
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of chiral α-alkynyl-α-hydroxyamides by enantioselective alkynylation of α-keto amides
Elena
Prieto
 ,
Celia
Andrés
* and
Javier
Nieto
*
,
Celia
Andrés
* and
Javier
Nieto
*
Instituto CINQUIMA and Departamento de Química Orgánica, Facultad de Ciencias, Universidad de Valladolid, Paseo de Belén, 7, 47011 Valladolid, Spain. E-mail: franciscojavier.nieto@uva.es
First published on 1st November 2025
Abstract
The α-hydroxyamide moiety is an important structural component in a wide variety of biologically active compounds and natural products. Herein, we describe a highly efficient and practical approach towards chiral quaternary α-alkynyl-α-hydroxyamides by Me2Zn-mediated addition of terminal alkynes to α-keto amides. The desired products are obtained in good yields and enantioselectivities with broad substrate and reagent scopes.
Introduction
α-Hydroxyamides are highly relevant core structures in chemical synthesis. Their structures appear in various natural products such as (+)-kopsihainanina A,1 aspergiline A2 or chimonamidine,3 and molecules that exhibit a wide range of biological activities, including anticonvulsant,4 antimicrobial,5 antifungal,6 anti-inflammatory,7 antipsoriatic8 or antiandrogen.9 α-Hydroxyamide derivatives are also known to behave as HIV protease inhibitors,10 matrix metalloproteinase inhibitors that induce tissue synthesis and degradation,11 or inhibitors of some kinases involved in the pathogenesis of autoimmune diseases.12 In addition, α-hydroxyamides are versatile intermediates in the synthesis of compounds with more complex structures13 and some of them are valuable ligands in asymmetric catalysis.14The synthesis of these core structures in an enantioselective manner is crucial when it comes to achieving the desired pharmacological properties. Among the different methods for synthesizing optically active α-hydroxyamides are the amidation of chiral α-hydroxy acids,15 the hydration of chiral cyanohydrins,16 the ring opening of chiral α,β-epoxy amides,17 the enantioselective Passerini-type reaction,18 the kinetic and enzymatic resolution of racemic mixtures of α-hydroxyamides19 and, the most used, the chemo- and enantioselective reduction of α-keto amides.20 Alternatively, the enantioselective addition of carbon nucleophiles to α-keto amides leads to α-hydroxy amides with a quaternary stereocenter. However, although this option has been frequently reported using cyclic α-keto amides as isatins,21 in acyclic keto amides, it has hardly been explored except for cyclization and annulation reactions.22 Examples of enantioselective nucleophilic addition leading to chiral acyclic α-hydroxy amides with a quaternary stereocenter are very rare. To the best of our knowledge, only four cases have been reported23 and neither involves the enantioselective addition of organometallic reagents to α-keto amides.
Recently, we have gained interest in the synthesis of functionalized chiral tertiary propargylic alcohols through the addition of acetylides to activated ketones24 and in this context, we decided to embark on the synthesis of α-alkynyl-α-hydroxyamides by enantioselective alkynylation of α-keto amides with alkynylzinc derivatives.25
Results and discussion
We first scanned a series of chiral ligands L1–15 (20 mol%), some of which had provided good enantioselectivities on the addition of organozinc derivatives to carbonyl compounds in previous works,26 in the model reaction of 1-phenyl-2-(pyrrolidine-1-yl)ethane-1,2-dione 1a, with the alkynylzinc derivative prepared from phenylacetylene and dimethylzinc in a dichloromethane–toluene 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture at rt for 24 hours and the results are collected in Table 1.
a
:1 mixture at rt for 24 hours and the results are collected in Table 1.
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Solvent | t (h) | Yieldb (%) | erc |
| a Reaction conditions: 0.1 mmol of 1a, 0.4 mmol of dimethylzinc, 0.4 mmol of phenylacetylene and 0.02 mmol of ligands L1–15, solvent (3.3 mL). b Yield of the isolated product after purification by flash column chromatography. c Enantiomeric ratio was determined by HPLC on a chiral stationary phase. d The reaction was carried out at 0 °C. | |||||
| 1 | L1 | CH2Cl2–toluene 3:1 |
24 | 45 | 38:62 |
| 2 | L2 | CH2Cl2–toluene 3:1 |
24 | 34 | 19:81 |
| 3 | L3 | CH2Cl2–toluene 3:1 |
24 | 51 | 23:77 |
| 4 | L4 | CH2Cl2–toluene 3:1 |
24 | 35 | 45:55 |
| 5 | L5 | CH2Cl2–toluene 3:1 |
24 | 17 | — |
| 6 | L6 | CH2Cl2–toluene 3:1 |
24 | 5 | — |
| 7 | L7 | CH2Cl2–toluene 3:1 |
24 | 60 | 86:14 |
| 8 | L8 | CH2Cl2–toluene 3:1 |
24 | 67 | 57:43 |
| 9 | L9 | CH2Cl2–toluene 3:1 |
24 | 72 | 85:15 |
| 10 | L10 | CH2Cl2–toluene 3:1 |
24 | 81 | 95:5 |
| 11 | L11 | CH2Cl2–toluene 3:1 |
24 | 66 | 87:13 |
| 12 | L12 | CH2Cl2–toluene 3:1 |
24 | 35 | 89:11 |
| 13 | L13 | CH2Cl2–toluene 3:1 |
24 | 76 | 81:19 |
| 14 | L14 | CH2Cl2–toluene 3:1 |
24 | 65 | 64:36 |
| 15 | L15 | CH2Cl2–toluene 3:1 |
24 | 71 | 34:66 |
| 16 | L10 | Toluene | 24 | 62 | 84:16 |
| 17 | L10 | AcOEt–toluene 3:1 |
24 | 26 | 70:30 |
| 18 | L10 | THF–toluene 3:1 |
24 | 28 | 68:32 |
| 19 | L10 | Et2O–toluene 3:1 |
24 | 48 | 80:20 |
| 20d | L10 | CH2Cl2–toluene 3:1 |
33 | 34 | 93:7 |
| 21 | L10 | CH2Cl2–toluene 3:1 |
1 | 53 | 95:5 |
| 22 | L10 | CH2Cl2–toluene 3:1 |
15 | 72 | 95:5 |
| 23 | L10 | CH2Cl2–toluene 3:1 |
30 | 77 | 94:6 |
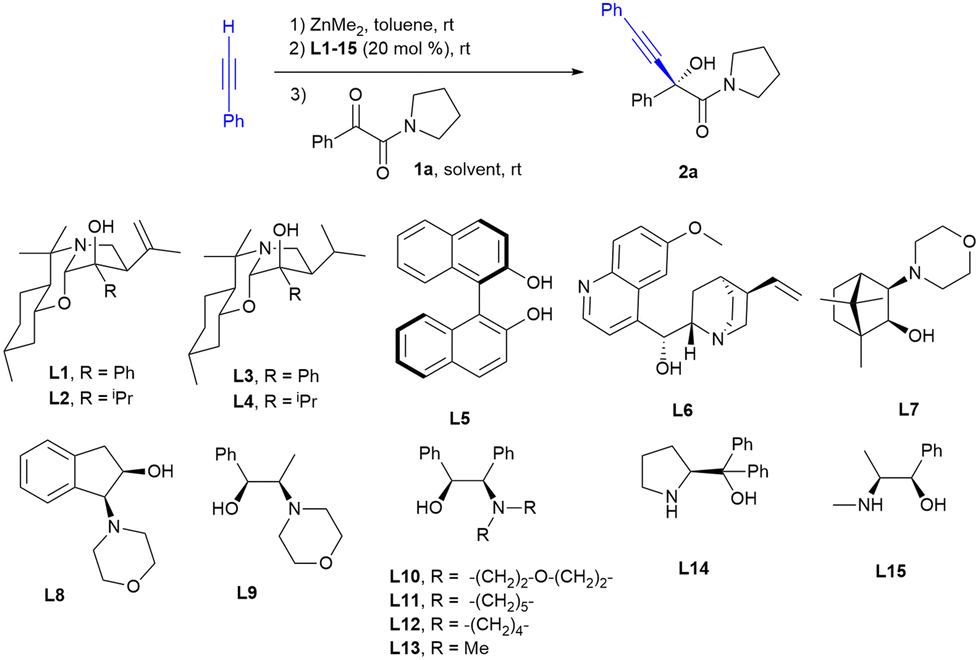
1,3-Benzoxazine L2 that had given us good results in previous works as a chiral ligand in the alkynylation of α-keto esters and α-diketones24 provided poor results in terms of enantioselectivity and chemical yield (entry 2 in Table 1). The highest enantioselectivities were achieved with ligand L10 (er = 95:5, entry 10 in Table 1), also achieving a good chemical yield. The use of only toluene as a solvent or a mixture of toluene and THF, diethyl ether or ethyl acetate did not improve either the chemical yield or the enantioselectivity of the reaction (entries 17–19 in Table 1). On the other hand, when lowering the reaction temperature from rt to 0 °C, a slight loss of enantioselectivity was observed in addition to a marked decrease in chemical yield due to a marked decrease in the rate of reaction (entry 20 in Table 1).
In previous works on the alkynylation of α-keto esters27 and α-diketones,24b a kinetic resolution of the resulting tertiary propargylic alcohols has been observed due to the over-addition of the organometallic reagent; however, in our case, no appreciable variation of the enantioselectivity with the reaction time was observed (entries 10 and 21–23 in Table 1).
After the optimal reaction conditions were determined (4 equiv. of phenylacetylene and dimethylzinc, 20 mol% of L10 as the ligand, at rt in a mixture of CH2Cl2:toluene 3:1 for 24 h), the substrate scope for the alkynylation of a series of aromatic and aliphatic α-keto amides was explored and the results are shown in Scheme 1.
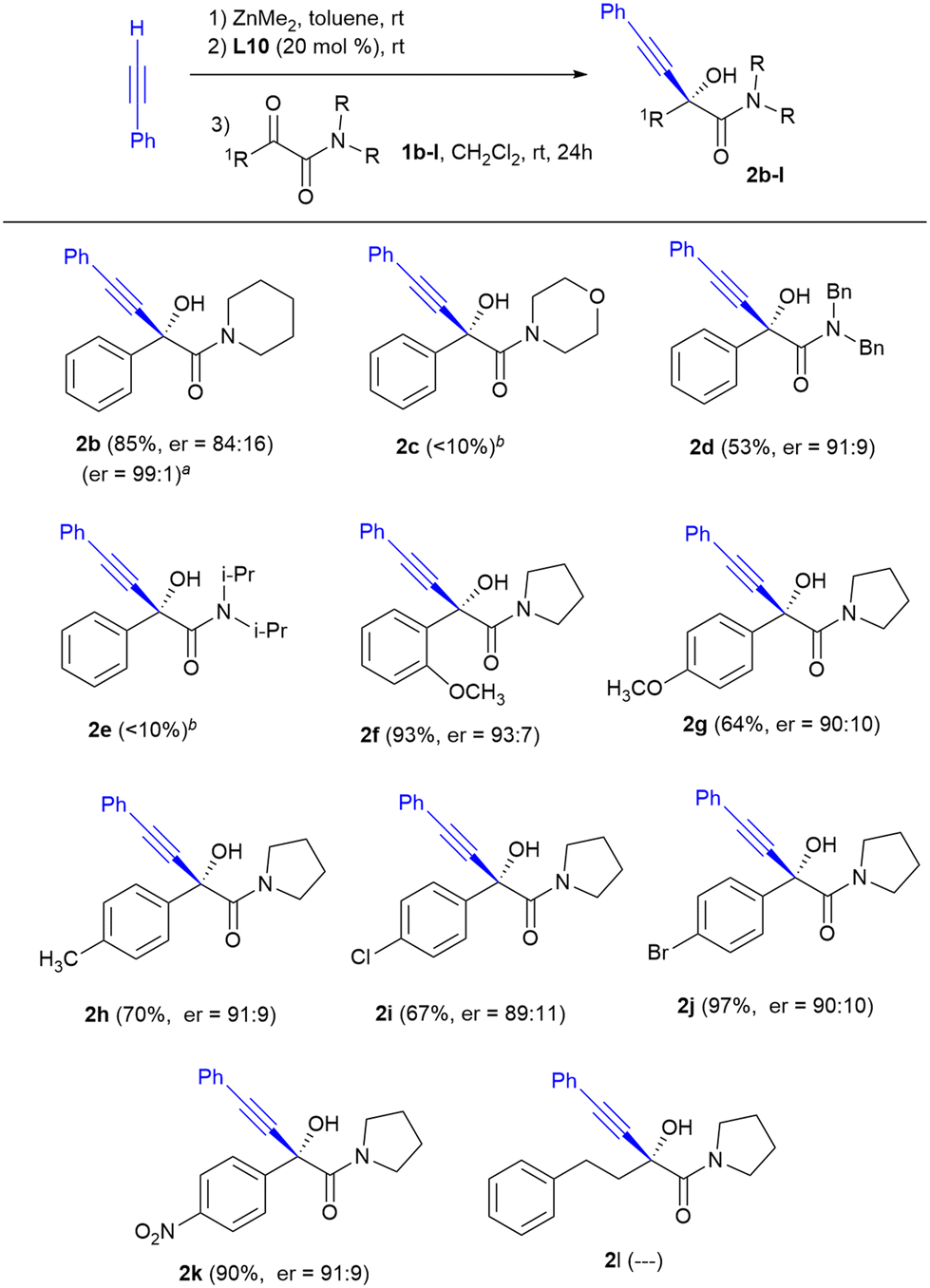 | ||
| Scheme 1 Substrate scope of the alkynylation of α-keto amides. Reaction conditions: 0.25 mmol of 1b–l, 1.0 mmol of dimethylzinc, 1.0 mmol of phenylacetylene and 0.05 mmol of ligand L10, in a CH2Cl2–toluene 3:1 mixture (3.3 mL) at rt for 24 h. Yield of the isolated product after purification by flash column chromatography. The enantiomeric ratio was determined by HPLC on a chiral stationary phase. aEnantiomeric ratio after recrystallization in hexane. bDetected by 1H NMR of the reaction mixtures but not isolated. | ||
The nitrogen substitution of the amide function appears to be a critical factor that drastically affects the chemical yield of the reaction. The morpholine-derived ketoamide 1c, as well as ketoamide 1e with two bulky isopropyl substituents, underwent little conversion to the alkynylation products 2c and 2e, recovering in both cases the starting ketoamide as the only by-product. In contrast, when using the ketoamides derived from piperidine 1b and dibenzylamine 1d, the corresponding hydroxyketones 2b and 2d were obtained with enantioselectivities comparable to those achieved with the aromatic amides derived from pyrrolidine 1a and 1f–k. In addition, the recrystallization of product 2b allowed the enantioselectivity to increase up to an excellent ratio of enantiomers of 99:1.
The enantiocontrol in the alkynylation of diverse aromatic α-keto amides derived from pyrrolidine seemed not to be influenced by electronic effects. The presence of both electron-donating substituents such as the methyl or methoxy groups (products 2f–h) or electron-withdrawing substituents such as Cl, Br or –NO2 (products 2i–k) in the ortho or para positions of the aromatic ring was tolerated without significant changes of enantioselectivity. All the reactions proceeded with good chemical yields. The decrease in chemical yield observed in some products was due to losses during the chromatographic purification. In contrast, when the reaction was performed with an alkyl-substituted α-keto amide namely 4-phenyl-1-(pyrrolidin-1-yl)butane-1,2-dione 1l, formation of α-hydroxyamide 2l was not observed.
To test the generality of this reaction with respect to the alkyne, we studied the influence of the electronic effects of some arylacetylenes, as well as the addition of aliphatic alkynes to α-keto amides 1a and 1f (Scheme 2).
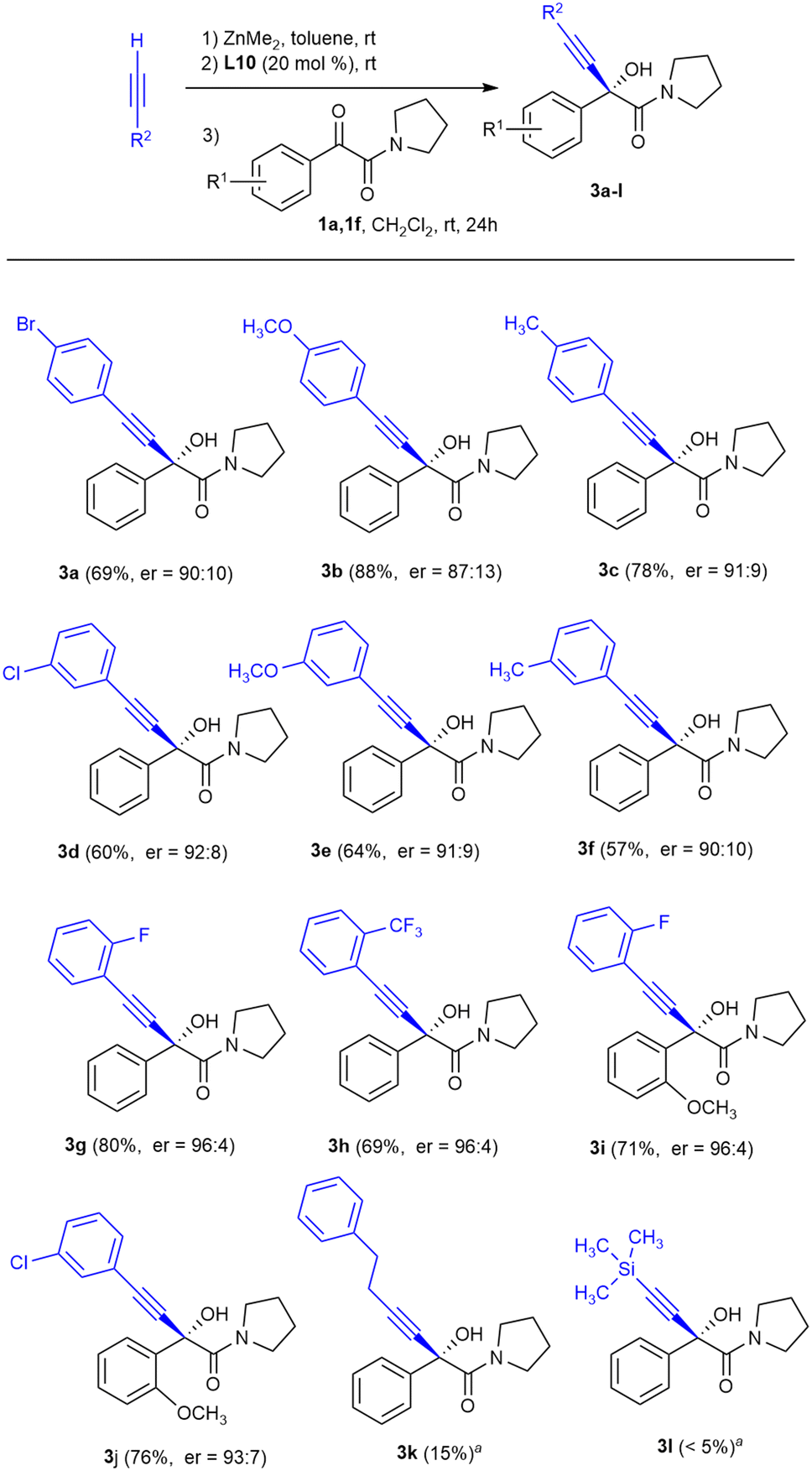 | ||
| Scheme 2 Scope of the reaction of α-keto amides 1a and 1f with terminal alkynes. Reaction conditions: 0.25 mmol of 1a or 1f, 1.0 mmol of dimethylzinc, 1.0 mmol of alkyne and 0.05 mmol of ligand L10, in a CH2Cl2–toluene 3:1 mixture (3.3 mL) at rt for 24 h. Yield of the isolated product after purification by flash column chromatography. The enantiomeric ratio was determined by HPLC on a chiral stationary phase. aUndetermined enantiomeric ratio. | ||
For arylacetylenes the corresponding α-alkynyl-α-hydroxyamides were obtained with good enantioselectivities (Scheme 2, compounds 3a–j), although the best results in terms of enantioselectivity were obtained when the aromatic ring in the alkyne was substituted in the ortho position by an electro-attracting fluorine or by a trifluoromethyl group (products 3g–i, er = 96:4). Unfortunately, the reactions of 1a with the aliphatic alkyne 4-phenyl-1-butyne and trimethylsilylacetylene did not provide appreciable amounts of alkynylation products 3k and 3l, recovering most of the untransformed ketoamide 1a.
In order to solve this lack of reactivity, the addition of 4-phenyl-1-butyne to ketoamide 1a in the presence of L10 was studied. Heating to 35 °C in a conventional oil bath for one hour allowed the isolation of hydroxyamide 3k with an enantioselectivity of er = 87:13 and a chemical yield of 62%. To our delight, the chemical yield improved up to 93% and enantioselectivity remained similar when heating was carried out under microwave irradiation. Under these conditions, in the absence of L10, formation of less than 5% of the racemate of 3k is observed.
Therefore, heating at 35 °C under microwave irradiation for 1 hour were chosen as the ideal conditions for the enantioselective addition of aliphatic terminal alkynes to α-ketoamides and the scope of the reaction for different aliphatic alkynes was studied (Scheme 3).
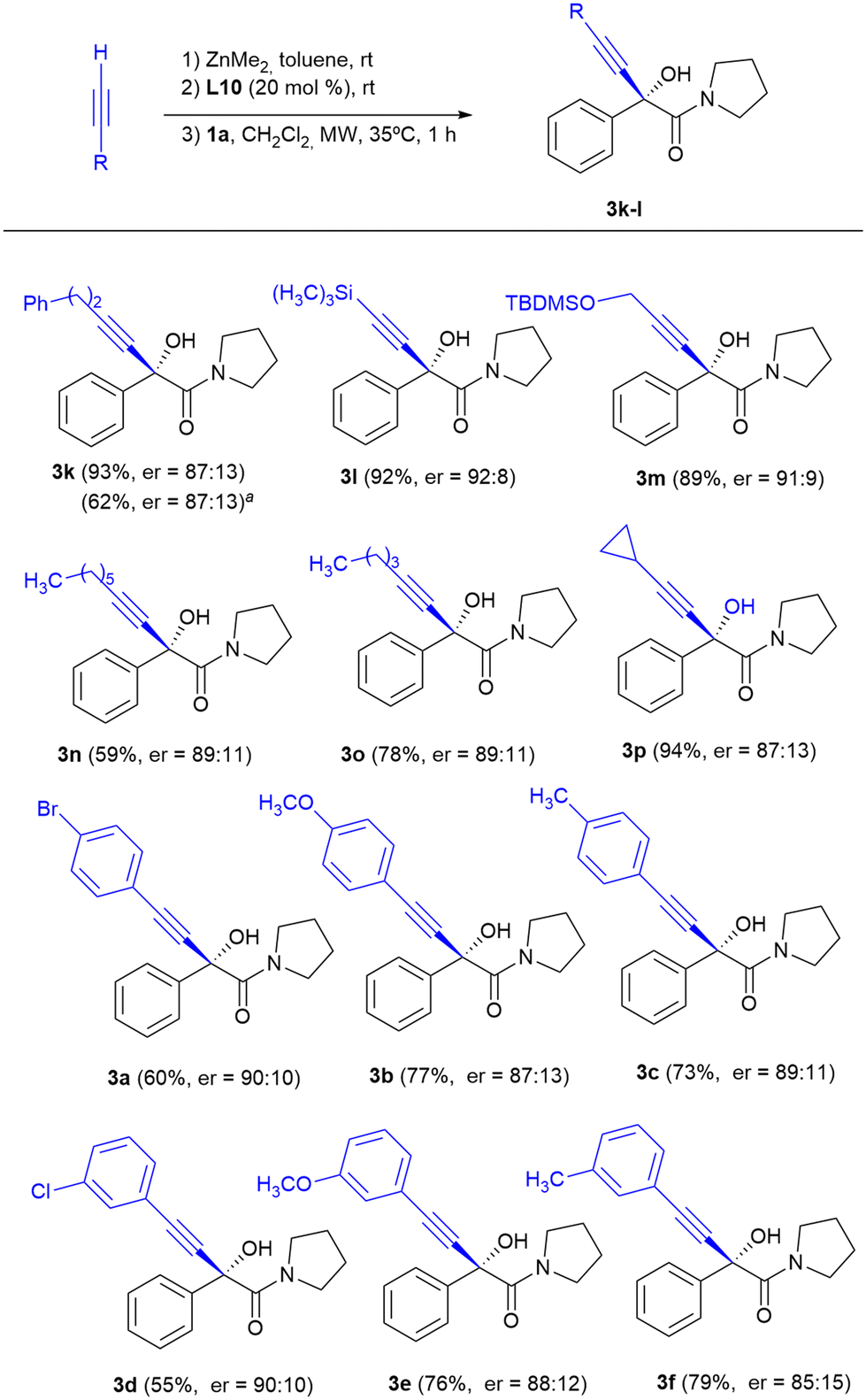 | ||
| Scheme 3 Scope of the reaction of α-keto amide 1a with terminal alkynes under microwave irradiation. Reaction conditions: 0.25 mmol of 1a, 1.0 mmol of dimethylzinc, 1.0 mmol of alkyne and 0.05 mmol of ligand L10, in a CH2Cl2–toluene 3:1 mixture (3.3 mL) at 35 °C for 1 h. The reaction was carried out in a CEM Discover 300 W microwave reactor, applying an initial power of 80 W. Yield of the isolated product after purification by flash column chromatography. The enantiomeric ratio was determined by HPLC on a chiral stationary phase. aThe reaction was carried out at 35 °C in a conventional oil bath for one hour. | ||
The addition of aliphatic terminal alkynes under these conditions occurred with moderate to good chemical yields and enantioselectivities slightly lower than those provided with the terminal aromatic alkynes at room temperature. The best results in terms of enantiocontrol were obtained upon the addition of alkynes with trimethylsilyl or tert-butyl(dimethyl)silyloxy substituents, with enantioselectivities comparable to those obtained upon the addition of arylacetylenes (compounds 3l, er = 92:8 and 3m, er = 91:9). Alkynylation of 1a with arylacetylene derivatives in the presence of 10L under microwave irradiation at 35 °C for 1 hour was also explored, and α-alkynyl-α-hydroxyamides 3a–f were obtained with yields similar to those achieved when the alkynylations were carried out at room temperature for 24 h; however, the enantioselectivities were slightly lower (compare the results of Scheme 2 with those of Scheme 3 for 3a–f).
The configuration of the newly formed stereogenic center of 2a was established by X-ray diffraction analysis and has been extended to all of the other α-alkynyl-α-hydroxyamides 2b–k and 3a–p based on mechanistic analogy.
Conclusions
In summary, we have developed an efficient method for the preparation of enantioenriched α-alkynyl-α-hydroxyamides with a quaternary stereocenter via Me2Zn-mediated addition of terminal alkynes to α-keto amides using the 1,2-amino alcohol L10 as a chiral ligand. A variety of aromantic α-keto amides bearing electron-withdrawing or electron-donating substituents on the aromatic ring and aromatic and aliphatic terminal alkynes were investigated, which gave rise to 1,2-addition products in moderate to good chemical yields and enantioselectivities.Experimental section
General information
All reactions were carried out in anhydrous solvents under a nitrogen atmosphere in dried glassware by means of Schlenk techniques. 1H NMR (400 or 500 MHz) and 13C NMR (100 or 126 MHz) spectra were recorded in CDCl3. Chemical shifts for protons are reported in ppm from tetramethylsilane with the residual CHCl3 resonance as an internal reference. Chemical shifts for carbons are reported in ppm from tetramethylsilane and are referenced to the carbon resonance of the solvent. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constants in hertz, and integration. Specific rotations were measured using a 2 mL cell with a 1 dm path length and a sodium lamp, and concentration is given in g per 100 mL. High resolution mass spectrometry analysis (HRMS) was performed using a quadrupole spectrometer with a TOF analyzer. Infrared spectra are reported in frequency of absorption and only the structurally most important peaks are given. Flash chromatography was carried out using silica gel (230–240 mesh). TLC analysis was performed on glass-backed plates coated with silica gel 60 and an F254 indicator and visualized by either UV irradiation or staining with I2 or phosphomolybdic acid solution. Chiral HPLC analysis was performed using Chiralpak AD-H, Phenomenex Lux Amylose-1, Phenomenex Lux i-Amylose-3, Phenomenex Lux i-Amylose-1 and Phenomenex Lux Cellulose-2 columns. UV detection was monitored at 254 nm.Microwave-assisted synthesis was carried out in a CEM Discover microwave reactor containing a single-mode microwave cavity in sealed reaction vessels and operating at a maximal microwave power of up to 80 W. The temperature at the bottom of the reaction vessel was automatically controlled with an IR sensor located below the microwave cavity floor. Reaction times refer to the hold times at the temperatures indicated and not to the total irradiation times.
Commercially available reagents were used as purchased without further treatment.
General procedure for the catalytic enantioselective addition of phenyl acetylene derivatives to α-keto amides
To a solution of 1.2 M of ZnMe2 in toluene (1.0 mmol, 0.83 mL) was added the corresponding alkyne (1.0 mmol) and the mixture was stirred under a nitrogen atmosphere at rt for 1 h. Then, a solution of 0.05 M of ligand L10 in CH2Cl2 (0.05 mmol, 1.0 mL) was added and the reaction was stirred for another 30 min, whereupon a solution of the keto amide (0.25 mmol) in CH2Cl2 (1.5 mL) was added dropwise and the reaction was stirred at rt for 24 h. The reaction was quenched with a saturated aqueous solution of NH4Cl, extracted with CH2Cl2, dried over MgSO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gel using a hexane/ethyl acetate mixture as the eluent.General procedure for the microwave assisted catalytic enantioselective addition of aliphatic terminal alkynes to α-keto amides
In an oven-dried microwave tube equipped with a magnetic stir bar, containing the aliphatic alkyne (1.0 mmol), a solution of 1.2 M of ZnMe2 in toluene (1.0 mmol, 0.83 mL) was added. The mixture was stirred under a nitrogen atmosphere at rt for 1 h and then a solution of 0.05 M of ligand L10 in CH2Cl2 (0.05 mmol, 1.0 mL) was added and the reaction was stirred for another 30 min, whereupon a solution of the keto amide (0.25 mmol) in CH2Cl2 (1.5 mL) was added, and the reaction mixture was irradiated in a CEM Discover microwave reactor operating at a microwave power of 80 W at 35 °C for 1 h. After cooling, the reaction was quenched with a saturated aqueous solution of NH4Cl, extracted with CH2Cl2, dried over MgSO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gel using a hexane/ethyl acetate mixture as the eluent.:iPrOH = 60:40, flow rate = 1 mL min−1, λ = 254 nm, tR = 19.4 min R enantiomer, tR = 22.7 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 13.7 min R enantiomer, tR = 20.0 min S enantiomer.
:iPrOH 85:15, flow rate = 1 mL min−1, λ = 254, tR = 10.7 min R enantiomer, tR = 13.5 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 18.8 min S enantiomer, tR = 28.0 min R enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 44.8 min R enantiomer, tR = 29.5 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 32.6 min R enantiomer, tR = 24.7 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 16.5 min R enantiomer, tR = 13.8 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 17.3 min R enantiomer, tR = 13.8 min S enantiomer.
:iPrOH 80:20, 1 mL min−1, λ = 254 nm, tR = 25.8 min R enantiomer, tR = 29.1 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254, tR = 22.2 min R enantiomer, tR = 30.4 min S enantiomer.
:iPrOH 60:40, flow rate = 1 mL min−1, λ = 254, tR = 34.6 min R enantiomer, tR = 41.8 min S enantiomer.
:iPrOH 60:40, flow rate = 1 mL min−1, λ = 254, tR = 26.3 min R enantiomer, tR = 32.9 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254, tR = 17.4 min R enantiomer, tR = 25.5 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254, tR = 28.8 min R enantiomer, tR = 37.2 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254, tR = 20.7 min R enantiomer, tR = 29.5 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254, tR = 20.2 min R enantiomer, tR = 29.1 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 14.1 min R enantiomer, tR = 22.7 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 21.8 min S enantiomer, tR = 29.0 min R enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 18.3 min S enantiomer, tR = 21.1 min R enantiomer.
:iPrOH 85:15, flow rate = 1 mL min−1, λ = 254 nm, tR = 17.3 min R enantiomer, tR = 15.0 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 12.5 min R enantiomer, tR = 17.2 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 27.2 min R enantiomer, tR = 15.7 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 11.5 min R enantiomer, tR = 8.5 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 13.6 min R enantiomer, tR = 8.8 min S enantiomer.
:iPrOH 80:20, flow rate = 1 mL min−1, λ = 254 nm, tR = 17.6 min R enantiomer, tR = 12.0 min S enantiomer.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article are available within the article and its supplementary information (SI). Supplementary information: preparation and characterization data of starting α-keto amides, copies of NMR spectra and chromatograms of new compounds and crystallographic data for 2a. See DOI: https://doi.org/10.1039/d5ob01546d.CCDC 2391708 (2a) contains the supplementary crystallographic data for this paper.30
Acknowledgements
The authors thank Junta de Castilla y León (projects FEDER-VA115P17 and VA149G18) for financial support. The authors also thank Dr José M. Martín-Álvarez for his assistance in the determination of the X-ray structure.References
- M. Mizutani, S. Yasuda and C. Mukai, Chem. Commun., 2014, 50, 5782–5785 RSC.
- M. C. Nakhla and J. L. Wood, J. Am. Chem. Soc., 2017, 139, 18504–18507 CrossRef CAS PubMed.
- H. Takayama, Y. Matsuda, K. Masubuchi, A. Ishida, M. Kitajima and N. Aimi, Tetrahedron, 2004, 60, 893–900 CrossRef CAS.
- (a) S. L. Shapiro, I. M. Rose and L. Freedman, J. Am. Chem. Soc., 1959, 81, 6322–6329 CrossRef CAS; (b) P. J. Jones, Y. Wang, M. D. Smith, N. J. Hargus, H. S. Eidam, H. S. White, J. Kapur, M. L. Brown and M. K. Patel, J. Pharmacol. Exp. Ther., 2007, 320, 828–836 CrossRef CAS PubMed.
- (a) D. Chen, H. Y. Chow, K. H. L. Po, W. Ma, E. L. Y. Leung, Z. Sun, M. Liu, S. Chen and X. Li, Org. Lett., 2019, 21, 5639–5644 CrossRef CAS PubMed; (b) M. Terreni, J. G. Tchamkam, U. Sarnataro, S. Rocchietti, R. Fernández-Lafuente and J. M. Guisán, Adv. Synth. Catal., 2005, 347, 121–128 CrossRef CAS; (c) T. Komori and H. Sakaguchi, WO2004016594, 2004, Sumitomo Chemical Company; (d) N. Matsumoto, I. Momose, M. Umekita, N. Kinoshita, M. Chino, H. Iinuma, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1998, 51, 1087–1092 CrossRef CAS PubMed.
- S.-J. Yu, C. Zhu, Q. Bian, C. Cui, X.-J. Du, Z.-M. Li and W.-G. Zhao, ACS Comb. Sci., 2014, 16, 17–23 CrossRef CAS PubMed.
- H. Schäcke, A. Schottelius, W.-D. Döcke, P. Strehlke, S. Jaroch, N. Schmees, H. Rehwinkel, H. Hennekes and K. Asadullah, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 227–232 CrossRef PubMed.
- (a) B. P. Klaholz, J.-P. Renaud, A. Mitschler, C. Zusi, P. Chambon, H. Gronemeyer and D. Moras, Nat. Struct. Biol., 1998, 5, 199–202 CrossRef CAS PubMed; (b) M. Belema, F. C. Zusi and K. M. Tramposch, WO0016769, 2000, Bristol Myers Squibb Co.
- (a) D. Masiello, S. Cheng, G. J. Bubley, M. L. Lu and S. P. Balk, J. Biol. Chem., 2002, 277, 26321–26326 CrossRef CAS PubMed; (b) M. C. Hodgson, I. Astapova, A. N. Hollenberg and S. P. Balk, Cancer Res., 2007, 67, 8388–8395 CrossRef CAS PubMed.
- T. Punniyamurthy and J. Iqbal, Tetrahedron Lett., 1997, 38, 4463–4466 CrossRef CAS.
- T. Hiruma, K. Kobayashi and S. Inomata, WO03020711, 2003.
- (a) N. Blaquiere, G. M. Castanedo, J. D. Burch, L. M. Berezhkovskiy, H. Brightbill, S. Brown, C. Chan, P. C. Chiang, J. J. Crawford, T. Dong, P. Fan, J. Feng, N. Ghilardi, R. Godemann, E. Gogol, A. Grabbe, A. J. Hole, B. Hu, S. G. Hymowitz, M. H. A. Ismaili, H. Le, P. Lee, W. Lee, X. Lin, N. Liu, P. A. McEwan, B. McKenzie, H. L. Silvestre, E. Suto, S. Sujatha-Bhaskar, G. Wu, L. C. Wu, Y. Zhang, Z. Zhong and S. T. Staben, J. Med. Chem., 2018, 61, 6801–6813 CrossRef CAS PubMed; (b) N. Blaquiere, J. Burch, G. Castanedo, J. A. Feng, B. Hu, S. Staben, G. Wu and P. Yen, WO2015025025, 2015, Hoffmann-La Roche; Genentech, Inc.
- (a) V. Pastore, L. Sabatier, A. Enrique, M. Marder and L. E. Bruno-Blanch, Bioorg. Med. Chem., 2013, 21, 841–846 CrossRef CAS PubMed; (b) C. Lamberth, A. Jeanguenat, F. Cederbaum, A. De Mesmaeker, M. Zeller, H.-J. Kempf and R. Zeun, Bioorg. Med. Chem., 2008, 16, 1531–1545 CrossRef CAS PubMed; (c) J. H. George and R. M. A. Adlington, Synlett, 2008, 2093–2096 CAS; (d) R. Peters, M. Althaus and A.-L. Nagy, Org. Biomol. Chem., 2006, 4, 498–509 RSC.
- (a) G. Blay, I. Fernandez, V. Hernández-Olmos, A. Marco-Aleixandre and J. R. Pedro, Tetrahedron: Asymmetry, 2005, 16, 1953–1958 CrossRef CAS; (b) G. Blay, L. Cardona, I. Fernández, A. Marco-Aleixandre, M. C. Muñoz and J. R. Pedro, Org. Biomol. Chem., 2009, 7, 4301–4308 RSC; (c) P. Geoghegan and P. O'Leary, ACS Catal., 2012, 2, 573–591 CrossRef CAS.
- (a) M. Huang, S. Zhong, M. Xu and Y. Liu, J. Chem. Res., 2015, 39, 274–276 CrossRef CAS.
- (a) T. Kanda, A. Naraoka and H. Naka, J. Am. Chem. Soc., 2019, 141, 825–830 CrossRef CAS PubMed; (b) P. Crochet and V. Cadierno, Eur. J. Inorg. Chem., 2021, 3225–3238 CrossRef CAS.
- (a) C. Sappino, A. Mari, A. Mantineo, M. Moliterno, M. Palagri, C. Tatangelo, L. Suber, P. Bovicelli, A. Ricelli and G. Righi, Org. Biomol. Chem., 2018, 16, 1860–1870 RSC; (b) H. Kakei, T. Nemoto, T. Ohshima and M. Shibasaki, Angew. Chem., Int. Ed., 2004, 43, 317–320 CrossRef CAS PubMed; (c) V. K. Aggarwal, G. Hynd, W. Picoul and J.-L. Vasse, J. Am. Chem. Soc., 2002, 124, 9964–9965 CrossRef CAS PubMed; (d) T. Nemoto, H. Kakei, V. Gnanadesikan, S.-Y. Tosaki, T. Ohshima and M. Shibasaki, J. Am. Chem. Soc., 2002, 124, 14544–14545 CrossRef CAS PubMed.
- (a) S. E. Denmark and Y. Fan, J. Org. Chem., 2005, 70, 9667–9676 CrossRef CAS PubMed; (b) L. Moni, L. Banfi, D. Cartagenova, A. Cavalli, C. Lambruschini, E. Martino, R. V. A. Orru, E. Ruijter, J. M. Saya, J. Sgrignani and R. Riva, Org. Chem. Front., 2020, 7, 380–398 RSC; (c) S. E. Denmark and Y. Fan, J. Am. Chem. Soc., 2003, 125, 7825–7827 CrossRef CAS PubMed.
- (a) A. Westerbeek, W. Szymański, B. L. Feringa and D. B. Janssen, ACS Catal., 2011, 1, 1654–1660 CrossRef CAS; (b) T. Murata, T. Kawanishi, A. Sekiguchi, R. Ishikawa, K. Ono, K. Nakata and I. Shiina, Molecules, 2018, 23, 2003 CrossRef PubMed; (c) W. Szymanski and R. Ostaszewski, J. Mol. Catal. B: Enzym., 2007, 47, 125–128 CrossRef CAS; (d) S.-S. Weng, M.-W. Shen, J.-Q. Kao, Y. S. Munot and C. T. Chen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3522–3527 CrossRef CAS PubMed.
- (a) F.-H. Zhang, C. Wang, J.-H. Xie and Q.-L. Zhou, Adv. Synth. Catal., 2019, 361, 2832–2835 CrossRef CAS; (b) R. Agarwal, Y. Liao, D.-J. Lin, Z.-X. Yang, C.-F. Lai and C.-T. Chen, Org. Chem. Front., 2020, 7, 2505–2510 RSC; (c) S. K. Gediya, V. K. Vyas, G. J. Clarkson and M. Wills, Org. Lett., 2021, 23, 7803–7807 CrossRef CAS PubMed; (d) N. C. Mamillapalli and G. Sekar, Chem. – Eur. J., 2015, 21, 18584–11858 CrossRef CAS PubMed; (e) B. Ma, T. Miao, Y. Sun, Y. He, J. Liu, Y. Feng, H. Chen and Q.-H. Fan, Chem. – Eur. J., 2014, 20, 9969–9978 CrossRef CAS PubMed; (f) S. Stella and A. Chadha, Catal. Today, 2012, 198, 345–352 CrossRef CAS.
- (a) P. Brandão and A. J. Burke, Tetrahedron, 2018, 74, 4927–4957 CrossRef; (b) Z.-Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443–1454 CrossRef CAS PubMed; (c) A. Kumar and S. S. Chimni, RSC Adv., 2012, 2, 9748–9762 RSC.
- (a) R. Di Sanza, T. L. N. Nguyen, N. Iqbal, S. P. Argent, W. Lewis and H. W. Lam, Chem. Sci., 2020, 11, 2401–2406 RSC; (b) L. Wang, Q. Ni, M. Blümel, T. Shu, G. Raabe and D. Enders, Chem. – Eur. J., 2015, 21, 8033–8037 CrossRef CAS PubMed; (c) A.-B. Xia, G.-J. Pan, C. Wu, X.-L. Liu, X.-L. Zhang, Z.-B. Li, X. H. Du and D.-Q. Xu, Adv. Synth. Catal., 2016, 358, 3155–3160 CrossRef CAS; (d) M. Hatano and T. Nishimura, Angew. Chem., Int. Ed., 2015, 54, 10949–10952 CrossRef CAS PubMed; (e) C. Joie, K. Deckers, G. Raabe and D. Ender, Synthesis, 2014, 1539–1546 Search PubMed; (f) C. Joie, K. Deckers and D. Enders, Synthesis, 2014, 799–808 Search PubMed; (g) S. Goudedranche, D. Pierrot, T. Constantieux, D. Bonne and J. Rodriguez, Chem. Commun., 2014, 50, 15605–15608 RSC; (h) T. Shirai, H. Ito and Y. Yamamoto, Angew. Chem., Int. Ed., 2014, 53, 2658–2661 CrossRef CAS PubMed; (i) L. Yin, M. Kanai and M. A. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7620–7623 CrossRef CAS PubMed; (j) L. Zhen, S. Tong, J. Zhu and M.-X. Wang, J. Org. Chem., 2020, 85, 13211–13219 CrossRef CAS PubMed; (k) H. Yang, W. Li, X.-M. Xu and Z.-L. Wang, Org. Lett., 2020, 22, 8814–8818 CrossRef CAS PubMed; (l) X.-M. Xu, L. Zhao, J. Zhu and M.-X. Wang, Angew. Chem., Int. Ed., 2016, 55, 3799–3803 CrossRef CAS PubMed; (m) L. Zhen, S. Tong, J. Zhu and M.-X. Wang, Chem. – Eur. J., 2020, 26, 401–405 CrossRef CAS PubMed; (n) X.-M. Xu, M. Xie, J. Li and M.-X. Wang, Org. Chem. Front., 2021, 8, 721–726 RSC; (o) L. Yang, D.-X. Wang, Z.-T. Huang and M.-X. Wang, J. Am. Chem. Soc., 2009, 131, 10390–10391 CrossRef CAS PubMed.
- (a) H. Echave, R. López and C. Palomo, Angew. Chem., Int. Ed., 2016, 55, 3364–3368 CrossRef CAS PubMed; (b) K. Kon, H. Takai, T. Kobayashi, Y. Kohari and M. Murata, Synlett, 2021, 829–832 CAS; (c) W. Luo, J. Zhao, J. Ji, L. Lin, X. Liu, H. Mei and X. Feng, Chem. Commun., 2015, 51, 10042–10045 RSC; (d) H. Xu and C. Wolf, Angew. Chem., Int. Ed., 2011, 50, 12249–12252 CrossRef CAS PubMed.
- (a) R. Infante, A. Gago, J. Nieto and C. Andrés, Adv. Synth. Catal., 2012, 354, 2797–2804 CrossRef CAS; (b) R. Infante, J. M. Martín-Alvarez, C. Andrés and J. Nieto, Org. Lett., 2017, 19, 1516–1519 CrossRef CAS PubMed; (c) E. Prieto, J. D. Martín, J. Nieto and C. Andrés, Org. Biomol. Chem., 2023, 21, 6940–6948 RSC.
- The diastereoselective version of this reaction has been previously described: S. W. Youn, Y. H. Kim, J.-W. Hwang and Y. Do, Chem. Commun., 2001, 996–997 RSC.
- For reviews of enantioselective alkynylation of carbonyl compounds, see: (a) B. M. Trost and A. H. Weiss, Adv. Synth. Catal., 2009, 351, 963–983 CrossRef CAS PubMed; (b) V. Bisai and V. K. Singh, Tetrahedron Lett., 2016, 57, 4771–4784 CrossRef CAS.
- H.-B. Chen, W.-H. Lai, Y. Zhao, D.-D. Qin, Y.-P. Ruan and Z.-H. Zhou, Synlett, 2014, 809–812 CrossRef.
- Racemic 2b has been previously described: K. Dhara, A. Kapat, T. Ghosh and J. Dash, Synthesis, 2016, 4260–4268 CAS.
- Racemic 2d has been previously described: D. Tejedor, L. Cotos and F. García-Tellado, Org. Lett., 2011, 13, 4422–4425 CrossRef CAS PubMed.
- CCDC 2391708: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2l8rwj.
| This journal is © The Royal Society of Chemistry 2025 |