
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhthalimide-based sulfur transfer reagents for the preparation of biologically relevant trisulfides
Conrad N. A.
Du
 ,
Austin
Sarker-Young
,
Meg
Shieh
,
Stephen
Lindahl
and
Ming
Xian
*
,
Austin
Sarker-Young
,
Meg
Shieh
,
Stephen
Lindahl
and
Ming
Xian
*
Department of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: ming_xian@brown.edu
First published on 20th October 2025
Abstract
Reactive sulfur species (RSS) are a class of biological sulfur compounds that play significant roles in various physiological processes. Among these, trisulfides (RSSSR) have been recognized for their unique biological activities such as antioxidation. However, there is still a lack of suitable methods for the preparation of biothiol and protein trisulfides that will enable a better understanding of trisulfide's roles in RSS biology. In this study, we developed and analyzed the use of phthalimide-based disulfide reagents to generate trisulfides from thiols found abundantly in physiologically relevant environments. This approach was tested on both small molecule and larger protein thiols. The results demonstrated that this method provides access to both biothiol- and protein-based trisulfides and may facilitate further exploration of the functions of trisulfides in biological systems.
Introduction
Reactive sulfur species (RSS) are a class of biologically existing sulfur containing compounds that play regulatory roles in various physiological processes. Representative RSS include hydrogen sulfide (H2S), thiols (RSH), disulfides (RSSR), polysulfides (RSSnSR), hydropersulfides (RSSH), S-nitrosothiols (RSNO), sulfenic acids (RSOH), etc. Among these species, organic polysulfides (R–S–Sn–S–R, n ≥ 1, R ≠ H), especially trisulfides (R–S–S–S–R), are naturally occurring in plants of the Allium genus (e.g. garlic). These trisulfides have been demonstrated to have antimicrobial, antioxidation, and anti-cancer activities and act as mediators in cellular signaling.1–4 Additionally, some natural products (e.g. calicheamicin and shishijimicin A) contain trisulfide linkages.5,6 In these molecules, the trisulfide moiety is critical for their biological activities. Trisulfides can also serve as the effective precursor of H2S and RSSH, two established redox signalling compounds. It should be noted that only a small number of proteins are currently known to contain the trisulfide motif.7,8 However, the increased understanding of the roles of polysulfides in cellular signalling has suggested that the prevalence of protein trisulfides may be underestimated.8 For example, sulfide quinone oxidoreductase couples the oxidation of H2S to the reduction of coenzyme Q10 in the mitochondrial electron transport chain. Considering its active site contains two cysteines linked by a trisulfide that participates in the mechanism of oxidizing H2S,9 methods that can be used to construct protein trisulfides are necessary for better understanding their roles in RSS biology. However, such methods are still very limited.10In organic synthesis, methods for the preparation of small molecule trisulfides have been reported.11 Representative examples (Scheme 1) include (a) the reaction of Na2S with two equivalents of Bunte salts;12 (b) the nucleophilic reaction of persulfides with mono-sulfur-based electrophiles (such as RS-Ts, RS-Cl, etc.);13–15 and (c) the reaction of thiols with disulfide-based electrophiles (e.g. RSSCl generated in situ from another thiol).16,17 While these methods have been successfully used in the preparation of many organic trisulfides, the formation of trisulfides with small biologically relevant thiols, such as cysteine and glutathione (GSH), and –SH containing proteins in aqueous conditions are still lacking. A report by Nicolaou et al. revealed that a phthalimide-based disulfide reagent could be used to install a methyl trisulfide in the synthesis of complex natural products such as shishijimicin A (Scheme 1).6 Inspired by the fact that this reagent can install trisulfides on complex natural products, we hypothesized that phthalimide-based disulfides could be applied under biologically relevant conditions to react with biothiols and proteins. Additionally, cysteine-based phthalimide reagents would be useful for the preparation of cysteine-based trisulfides that would be suitable for biological investigations with trisulfides. Herein, we report the preparation of such reagents and their applications in the preparation of both small molecule trisulfides and protein trisulfides.
 | ||
| Scheme 1 Representative synthetic methods for trisulfides (a–c) and our idea of using phthalimide disulfides to prepare biothiol trisulfides. | ||
Results and discussion
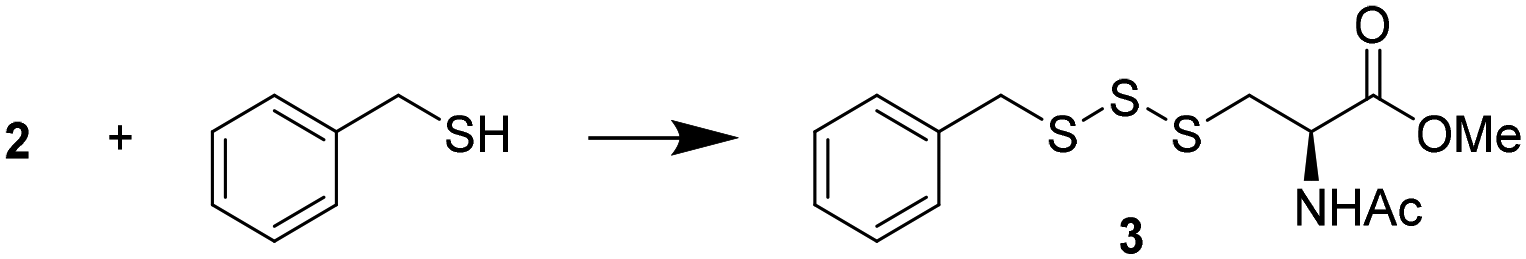
Preparation of cysteine–phthalimide disulfide 2
N-Acetyl-L-cysteine methyl ester (NACMe) was chosen to prepare the phthalimide disulfide reagent 2, because 2 could be used to construct cysteine-based trisulfides. Synthesis of 2 (Scheme 2) began with treating phthalimide with sulfur monochloride (S2Cl2) to give N,N′-thiobisphthalimide 1 in a 74% yield. The treatment of N,N′-thiobisphthalimide with NACMe then produced 2 with a 72% yield. Reagent 2 is a white solid that can be stored at low temperatures (such as 0 °C) without noticeable degradation for months. | ||
| Scheme 2 Preparation of cysteine–phthalimide disulfide 2. | ||
Optimization of conditions for trisulfide synthesis
Benzyl mercaptan (BnSH) was chosen as a model thiol to optimize trisulfide formation with reagent 2. As shown in Table 1, when a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 2 and BnSH was used, the desired trisulfide 3 was obtained in a 60% yield (entry 1). If excess thiol was used (entry 2), the yield dropped to 41%. This is likely because the unreacted BnSH can react with and consume the trisulfide product. On the other hand, the use of excess reagent 2 in the reaction was found to increase the yield of 3 to 85% (entries 3 and 4). Since both 2 equiv. and 1.5 equiv. of 2 gave similar results, we decided to use 1.5 equiv. of 2 in the following studies to examine the effect of different solvents on the reaction (entries 4–7). All organic solvents, including dichloromethane (DCM), acetonitrile (ACN), and methanol (MeOH), were found to work well, while the use of water led to a lower yield (60%). This could be attributed to 2 having low solubility in water. Aqueous miscible organic solvents, such as ACN, could be used to offset the solubility issue. Nevertheless, the reaction was still fairly productive in water, suggesting that trisulfide formation could occur with water soluble biothiols.
:1 ratio of 2 and BnSH was used, the desired trisulfide 3 was obtained in a 60% yield (entry 1). If excess thiol was used (entry 2), the yield dropped to 41%. This is likely because the unreacted BnSH can react with and consume the trisulfide product. On the other hand, the use of excess reagent 2 in the reaction was found to increase the yield of 3 to 85% (entries 3 and 4). Since both 2 equiv. and 1.5 equiv. of 2 gave similar results, we decided to use 1.5 equiv. of 2 in the following studies to examine the effect of different solvents on the reaction (entries 4–7). All organic solvents, including dichloromethane (DCM), acetonitrile (ACN), and methanol (MeOH), were found to work well, while the use of water led to a lower yield (60%). This could be attributed to 2 having low solubility in water. Aqueous miscible organic solvents, such as ACN, could be used to offset the solubility issue. Nevertheless, the reaction was still fairly productive in water, suggesting that trisulfide formation could occur with water soluble biothiols.
|
|
||||
|---|---|---|---|---|
| Entry | Equiv. of 2 | Equiv. BnSH | Solvent | Yield of 3 |
| Reaction conditions: BnSH (40 mM); room temperature; 2 hours. | ||||
| 1 | 1 | 1 | DCM | 60% |
| 2 | 1 | 1.5 | DCM | 41% |
| 3 | 2 | 1 | DCM | 85% |
| 4 | 1.5 | 1 | DCM | 80% |
| 5 | 1.5 | 1 | ACN | 83% |
| 6 | 1.5 | 1 | MeOH | 80% |
| 7 | 1.5 | 1 | H2O | 60% |
Scope of thiol substrates for trisulfide synthesis
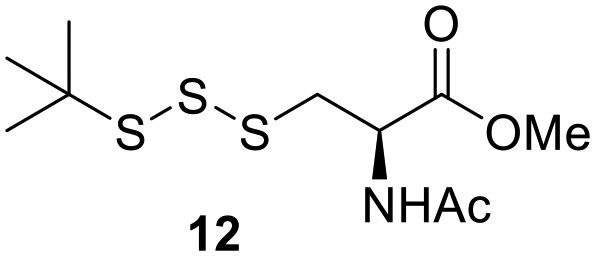
After determining the optimal conditions for trisulfide synthesis, the scope of applicable thiols was evaluated (Table 2). In all cases with primary, secondary, tertiary, and aryl thiols, the treatment of 2 with the thiols produced trisulfide products in excellent yields. Interestingly with tertiary thiols, 0.5 equivalents of triethylamine (TEA) was necessary for >60% yields. We also tested two water soluble thiols, GSH and thioglucose, in this reaction. For these substrates, a solvent mixture of ACN–H2O was needed, and the reactions afforded the desired trisulfide products in >70% yields.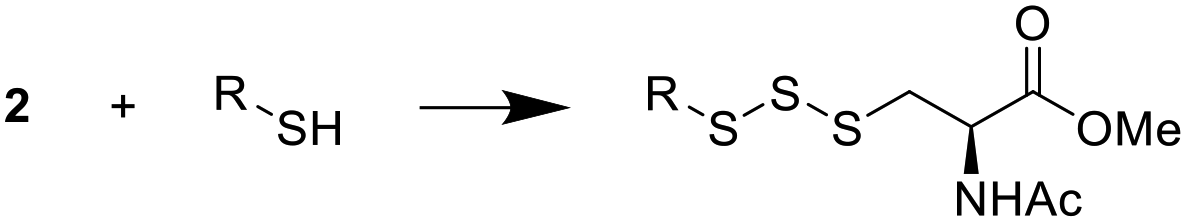


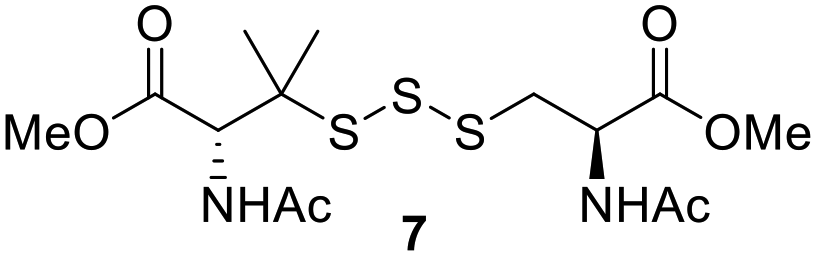

Preparation of protein-based trisulfides
Having successfully prepared trisulfides with water soluble thiols GSH and thioglucose, we then investigated reagent 2's ability to form trisulfides with thiol-containing proteins. Bovine serum albumin (BSA) was freshly reduced and treated with 2 (10 equiv.) in phosphate buffer (pH 7.4) at 4 °C overnight. Next, the protein was purified by PD-10 column and subjected to MS analysis. As seen in Fig. 1, the cysteine-based trisulfide adduct was detected on the intact protein. We also observed up to five trisulfide adducts on BSA, as reduced BSA is known to contain up to five free thiols.18 Besides BSA, we also tested papain in this reaction, and the trisulfide modification was also observed (see Fig. S1 in the SI). This new method for the preparation of trisulfide-containing proteins may be used to enhance our understanding of protein polysulfides and their roles in biological systems. | ||
| Fig. 1 (a) The reaction scheme of BSA trisulfide formation. Deconvoluted mass spectra of (b) reduced BSA and (c) reduced BSA treated with 10 equiv. of 2. | ||
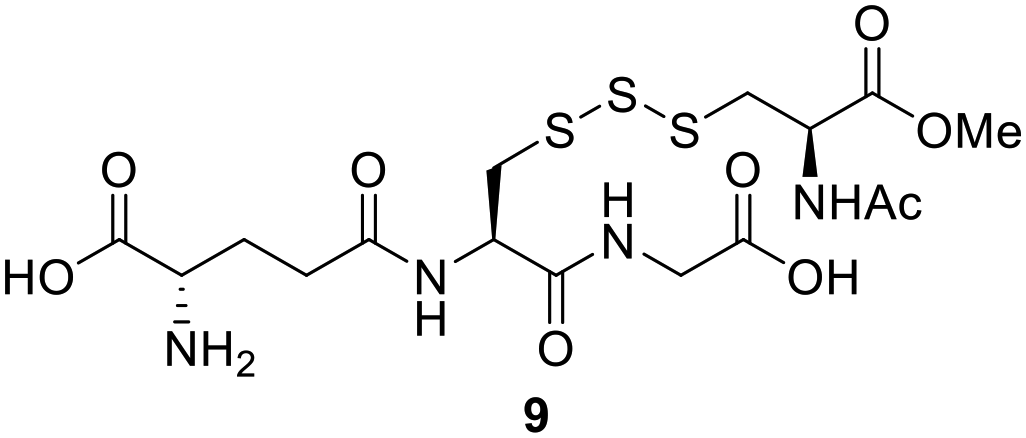
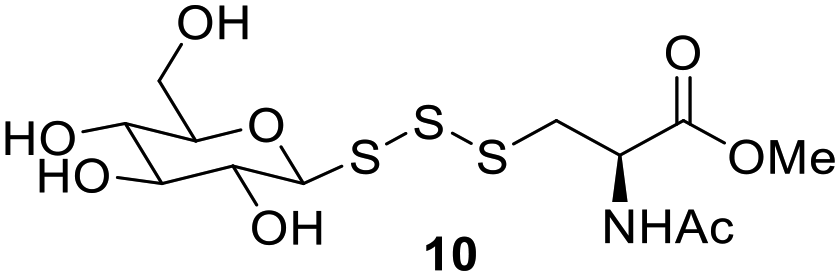
Trisulfides as sulfane sulfur precursors in cells
Polysulfides belong to the sulfane sulfur family and are known precursors of persulfides and H2S. Glutathione trisulfide and glucose tetrasulfide have previously been used to induce H2S and persulfide formation in biologically relevant conditions.18,19 Considering trisulfides 9 and 10 are NACMe hybrids of the aforementioned polysulfide reagents, we wondered if these trisulfides could also be used to increase sulfane sulfur levels in cells. To test this, HeLa cells were incubated with 9 or 10 (50 μM) for 30 min and then washed with PBS to remove excess trisulfides. Cells were then incubated with SSP4, a known fluorescent probe for sulfane sulfur, for 50 min and washed before being subjected to fluorescence imaging.20 As shown in Fig. 2, a significant increase in fluorescence was observed in cells treated with 9 or Na2S3 (positive control) compared to cells with no treatment. Surprisingly, treatment with 10 did not result in a significant increase in fluorescence. This may be due to compound 10's weaker cell permeability or its weaker reactivity toward –SH nucleophiles as observed in its weaker H2S release ability compared to compound 9 (as shown in Fig. S2). These results demonstrate that 9 can increase intracellular sulfane sulfur levels and may potentially be used for additional cellular applications in the future.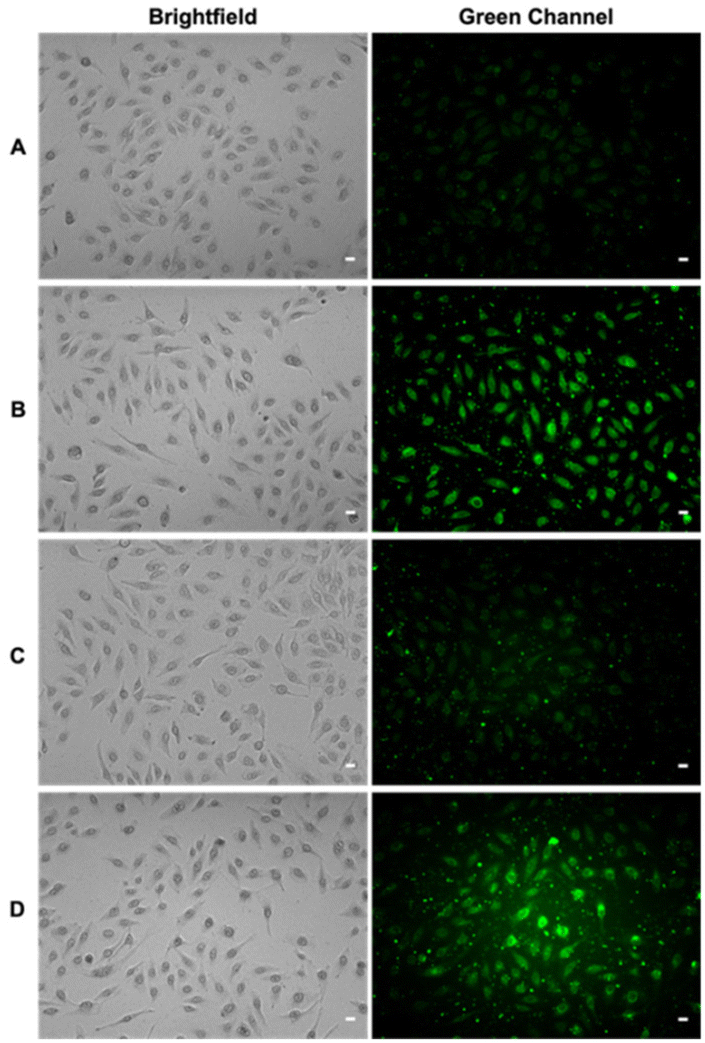 | ||
| Fig. 2 Representative fluorescence images of HeLa cells treated with (A) no treatment or 50 μM of (B) 9, (C) 10, or (D) Na2S3 for 30 min at 37 °C, 5% CO2. After incubation, cells were washed 1× with PBS and then incubated with SSP4 (20 μM) and CTAB (100 μM) for 50 min at 37 °C, 5% CO2. Cells were then washed 3× with PBS and suspended in Fluorobrite DMEM before fluorescence imaging. Scale bars = 20 μm. | ||
Comparing trisulfides’ sulfur transfer ability
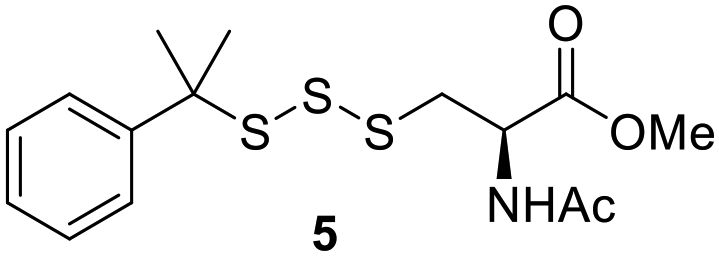
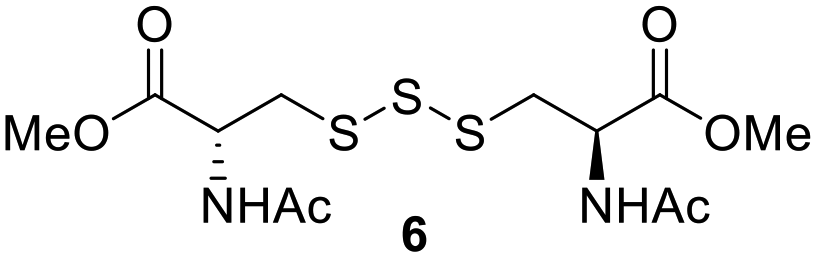
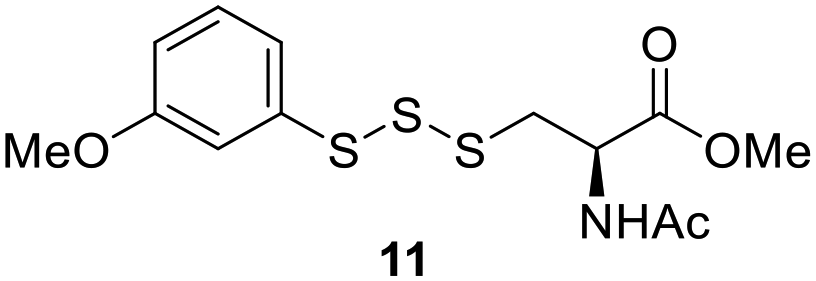
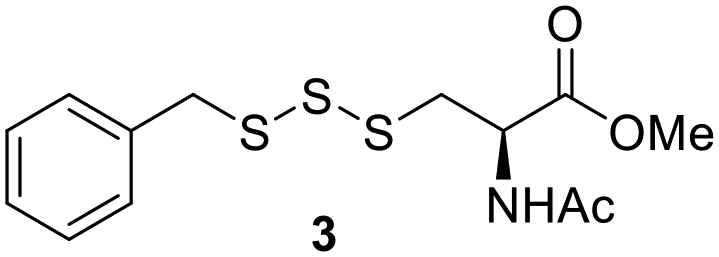
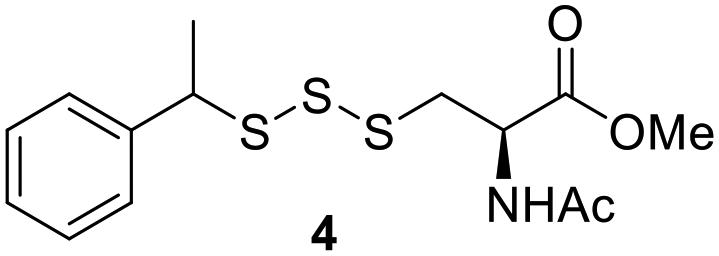
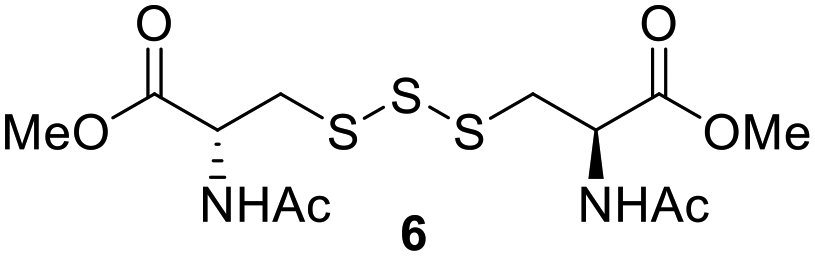
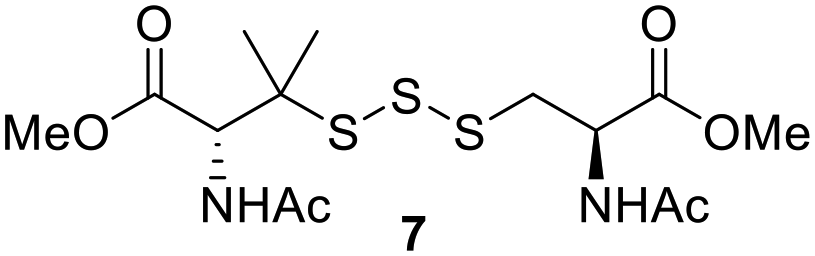
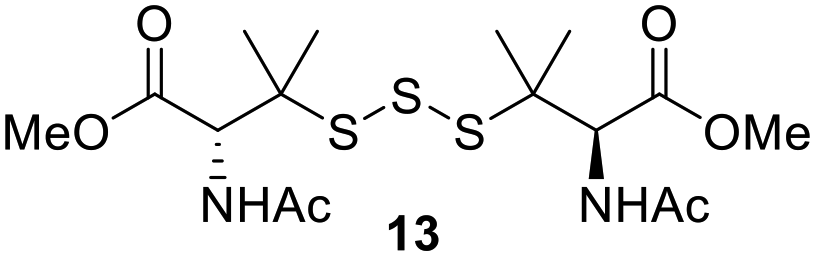
One of the pathways in which trisulfides can increase cellular sulfane sulfur levels is by inducing persulfidation. Essentially, trisulfides can undergo sulfur transfer reactions to release the S0 atom (i.e. the middle S atom) to the acceptor thiol and form persulfides such as GSSH. With a series of trisulfide compounds (3–7 & 13) in hand, we set to understand their structure–activity relationships as sulfur transfer reagents.To model the sulfur transfer ability of trisulfides, we employed their reaction with phosphine compounds, which is known to reduce trisulfides to their respective disulfide.21 We also found that trisulfide 3 treated with triphenylphosphine (PPh3) formed the disulfide and triphenylphosphine sulfide (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PPh3), further demonstrating that the middle S atom was transferred. To determine the sulfur transfer ability of different trisulfides, the initial rate of SPPh3 formation was measured. Trisulfide (2 mM) was treated with PPh3 (20 mM) in CDCl3 and the formation of SPPh3 was monitored by 31P NMR. As seen in Table 3, there was a decrease in the initial rate of SPPh3 formation from compounds 3 to 5 for every methyl group added adjacent to the trisulfide linkage. In compounds 6, 7, and 13, where the cysteine moiety was replaced with a penicillamine and thus adding a gem-dimethyl proximal to the trisulfide linkage, also decreased the initial rate of SPPh3 formation. Overall, as the steric bulk increased around the trisulfide linkage, the rate of SPPh3 formation also decreased, indicating that the sulfur transfer ability of the trisulfide was decreased. Unlike 3–7 where only one side of the trisulfide was modified, 13 contained a gem-dimethyl moiety on both sides of the trisulfide linkage and was expected to exhibit a larger decrease in the initial rate of SPPh3 formation. However, the addition of the second gem-dimethyl group only resulted in about a 50% decrease in the initial rate. This was similar to the percent decrease observed for each methyl ground addition on compounds 3–5.
PPh3), further demonstrating that the middle S atom was transferred. To determine the sulfur transfer ability of different trisulfides, the initial rate of SPPh3 formation was measured. Trisulfide (2 mM) was treated with PPh3 (20 mM) in CDCl3 and the formation of SPPh3 was monitored by 31P NMR. As seen in Table 3, there was a decrease in the initial rate of SPPh3 formation from compounds 3 to 5 for every methyl group added adjacent to the trisulfide linkage. In compounds 6, 7, and 13, where the cysteine moiety was replaced with a penicillamine and thus adding a gem-dimethyl proximal to the trisulfide linkage, also decreased the initial rate of SPPh3 formation. Overall, as the steric bulk increased around the trisulfide linkage, the rate of SPPh3 formation also decreased, indicating that the sulfur transfer ability of the trisulfide was decreased. Unlike 3–7 where only one side of the trisulfide was modified, 13 contained a gem-dimethyl moiety on both sides of the trisulfide linkage and was expected to exhibit a larger decrease in the initial rate of SPPh3 formation. However, the addition of the second gem-dimethyl group only resulted in about a 50% decrease in the initial rate. This was similar to the percent decrease observed for each methyl ground addition on compounds 3–5.
PPh3 formation from the trisulfides
|
|
|
|---|---|
| Trisulfide | Initial rate (M s−1) |
|
10 × 10−8 |
|
7 × 10−8 |

|
3 × 10−8 |
|
7 × 10−8 |
|
4 × 10−8 |
|
2 × 10−8 |
To further understand the structure–activity relationship between the addition of a methyl group proximal to the trisulfide linkage and the sulfur transfer ability of the trisulfides, density functional theory (DFT) calculations were carried out to identify the lowest energy conformer of trisulfides 3–5, as it may represent each trisulfide's conformer in solutions. As seen in Fig. 3, the lowest energy conformer of trisulfides 3–5 adopted a folded structure with the central sulfur atom of the trisulfide linkage exposed to solution. In the cases of 4 and 5, the methyl and gem-dimethyl groups may not be bulky enough to significantly hinder the exposed trisulfide linkage. This may explain why there is only a moderate decrease observed for the initial rate of SPPh3 formation. Thus, a larger moiety proximal to the trisulfide linkage is necessary to significantly impact the trisulfide's sulfur transfer ability.
 | ||
| Fig. 3 DFT predictions of the most stable conformers for trisulfides 3–5. | ||
Experimental
Materials and methods
Reagents and solvents were of the highest grade available and used without purification. All reactions were stirred using Teflon-coated magnetic stir bars. Reactions were monitored by thin layer chromatography (TLC) with 0.25 mm pre-coated silica gel plates. TLC plates were visualized by ultraviolet light and/or treatment with p-anisaldehyde stain followed by gentle heating. Purification of products was accomplished by flash column chromatography and purified compounds showed a single spot on TLC. NMR spectra were measured on a Bruker high field NMR spectrometer (1H at 400 MHz or 600 MHz, 13C at 151 MHz) and are reported in parts per million (ppm) on the δ scale relative to CDCl3 (δ 7.26 for 1H, δ 77.16 for 13C) and D2O (δ 4.79 for 1H). High resolution mass spectrometry analysis was performed by the Agilent 6230 TOF LC/MS (ESI), and the deconvoluted mass spectra of the proteins was obtained through Agilent Mass Hunter BioConfirm software.General procedure for the synthesis of trisulfides
1.5 equivalents of reagent 2 was dissolved in DCM. Thiol (1 equivalent) was dissolved in DCM and added dropwise to the reaction solution for a final concentration of 40 mM with respect to the thiol. The reaction was stirred at room temperature for 2 hours and monitored by TLC. After 2 hours, solvent was removed by rotary evaporator, and crude material was purified by flash column chromatography. Pure trisulfide was dried under high vacuum and analysed by 1H, 13C NMR, and ESI-MS.Bovine serum albumin trisulfide formation
Reduced BSA was prepared according to literature procedures.17 Briefly, BSA (150 mg) was added to 2.3 mL phosphate buffer (100 mM, 0.1 mM pentetic acid, pH 7.4). Mercaptoethanol (23 μL) was added to the mixture, and the mixture was incubated overnight in the dark at 4 °C. The mixture was then purified by PD-10 column, and protein-containing fractions were quantified by Nanodrop. The most concentrated fraction yielded 0.64 mM of reduced BSA. A 28 mM stock solution of 2 in ACN was freshly prepared. 78 μL of the most concentrated fraction of reduced BSA was diluted in 404 μL phosphate buffer and treated with 18 μL of 2 stock solution for a final concentration of reduced BSA (100 μM) and 2 (1.0 mM). The reaction solution was incubated overnight at 4 °C in the dark in an Eppendorf tube. The reaction mixture was then purified by PD-10 column, with an aliquot from the most concentrated fraction (44 μM), diluted to 16 μM for MS analysis. An aliquot of the most concentrated fraction of reduced BSA was also diluted to 16 μM for MS analysis. Both samples were passed through a Jupiter 5 μM C4 300 Å column with mobile phase (H2O/ACN, 0.1% formic acid) for desalting during LC-MS-TOF analysis, and the deconvoluted mass spectra were obtained with the Agilent BioConfirm software.Cell imaging
HeLa cells were cultured in DMEM/F12 (1:1) (Gibco, Invitrogen, #11330-032) medium supplemented with 10% fetal bovine serum (FBS) at 37 °C, 5% CO2 and seeded in a 48-well plate (Thermofisher Scientific, #150687). After 48 hours (∼85% confluency), media was aspirated, and cells were washed 2× with PBS. 9, 10, and Na2S3 were freshly prepared at 10 mM stocks in sterile PBS (Gibco, ThermoFisher Scientific, #10010072) and then diluted to 50 μM in Fluorobrite DMEM before being added to the cells. Cells were incubated for 30 min at 37 °C, 5% CO2. Afterwards, the cells were washed once with PBS and incubated with SSP4 (20 μM, 0.2% DMSO) and CTAB (100 μM, 0.5% EtOH) in Fluorobrite DMEM for 50 min at 37 °C, 5% CO2. Cells were then washed 3× with PBS, suspended in Fluorobrite DMEM, and subjected to fluorescence imaging on the Keyence All-in-One Fluorescence Microscope (BZ-X810).
Initial rate kinetics of triphenylphosphine sulfide formation
Fresh stock solutions of trisulfide and triphenylphosphine (PPh3) were prepared in CDCl3. In an NMR tube was added CDCl3, an aliquot of trisulfide stock solution, and an aliquot of PPh3 stock solution for a final volume of 500 μL with a final concentration of trisulfide (2 mM) and PPh3 (20 mM). The formation of triphenylphosphine sulfide was monitored by 31P NMR over 1.5 hours, with an almost complete disappearance of trisulfide after 24 hours. The initial rate of formation of triphenylphosphine sulfide was calculated from the change in concentration of triphenylphine sulfide over the initial 1.5 hours.Computational method for trisulfide conformer
Trisulfides of interest (3–5) were modeled by Avogadro 1.2.0. A systematic rotor search was performed on each trisulfide using Avogadro generating 243 conformers. The resulting conformers were analysed by molecular mechanics method MMFF94 and ranked by predicted energy levels. The 30 conformers with the predicted lowest energy by MMFF94 were each optimized using Gaussian 09. Their energy levels were then determined by DFT in the gas phase at the ωB97XD/6-311G(2d,p) level of theory to determine the lowest energy. Frequency calculated were performed using keywords freq. It has been previously demonstrated that ωB97XD/6-311G(2d,p) is a rigorous level of theory for polysulfide compounds.22Conclusions
Herein, we reported the preparation and evaluation of compound 2, a phthalimide-based sulfur transfer reagent for the synthesis of cysteine trisulfides. Thiols applicable for trisulfide synthesis involve primary, secondary, and tertiary organic thiols and biothiols, including proteins. Considering trisulfides are sulfane sulfur transfer reagents, the effects of sterics on their persulfide donating ability was investigated by experimental and computational methods. This efficient method of using phthalimide-based disulfide reagents to induce trisulfide formation on both small molecules and proteins may help increase the understanding of the roles of these polysulfides in RSS biology.Author contributions
Conrad N. A. Du: writing – original draft; investigation; formal analysis. Austin Sarker-Young: writing – review & editing; methodology; investigation; data curation; formal analysis. Meg Shieh: writing – review & editing; investigation; visualization. Stephen Lindahl: writing – review & editing; conceptualization. Ming Xian: writing – review & editing; writing – original draft; funding acquisition; conceptualization.Conflicts of interest
The authors have no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental protocols, compound characterization data. See DOI: https://doi.org/10.1039/d5ob01481f.Acknowledgements
This work is supported by the NIH (R35GM149170). M. S. is supported by a NIH F31 Predoctoral Fellowship (F31HL170516).References
- L. D. Lawson, in Phytomedicines of Europe, American Chemical Society, 1998, vol. 691, ch. 14, pp. 176–209. DOI:10.1021/bk-1998-0691.ch014.
- S. Lindahl and M. Xian, Curr. Opin. Chem. Biol., 2023, 75, 102325, DOI:10.1016/j.cbpa.2023.102325.
- C.-Y. Tsai, C.-C. Wang, T.-Y. Lai, H.-N. Tsu, C.-H. Wang, H.-Y. Liang and W.-W. Kuo, Int. J. Cardiol., 2013, 168, 1286–1297, DOI:10.1016/j.ijcard.2012.12.004.
- R. Malla, R. Marni, A. Chakraborty and M. A. Kamal, J. Pharm. Anal., 2022, 12, 221–231, DOI:10.1016/j.jpha.2021.11.004.
- K. C. Nicolaou, C. W. Hummel, M. Nakada, K. Shibayama, E. N. Pitsinos, H. Saimoto, Y. Mizuno, K. U. Baldenius and A. L. Smith, J. Am. Chem. Soc., 1993, 115, 7625–7635, DOI:10.1021/ja00070a006.
- K. C. Nicolaou, R. Li, Z. Lu, E. N. Pitsinos, L. B. Alemany, M. Aujay, C. Lee, J. Sandoval and J. Gavrilyuk, J. Am. Chem. Soc., 2018, 140, 12120–12136, DOI:10.1021/jacs.8b06955.
- E. Canova-Davis, I. P. Baldonado, R. C. Chloupek, V. T. Ling, R. Gehant, K. Olson and B. L. Gillece-Castro, Anal. Chem., 1996, 68, 4044–4051, DOI:10.1021/ac9605915.
- R. W. Nielsen, C. Tachibana, N. E. Hansen and J. R. Winther, Antioxid. Redox Signaling, 2011, 15, 67–75, DOI:10.1089/ars.2010.3677.
- A. P. Landry, D. P. Ballou and R. Banerjee, ChemBioChem, 2021, 22, 949–960, DOI:10.1002/cbic.202000661.
- G. J. L. Bernardes, J. P. Marston, A. S. Batsanov, J. A. K. Howard and B. G. Davis, Chem. Commun., 2007, 3145–3147, 10.1039/B706682A.
- D. Ali, Y. Amer, W. F. Petersen, C. H. Kaschula and R. Hunter, SynOpen, 2021, 05, 49–64, DOI:10.1055/s-0040-1706018.
- D. Bhattacherjee, A. Sufian, S. K. Mahato, S. Begum, K. Banerjee, S. De, H. K. Srivastava and K. P. Bhabak, Chem. Commun., 2019, 55, 13534–13537, 10.1039/c9cc05562b.
- D. Ali, R. Hunter, C. H. Kaschula, S. De Doncker and S. C. M. Rees-Jones, J. Org. Chem., 2019, 84, 2862–2869, DOI:10.1021/acs.joc.8b03262.
- T. Nakabayashi and J. Tsurugi, J. Org. Chem., 1961, 26, 2482–2486, DOI:10.1021/jo01351a082.
- S. Xu, Y. Wang, M. N. Radford, A. J. Ferrell and M. Xian, Org. Lett., 2018, 20, 465–468, DOI:10.1021/acs.orglett.7b03846.
- H. Böhme and M. Clement, Justus Liebigs Ann. Chem., 1952, 576, 61–69, DOI:10.1002/jlac.19525760107.
- G. Derbesy and D. N. Harpp, Tetrahedron Lett., 1994, 35, 5381–5384, DOI:10.1016/s0040-4039(00)73505-2.
- S. Lindahl, M. Shieh, T. Zhang, C. Guo, J. R. Robinson, T. Sawa and M. Xian, Redox Biol., 2024, 70, 103045, DOI:10.1016/j.redox.2024.103045.
- M. Maemura, M. Morita, S. Ogata, Y. Miyamoto, T. Ida, K. Shibusaka, S. Negishi, M. Hosonuma, T. Saito, J. Yoshitake, T. Takata, T. Matsunaga, E. Mishima, U. Barayeu, T. Akaike and F. Yano, Redox Biol., 2025, 81, 103545, DOI:10.1016/j.redox.2025.103545.
- M. Shieh, X. Ni, S. Xu, S. P. Lindahl, M. Yang, T. Matsunaga, R. Flaumenhaft, T. Akaike and M. Xian, Redox Biol., 2022, 56, 102433, DOI:10.1016/j.redox.2022.102433.
- K. Cumnock, T. Tully, C. Cornell, M. Hutchinson, J. Gorrell, K. Skidmore, Y. Chen and F. Jacobson, Bioconjugate Chem., 2013, 24, 1154–1160, DOI:10.1021/bc4000299.
- S. Nikoo, P. J. Meister, J. J. Hayward and J. W. Gauld, Molecules, 2018, 23, 3323, DOI:10.3390/molecules23123323.
| This journal is © The Royal Society of Chemistry 2025 |