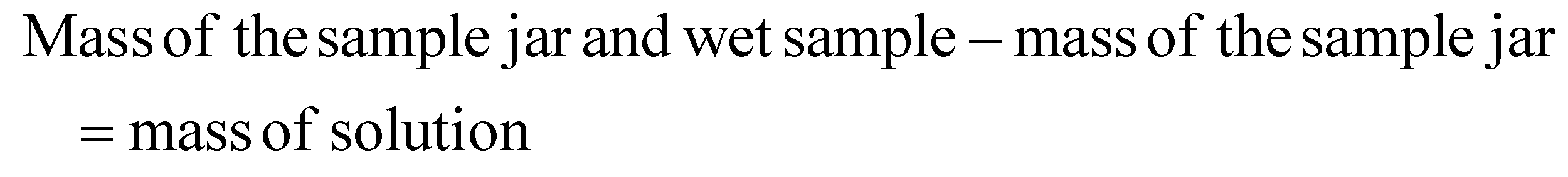DOI:
10.1039/D4PM00286E
(Paper)
RSC Pharm., 2025,
2, 398-412
Controlled isolation of a novel polymorphic form of chlorothiazide by spray drying†
Received
3rd October 2024
, Accepted 8th January 2025
First published on 16th January 2025
Abstract
This study outlines a route to producing a novel polymorphic form of chlorothiazide (CTZ). CTZ was spray dried using three different atomising gas flowrate settings to determine whether it has any effect on the solid-state of CTZ. At a lower atomising gas flowrate, a new form of CTZ, CTZ form IV, was obtained in pure form, whereas at the highest atomising gas flowrate, a mixture of CTZ form I and CTZ form IV was obtained. The morphology of CTZ form I was prism-shaped, and the new form, CTZ form IV, consisted of spherical clusters, some of which were porous and some non-porous. As a result of the rapid drying process, acetone was trapped within the porous clusters and could be released by milling. CTZ form IV has been shown to be stable at room temperature and below 40% relative humidity (RH); however, after 1 week of stability under accelerated conditions of 40 °C/75% RH, CTZ form IV converted to CTZ form I. Also, at high temperatures between 150 °C and 175 °C, CTZ form IV converted to form I, with the simultaneous release of acetone upon its morphology change. This study demonstrates how spray drying can be useful to discover new forms of APIs by a controlled drying process.
Introduction
The late discovery of new polymorphs of active pharmaceutical ingredients (APIs) is a recurring issue in the development of pharmaceuticals. Notable examples include the polymorphism of ranitidine hydrochloride, rotigotine, and acetylsalicylic acid.1 Polymorphs have distinct and characteristic physicochemical properties such as solubility, bioavailability and stability;2 therefore, if the polymorphic forms of an API are not fully explored, this can become an issue when commercialising the API. Ritonavir is a well-documented example of a pharmaceutical that was marketed before another polymorph ‘appeared’, which had a lower solubility and bioavailability, leading to product withdrawal.3,4 Thorough polymorph screening is essential when applying for a patent, and it is a requirement by the U.S. Food and Drug Administration that the polymorphism of all APIs is investigated before clinical trials and continually investigated while being produced at scale.5,6 It is understood that the number of polymorphs discovered is related to the time and money spent searching for them, and Neumann and van de Streek raise the question of whether any commercial drugs currently being sold still have more stable polymorphs that are not yet discovered.7
New polymorphic forms generally appear upon changes in crystallisation conditions such as mechanochemical methods, crystallisation from liquid solutions,8 variation in pressure and temperature,9 seeding8,10 and more. Spray drying has also been shown to have the ability to control the polymorphic form of an API.11 Such control can be due to confinement within the droplet and the consequent supersaturated state being rapidly isolated and dried. In 1982, spray drying was used to isolate the fifth polymorphic form of phenylbutazone, which was shown to be dependent on the set temperature of the spray dryer.12 Since then, however, it has not been used as a primary method to isolate novel polymorphic forms of APIs. Spray drying has been increasingly used for particle engineering purposes,13,14 it is commonly used in the pharmaceutical industry and it is a scalable technique.15 Applying spray drying to pharmaceutical materials can lead to the discovery of additional crystal forms, which have not been isolated by traditional screening methods. This study aims to spray dry chlorothiazide (CTZ) without the presence of additives to determine whether there is any change in the solid-state form of CTZ under the set spray drying conditions.
CTZ is a diuretic and has a secondary function as an antihypertensive.16,17 It is a BCS class IV API and has a low solubility in water and many organic solvents.18 It was the first thiazide diuretic to be introduced to the American market as its stable polymorphic form I by Merck in 1958.17 CTZ has previously been extensively screened for new polymorphic forms under ambient conditions. Several new solvates were identified; however, no new polymorphic forms were identified.19–25 In 2010, a second polymorph of CTZ was discovered through high pressure recrystallisation.9 CTZ form II was obtained at 4.4 GPa and 293 K and is not stable under ambient conditions. In June 2024, a third polymorphic form of CTZ was discovered.26 CTZ form III can be crystallised from various basic aqueous solutions and was found to be less thermodynamically favoured than form I.
Previous studies on spray drying chlorothiazide have investigated controlling the physicochemical properties of salts of chlorothiazide. Paluch et al. evaluated the impact of various parameters on the properties of CTZ sodium (CTZNa) and CTZ potassium (CTZK).27–30 Only two studies have investigated spray drying pure CTZ and not a salt of CTZ. Corrigan et al. investigated whether an amorphous form of CTZ could be obtained via spray drying; however, its amorphous form was too unstable.31 Another study by Paluch et al. investigated spray drying pure chlorothiazide and only obtained CTZ form I, using a mixture of acetone and water as the solvent.32 In this work, spray drying is used as a method to control the polymorphism of an already commercial active pharmaceutical ingredient (API), CTZ, without the use of additives. The discussions herein report how spray drying is applied to isolate a novel polymorphic form of CTZ, with a focus on characterising the particle properties of the material obtained.
Materials
Chlorothiazide 98% (Thermo Fisher Scientific, USA), acetone (Thermo Fisher Scientific, USA), deuterated dimethyl sulfoxide (DMSO-d6) 99.9% with TMS (0.1 vol%) (Merck Life Science, USA), deionised water (ELGA LabWater, UK), and sodium chloride (Merck Life Science, USA) were used in this study.
Methods
Solubility screening
Saturated solutions of CTZ were prepared in methanol, acetone and isopropyl alcohol. A Polar Bear Plus (PBP) (Cambridge Reactor Design, UK) was used to keep the solutions stirring at 25 °C for 24 h. After the solutions were removed from the PBP, they were allowed to settle. A clean, dry sample jar was weighed and labelled. Once the excess solute material settled at the bottom of the jars taken from the PBP, an aliquot was taken from the liquid portion above the solute. The aliquot was dispensed through a 0.45 μm PTFE membrane filter (VWR International Ltd, Ireland) into the clean, dry sample jar, which was reweighed (‘wet sample’). The jar was then placed in an oven at 50 °C for 24 h to ensure all the solvent evaporated. The jar was then re-weighed (‘dry sample’). This was performed in triplicate for each solvent. The following calculation was used to determine the solubility of CTZ in each solvent:
| Mass of solution − mass of solute = mass of solvent |
Spray drying
A B-290 Mini Spray Dryer coupled with the Büchi Inert Loop B-295 (Büchi Labortechnik AG, Switzerland) was used in this investigation. A closed loop was set up as an organic solvent, acetone, was used. A two-fluid nozzle was used for each run, and the experiments were conducted using 3.85 mg ml−1 chlorothiazide in acetone. The condenser temperature was fixed at −20 °C. The aspirator was set at 100% (35.0 m3 h−1). The inlet temperature was adjusted to keep the outlet temperature fixed at 50 °C. The feed flowrate was set at 1.5 ml min−1. For each run, the atomising gas flowrate was varied between 473 and 742 L h−1.
Powder X-ray diffraction (PXRD)
Diffractograms were recorded at room temperature using an Empyrean diffractometer (Malvern Panalytical, UK) in reflection mode with an incident beam of Cu Kα radiation (λ = 1.5406 Å) in the 2θ range between 5° and 40°. The tube voltage was set at 45 kV, and the current was set at 40 mA. Samples were lightly pressed on a silicon zero-background disc before analysis.
Structural characterization from powder data
The initial unit cell metrics of CTZ form IV were determined using DICVOL 06, used via the Panalytical X-Pert HighScore Plus Suite. The cell metrics were then refined against the measured PXRD data using the Pawley method in EXPO (v 1.21.09),33 and the structure was solved by simulated annealing in the same software starting from the molecular structure and conformation of CTZ form I. The early solution was refined using the Rietveld method.
Variable temperature powder X-Ray diffraction (VT-PXRD)
Diffractograms at different temperatures were recorded using an X'Pert MPD Pro diffractometer (Malvern PANalytical, UK) equipped with an X'Celerator detector, operating in scanning line detector mode, and an incident beam of Cu Kα radiation (λ = 1.5406 Å) in the 2θ range between 5° and 40°. An Anton Paar TTK 450 stage coupled with the Anton Paar TCU 110 Temperature Control Unit (Anton Paar GmbH, Austria) was used to record the variable temperature diffractograms. The powder was loaded onto a zero-background sample holder made for the Anton Paar TTK 450 chamber. Measurements were performed under a nitrogen stream between 25 °C and 250 °C, at a 10 °C min−1 heating rate, and the sample was then cooled back to 25 °C.
Thermogravimetric analysis (TGA)
A TGA Q50 (TA Instruments, USA) was used to carry out TGA analysis. Approximately 2–5 mg of the samples were weighed into the crucible. The samples were heated to 400 °C at a heating rate of 10 °C min−1 under nitrogen gas.
Differential scanning calorimetry (DSC)
A DSC 214 Polyma (NETZSCH, Germany) was used to perform DSC analysis on each of the samples. Approximately 3.5 mg of the sample were loaded into a hermetically sealed aluminium pan. Spray dried samples were heated at 10 °C min−1 under a nitrogen flowrate of 30 ml min−1. Powders of CTZ Run B were subsequently heated at accelerated heating rates of 100 °C min−1, 200 °C min−1, and 300 °C min−1 from 20 °C to 400 °C, 420 °C and 447 °C, respectively. Two samples of CTZ Run B were heated to 260 °C at 200 °C min−1 and 500 °C min−1.
Fourier transform infrared (FTIR) spectroscopy
A Nicolet iS50 FTIR spectrometer (Thermo Fisher Scientific, USA) was used in transmission mode to produce the FTIR results. Spectra were obtained across a wavenumber range of 500 cm−1 to 4000 cm−1 using 64 scans per spectrum.
Scanning electron microscopy (SEM)
A Hitachi SU-70 scanning electron microscope (Hitachi Inc., Japan) was used to obtain the SEM images presented herein. An accelerating voltage of 5 kV was applied. A small amount of the powder was placed onto adhesive carbon tape, which had been previously attached to a 15 mm cylindrical aluminium SEM stub. The samples were coated with gold (∼93.8 Å) using an Emitech K550 (Emitech, United Kingdom) sputter coater at 20 mA for 90 s.
Focused ion beam (FIB)-SEM
An FEI Helios G4 CX dual-beam microscope (Thermo Fisher Scientific, USA) was used to obtain a cross-sectional view of the particles. Sample preparation was the same as for SEM analysis. The part of the particle to be protected from the beam was coated with an additional 1 μm layer of platinum.
Particle size analysis (PSA)
The ImageJ java-based image processing program was used to analyse the average particle size from the obtained SEM images. To determine the average particle size for the sample, the diameter of 150 particles was measured.
Nuclear magnetic resonance (NMR)
A Varian 500 MHz spectrometer (Agilent Technologies, USA) was used to carry out NMR analysis. For proton NMR, the number of scans was 16, the pulse angle was set at 45°, the relaxation delay was 1 s, the spectral width was −2 to 14 ppm and the acquisition time was 2.045 s. For carbon-13 NMR, the number of scans was 256, the pulse angle was set at 45°, the relaxation delay was 1 s, the spectral width was −14.3 to 234.3 ppm and the acquisition time was 1.049 s. The samples were prepared in high-throughput NMR tubes with an outer diameter of 0.019 mm. 10 mg ml−1 CTZ form I and CTZ Run B samples were prepared in DMSO-d6. An additional CTZ form I sample was prepared and spiked with 0.125 μL of acetone. An additional CTZ Run B sample was prepared and a small quantity of D20 was added.
Stability testing
An Amebis U062 temperature-controlled cabinet was used to store samples for stability analysis. The stability conditions were set at ICH standard accelerated stability conditions of 40 °C/75% relative humidity (RH).34 A saturated solution of sodium chloride was used to set the humidity at 75%.35 Stability testing was also carried out on samples stored in a desiccator at room temperature (18.6–20.9 °C) with 37–39% RH. The temperature and humidity of the desiccator were measured using a ThermoPro TP357 Hygrometer and Thermometer Sensor (ThermoPro, USA). Stability testing was carried out for 3 months.
Vacuum experiment
A VACUtherm vacuum oven (Thermo Scientific, USA) was used to store the powder sample in an open vial overnight and a vacuum was reached at 0 bar. Table 1 outlines the four vacuum conditions used.
Table 1 Vacuum experiment conditions
| Condition |
Sample |
Pressure |
Temperature |
Time |
|
Condt. 1
|
CTZ Run B
|
Vacuum |
Room temperature |
1 h |
|
Condt. 2
|
CTZ Run B
|
Vacuum |
Room temperature |
Overnight |
|
Condt. 3
|
CTZ Run B
|
Vacuum |
70 °C |
Overnight |
|
Condt. 4
|
CTZ Run B
|
Vacuum |
100 °C |
Overnight |
Milling experiment
A Mixer Mill MM 400 (RESTCH, Germany) was used to mill the sample. A 5 ml agate milling jar and a 10 mm diameter agate ball were used to mill 40 mg of the sample for 1 hour at a frequency of 25 Hz.
Hirshfeld surface analysis
The intermolecular interactions within the CTZ crystal structures were identified using molecular Hirshfeld surface and fingerprint analysis, and the plots were generated using the CrystalExplorer21 software.36 The crystallographic data for CTZ form I (CSD Refcode QQQAUG09) and form IV (this work) were used as structural models.
Results and discussion
Solubility screening
The solubility of CTZ was analysed in three solvents that are regarded as green solvents37 and are commonly used for spray drying:38,39 methanol, acetone and isopropyl alcohol (IPA). CTZ had the highest solubility in acetone, 10 mg ml−1, and therefore was chosen as the solvent for spray drying (Table 2).
Table 2 Solubility screening of chlorothiazide in methanol, acetone and IPA
| Solvent |
Result 1 |
Result 2 |
Result 3 |
Average [Std. Dev.] |
| Methanol |
0.00293 g ml−1 |
0.00273 g ml−1 |
0.00276 g ml−1 |
0.00281 g ml−1 [9 × 10−5] |
| Acetone |
0.010 g ml−1 |
0.011 g ml−1 |
0.010 g ml−1 |
0.010 g ml−1 [5 × 10−3] |
| IPA |
0.00069 g ml−1 |
0.00073 g ml−1 |
0.00060 g ml−1 |
0.00067 g ml−1 [5 × 10−5] |
Spray drying
Three different sets of parameters were used to spray dry a solution of CTZ in acetone and are listed in Table 3. The only varying parameter was the atomising gas flowrate. A low concentration of 3.85 mg ml−1 was used due to CTZ having a solubility of 10 mg ml−1 in acetone. For efficient spray drying, the concentration must be lower than the maximum solubility of the material in the solvent and, therefore, 3.85 mg ml−1 was chosen. The feed flowrate was fixed at 1.5 ml min−1 for each run. The inlet temperature was set to keep the outlet temperature at approximately 50 °C. There was a 1–2 °C fluctuation in the outlet temperature throughout each run. Spray dried samples were transferred from the sample collection vessel to a sample vial and stored in a desiccator at room temperature.
Table 3 Spray dried sample information and spray dryer parameters
| Sample information |
Set parameters |
Results |
| Sample ID |
Solid concentration (w/v%) |
Inlet (°C) |
Feed flowrate (ml min−1) |
Atomising gas (L h−1) |
Outlet (°C) |
Yield (%) |
| CTZ Run A |
0.4 |
65 |
1.5 |
742 |
49–50 |
79.1 |
| CTZ Run B |
0.4 |
65 |
1.5 |
601 |
49–51 |
61.1 |
| CTZ Run C |
0.4 |
65 |
1.5 |
473 |
50–51 |
41.8 |
From Table 3, it can be observed that the yield increased with increasing atomising gas flowrate. This is due to higher atomising gas flowrates, which atomise the feed solution into smaller droplets. The smaller droplets have a higher surface area, allowing for evaporation to occur quicker than at low atomising gas flowrates. As the droplets dry quicker, they are more likely to reach the collection vessel instead of sticking to the glass walls of the spray dryer.
Powder X-ray diffraction
PXRD analysis was carried out on each of the spray dried samples, and the obtained diffractograms are displayed in Fig. 1. From the results, it can be observed that the PXRD pattern of CTZ Run A presents the characteristic peaks of CTZ form I, with additional peaks from an unknown phase. Characteristic peaks of form I include those with the highest intensity at 14.52°, 19.92°, 20.58°, 21.91°, and 26.40°. Characteristic peaks of the unknown phase include those with the highest intensity at 17.94°, 18.96°, 21.07°, 21.67°, and 25.56°. No characteristic peaks of CTZ form II or CTZ form III were identified. CTZ Run B shows no characteristic peaks of CTZ form I and only displays peaks from the unknown phase. CTZ Run C also shows characteristic peaks of the unknown phase and one small peak of CTZ form I at 20.58°. The peaks of the unknown phase were compared to those of all known crystalline forms of chlorothiazide in the CSD and did not correspond to any previously identified characteristic peaks of chlorothiazide forms; therefore, a new crystal form was obtained.
 |
| | Fig. 1 Comparison of experimental PXRD diffractograms for spray dried samples and calculated diffractograms for CTZ form I (CSD Refcode: QQQAUG09). The characteristic peaks of the unknown phase, include the peaks with the highest intensity, are annotated (*). | |
Crystal structure determination
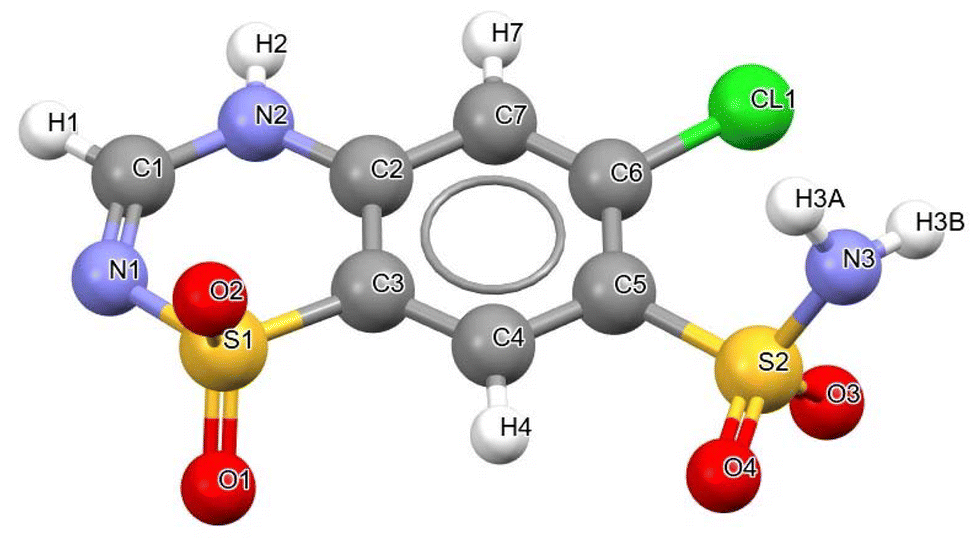
Since all attempts to obtain single crystals of the new form were unsuccessful (see the ESI†) and resulted in the formation of CTZ form I, similar to the extensive crystallisation screening carried out by Johnston et al.,25 the structure of the new phase was solved from powder data of CTZ Run B (see ESI Fig. S1†). The new phase was identified to be a new polymorphic form of CTZ: CTZ form IV. Form IV crystallises in the monoclinic P21/c space group with a = 7.33883 Å, b = 14.75397 Å, c = 10.87998 Å and β = 114.951° (Table 4). The unit cell contains four CTZ molecules (Z = 4), with one molecule in the asymmetric unit. The crystal structure data of the reported CTZ form I, form II, form III and the new form IV are presented in Table 4. The structure of all CTZ crystal forms is sustained by a dense network of hydrogen bonds due to the numerous H-bond donors and acceptors present in the CTZ molecule (Fig. 2). In form I, form II (isostructural to form I), and form III, the hydrogen bonds lead to the formation of a tilted column structure of CTZ molecules (although in form III, the columns are arranged in a different manner).9,26 In form IV, however, the CTZ molecules form dimers arranged in a double herringbone pattern (Fig. 3[A]).
 |
| | Fig. 2 Chemical structure of CTZ and atomic numbering. | |
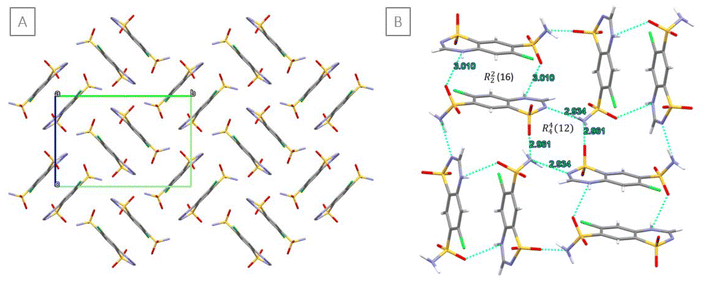 |
| | Fig. 3 [A] Crystal packing arrangement of CTZ form IV, view down the a-axis; H atoms are not shown for clarity. [B] Representation of the hydrogen-bonded ring motifs R22(16) and R44(12) formed by dimers in the double herringbone pattern (H-bonds are shown as light blue dashed lines). | |
Table 4 Crystallographic parameters of polymorphic forms of CTZ
| |
CTZ form I
|
CTZ form II
|
CTZ form III
|
CTZ form IV
|
|
Methods of polymorph discovery: SC, solution crystallisation; HP, high-pressure recrystallisation; and SP, spray drying crystallisation.
|
| CSD refcode |
QQQAUG099 |
QQQAUG159 |
QQQAUG2226 |
This work |
| Chemical formula |
C7H6ClN3O4S2 |
C7H6ClN3O4S2 |
C7H6ClN3O4S2 |
C7H6ClN3O4S2 |
|
M
w/g mol−1 |
29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 572 572 |
29572 |
29572 |
29572 |
| Methoda |
SC |
HP |
SC |
SP |
|
T/K |
293 |
293 |
100 |
293 |
|
P/GPa |
0 |
4.4 |
0 |
0 |
| Crystal system |
Triclinic |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P1 |
P1 |
P21 |
P21/c |
|
a/Å |
4.8746(14) |
4.5100(5) |
4.8296(1) |
7.3334(17) |
|
b/Å |
6.4011(10) |
5.9287(6) |
6.2703(1) |
14.741(3) |
|
c/Å |
8.980(3) |
8.503(3) |
16.9551(2) |
10.871(2) |
|
α/° |
74.05(2) |
76.528(16) |
90 |
90 |
|
β/° |
83.54(2) |
85.624(17) |
92.214(1) |
114.959(5) |
|
γ/° |
80.468(18) |
83.203(8) |
90 |
90 |
|
V/Å3 |
265.03 |
219.267 |
513.069 |
1065.42 |
|
Z, Z′ |
1, 1 |
1, 1 |
2, 1 |
4, 1 |
|
d/g cm−3 |
1.853 |
2.239 |
1.914 |
1.844 |
The dimer is the basic structural unit of CTZ form IV: two CTZ molecules are connected via two hydrogen bonds between the protonated nitrogen of the thiadiazine ring and a sulfonamide oxygen (N2–H2⋯O3 3.010 Å), forming a bimolecular hydrogen-bonded ring motif R22(16). The two CTZ molecules are related by an inversion centre. This dimer resembles type 3 of the four bimolecular CTZ⋯CTZ pairwise face–face motifs described by Johnston et al., which are commonly observed in the structure of CTZ compounds.25 The dimers are interconnected through a hydrogen-bonded ring motif R44(12) between four CTZ molecules. These H-bonds connect the nitrogen of the sulfonamide group with the sulfonyl oxygen (N3–H3B⋯O2 2.961 Å) and with the π nitrogen of the thiadiazine ring (N3–H3A⋯N1 2.934 Å) (Fig. 3[B]).
Parallel dimers interact through close contacts between the chlorine and the sulfonamide oxygen (Cl1⋯O4 3.002 Å), and between a sulfonyl oxygen and a phenyl carbon (C7–H7⋯O1 3.150 Å), as observed also in CTZ form I (Fig. S2†). Form IV differs from forms I, II and III not only in the crystal packing arrangement but also in the molecular conformation that CTZ assumes, specifically in the torsional angle of the sulfonamide group and the position of the sulfonyl group in the thiadiazine ring (Fig. S3†). In CTZ form I, the torsional angle (N3–S2–C5–C4) measures 109.98° (108.21° in form II, 106.7° in form III), while in form IV, it increases to 120.25°, resulting in a slightly more staggered conformation (Fig. S3†). The other characteristic that distinguishes form IV from the other forms is the orientation of the sulfonyl group relative to the sulfonamide side chain. This difference can be described using the CTZ conformational minima presented by Johnston et al.:25 in forms I, II and III, the SO2 group and the sulfonamide chain point in opposite directions (minot conformation). In CTZ form IV, however, the sulfonyl group points in the same direction as the sulfonamide (minst conformation), as shown in the superimposition of form I and IV in Fig. S4.†
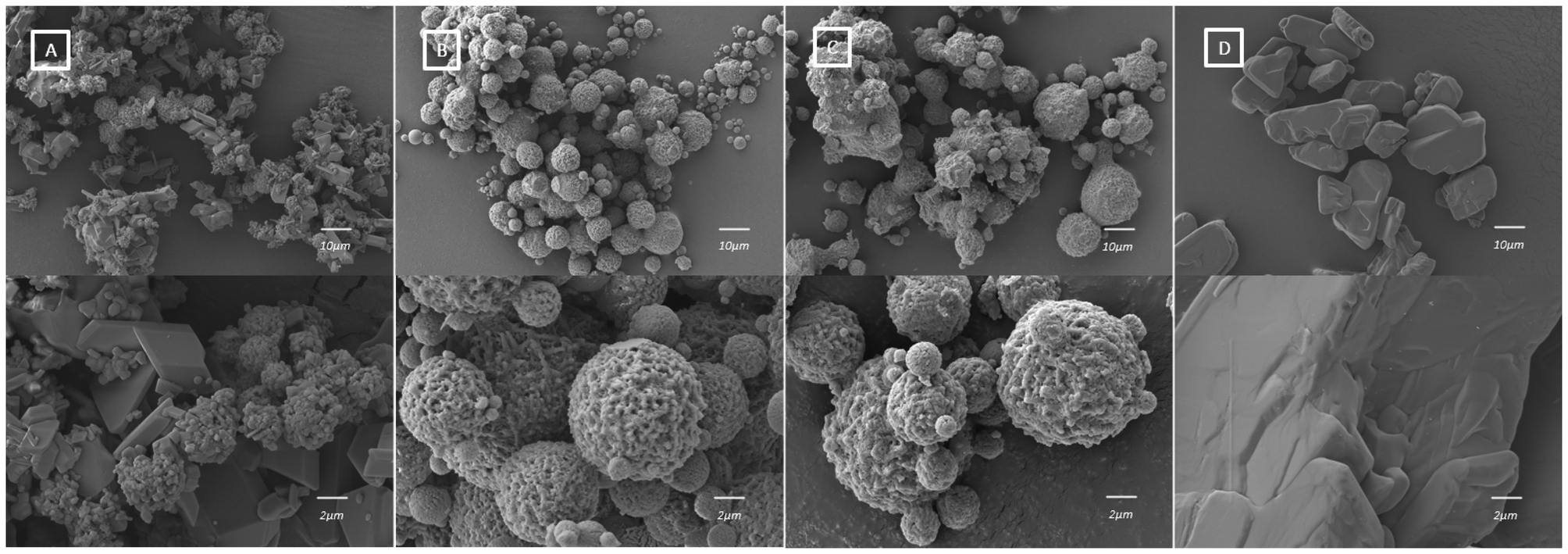
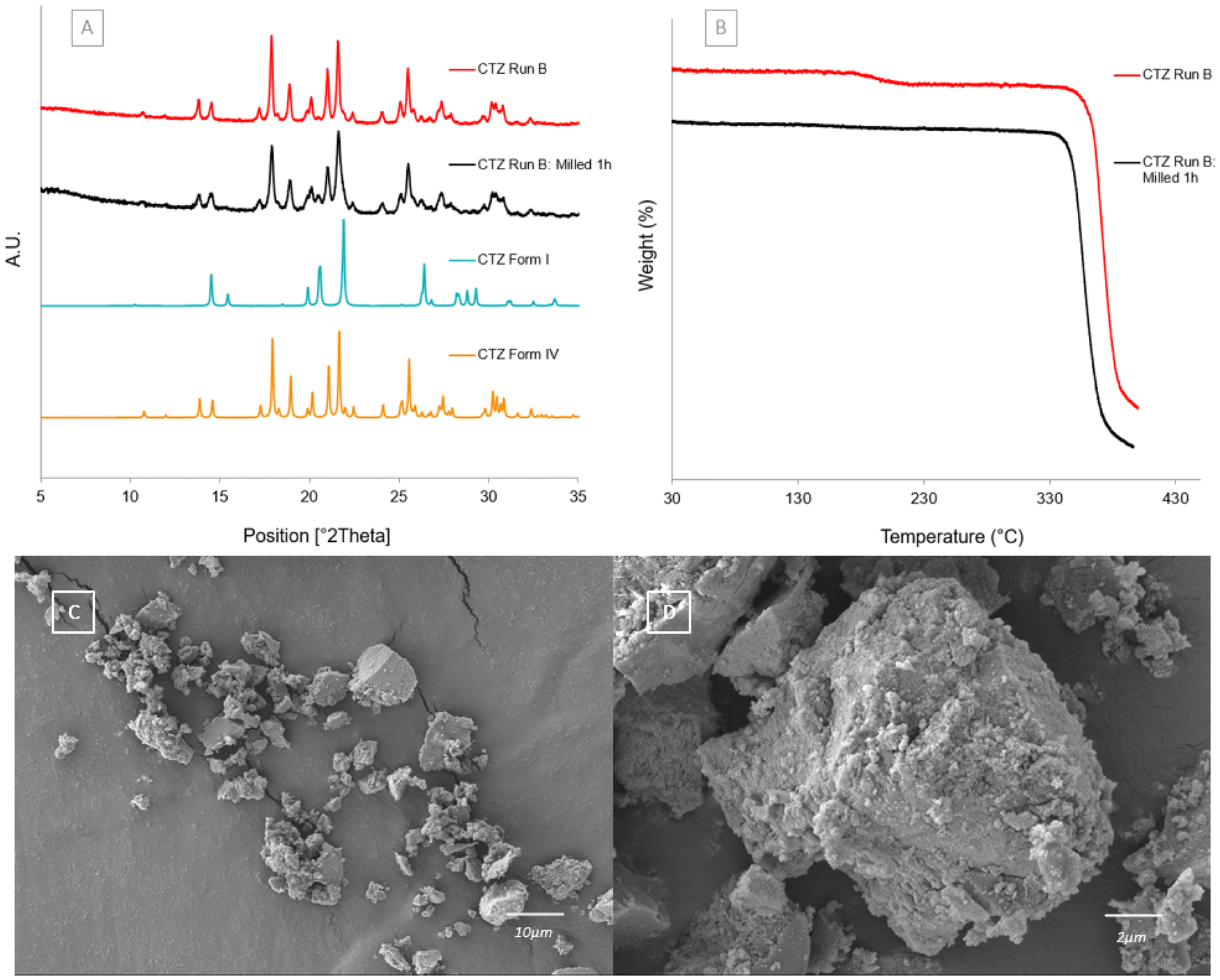
Scanning electron microscopy and particle size analysis
The SEM images of each sample are displayed in Fig. 4. CTZ form I is shown in Fig. 4[D] and is block-like in shape. The particles of CTZ Run B, which was found by PXRD to consist of pure CTZ form IV, form spherical clusters (Fig. 4[B]). As the block-like particles represent CTZ form I, the spherical clusters likely represent CTZ form IV in the sample. Fig. 4[A] shows the crystal habit of CTZ Run A, which was found by PXRD to be a mixture of CTZ form I and CTZ form IV. This is confirmed by the crystal habit, which is a mixture of both block-like particles and spherical clusters. Fig. 4[C] shows the crystal habit of CTZ Run C, which appears as more agglomerated clusters. Agglomerated clusters have a lower surface area, which can lead to issues with dissolution.40 These images support the findings by PXRD and show that the parameters used to produce CTZ Run B, consisting of CTZ form IV, are optimal. The particle size of CTZ Run B was analysed from the SEM images; the Dn10 was 2.384 μm, the Dn50 was 5.267 μm and the Dn90 was 9.085 μm.
 |
| | Fig. 4 SEM images of [A] CTZ Run A, [B] CTZ Run B, [C] CTZ Run C, and [D] CTZ form I. | |
Thermal analysis and variable-temperature PXRD
DSC and TGA were carried out on each of the spray dried samples, and the results are displayed in Fig. 5 and 6. Fig. 5[B] displays a magnified version of Fig. 5[A]. The DSC thermogram of CTZ form I shows a large endotherm at 362.0 °C with an onset at 358.6 °C corresponding to the melting point of form I. The DSC thermogram of CTZ Run A shows a small endotherm at 213.6 °C with an unclear onset and a larger endotherm at 366.8 °C with an onset at 362.8 °C. The DSC thermogram of CTZ Run B consists of a small endotherm at 195.8 °C with an onset at 166.8 °C and a larger endotherm at 365.5 °C with an onset at 359.5 °C. The DSC thermogram of CTZ Run C consists of a small endotherm at 154.3 °C with an onset at 130.9 °C and a larger endotherm at 366.6 °C with an onset at 364.2 °C.
 |
| | Fig. 5 [A] DSC thermograms of each spray dried sample and CTZ form I heated at 10 °C min−1. [B] Magnified DSC thermograms of ‘[A]’, with a closer view between 40 and 300 °C. | |
 |
| | Fig. 6 [A] TGA thermograms of spray dried sample CTZ Run B and CTZ form I heated at 10 °C min−1. [B] Magnified TGA thermograms of ‘[A]’, with a closer view between 80 and 240 °C. | |
The TGA thermogram of CTZ Run B in Fig. 6 shows a small weight loss of 2.46% with an onset at 164.10 °C and decomposition with an onset at 368.58 °C. This weight loss occurs in the same temperature range in which the small endotherm appears in DSC (166.8 °C); the decomposition occurs when the sample begins to melt (which coincides with the temperature range of the large endothermic peak in DSC).
The small weight loss of 2.46% cannot be related to material being removed from the crystal structure and therefore is likely due to a residual solvent being removed. The only solvent used in this study was acetone, with a boiling point of 56 °C; therefore, acetone molecules must be trapped within the spray dried particles and are not released until the samples are heated above 150 °C.
A VT-PXRD analysis was carried out on the powder of CTZ Run B, which consists of CTZ form IV in pure form, to investigate the behaviour of the new form upon heating. The results from the analysis shown in Fig. 7 show that in the temperature range of 150–200 °C, a polymorphic transition takes place and CTZ form IV transforms into CTZ form I.
 |
| | Fig. 7 VT-PXRD of CTZ Run B heated to 250 °C at a heating rate of 10 °C min−1 and cooled back to 25 °C. | |
Therefore, as confirmed by VT-PXRD, the small endothermic peak in the DSC corresponds to the polymorphic transition taking place between CTZ form IV and CTZ form I, and the release of acetone. The polymorphic transition occurs between 150 °C and 175 °C in the VT-PXRD diffractogram; accordingly, the DSC thermogram shows the onset for this thermal event at 166.8 °C. The VT-PXRD also confirms that the large endothermic peak at 365.5 °C in the DSC thermogram of CTZ Run B corresponds to the melting point of form I.
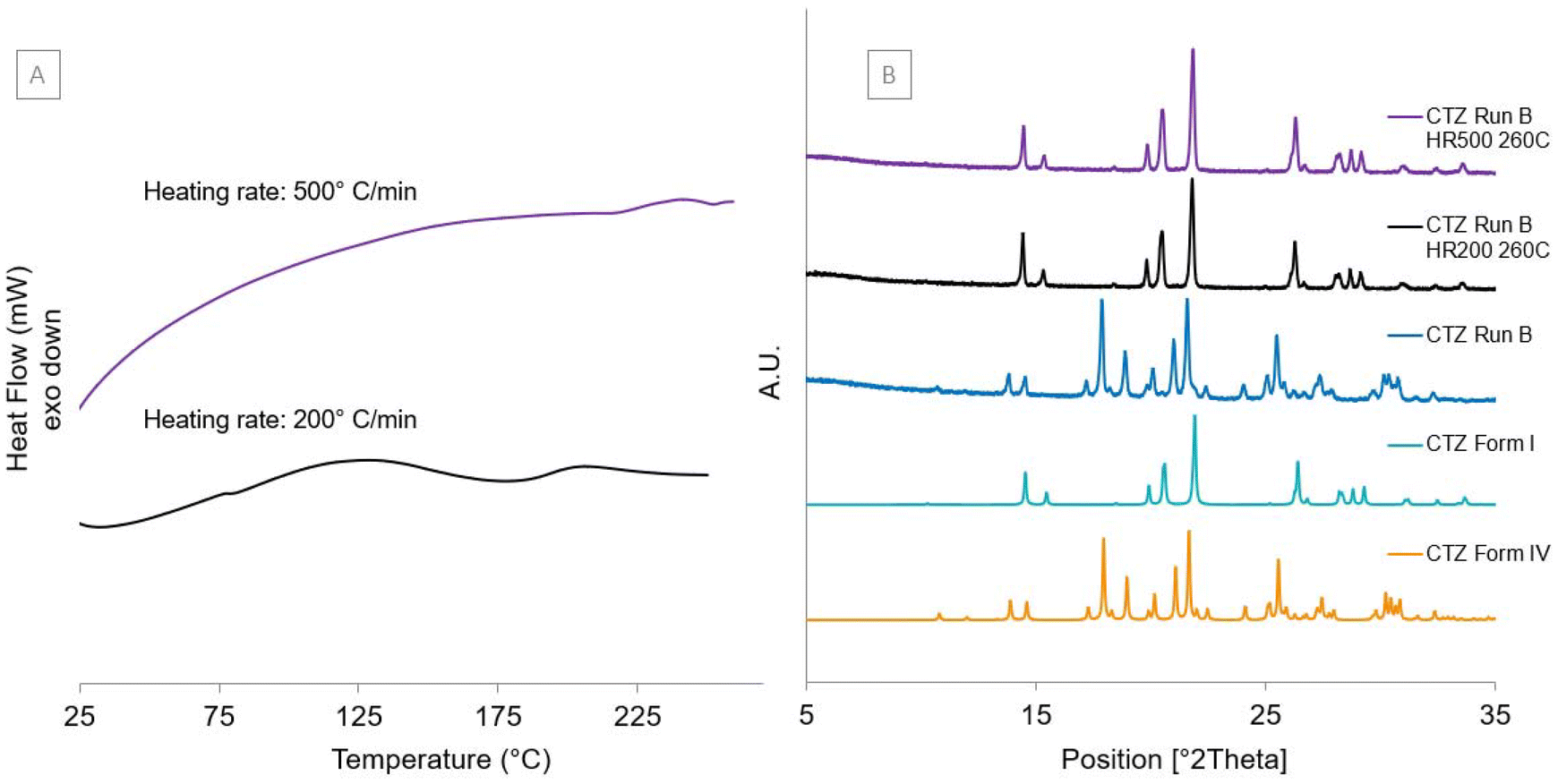
CTZ form IV was analysed using DSC at faster heating rates to see if it was possible to identify the melting temperature of form IV before it converts to CTZ form I. Powders of CTZ Run B were heated at three different accelerated heating rates, 100 °C min−1, 200 °C min−1 and 300 °C min−1. From these results, it was still unclear whether the melting observed was for CTZ form IV or CTZ form I as the small endotherm peak is still present at accelerated heating rates (Fig. S5†). DSC analysis was also carried out at 200 °C min−1 and at the maximum heating rate of 500 °C min−1 from 25 °C to 260 °C, which is after the polymorphic transition but before CTZ melts (Fig. 8[A]). The pan was then retrieved from the DSC, reopened and the powder was analysed using PXRD. The PXRD diffractograms in Fig. 8[B] show that the polymorphic transition from CTZ form IV to form I has occurred, as the samples retrieved from the DSC were identified to be CTZ form I. This shows that it is not possible to identify the melting temperature of CTZ form IV with the DSC used as it can only reach a heating rate of 500 °C min−1.
 |
| | Fig. 8 [A] DSC thermograms of the powders of CTZ Run B heated to 260 °C at heating rates (HR) of 200 °C min−1 and 500 °C min−1. [B] Experimental PXRD diffractograms of CTZ Run B after being heated to 260 °C at 200 °C min−1 [CTZ Run B HR200 260C] and 500 °C min−1 [CTZ Run B HR500 260C] and calculated diffractograms for CTZ form IV and CTZ form I. | |
Focused ion beam scanning electron microscopy
In order to determine whether acetone could be trapped inside the spherical particles of CTZ form IV, FIB-SEM analysis was performed. The analysis was carried out on four particles of CTZ Run B powders. The images produced are displayed in Fig. 9. These images show that the sample consists of both porous and non-porous particles. This confirms that it is possible for acetone to be trapped within the internal structure of the spherical particles and that the weight loss appearing in TGA is a result of the loss of acetone molecules. The presence of a mixture of porous and non-porous particles explains why there is a low percentage of acetone loss from the sample in TGA as there is limited space for it to be trapped.
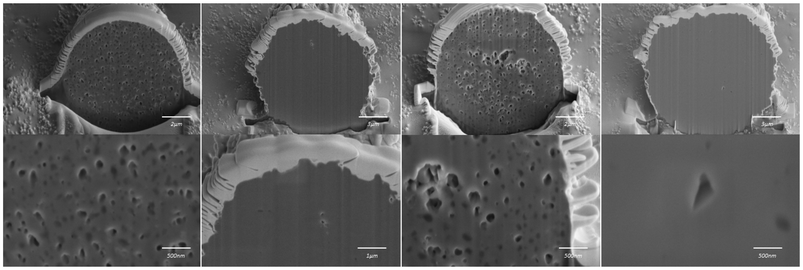 |
| | Fig. 9 FIB-SEM images of four particles from CTZ Run B. | |
As the polymorphic transition and the loss of acetone occur between 150 °C and 200 °C, it can be proposed that the transition causes a morphological rearrangement within the particles, allowing for the acetone trapped within the particle's pores to be released. To confirm this hypothesis, both vacuum and milling experiments were carried out to see if the acetone could be removed from within the pores.
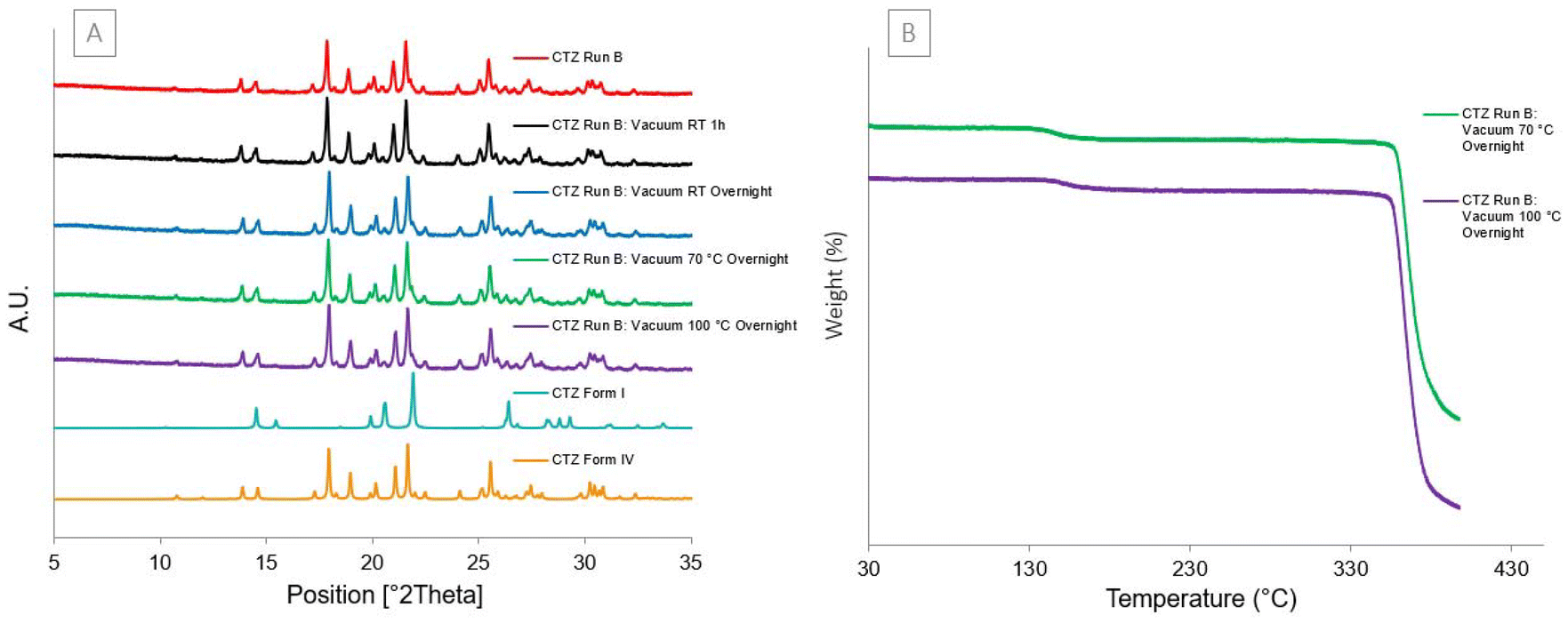
Vacuum experiment
Powders of CTZ Run B (CTZ form IV) were placed under vacuum conditions, as listed in Table 1. As the sample did not change after condt. 1, the temperature was increased to 100 °C and the time period was extended to overnight. After the sample experienced each of these four conditions, there was no change in the PXRD patterns (Fig. 10[A]) or the TGA thermogram of the sample (Fig. 10[B]). This shows that the sample remained in CTZ form IV and the acetone remained within the particles. The temperature was not increased again as it would approach the polymorphic transition temperature, causing the acetone to be released due to the polymorphic change instead of due to the vacuum conditions. This shows the robustness of the morphology of the porous particles to withstand vacuum conditions and retain the trapped acetone at 100 °C overnight.
 |
| | Fig. 10 [A] Experimental PXRD pattern of each vacuum experiment and calculated diffractograms for CTZ form I and IV. [B] TGA thermograms of the final two vacuum conditions. | |
Milling experiment
Milling the powders of CTZ Run B (CTZ form IV) for 1 hour resulted in the breaking of the particle clusters’ shape, allowing for the trapped acetone to be released. PXRD analysis shows that after milling, the powders consist of a mixture of CTZ form IV and a trace of form I (Fig. 11[A]). Fig. 11[B] confirms that the acetone has been released as there is no longer a weight loss present at 164.10 °C. This shows that CTZ form IV is not reliant on the presence of acetone as it is still present after the acetone is released. There is no solvent-mediated transformation occurring due to the presence of acetone. Fig. 11[C] shows an SEM image of the milled sample, in which it is clear that the clusters have all broken apart, and Fig. 11[D] shows a closer view of the milled sample.
 |
| | Fig. 11 [A] Experimental PXRD patterns of CTZ Run B, CTZ Run B milled for 1 hour and calculated for CTZ forms I and IV. [B] TGA thermograms of CTZ Run B and CTZ Run B milled for 1 hour. [C] SEM image of CTZ Run B milled for 1 hour. [D] Magnified SEM image of CTZ Run B milled for 1 hour. | |
Nuclear magnetic resonance
To confirm that the weight loss was due to the loss of acetone, NMR analysis was performed, as FTIR analysis did not detect the presence of acetone (Fig. S6†). Spectra of DMSO-d6, CTZ form I in DMSO-d6, CTZ form I in DMSO-d6 spiked with 0.125 μL of acetone and CTZ Run B in DMSO-d6 are shown in Fig. 12. In the proton NMR spectra, the peak at 0 ppm corresponds to TMS, the peak at 1.2 ppm corresponds to an impurity in the CTZ, the peaks at 2.4–2.5 ppm correspond to the protons in the sulphur methyl groups in DMSO-d6, the peak at 3.3 ppm corresponds to the D2O present in DMSO-d6 and the peaks between 7.5 and 8.2 ppm represent the three protons on the aromatic rings of CTZ. In Fig. 12[A], two additional peaks are observed for DMSO-d6 + CTZ Run B and DMSO-d6 + CTZ form I + Acetone at ∼1 and 2 ppm. The peak at 2 ppm (ref. 41) is attributed to the protons present in acetone, and the peak at ∼1 ppm is due to an impurity in the acetone used as it is 99.8% pure. There is a peak present at 12.6 ppm for both samples, including CTZ form I, which is due to the presence of exchangeable protons (see the ESI†). The results confirm that acetone is present in CTZ Run B.
 |
| | Fig. 12 [A] Proton NMR of CTZ Run B, CTZ form I spiked with acetone, CTZ form I, and the solvent used in each sample, deuterated DMSO. [B] Carbon-13 NMR of CTZ Run B, CTZ form I spiked with acetone, CTZ form I, and the solvent used in each sample, deuterated DMSO. | |
In the carbon-13 NMR spectra, Fig. 12[B], the four carbons on TMS are present at 0 ppm. The carbons in DMSO-d6 are present between 38 and 40 ppm, and the six carbons on CTZ are present between 120 and 150 ppm. The additional peak present in DMSO-d6 + CTZ Run B and DMSO-d6 + CTZ form I + Acetone is at 31.18 ppm, further confirming the presence of acetone.41
Stability testing
The stability of CTZ form IV produced in CTZ Run B was analysed over 3 months under both accelerated stability conditions and in a desiccator at room temperature. These conditions were chosen in order to assess how stable the polymorphic form is under low humidity (Fig. 13[A]) and high humidity (Fig. 13[B]) conditions. The change in polymorphic form is not due to the temperature, as confirmed by the vacuum experiment. At low humidity, CTZ form IV remained stable and pure. At high humidity, CTZ form IV converted to CTZ form I within the first 7 days. The SEM images in Fig. 13[C] and [D] show that at high humidity the crystal habit of the particles has changed. As mentioned above, in a study by Paluch et al., CTZ was spray dried under similar spray drying conditions; however, a mixture of acetone and water was used as the solvent.32 These stability results may explain why CTZ form I was obtained in that study rather than CTZ form IV obtained in this study. When CTZ form IV is exposed to high humidity or in the presence of water, it converts into CTZ form I.
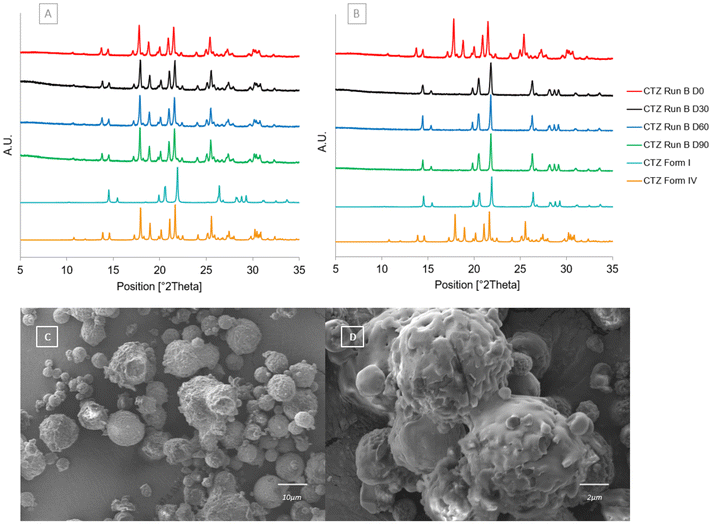 |
| | Fig. 13 [A] PXRD patterns of CTZ Run B at each stability timepoint after being stored in a desiccator with less than 40% RH at room temperature and calculated diffractograms for forms I and IV. [B] PXRD patterns of CTZ Run B at each stability timepoint after being stored at 40 °C, 75% RH, and calculated diffractograms for forms I and IV. [C] SEM image of CTZ Run B after 30 days of stability at 40 °C, 75% RH. [D] Magnified SEM image of CTZ Run B after 30 days of stability at 40 °C, 75% RH. | |
Hirshfeld surface analysis
The comparison of the different intermolecular interactions in the crystal structure between CTZ form I and form IV can be visualised by plotting the 2D-fingerprint of the Hirshfeld surface (Fig. 14 and Fig. S8†).
 |
| | Fig. 14 Hirshfeld 2-D fingerprint plots for [A] CTZ form I and [B] CTZ form IV. | |
The overall fingerprint plot of form IV resembles that of form I; this is because the O⋯H contacts, represented by the two longest tails in the plot, are the main interactions in the crystal structure of both forms (≈41% of the total surface area) and involve the same donors and acceptors, although in different combinations due to the different molecular arrangements. For example, in form IV, O3 forms a H-bond with N2, while, in form I, it interacts with N3 through a H-bond of the same length. The polymorphism is more evident by plotting the N⋯H contacts: form IV presents shorter contacts (longer tails in the plot) and less extended surface area in comparison with form I (7.1% vs. 10.6%). In form IV, N1 forms a H-bond of 2.934 Å with N3, whereas in form I, this distance increases to 3.221 Å. This is the only N⋯H interaction in form IV, whereas in form I, an additional mild interaction, N2–H2⋯N3, is present (Fig. 15). A comparison of the C⋯C contact fingerprint plot also highlights some differences: form IV shows a much closer distance, which is due to the greater overlap of the phenyl rings between the CTZ molecules in the dimer of form IV in comparison with the tilted column structure of form I (Fig. 15).
 |
| | Fig. 15 Hirshfeld surfaces for the contacts N⋯H in [A] CTZ form I and [B] CTZ form IV, and the contacts C⋯C in [C] CTZ form I and [D] CTZ form IV. | |
Effect of spray drying on the solid form
This study has demonstrated a route to producing a novel polymorphic form of chlorothiazide. CTZ form IV has been isolated in pure form in a controlled manner by tuning the atomising gas flowrate to optimal conditions of 601 L h−1. When the liquid feed enters the drying chamber, the set atomising gas flowrate is responsible for atomising the droplets into the drying chamber. As a result, the droplet sizes and subsequent particle sizes are dependent on the flowrate of the atomising gas. As the droplets disperse in the drying chamber, set at a high temperature, the temperature of the droplets begins to rise, and the rate of solvent transport from the centre of the droplet can no longer match the rate of evaporation. The droplets become supersaturated, and nuclei form spontaneously, initiating crystallisation. As the droplets begin to dry from the outside in, a crust begins to form around the wet bulb centre during this falling-rate period.42 For high atomising gas flowrates and, subsequently, smaller droplets, this process occurs more rapidly. A previous study highlighted that the highest atomising gas flowrate produces metastable forms of APIs.11 In this case, at the highest atomising gas flowrate, a mixture of CTZ form I and CTZ form IV was obtained and pure CTZ form IV was isolated at a lower flowrate of 601 L h−1. Stability studies confirm that CTZ form IV is stable at room temperature below 40% RH. An explanation for this is that CTZ form IV could be the kinetically favoured form when an atomising gas flowrate of 601 L h−1 is used. It is also known that rapid drying can also lead to the formation of hollow or porous particles.30 To the best of our knowledge, this is a rare example of solvent being trapped in a porous spherical cluster, spray dried using only a single API and solvent. Previous studies have shown solvent entrapment within a non-porous surface, leading to the formation of hollow particles.43–46
Conclusion
In this study, a novel polymorphic form of CTZ, CTZ form IV, was isolated without any additives present via spray drying. The study demonstrates that the atomising gas flowrate can control which polymorphic form is obtained. CTZ form IV is stable below 40% RH under room temperature conditions. Upon heating, it converts to CTZ form I between 150 °C and 200 °C. While transitioning from a porous morphology to a prism structure, it releases trapped acetone vapours, confirmed by NMR, which appear as a weight loss of 2.46% in TGA. Using spray drying to identify novel polymorphic forms of APIs is a promising new route to polymorph discovery. This unique polymorphic form, with its unique shape and ability to trap the solvent, highlights the advantageous capabilities of spray drying over common crystallisation methods. This study demonstrated that spray drying can expand the potential of pharmaceutical materials, both commercial and in development, by improving their properties and identifying new polymorphic forms that have not yet been discovered.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded by the Irish Research Council under grant number GOIPG/2020/1648. This work was supported by SSPC, the SFI Research Centre for Pharmaceuticals, under grant number 12/RC/2275_P2. We thank Dr Róisín Doohan, Chief Technical Officer, Mr James Donnellan, Senior Technical Officer, and Dr Paulsi Selvakumaran, Senior Technical Officer (NMR Facility SBCS, University of Galway), for NMR analysis and Dr Sergey Beloshapkin, Instrument Scientist (Bernal Institute, University of Limerick), for FIB-SEM analysis.
References
- D.-K. Bučar, R. W. Lancaster and J. Bernstein, Disappearing Polymorphs Revisited, Angew. Chem., Int. Ed., 2015, 54(24), 6972–6993 CrossRef PubMed.
- A. J. Cruz-Cabeza and J. Bernstein, Conformational polymorphism, Chem. Rev., 2014, 114(4), 2170–2191 CrossRef CAS PubMed.
- P. Sacchi,
et al., Crystal size, shape, and conformational changes drive both the disappearance and reappearance of ritonavir polymorphs in the mill, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(15), e2319127121 CrossRef CAS PubMed.
- S. R. Chemburkar,
et al., Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development, Org. Process Res. Dev., 2000, 4(5), 413–417 CrossRef CAS.
-
J. Bernstein and J. MacAlpine, Pharmaceutical Crystal Forms and Crystal–Form Patents: Novelty and Obviousness1, in Polymorphism in the Pharmaceutical Industry: Solid Form and Drug Development, 2018, pp. 469–483 Search PubMed.
-
C. f. D. Evaluation and R. O. o. G. Drugs, Guidance for industry: ANDAs, pharmaceutical solid polymorphism, chemistry, manufacturing and controls information, U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, 2007 Search PubMed.
- M. A. Neumann and J. van de Streek, How many ritonavir cases are there still out there?, Faraday Discuss., 2018, 211, 441–458 RSC.
- P. c. C. Cruz, F. A. Rocha and A. M. Ferreira, Application of selective crystallization methods to isolate the metastable polymorphs of paracetamol: A review, Org. Process Res. Dev., 2019, 23(12), 2592–2607 CrossRef CAS.
- I. D. Oswald,
et al., High-pressure structural studies of the pharmaceutical, chlorothiazide, CrystEngComm, 2010, 12(9), 2533–2540 RSC.
- Y. Mori,
et al., Metastable crystal growth of acetaminophen using solution-mediated phase transformation, Appl. Phys. Express, 2016, 10(1), 015501 CrossRef.
- A. Parkes,
et al., Controlled isolation and stabilisation of pure metastable carbamazepine form IV by droplet-confinement via a continuous manufacturing route, CrystEngComm, 2022, 24, 6825–6829 RSC.
- Y. Matsuda,
et al., Physicochemical characterization of spray–dried phenylbutazone polymorphs, J. Pharm. Sci., 1984, 73(2), 173–179 CrossRef CAS PubMed.
- R. Vehring, Pharmaceutical particle engineering via spray drying, Pharm. Res., 2008, 25(5), 999–1022 CrossRef CAS PubMed.
- F. Lyu,
et al., Combined control of morphology and polymorph in spray drying of mannitol for dry powder inhalation, J. Cryst. Growth, 2017, 467, 155–161 CrossRef CAS.
- A. Ziaee,
et al., Spray drying of pharmaceuticals and biopharmaceuticals: Critical parameters and experimental process optimization approaches, Eur. J. Pharm. Sci., 2019, 127, 300–318 CrossRef CAS PubMed.
- E. D. Freis,
et al., Treatment of essential hypertension with chlorothiazide (Diuril): its use alone and combined with other antihypertensive agents, JAMA, J. Am. Med. Assoc., 1958, 166(2), 137–140 CrossRef CAS PubMed.
-
R. M. Kaiser, The Introduction of the Thiazides: A Case Study in Twentieth-Century, in The Inside Story of Medicines: A Symposium, American Institute of the History of Pharmacy, 1997 Search PubMed.
- M. Majrashi,
et al., Experimental measurement and thermodynamic modeling of Chlorothiazide solubility in supercritical carbon dioxide, Case Stud. Therm. Eng., 2023, 41, 102621 CrossRef.
- P. Fernandes,
et al., Powder study of chlorothiazide N, N-dimethylformamide solvate, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2006, 62(6), 2216–2218 CrossRef.
- P. Fernandes,
et al., Solving molecular crystal structures from X-ray powder diffraction data: The challenges posed by γ-carbamazepine and chlorothiazide N, N,-dimethylformamide (1/2) solvate, J. Pharm. Sci., 2007, 96(5), 1192–1202 CrossRef CAS PubMed.
- A. Johnston,
et al., Chlorothiazide N, N-dimethylacetamide disolvate, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2007, 63(5), 2422 CrossRef.
- A. Johnston,
et al., Chlorothiazide dimethyl sulfoxide solvate, Acta Crystallogr., Sect. E:Struct. Rep. Online, 2007, 63(5), 2423 CrossRef.
- A. Johnston, A. J. Florence and A. R. Kennedy, Chlorothiazide formic acid solvate (1/2), Acta Crystallogr., Sect. E:Struct. Rep. Online, 2007, 63(10), 4021 CrossRef.
- A. Johnston, A. J. Florence and A. R. Kennedy, Chlorothiazide–pyridine (1/3), Acta Crystallogr., Sect. E:Struct. Rep. Online, 2008, 64(6), 1105–1106 CrossRef PubMed.
- A. Johnston,
et al., Experimental and predicted crystal energy landscapes of chlorothiazide, Cryst. Growth Des., 2011, 11(2), 405–413 CrossRef CAS.
- R. K. Brydson and A. R. Kennedy, A monoclinic polymorph of chlorothiazide, Struct. Rep., 2024, 80(7), 806–810 CAS.
- K. J. Paluch,
et al., Impact of alternative solid state forms and specific surface area of high-dose, hydrophilic active pharmaceutical ingredients on tabletability, Mol. Pharm., 2013, 10(10), 3628–3639 CrossRef CAS PubMed.
- K. J. Paluch,
et al., Preparation and solid state characterisation of chlorothiazide sodium intermolecular self-assembly suprastructure, Eur. J. Pharm. Sci., 2010, 41(5), 603–611 CrossRef CAS PubMed.
- K. J. Paluch,
et al., Impact of process variables on the micromeritic and physicochemical properties of spray-dried microparticles–Part II. Physicochemical characterisation of spray-dried materials, J. Pharm. Pharmacol., 2012, 64(11), 1583–1591 CrossRef CAS PubMed.
- K. J. Paluch,
et al., Impact of process variables on the micromeritic and physicochemical properties of spray-dried porous microparticles, part I: introduction of a new morphology classification system, J. Pharm. Pharmacol., 2012, 64(11), 1570–1582 CrossRef CAS PubMed.
- O. Corrigan, E. Holohan and K. Sabra, Amorphous forms of thiazide diuretics prepared by spray-drying, Int. J. Pharm., 1984, 18(1–2), 195–200 CrossRef CAS.
- K. J. Paluch,
et al., A novel approach to crystallisation of nanodispersible microparticles by spray drying for improved tabletability, Int. J. Pharm., 2012, 436(1–2), 873–876 CrossRef CAS PubMed.
- A. Altomare,
et al., EXPO2013: a kit of tools for phasing crystal structures from powder data, J. Appl. Crystallogr., 2013, 46(4), 1231–1235 CrossRef CAS.
-
EMA, Stability testing of new drug substances and products. ICH Q1A (R2) 2003 1-24]; available from: https://www.ema.europa.eu/en/ich-q1a-r2-stability-testing-new-drug-substances-drug-products-scientific-guideline.
- L. Greenspan, Humidity fixed points of binary saturated aqueous solutions, J. Res. Natl. Bur. Stand., 1977, 81(1), 89–96 CrossRef.
- P. R. Spackman,
et al., CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54(3), 1006–1011 CrossRef CAS PubMed.
- M. Tobiszewski, Metrics for green analytical chemistry, Anal. Methods, 2016, 8(15), 2993–2999 RSC.
- B. B. Patel,
et al., Revealing facts behind spray dried solid dispersion technology used for solubility enhancement, Saudi Pharm. J., 2015, 23(4), 352–365 CrossRef PubMed.
- D. Walsh,
et al., Engineering of pharmaceutical cocrystals in an excipient matrix: Spray drying versus hot melt extrusion, Int. J. Pharm., 2018, 551(1), 241–256 CrossRef CAS PubMed.
- K. Kale, K. Hapgood and P. Stewart, Drug agglomeration and dissolution – What is the influence of powder mixing?, Eur. J. Pharm. Biopharm., 2009, 72(1), 156–164 CrossRef CAS PubMed.
- N. R. Babij,
et al., NMR Chemical Shifts of Trace Impurities: Industrially Preferred Solvents Used in Process and Green Chemistry, Org. Process Res. Dev., 2016, 20(3), 661–667 CrossRef CAS.
- E. Boel,
et al., Unraveling particle formation: from single droplet drying to spray drying and electrospraying, Pharmaceutics, 2020, 12(7), 625 CrossRef CAS PubMed.
- A. Haser,
et al., Melt extrusion vs. spray drying: The effect of processing methods on crystalline content of naproxen-povidone formulations, Eur. J. Pharm. Sci., 2017, 102, 115–125 CrossRef CAS PubMed.
-
M. Lowinger, et al., Practical considerations for spray dried formulation and process development, in Discovering and developing molecules with optimal drug-like properties, Springer, 2014, pp. 383–435 Search PubMed.
- A. Singh and G. Van den Mooter, Spray drying formulation of amorphous solid dispersions, Adv. Drug Delivery Rev., 2016, 100, 27–50 CrossRef CAS PubMed.
-
J.-C. Lin and J. W. Gentry, Spray drying drop morphology: experimental study. Aerosol Science & Technology, 2003, 371, pp. 15–32 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Enrico
Spoletti
b,
John
O'Reilly
b,
Matteo
Lusi
a,
Enrico
Spoletti
b,
John
O'Reilly
b,
Matteo
Lusi