanisms such as water diffusion into the polymeric matrix, swelling of the matrix and the dissolution of the polymer itself.48,62,63 In this model, Mt/M∞ represents the fraction of drug released at time t, while k is a constant related to the drug release that accounts for the geometrical characteristics of the matrix tablet, and n is the diffusional exponent of drug release. For cylindrical tablets, like those used in this study, an n value up to 0.45 indicates Fickian diffusion, while an n value greater than 0.89 indicates case-II transport. Values between these two suggest the occurrence of anomalous transport, as reported in various studies.38,64–66
| | 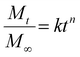 | (8) |
2.2.8 Statistical analysis.
The dissolution data were statistically analysed using a one-way ANOVA test in Minitab 19 to evaluate significant differences between the mean values of the formulations. If ANOVA indicated a significant difference (p < 0.05), Tukey's post hoc test was conducted at a 5% confidence level to identify specific differences between groups. A p-value less than 0.05 was considered statistically significant, whereas values equal to or greater than 0.05 were regarded as not significant.
3. Results and discussion
3.1. Porosity and hardness values
The porosity and hardness data for the PEO-based compacts are depicted in Table 2. An inverse correlation is observed between the hardness and porosity data of the compacts. All the PEO-excipient compacts displayed higher porosity values than the pure polymer. A higher amount of PEO in the compacts leads to an increase in the strength of the tablets. In addition, the characteristics of the excipients further influenced the final compact properties. MCC, due to its plastic deformation, produces compacts with higher hardness, whereas lactose and DCP, which tend to fragment, result in less dense and softer compacts.67,68 This is evident in the 1:3 ratio compacts with lactose, which showed the highest porosity (20.6 ± 2.2%) and the lowest hardness (72 ± 3.0 N), while compacts with MCC in a 3:1 ratio had the lowest porosity (9.6 ± 1.2%) and the highest hardness (297 ± 0.1 N).
Table 2 Porosity and hardness values for PEO polymer excipient blends
| Polymer |
Porosity (%) |
Hardness (N) |
| Pure PEO |
4.8 ± 1.2 |
297.1 ± 0a |
| Excipient |
Porosity (%) |
Hardness (N) |
|
This was recorded as the highest hardness level of the instrumentation as the compacts failed to break with triplicate measurements. This suggests the pure PEO compacts have hardness values greater than that recorded here.
|
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 ratio :1 ratio
|
| Lactose |
17.7 ± 1.8 |
150.8 ± 19.7 |
| DCP |
12.8 ± 2.5 |
206.4 ± 17.7 |
| MCC |
13.3 ± 0.9 |
295.0 ± 4.3 |
|
1:3 ratio
|
| Lactose |
20.6 ± 2.2 |
72 ± 3.0 |
| DCP |
17.0 ± 2.3 |
89.2 ± 25.1 |
| MCC |
16.4 ± 0.6 |
210.7 ± 7.6 |
|
3:1 ratio
|
| Lactose |
13.7 ± 0.7 |
272.7 ± 10.7 |
| DCP |
11.1 ± 1.5 |
297.0 ± 0.1 |
| MCC |
9.6 ± 1.2 |
297 ± 0.1 |
The porosity and hardness values for the XG-based compacts are shown in Table 3. The porosity values are higher in all of the lactose, DCP, and MCC-containing compacts than those for the pure XG compacts. An exception was the 1:3 MCC compact, which can be due to the high amount of MCC in the formulations. The data displayed a rise in porosity from DCP to lactose to MCC, revealing the influence of different excipients on the compression characteristics of XG compacts. The XG and DCP compacts demonstrated the highest porosity (23.1 ± 1.1%) and the lowest hardness (44.7 ± 3.1 N) at the 1:3 ratio. On the other hand, the MCC blends in the same ratio exhibited the highest hardness (239.2 ± 33.2 N) and the lowest porosity (15.0 ± 0.5%).
Table 3 Porosity and hardness values for XG polymer excipient blends
| Polymer |
Porosity (%) |
Hardness (N) |
| Pure XG |
16.9 ± 0.2 |
265.2 ± 14.2 |
| Excipient |
Porosity (%) |
Hardness (N) |
|
1:1 ratio
|
| Lactose |
17.4 ± 1.0 |
109.8 ± 7.9 |
| DCP |
19.4 ± 1.0 |
101.2 ± 10.5 |
| MCC |
18.7 ± 0.7 |
170.8 ± 10.3 |
|
1:3 ratio
|
| Lactose |
20.8 ± 0.4 |
48.8 ± 1.3 |
| DCP |
23.1 ± 1.1 |
44.7 ± 3.1 |
| MCC |
15.0 ± 0.5 |
239.2 ± 33.2 |
|
3:1 ratio
|
| Lactose |
20.4 ± 0.4 |
106.2 ± 4.7 |
| DCP |
22.2 ± 1.2 |
99.2 ± 8.1 |
| MCC |
18.9 ± 2.8 |
170.4 ± 35.8 |
3.2. Contact angle measurements
Contact angle measurements were conducted for all the PEO-based compacts, and the findings revealed the highest contact angle for the pure PEO compacts at 54.4° (Fig. 1). The analysis showed that a decrease in the contact angle of the compacts with increasing excipient content, suggesting a greater hydrophilicity. The 1:3 ratios of PEO:lactose and PEO:MCC had the lowest contact angles of 29.6° and 29.8°, respectively. This behaviour might affect the initial wetting and hydration of the compacts and might therefore result in faster initial drug release (Fig. 1).
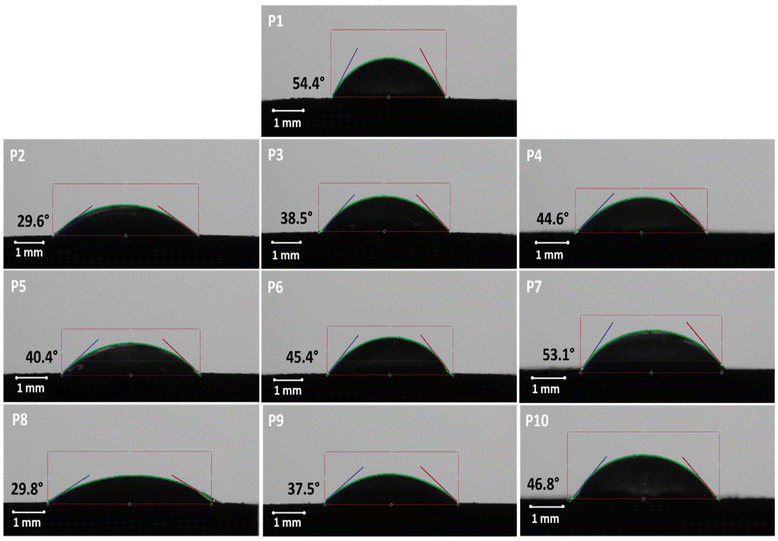 |
| | Fig. 1 Contact angle testing results for pure PEO and PEO:excipient compacts at 1:3, 1:1 and 3:1 ratios. Note: pure PEO (P1) and PEO:lactose compacts at 1:3, 1:1 and 3:1 ratio P2–P4 respectively; PEO:DCP compacts at 1:3, 1:1 and 3:1 ratio P5–P7 respectively; PEO:MCC compacts at 1:3, 1:1 and 3:1 ratio P8–P10 respectively. | |
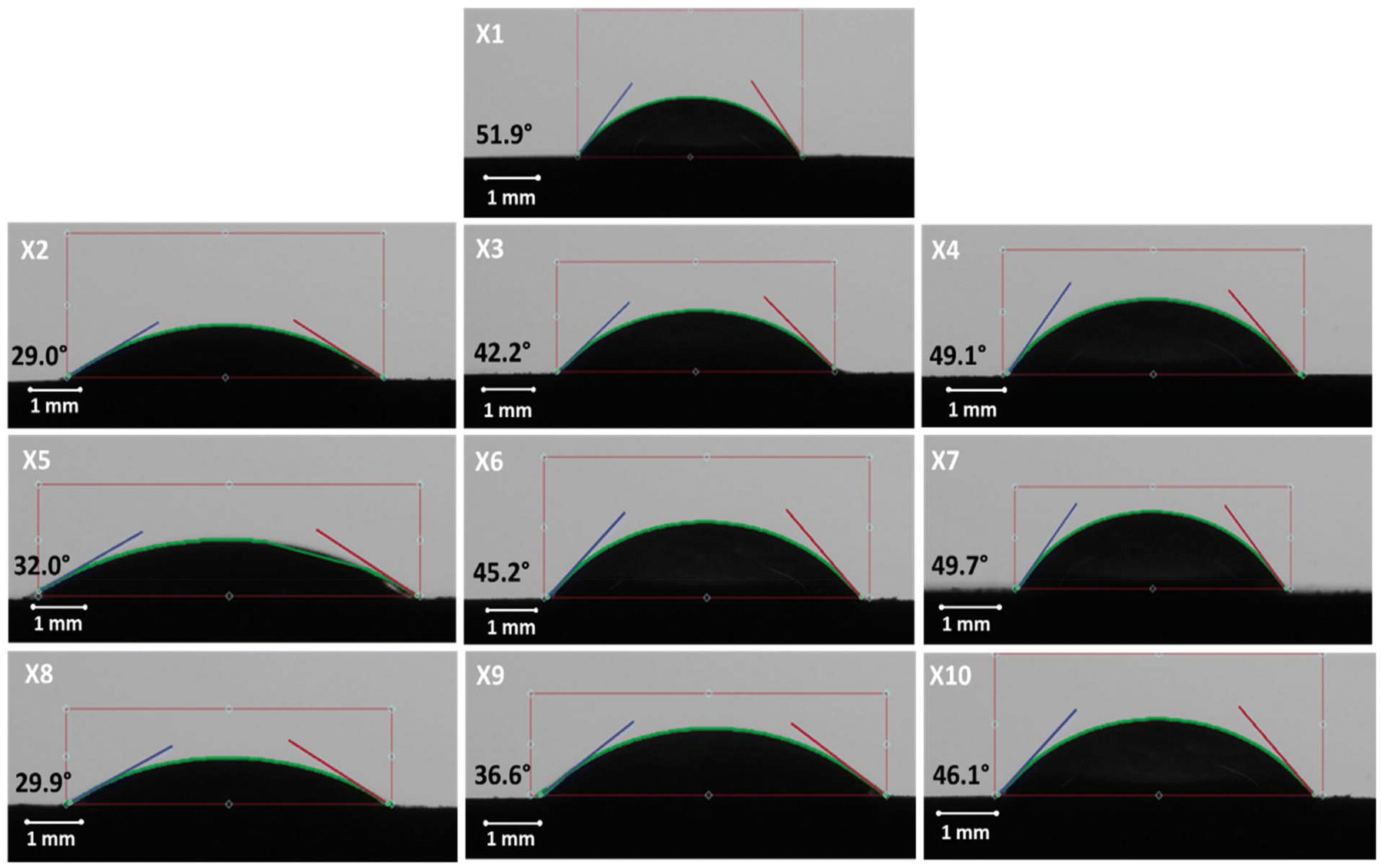
The contact angle analysis for the XG-based compacts is shown in Fig. 2. A very high contact angle was observed for the pure XG formulations at 51.9°. A similar trend to the PEO formulations was observed regarding the effect of excipients on surface hydrophilicity. The 1:3 XG:lactose formulation revealed a contact angle of 29.0°, indicating a greater potential for water absorption.
 |
| | Fig. 2 Results of contact angle tests at 1:1, 1:3, and 3:1 ratio for pure XG and XG excipient compacts. Note: pure XG (X1) and XG:lactose compacts at 1:3, 1:1 and 3:1 ratio X2–X4 respectively; XG:DCP compacts at 1:3, 1:1 and 3:1 ratio X5–X7 respectively; XG:MCC compacts at 1:3, 1:1 and 3:1 ratio X8–X10 respectively. | |
3.3.
In vitro release studies
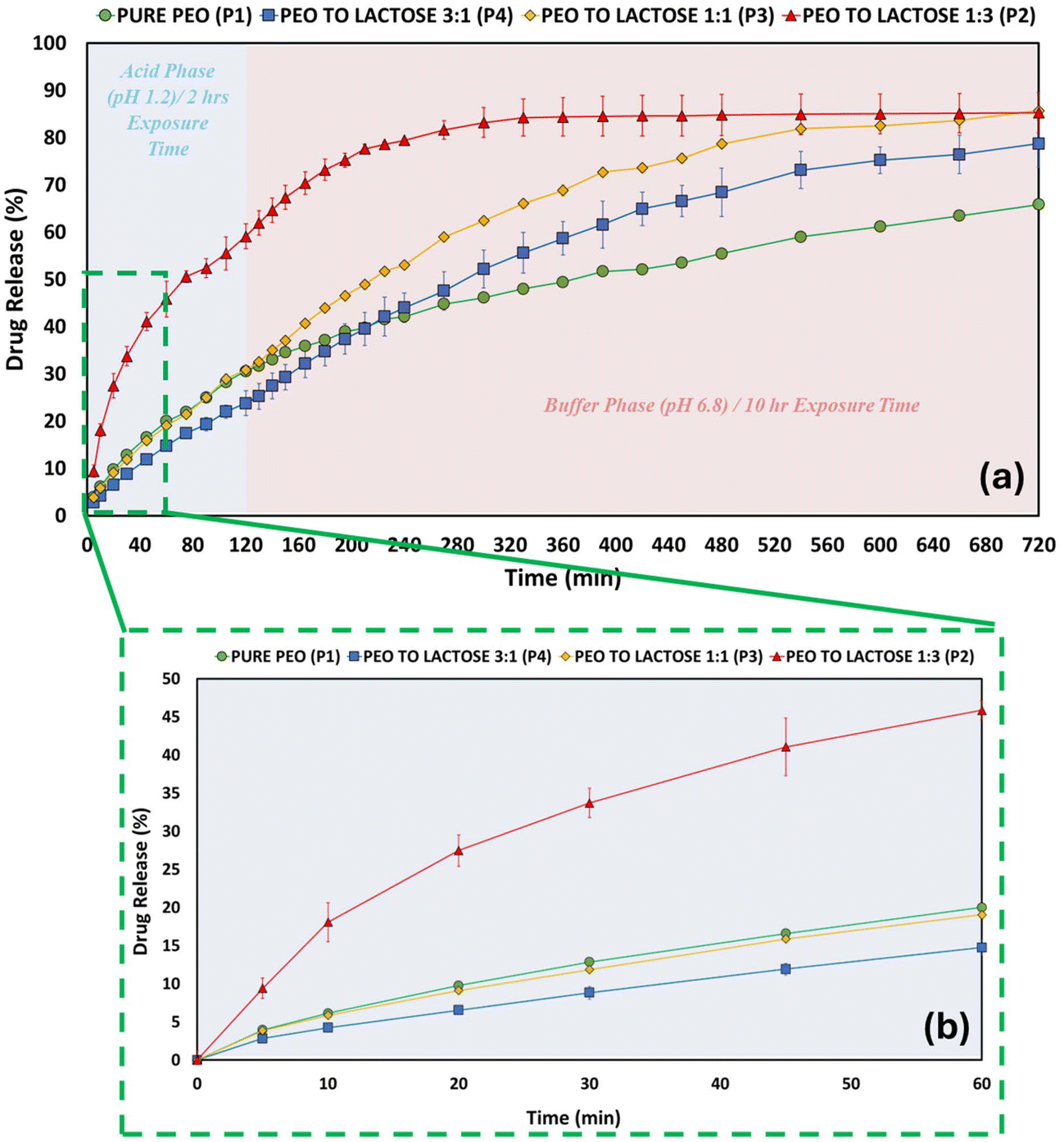
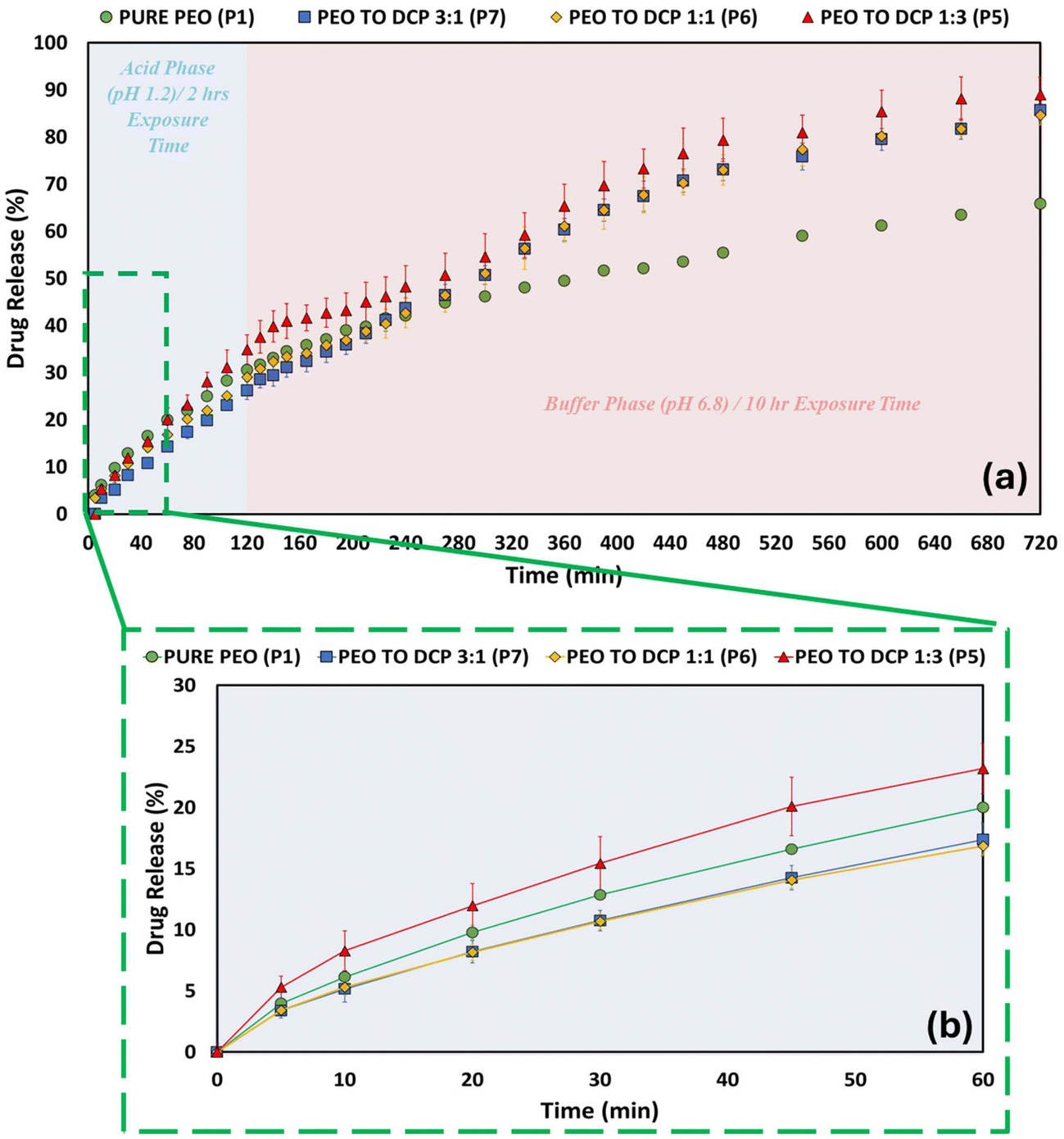
Fig. 3a shows the 12 h in vitro dissolution profiles of PPN from PEO and PEO:lactose compacts in three ratios (1:3, 1:1, 3:1). In comparison to the “pure” PEO compact, all the PEO:excipient compacts demonstrated a higher percentage of drug release. An increased amount of PEO in the formulation leads to decreased drug release, thus playing a crucial role in determining the drug retention capability of the matrix. The 1:3 PEO:lactose ratio demonstrated the highest percentage of drug release in the 12 h investigation, with 86% of the drug in the compact being released. In contrast, PEO compacts showed only 66% drug release. Fig. 3b, 4b and 5b focus on the first 60 min of drug release, showcasing the initial burst effect commonly observed in extended-release formulations.8,69–71 In the initial drug release phase (first 60 min), the influence of the different excipients on the drug release is evident. The ratio with the highest amount of excipient (3:1 polymer to excipient) showed a higher drug release in the following order: lactose > MCC > DCP. This trend can be linked to the solubility of the excipients, which is also evident in the contact angle values obtained for the compacts. Lactose, being water-soluble and exhibiting a lower contact angle, creates diffusion pathways and increases porosity within the matrix, leading to the highest drug release.72 MCC, which is water-dispersible, also facilitates drug release but to a lesser extent than lactose.43 DCP, as a water-insoluble excipient, shows the slowest drug release.73 Modifying the amount of polymer in a formulation or changing the excipient can act as drug-release modifiers in dosing systems. A one-way ANOVA followed by Tukey's test was employed to assess the statistical significance of the differences among the samples. The null hypothesis stated that the profiles were identical, and a 95% confidence interval was used for testing. However, the results revealed a significant difference (p < 0.05), indicating that the excipient type and ratio influence the drug release. Tukey's post hoc analysis revealed that the 1:3 ratio of lactose exhibited significantly higher drug release compared to the 3:1 and 1:1 ratios of all other formulations apart from the 1:1 lactose ratio. This indicates that increasing the lactose content to a 1:3 ratio significantly enhances drug release.
 |
| | Fig. 3 Dissolution profiles PEO compacts and the three ratios (1:3, 1:1 and 3:1) PEO:lactose compacts. (a) Drug release profiles over the 12 h period. (b) Zoomed-in view of the initial 60 min showing the early drug release. | |
 |
| | Fig. 4 Dissolution profiles PEO compacts and the three ratios (1:3, 1:1 and 3:1) PEO:DCP compacts. (a) Drug release profiles over the 12 h period. (b) Zoomed-in view of the initial 60 min showing the early drug release. | |
 |
| | Fig. 5 Dissolution profiles PEO compacts and the three ratios (1:3, 1:1 and 3:1) PEO:MCC compacts. (a) Drug release profiles over the 12 h period. (b) Zoomed-in view of the initial 60 min showing the early drug release. | |
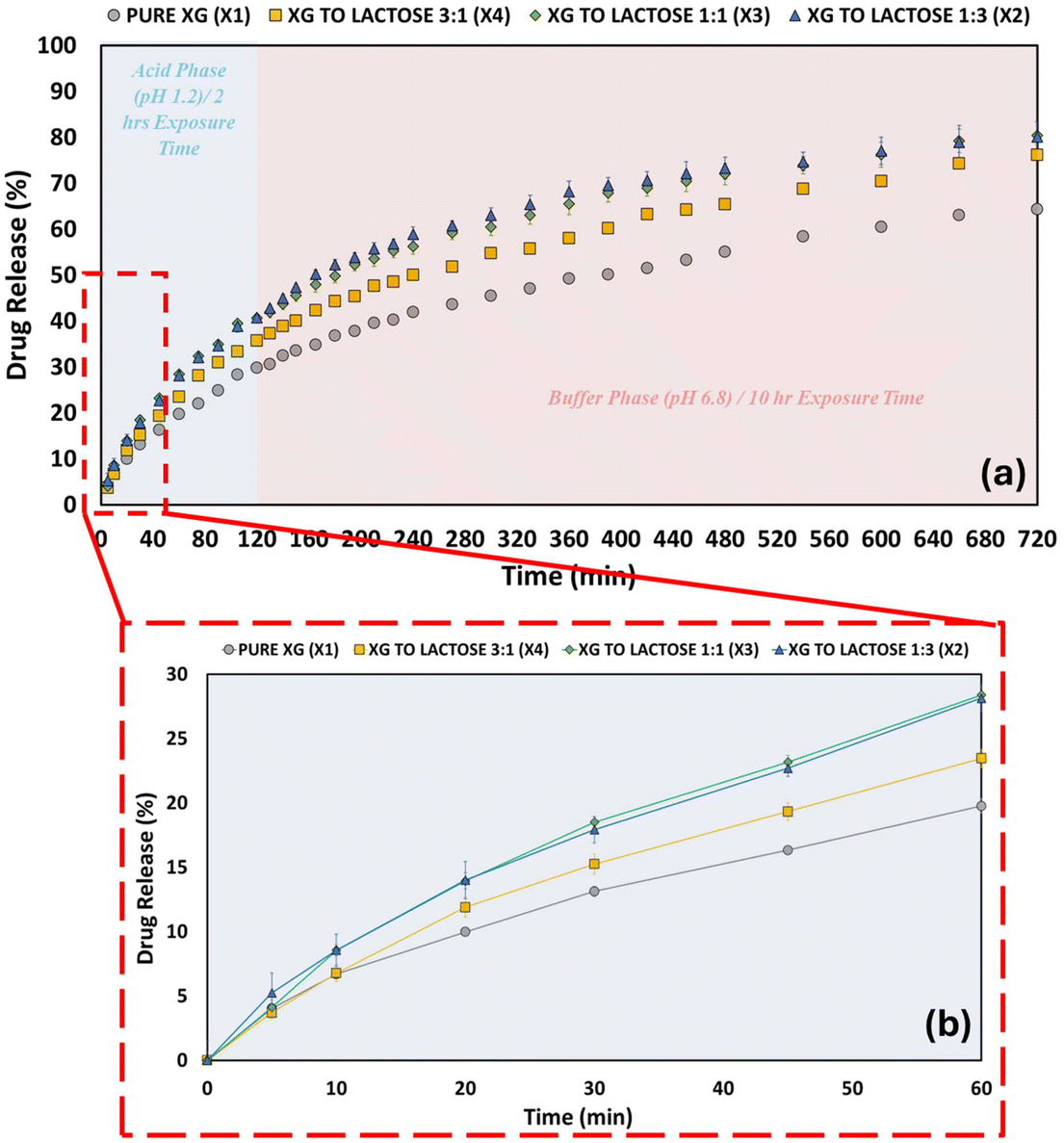
Fig. 6 shows the 12 h in vitro dissolution profiles of PPN from XG and XG compacts in three ratios (1:3, 1:1, 3:1) with lactose. The extent of drug release is affected by the amount of XG present in the composition of the compact and is a key factor in governing the ability of the matrix to retain drugs. The polymer-to-lactose ratio of 1:3 showed the highest percentage (80%) of drug release throughout the analysis. XG compacts, on the other hand, displayed a lower drug release of only 64%. Adjusting the polymer content or varying the excipient can effectively modify drug release in pharmaceutical dosage forms. For instance, an increase in drug release percentage was reported in a previous study when lactose was incorporated into gum karaya matrix tablets. This change is associated with the diffusion of lactose into the gel layer, which increases the porosity of the matrix.74 The performance of gum olibanum with XG and HPMC in tramadol sustained-release matrix tablets has been reported and compared.75 When lactose was used as a bulking agent, the drug released more quickly than when DCP and MCC were used. This is because lactose creates diffusion pathways within the polymeric matrix.75 Similarly, the effects of the excipients (lactose, DCP and MCC) and polymers (XG and HPMC) in the preparation of ibuprofen-polymer matrix tablets was investigated.76 The excipient containing tablets had significantly higher drug release profiles than those without excipients. Lactose and MCC had shorter t25 values than that of the DCP. The authors reported that despite being insoluble in water, MCC might have had a major effect due to its ability to disintegrate, which might have aided in the disintegration of the matrix tablet.63 Another study assessed losartan potassium sustained release matrix tablets using natural polymers (locust bean gum, karaya gum and Tara gum) as release retardant polymers. In every formulation, drug release was correspondingly delayed as the polymer content increased. DCP was the diluent that controlled the drug release the most out of the three that were used. The order of release retardation was DCP > MCC > lactose. The observed behaviour was attributed to the diluent's solubility.77 Unlike PEO-based formulations, which were sensitive to excipient changes, XG-based formulations displayed no significant differences in drug release profiles, as confirmed by the statistical analysis (one-way ANOVA, p > 0.05) (Fig. 7 and 8).
 |
| | Fig. 6 Dissolution profiles XG compacts and the three ratios (1:3, 1:1 and 3:1) of XG:lactose compacts. (a) Drug release profiles over the 12 h period. (b) Zoomed-in view of the initial 60 min showing the early drug release. | |
 |
| | Fig. 7 Dissolution profiles XG compacts and the three ratios (1:3, 1:1 and 3:1) of XG:DCP compacts. (a) Drug release profiles over the 12 h period. (b) Zoomed-in view of the initial 60 min showing the early drug release. | |
 |
| | Fig. 8 Dissolution profiles XG compacts and the three ratios (1:3, 1:1 and 3:1) of XG:MCC compacts. (a) Drug release profiles over the 12 h period. (b) Zoomed-in view of the initial 60 min showing the early drug release. | |
Another study evaluated Sterculia foetida gum as a hydrophilic matrix polymer. The influence of excipients on soluble APIs like diltiazem hydrochloride release was investigated. Adding swelling, insoluble excipients such as MCC significantly (p < 0.05) altered the release profile by modifying gel layer swelling. When the insoluble filler DCP was used, diltiazem hydrochloride became trapped, potentially generating “stress cracks” leading to dose dumping or matrix failure. Soluble fillers like lactose hydrate dissolve and diffuse simultaneously, improving drug solubility by reducing the diffusion path.78
3.4. DE, MDT, MDR and similarity factor
To investigate the differences in drug release from PEO tablets, DE, MDT, mean dissolution rate (MDR), and f2 were calculated and presented in Table 4. The DE values for the formulations containing the excipients were higher than those of pure PEO (46.1 ± 0.1%). The highest DE calculated was for the PEO:lactose 1:3 (64.2 ± 7.7%), showing an enhancement in drug release compared to pure PEO. In addition, PEO:lactose 1:3 had the lowest MDT (77.1 ± 9.6 min), indicating a much faster dissolution compared to pure PEO (216.3 ± 2.1 min). The MDR also increased with the addition of the excipients. The highest MDR was seen in PEO:lactose 1:3 (0.37 ± 0.08% min−1), indicating that higher lactose content resulted in faster drug release compared to the much slower rate of pure PEO (0.15 ± 0.00% min−1). The effect of the addition of excipients at different ratios was also investigated, and it was shown that DE increased with higher lactose content, from 52.2 ± 3.3% for the PEO:lactose 3:1 to 64.2 ± 7.7% for PEO:lactose 1:3. Similarly, the MDR was highest in PEO:lactose 1:3, while the MDT showed a reduction, indicating that higher lactose ratios led to quicker drug release. The DE values for the PEO:DCP formulations changed less drastically than those for the lactose. PEO:DCP 1:1 ratio had a DE of 54.2 ± 1.1%, which was slightly higher than that of the PEO:DCP 3:1 (52.5 ± 1.4). The formulations containing MCC showed a steady increase in DE, with PEO:MCC 1:3 reaching 59.7 ± 0.7%. The MDR for PEO:MCC 1:3 (0.21 ± 0.01% min−1) was higher than that for PEO:MCC 3:1 (0.14 ± 0.00% min−1), indicating a faster drug release in higher MCC content compacts. Interestingly, the MDT for PEO:MCC 3:1 was the longest among all formulations (249.5 ± 6.7 min), reflecting the slowest dissolution rate, likely due to the swelling effect of the MCC. To evaluate the effect of different excipients at the same 1:3 ratio, PEO:lactose 1:3 showed the highest DE at 64.2 ± 7.7%, higher than PEO:MCC 1:3 (59.7 ± 0.7%) and PEO:DCP 1:3 (51.9 ± 1.4%). The MDT was shorter for PEO:lactose 1:3 (77.1 ± 9.6 min) than the MDT values for PEO:DCP 1:3 (184.5 ± 10.1 min) and PEO:MCC 1:3 (182.2 ± 8.8 min), suggesting a slower release rate. This suggests that lactose in the 1:3 ratio facilitated the most rapid drug release, while DCP and MCC produced a slower, more controlled release. These results are supported by the dissolution results.
Table 4 Overview of DE (%), MDT (min), MDR (% min−1), and similarity factors (f2) for PPN release in PEO-based tables. The formulations feature various ratios of lactose, DCP and MCC
| Formulation |
DE (%) |
MDT (min) |
MDR (% min−1) |
Similarity factor (f2) |
| Pure PEO |
46.1 ± 0.1 |
216.3 ± 2.1 |
0.15 ± 0.00 |
— |
| PEO lactose 3:1 |
52.2 ± 3.3 |
242.3 ± 2.3 |
0.16 ± 0.01 |
55.4 ± 5.2 |
| PEO lactose 1:1 |
58.9 ± 0.7 |
204.4 ± 4.3 |
0.19 ± 0.01 |
44.7 ± 0.8 |
| PEO lactose 1:3 |
64.2 ± 7.7 |
77.1 ± 9.6 |
0.37 ± 0.08 |
27.1 ± 0.4 |
| PEO DCP 3:1 |
52.5 ± 1.4 |
228.7 ± 35.8 |
0.18 ± 0.02 |
49.7 ± 2.9 |
| PEO DCP 1:1 |
54.2 ± 1.1 |
246.1 ± 18.2 |
0.17 ± 0.01 |
52.1 ± 3.2 |
| PEO DCP 1:3 |
51.9 ± 1.4 |
184.5 ± 10.1 |
0.22 ± 0.01 |
45.6 ± 2.3 |
| PEO MCC 3:1 |
48.1 ± 0.3 |
249.5 ± 6.7 |
0.14 ± 0.00 |
59.5 ± 1.6 |
| PEO MCC 1:1 |
57.4 ± 0.5 |
219.3 ± 4.0 |
0.18 ± 0.00 |
49.9 ± 1.2 |
| PEO MCC 1:3 |
59.7 ± 0.7 |
182.2 ± 8.8 |
0.21 ± 0.01 |
43.3 ± 1.0 |
The similarity factor f2 was determined for the different PEO formulations to compare the dissolution profiles (Table 4). Pure PEO formulations served as the reference for all the other formulations. The PEO formulations containing lactose showed different similarity values dependent on the ratio of lactose in the tablet. The PEO lactose 3:1 formulation showed a similarity factor of 55, whereas the PEO lactose 1:1 formulation had a slightly lower f2 value of 45, suggesting moderate variation in the dissolution behavior. The PEO lactose 1:3 had a similarity factor of 27, suggesting differences in the dissolution profile, likely due to the high ratio of lactose in the formulation. For the formulations containing DCP as the main excipient, the PEO:DCP 3:1 and PEO:DCP 1:1 had f2 values of 50 and 52, respectively. However, the PEO DCP 1:3 had a lower f2 value of 46, indicating differences in the dissolution behavior. The formulations with MCC generally displayed higher similarity factors, particularly PEO MCC 3:1, with an f2 value of 59. This high correlation might be due to the swelling nature of the MCC excipient, which resembles the swelling of the PEO polymer. Meanwhile, the PEO MCC 1:3 formulation exhibited a lower f2 value of 43. In conclusion, the f2 values suggest that while most of the compacted formulations hold a degree of similarity in their dissolution profiles, high ratios (1:3) of the excipients lead to dissimilarity in the dissolution behavior.
The influence of the excipient type and ratio was also investigated for the XG formulations (Table 5). As apparent in the PEO formulations, an increase in DE values with increasing lactose content was observed from the XG:lactose 3:1 (54.1 ± 0.8%) to the XG lactose 1:3 (60.8 ± 1.8%) compacts. However, the extent of the DE improvement was less than what was observed in the PEO formulations. A similar increase in MDT and increase in MDR were noted with increasing lactose content. DCP resulted in less difference in DE than in the lactose formulations. XG:DCP 1:1 had a DE of 51.8 ± 1.6%, slightly higher than XG:DCP 3:1 (49.5 ± 0.9%). The formulations MCC showed an increase in DE, with XG:MCC 1:3 reaching 55.8 ± 0.6%. However, the XG:MCC 3:1 exhibited the longest MDT (209.1 ± 4.5 min), a trend also observed in the PEO:MCC formulations. The XG:lactose 1:3 achieved the highest DE (60.8 ± 1.8%) compared to the 1:3 ratio of excipient across the formulations, followed by XG:DCP 1:3 (57.2 ± 0.5%) and XG:MCC 1:3 (55.8 ± 0.6%).
Table 5 Overview of DE (%), MDT (min), MDR (% min−1), and similarity factors (f2) for PPN release in XG-based tables. The formulations feature various ratios of lactose, DCP and MCC
| Formulation |
DE (%) |
MDT (min) |
MDR (% min−1) |
Similarity factor (f2) |
| Pure XG |
45.5 ± 0.5 |
210.6 ± 2.8 |
0.16 ± 0.01 |
— |
| XG lactose 3:1 |
54.1 ± 0.8 |
208.7 ± 2.0 |
0.17 ± 0.01 |
55.6 ± 3.0 |
| XG lactose 1:1 |
59.7 ± 1.7 |
185.1 ± 3.2 |
0.20 ± 0.01 |
44.4 ± 3.4 |
| XG lactose 1:3 |
60.8 ± 1.8 |
173.0 ± 7.2 |
0.20 ± 0.01 |
42.5 ± 2.9 |
| XG DCP 3:1 |
49.5 ± 0.9 |
203.0 ± 8.7 |
0.16 ± 0.00 |
70.4 ± 6.7 |
| XG DCP 1:1 |
51.8 ± 1.6 |
199.4 ± 5.1 |
0.17 ± 0.01 |
61.2 ± 5.1 |
| XG DCP 1:3 |
57.2 ± 0.5 |
198.9 ± 1.1 |
0.18 ± 0.01 |
48.5 ± 1.9 |
| XG MCC 3:1 |
47.2 ± 0.6 |
209.1 ± 4.5 |
0.15 ± 0.00 |
84.0 ± 8.4 |
| XG MCC 1:1 |
49.1 ± 0.8 |
197.5 ± 0.9 |
0.16 ± 0.01 |
71.6 ± 6.9 |
| XG MCC 1:3 |
55.8 ± 0.6 |
181.3 ± 1.9 |
0.18 ± 0.01 |
50.1 ± 2.2 |
The similarity factor (f2) was also determined to compare dissolution profiles across the XG formulations (Table 5). The XG:lactose 3:1 ratio achieved a similarity value of 56, while XG:lactose 1:3 had a value of 42, indicating a difference in dissolution profiles due to the higher lactose content. A higher similarity factor was observed in the XG:DCP 3:1 ratio (f2 = 70). The MCC formulation (XG:MCC 3:1) displayed the highest similarity factor, achieving a value of f2 = 84. This is in line with the trend seen in PEO:MCC formulations, where MCC content displayed dissolution behavior more aligned with the polymer, likely due to the swelling nature of MCC. In conclusion, while both XG and PEO formulations showed an increase in drug release with increasing excipient content, the effects of lactose, DCP and MCC were more pronounced in the PEO formulations. An increase in lactose content resulted in more prominent deviations in the PEO formulations at higher excipient ratios. Our previous studies utilised dissolution imaging to visualise and quantify the swelling behaviour of hydrophilic polymers. The results revealed that XG-based compacts exhibited a greater swelling capacity compared to PEO, with the pure XG compacts reaching an average swelling percentage of 211% by the end of the 2 h time period. In comparison, PEO-only compacts achieved an average swelling percentage of 135% over the same time frame.79,80 The rapid hydration and significant swelling of XG resulted in the formation of a larger gel layer. This could be the result of the greater resistance that XG exhibits to the effects of excipients, causing the excipients to have a less pronounced impact on the drug release profiles when XG is used. In contrast, PEO has a lower swelling capacity, and the thinner gel layer is more influenced by excipient variations. This highlights the importance of polymer and excipient types in optimising extended-release formulations. A study examined the viscosity of XG and PEO solutions and the findings revealed that XG exhibited higher viscosity compared to PEO.81 This difference in viscosity supports the notion that XG forms a robust gel layer that exhibits greater resistance to excipient effects.
3.5. Drug release kinetics
The drug release profiles for the PEO:excipient compacts in various ratios (3:1, 1:1, and 1:3) were analysed using several kinetic models (zero-order, first-order, Higuchi and Korsmeyer-Peppas (power-law))66 to identify the most suitable model and understand the underlying mechanism of drug release. The data presented in Table 6 indicates that most of the PEO compact formulations followed the Peppas model for drug release. For instance, formulations such as PEO:DCP 3:1, PEO:DCP 1:1, and PEO:MCC 1:1 demonstrated strong fits to the Peppas model with R2 values of 0.999, 0.998, and 0.999, respectively. However, the PEO:lactose 3:1 and PEO:MCC 3:1 formulations displayed drug release profiles that fitted the first-order kinetics, with R2 values of 0.997 and 0.999, respectively. In these formulations, the % of drug release was proportional to the amount of the drug in the tablet, displaying a more concentration-dependent release mechanism. Notably, the PEO:lactose 1:3 formulation was best described by the Higuchi model of drug release with an R2 value of 0.979. The high amount of lactose in the compact might have resulted in a diffusion-driven release mechanism, resembling the characteristics of the Higuchi model. The n value in the Korsmeyer-Peppas (power-law) kinetic model can provide insights into the mechanisms of drug release for the compact formulations being studied. For cylindrical matrix tablets, an n value of ≤0.45 implies Fickian diffusion or case-I kinetics, where the release is primarily controlled by diffusion. A n value ≥0.89 indicates super case-II transport, which is regulated by the erosion of the hydrated layer and follows zero-order kinetics, meaning the drug release is time independent. For n values between (0.45 ≤ n ≤ 0.89), the mechanism suggests anomalous or non-Fickian transport, implying that both drug diffusion and polymer erosion play roles in the release process.82 The values of the release exponent “n” from the Peppas model ranged from 0.544 to 0.783, indicating non-Fickian (anomalous) transport for all formulations. This implies that both drug diffusion and polymer erosion mechanisms contribute to the drug release process.82
Table 6 Kinetic models of PPN release from PEO-based tablet formulations, showing R2 values and the release exponent (n)
| Formulation |
Zero order |
First order |
Higuchi |
Peppas (power low) |
| RSQ |
RSQ |
RSQ |
RSQ |
n
|
| The bolded R2 values represent the highest correlation coefficients within each row, identifying the best-fitting kinetic model for each formulation. |
| Pure PEO |
0.893 |
0.954 |
0.988 |
0.989
|
0.571 |
| PEO lactose 3:1 |
0.993 |
0.997
|
0.975 |
0.996 |
0.732 |
| PEO lactose 1:1 |
0.994 |
0.996 |
0.980 |
0.997
|
0.692 |
| PEO lactose 1:3 |
0.901 |
0.955 |
0.979
|
0.969 |
0.544 |
| PEO DCP 3:1 |
0.990 |
0.993 |
0.983 |
0.999
|
0.683 |
| PEO DCP 1:1 |
0.976 |
0.990 |
0.988 |
0.998
|
0.666 |
| PEO DCP 1:3 |
0.938 |
0.975 |
0.993 |
0.995
|
0.585 |
| PEO MCC 3:1 |
0.989 |
0.999
|
0.978 |
0.998 |
0.783 |
| PEO MCC 1:1 |
0.981 |
0.998 |
0.992 |
0.999
|
0.656 |
| PEO MCC 1:3 |
0.977 |
0.997 |
0.997 |
0.999
|
0.599 |
According to Table 7, most of the XG formulations displayed drug release profiles that followed the Higuchi and Peppas models, suggesting a combination of diffusion-controlled and matrix erosion mechanisms. The R2 values for the Higuchi model ranged from 0.978 to 0.998, suggesting that diffusion played a key role in drug release for the XG formulations. The Peppas model also showed a strong fit (R2 values) for formulations like pure XG (0.991), XG lactose 1:3 (0.997), and XG DCP 1:3 (0.981). The “n” values, ranging from 0.558 to 0.640, suggest anomalous (non-Fickian) transport. This suggests that drug release was driven by a combination of diffusion and polymer erosion mechanisms. The findings are similar to those of a study on PPN sustained release tablets, with the use of XG as the matrix-forming polymer. The study showed an inverse relationship between the amount of XG and the release rate of PPN. The drug release was best fitted with the Higuchi model. The formulations also displayed characteristics of Fickian diffusion and anomalous (non-Fickian) mechanisms.83
Table 7 Kinetic models of PPN release from XG-based tablet formulations, showing R2 values and the release exponent (n)
| Formulation |
Zero order |
First order |
Higuchi |
Peppas (power low) |
| RSQ |
RSQ |
RSQ |
RSQ |
n
|
| The bolded R2 values represent the highest correlation coefficients within each row, identifying the best-fitting kinetic model for each formulation. |
| Pure XG |
0.901 |
0.959 |
0.991
|
0.991
|
0.558 |
| XG lactose 3:1 |
0.906 |
0.962 |
0.991
|
0.985 |
0.624 |
| XG lactose 1:1 |
0.931 |
0.978 |
0.996
|
0.984 |
0.630 |
| XG lactose 1:3 |
0.955 |
0.989 |
0.998
|
0.997 |
0.619 |
| XG DCP 3:1 |
0.888 |
0.951 |
0.987
|
0.982 |
0.587 |
| XG DCP 1:1 |
0.866 |
0.934 |
0.978
|
0.978
|
0.578 |
| XG DCP 1:3 |
0.889 |
0.948 |
0.983
|
0.981 |
0.640 |
| XG MCC 3:1 |
0.885 |
0.951 |
0.986
|
0.981 |
0.590 |
| XG MCC 1:1 |
0.866 |
0.934 |
0.978
|
0.978
|
0.578 |
| XG MCC 1:3 |
0.889 |
0.948 |
0.983
|
0.981 |
0.640 |
In summary, the analysis of the drug release kinetics for the PEO and XG-based formulations showed that the majority of the PEO formulations followed the Korsmeyer-Peppas (power-law) model. The release exponent n values indicate non-Fickian (anomalous) transport, driven by both diffusion and polymer erosion mechanisms. However, most of the XG formulations followed the Higuchi model.
4. Conclusions
In this study, the drug release profiles of PPN from various PEO and XG-based formulations containing different ratios of excipients (lactose, DCP and MCC) were investigated. The aim was to examine the impact of excipient type and ratio on the drug release kinetics and mechanisms of these polymers. The dissolution profiles showed that all PEO:excipient compacts had a higher percentage of drug release compared to the pure PEO compact. There was an inverse relationship between the amount of PEO in the formulation and the extent of drug release, signifying PEO's capability for drug retention within the matrix. Lower amounts of PEO resulted in higher drug release levels. Drug release mechanisms were investigated using different kinetic models, including zero order, first order, Higuchi, and Peppas models. The findings suggested that the Peppas model provided the best fit for most PEO-based formulations. The release exponent n values ranged from 0.544 to 0.783, indicating non-Fickian (anomalous) transport mechanisms. This implies that both drug diffusion and polymer erosion contribute to the release process. The drug release profiles of the XG formulations followed Higuchi and Peppas models, suggesting a combination of diffusion-controlled and matrix erosion mechanisms. The R2 values for the Higuchi model were particularly high, ranging from 0.978 to 0.998, suggesting that diffusion played a key role in drug release. The results also showed that XG-based formulations were more capable of maintaining similar drug release profiles despite changes in excipient type and ratio. In contrast, PEO-based formulations were more sensitive to excipient variations. This could be due to the distinct swelling profiles of the polymers. XG forms a larger gel layer than PEO, resulting in a greater resistance to variations in excipients. The findings indicate that it is possible to tailor drug release profiles by selecting and adjusting the type and ratio of excipients in the formulation. For ER formulations that require robustness to excipient changes, XG polymer is better suited due to their ability to maintain consistent release profiles despite compositional changes. In contrast, PEO-based formulations require tighter control of excipient ratios due to their sensitivity to changes in excipients. These findings provide valuable insight into developing ER formulations.
Abbreviations
| ER | Extended-release |
| PEO | Polyethylene oxide |
| XG | Xanthan gum |
| DCP | Dicalcium phosphate |
| MCC | Microcrystalline cellulose |
| API | Active pharmaceutical ingredient |
| PPN | Propranolol hydrochloride |
| USP | United States Pharmacopeia |
| HME | Hot melt extrusion |
| HCl | Hydrochloric acid |
| KCl | Potassium chloride |
| NaOH | Sodium hydroxide |
| KH2PO4 | Potassium phosphate monobasic |
| DE | Dissolution efficiency |
| MDT | Mean dissolution time |
| MDR | Mean dissolution rate |
|
f
2
| Similarity factor |
Author contributions
Conceptualization: H. M., A. W., K. A. A.; formal analysis: H. M., A. W., N. M., S. D., K. A. A.; investigation: H. M., A. W., N. M., S. D., J. W., K. A. A.; methodology: H. M., A. W., N. M., S. D., J. W., K. A. A.; writing – original draft: H. M., N. M., K. A. A.; writing – review and editing: H. M., K. A. A.
Data availability
The authors confirm that the data supporting the finding of this study are available within the article.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Haja Muhamad acknowledges the University of Huddersfield for support.
References
- S. Adepu and S. Ramakrishna, Molecules, 2021, 26, 5905 CrossRef CAS PubMed.
- P. Colombo, R. Bettini, P. Santi, A. De Ascentiis and N. A. Peppas, J. Controlled Release, 1996, 39, 231–237 CrossRef CAS.
- D. G. Kanjickal and S. T. Lopina, Crit. Rev. Ther. Drug Carrier Syst., 2004, 21, 345–386 CrossRef CAS PubMed.
- G. Vilar, J. Tulla-Puche and F. Albericio, Curr. Drug Delivery, 2012, 9, 367–394 CrossRef CAS PubMed.
- T. D. Reynolds, S. H. Gehrke, A. S. Hussain and L. S. Shenouda, J. Pharm. Sci., 1998, 87, 1115–1123 CrossRef CAS PubMed.
- D. Caccavo, S. Cascone, G. Lamberti, A. A. Barba and A. Larsson, Smart drug delivery systems, 2016, 388 Search PubMed.
- L. Ma, L. Deng and J. Chen, Drug Dev. Ind. Pharm., 2014, 40, 845–851 CrossRef CAS PubMed.
- J. D. Vanza, R. B. Patel, R. R. Dave and M. R. Patel, Pharm. Dev. Technol., 2020, 25, 1169–1187 CrossRef CAS PubMed.
- A. Moroni and I. Ghebre-Sellassie, Drug Dev. Ind. Pharm., 1995, 21, 1411–1428 CrossRef CAS.
- S. Shojaee, I. Cumming, W. Kaialy and A. Nokhodchi, Colloids Surf., B, 2013, 111, 486–492 CrossRef CAS PubMed.
- B. N. Tukarama, I. V. Rajagopalana and P. S. I. Shartchandraa, Iran. J. Pharm. Res., 2010, 9, 349–358 Search PubMed.
- S. Shojaee, I. Cumming, W. Kaialy and A. Nokhodchi, Colloids Surf., B, 2013, 111, 486–492 CrossRef CAS PubMed.
- H. Kojima, K. Yoshihara, T. Sawada, H. Kondo and K. Sako, Eur. J. Pharm. Biopharm., 2008, 70, 556–562 CrossRef CAS PubMed.
- A. Butreddy, S. Sarabu, N. Dumpa, S. Bandari and M. A. Repka, Int. J. Pharm., 2020, 587, 119624 CrossRef CAS PubMed.
- S. Prodduturi, K. L. Urman, J. U. Otaigbe and M. A. Repka, AAPS PharmSciTech, 2007, 8, E152–E161 CrossRef PubMed.
- M. Gajdošová, D. Vetchý, J. Muselík, J. Gajdziok, J. Juřica, M. Vetchá, K. Hauptman and V. Jekl, Int. J. Pharm., 2021, 592, 120086 CrossRef PubMed.
- I. Kurćubić, U.-J. Vajić, S. Cvijić, M. Crevar-Sakač, N. Bogavac-Stanojević, Z. Miloradović, N. Mihajlović-Stanojević, M. Ivanov, D. Karanović and Đ. Jovović, Int. J. Pharm., 2021, 610, 121266 CrossRef PubMed.
- J. Pajander, A. Rensonnet, S. Hietala, J. Rantanen and S. Baldursdottir, Int. J. Pharm., 2017, 518, 203–212 CrossRef CAS PubMed.
- T. T.-D. Tran and P. H.-L. Tran, J. Drug Delivery Sci. Technol., 2013, 23, 269–274 CrossRef CAS.
- M. Jadav, D. Pooja, D. J. Adams and H. Kulhari, Pharmaceutics, 2023, 15, 402 CrossRef CAS PubMed.
- V. S. Verma, K. Sakure and H. R. Badwaik, Curr. Chem. Biol., 2017, 11, 10–20 CrossRef CAS.
- B. Wang, Y. Han, Q. Lin, H. Liu, C. Shen, K. Nan and H. Chen, J. Mater. Chem. B, 2016, 4, 1853–1861 RSC.
- A. Verma, A. Kumar, S. Ramasamy, A. Yadav and R. Bijauliya, J. Drug Delivery Ther., 2021, 11, 27–32 CrossRef CAS.
- P. Yeole, U. Galgatte, I. Babla and P. Nakhat, Indian J. Pharm. Sci., 2006, 68, 185–189 CrossRef CAS.
- E. Verhoeven, C. Vervaet and J. P. Remon, Eur. J. Pharm. Biopharm., 2006, 63, 320–330 CrossRef CAS PubMed.
- M. M. Yahoum, S. Toumi, H. Tahraoui, S. Lefnaoui, M. Kebir, A. Amrane, A. A. Assadi, J. Zhang and L. Mouni, Micromachines, 2023, 14, 609 CrossRef PubMed.
- T. Phaechamud and G. C. Ritthidej, Drug Dev. Ind. Pharm., 2007, 33, 595–605 CrossRef CAS PubMed.
- Y. E.-S. Hamza and M. H. Aburahma, AAPS PharmSciTech, 2009, 10, 1357–1367 CrossRef CAS PubMed.
- H. Santos, F. Veiga, M. E. Pina and J. J. Sousa, Int. J. Pharm., 2005, 295, 15–27 CrossRef CAS PubMed.
- R. C. Mundargi, S. A. Patil, S. A. Agnihotri and T. M. Aminabhavi, Drug Dev. Ind. Pharm., 2007, 33, 79–90 CrossRef CAS PubMed.
- G. Vasvári, J. Kalmár, P. Veres, M. Vecsernyés, I. Bácskay, P. Fehér, Z. Ujhelyi, Á. Haimhoffer, Á. Rusznyák and F. Fenyvesi, Drug Discovery Today:Technol., 2018, 27, 71–80 CrossRef PubMed.
- B. N. Tukaram, I. V. Rajagopalan and P. S. I. Shartchandra, Iran. J. Pharm. Res., 2010, 9, 349 CAS.
- C. Shi, H. Zhao, Y. Fang, L. Shen and L. Zhao, Drug Discovery Today, 2023, 28, 103696–103696 CrossRef CAS PubMed.
- H. Ruey-ching and G. R. Peck, Pharm. Technol. North Am., 2001, 25, 54 Search PubMed.
- G. Thoorens, F. Krier, B. Leclercq, B. Carlin and B. Evrard, Int. J. Pharm., 2014, 473, 64–72 CrossRef CAS PubMed.
- Y. Hattori and M. Otsuka, J. Near Infrared Spectrosc., 2014, 22, 199–204 CrossRef CAS.
- N. Damodharan, Res. J. Pharm. Technol., 2020, 13, 1339–1345 CrossRef.
- M. P. Paarakh, P. A. Jose, C. Setty and G. Peterchristoper, Int. J. Pharm. Res. Technol., 2018, 8, 12–20 CrossRef.
- M. A. Kalam, M. Humayun, N. Parvez, S. Yadav, A. Garg, S. Amin, Y. Sultana and A. Ali Cont, J. Pharm. Sci., 2007, 1, 30–35 Search PubMed.
- S. Vidyadhara, R. L. C. Sasidhar and R. Nagaraju, Indian J. Pharm. Sci., 2013, 75, 185–190 CAS.
- C.-J. Kim, Drug Dev. Ind. Pharm., 1998, 24, 645–651 CrossRef CAS PubMed.
- S. A. Bravo, M. C. Lamas and C. J. Salamón, J. Pharm. Pharm. Sci., 2002, 5, 213–219 CAS.
- M. Levina and A. R. Rajabi-Siahboomi, J. Pharm. Sci., 2004, 93, 2746–2754 CrossRef CAS PubMed.
- R. O. Williams, T. D. Reynolds, T. D. Cabelka, M. A. Sykora and V. Mahaguna, Pharm. Dev. Technol., 2002, 7, 181–193 CrossRef CAS PubMed.
- P. Yeole, U. Galgatte, I. Babla and P. Nakhat, Indian J. Pharm. Sci., 2006, 68, 185–189 CrossRef CAS.
- M. M. Talukdar, P. Rombaut and R. Kinget, Pharm. Dev. Technol., 1998, 3, 1–6 CrossRef CAS PubMed.
- M. A. Mughal, Z. Iqbal and S. H. Neau, AAPS PharmSciTech, 2011, 12, 77–87 CrossRef CAS PubMed.
- S. A. Chime, G. Onunkwo and I. Onyishi, Res. J. Pharm., Biol. Chem. Sci., 2013, 4, 97–103 CAS.
- H. Windolf, R. Chamberlain and J. Quodbach, Pharmaceutics, 2021, 13, 1453 CrossRef CAS PubMed.
- K. Tahara, K. Yamamoto and T. Nishihata, Int. J. Pharm., 1996, 133, 17–27 CrossRef CAS.
- F. Podczeck, Int. J. Pharm., 1993, 97, 93–100 CrossRef CAS.
- M. S. Reza, M. A. Quadir and S. S. Haider, J. Pharm. Pharm. Sci., 2003, 6, 282–291 CAS.
- K. A. Khan, J. Pharm. Pharmacol., 1975, 27, 48–49 CrossRef CAS PubMed.
- D. P. Vaughan and G. T. Tucker, J. Pharm. Pharmacol., 1976, 28, 932–933 CrossRef CAS PubMed.
- N. H. Anderson, M. Bauer, N. Boussac, R. Khan-Malek, P. Munden and M. Sardaro, J. Pharm. Biomed. Anal., 1998, 17, 811–822 CrossRef CAS PubMed.
- R. E. Stevens, V. Gray, A. Dorantes, L. Gold and L. Pham, AAPS J., 2015, 17, 301–306 CrossRef CAS PubMed.
- F. Zeeshan, P. Y. Lin and R. Sheshala, Indian J. Pharm. Educ., 2020, 54, 647–653 CrossRef CAS.
- P. Kok-Khiang and C. Wong, Drug Dev. Ind. Pharm., 2000, 26, 723–730 CrossRef PubMed.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm., 2010, 67, 217–223 CAS.
- P. Paarakh, P. Jose, C. Setty and P. Christoper, Int. J. Pharm. Res. Technol., 2018, 8(1), 12–20 Search PubMed.
- J. Siepmann and F. Siepmann, Int. J. Pharm., 2013, 453, 12–24 CrossRef CAS PubMed.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS PubMed.
- M. V. S. Varma, A. M. Kaushal, A. Garg and S. Garg, Am. J. Drug Delivery, 2004, 2, 43–57 CrossRef CAS.
- J. Siepmann and F. Siepmann, Int. J. Pharm., 2008, 364, 328–343 CrossRef CAS PubMed.
- E. Nep, K. Asare-Addo, M. U. Ghori, B. R. Conway and A. M. Smith, Int. J. Pharm., 2015, 496, 689–698 CrossRef CAS PubMed.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS PubMed.
- Y. Zhang, Y. Law and S. Chakrabarti, AAPS PharmSciTech, 2003, 4, E62–499 CrossRef PubMed.
- R. M. Iyer, S. Hegde, J. Dinunzio, D. Singhal and W. Malick, Pharm. Dev. Technol., 2014, 19, 583–592 CrossRef CAS PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS PubMed.
- J. S. Park, J. Y. Shim, J. S. Park, Y. W. Choi and S. H. Jeong, Drug Dev. Ind. Pharm., 2011, 37, 664–672 CrossRef CAS PubMed.
- K. P. Gharti, P. Thapa, U. Budhathoki and A. Bhargava, J. Young Pharm., 2012, 4, 201–208 CrossRef CAS PubMed.
- B. N. Tukaram, I. V. Rajagopalan and P. S. I. Shartchandra, Iran. J. Pharm. Res., 2010, 9, 349–358 CAS.
- R. O. Williams 3rd, T. D. Reynolds, T. D. Cabelka, M. A. Sykora and V. Mahaguna, Pharm. Dev. Technol., 2002, 7, 181–193 CrossRef PubMed.
- M. K. Senapati, A. Srinatha and J. K. Pandit, Indian J. Pharm. Sci., 2006, 68, 824–826 CrossRef CAS.
- A. B. S. Manjula, A. Srinatha and B. K. Sridhar, Indian J. Pharm. Educ. Res., 2014, 48, 48–59 Search PubMed.
- B. O. Patani, O. D. Akin-Ajani, A. Kumaran and O. A. Odeku, Polim. Med., 2022, 52, 67–76 CrossRef PubMed.
- V. Viswanath, U. Chandrasekhar, B. Rao and K. G. Prakash, Int. J. Appl. Pharm. Sci. Res., 2016, 3, 127–132 CrossRef.
- A. A. Chivate, S. S. Poddar, S. Abdul and G. Savant, AAPS PharmSciTech, 2008, 9, 197–204 CrossRef CAS PubMed.
- H. Muhamad, A. Ward, R. Abdulhussain, J. Williamson, L. Blunt, B. Conway, J. Østergaard and K. Asare-Addo, J. Drug Delivery Sci. Technol., 2024, 106538, DOI:10.1016/j.jddst.2024.106538.
- H. Muhamad, A. Ward, K. Patel, J. Williamson, L. Blunt, B. Conway, J. Østergaard and K. Asare-Addo, Int. J. Pharm., 2024, 666, 124850 CrossRef CAS PubMed.
- A. S. Pereira, R. M. Andrade and E. J. Soares, J. Non-Newtonian Fluid Mech., 2013, 202, 72–87 CrossRef CAS.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- A. Ali, M. Iqbal, N. Akhtar, H. M. Khan, A. Ullah, M. Uddin and M. T. Khan, Acta Pol. Pharm., 2013, 70, 283–289 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a


 represents the area under the dissolution curve (AUC) up to time t, where y100 is the maximum dissolution value (usually the percentage of drug released at equilibrium), and t is the total time of the dissolution test.
represents the area under the dissolution curve (AUC) up to time t, where y100 is the maximum dissolution value (usually the percentage of drug released at equilibrium), and t is the total time of the dissolution test.