
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2-Migratory ring expansion of a BN-naphthalene
Braden A.
Mediavilla
,
Maxime A.
Siegler
and
Rebekka S.
Klausen
 *
*
Department of Chemistry, Johns Hopkins University, 3400 N Charles St, Baltimore, MD, USA. E-mail: klausen@jhu.edu
First published on 23rd October 2025
Abstract
Organoboranes hold an undeniable value in modern organic chemistry and show ever-growing potential in polymer design and functionalization. Azaborines, in particular, are still being explored as a promising class of organoboron compounds. We investigated the reactivity of a 1,2-azaborine towards Matteson homologation, a cornerstone of organoborane transformations. The azaborine was shown to be reactive towards nucleophiles. Addition of n-BuLi led to exchange of the R group, while addition of chloromethyllithium formed the desired tetracoordinate intermediate. However, rather than homologation, the complex readily underwent ring expansion via migration of the adjacent C(sp2), followed by deborylation. Computational analysis revealed a stable geometry of the azaboronate in which the migrating carbon is antiperiplanar to the leaving group, and which we propose as being responsible for the observed reactivity. This finding suggests a unique role for azaborines in molecular transformations which may be unachievable with other organoboron frameworks.
Introduction
Organoboranes are a versatile functional group in the organic chemist's toolbox, due to the ability to convert the C–B bond into a variety of other functional groups including C–O,1,2 C–N,3,4 and C–C5 bonds, often with high stereospecificity. This arises from the ability of groups to migrate from a tetrasubstituted, anionic boronate in a 1,2-rearrangement, a mechanism with ongoing synthetic utility6–9 and implicated in such established reactions as stereoretentive oxidation of organoboranes and the Matteson homologation (Scheme 1). The Matteson homologation is the insertion of a single CH2 or CHX (X = halogen) group into a C–B bond, which can be employed iteratively and in combination with other organoborane functionalizations.10–14 | ||
| Scheme 1 1,2-Boronate rearrangements in organoborane oxidation and the Matteson homologation. | ||
While a variety of organoborane functional groups have been reported, an important emerging class are 1,2-azaborines, aromatic compounds in which one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond has been replaced with the isoelectronic B–N bond (Fig. 1a). These unusual aromatic heterocycles have been widely investigated as biological isosteres, in materials science, and in catalysis,15 but far less work has focused on their utility as synthetic intermediates. This is despite the exciting potential of azaborines to solve synthetic challenges, particularly in the context of polymer chemistry. Due to its aromatic character, the monomer BN 2-vinylnaphthalene (BN2VN, Fig. 1b) has styrene-like reactivity and versatility, allowing for controlled radical, free radical, and syndioselective coordination polymerization and co-polymerization.16–23
C double bond has been replaced with the isoelectronic B–N bond (Fig. 1a). These unusual aromatic heterocycles have been widely investigated as biological isosteres, in materials science, and in catalysis,15 but far less work has focused on their utility as synthetic intermediates. This is despite the exciting potential of azaborines to solve synthetic challenges, particularly in the context of polymer chemistry. Due to its aromatic character, the monomer BN 2-vinylnaphthalene (BN2VN, Fig. 1b) has styrene-like reactivity and versatility, allowing for controlled radical, free radical, and syndioselective coordination polymerization and co-polymerization.16–23
 | ||
| Fig. 1 (a) Previous research: applications of azaborines as benzene mimics, as synthetic intermediates in polymer synthesis, and as electronically modular ligands for Suzuki cross-coupling reactions, adapted from Sun et al. (2015).15 (b) This work: investigation of migratory aptitude in the rearrangement of BN naphthalene-derived azaboronate complexes. | ||
We became interested in whether polymeric materials derived from BN2VN could also undergo a Matteson homologation, considering the similarities in mechanism between hydrogen peroxide mediated oxidation and Matteson homologation. Achieving this functionalization would not only facilitate polymer synthesis but could also impact molecular synthesis. If Matteson homologation of 1,2-azaborine systems were possible, existing heterocycles24–32 could be rapidly further diversified without requiring de novo synthesis of the sometimes challenging heterocyclic core.
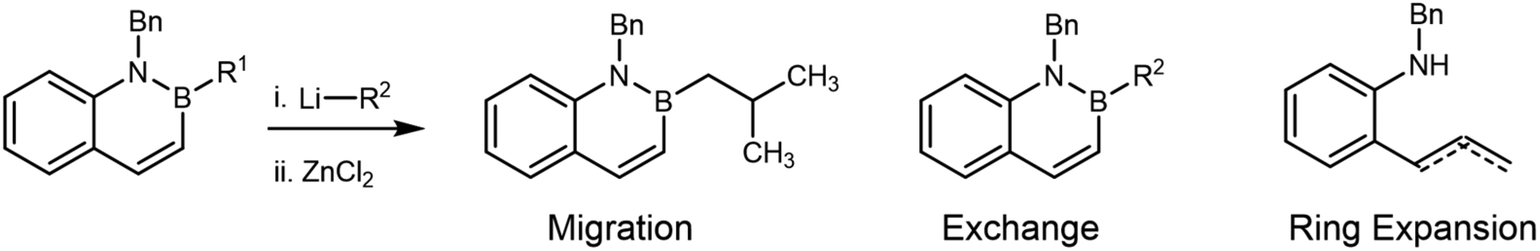
Therefore, in considering the feasibility of the proposed homologation of B-alkyl azaborine derivatives, two different steps must occur (Fig. 1b): (i) addition of chloromethyllithium (or related carbenoid) to the boron of an azaborine ring to form an azaboronate complex I and (ii) selective migration of the exocyclic bond labeled “a” from azaboronate complex I. Migration of either the amine or the bond labeled “b” would lead to ring expansion and loss of BN aromaticity. While prior work from our group supported the feasibility of organolithium addition to the boron of BN naphthalene,24 the relative migratory ability of varying groups on boron has been incompletely elucidated. Existing experimental and theoretical work suggests that endocyclic migration is uncommon due to conformational constraints of the migration.33–36 However, in the last 5 years, several methods for ring-contractive endocyclic migration have been developed.37–42 As such, and additionally considering the unique electronic and conformational context of these aromatic azaboronates, the expected decomposition pathway for complex I was unclear.
Here, we report our attempts to perform the Matteson homologation on B-isopropyl functionalized BN naphthalenes. We observed a novel ring-expansion via migration of the C(sp2) adjacent to the boron in the naphthalene core. Extensive characterization supports assignment of the major products to deborylated variants of this ring-expanded structure (Fig. 1b).
Results and discussion
We began by synthesizing examples of BN naphthalenes with different alkyl substituents on boron. We initially targeted compounds 2a and 2b (Scheme 2a), with an isopropyl and isobutyl side chain respectively, because Matteson homologation of 2a would yield 2b. Known 2-vinylaniline43 (1) was a common precursor via Molander's procedure29 for borylative annulation. Borylative annulation proceeded in high yields and an X-ray crystal structure confirmed the planar structure of 2a (Scheme 2b). | ||
| Scheme 2 (a) Synthesis of BN naphthalenes 2a–b. (b) X-ray crystal structure of 2a determined at 110.00(10) K. Thermal ellipsoids set at the 50% probability level with most hydrogen atoms omitted for clarity. Black = carbon, blue = nitrogen, purple = boron, pink = hydrogen. (c) Deprotonation of 2a observed instead of Matteson homologation. | ||
Attempted Matteson homologation of 2a did not yield the desired 2b (Scheme 2c). The one-pot reaction conditions employed lithium-halogen exchange of chloroiodomethane (I–CH2–Cl) and n-butyllithium (n-BuLi) in the presence of 2a. The only observed reaction was partial deprotonation (Scheme 2c and Fig. S1). Reaction with 2 equivalents of the carbenoid did not afford any additional products.
We therefore synthesized N-benzyl protected BN naphthalenes 4a–c (Scheme 3a), a common and facile derivatization of 1,2-azaborines which renders it unable to undergo the observed deprotonation.9,14,15 An X-ray crystal structure confirmed the structure of 4a (Scheme 3b). The synthesis of 4b has not previously been reported and could be prepared by similar reactions as have been described elsewhere.20,24,29 However, our 35% yield on preparative scale was significantly lower than observed for similar compounds. We attributed this to the formation of 1H NMR-silent inorganic byproducts which required multiple purification steps to fully remove, as described completely in the SI.
 | ||
| Scheme 3 (a) Synthesis of BN naphthalenes 4a–c. (b) X-ray crystal structures of 4a and 4c, determined at 243.00(10) and 110.00(10) K, respectively. Thermal ellipsoids set at the 30% probability level with hydrogen atoms omitted for clarity. Black = carbon, blue = nitrogen, purple = boron. | ||
Reaction of 4a with chloromethyllithium produced no measurable quantity of the homologation product 4b under the same conditions which transformed pinacol boronic ester i-PrBpin to the homologation product i-BuBpin in 79% yield (Scheme 4avs.Scheme 4b). Instead of cleanly forming 4b, a complex mixture of products was obtained, the assignment of which will be described later (vide infra). As a first hypothesis, we considered if the azaborine ring of 4a might inhibit the lithium–halogen exchange reaction of n-BuLi and ICH2Cl, preventing formation of chloromethyllithium. We therefore tested if i-PrBpin could still undergo homologation in the presence of 4a. Treatment of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4a and i-PrBpin with chloromethyllithium resulted in complete conversion of i-PrBpin to i-BuBpin with no formation of 4b detected by NMR or mass spectrometry (Fig. S2). This experiment indicates that the lack of homologation observed with 4a cannot be attributed to inhibition of the lithium–halogen exchange reaction between chloroiodomethane and n-BuLi.
:1 mixture of 4a and i-PrBpin with chloromethyllithium resulted in complete conversion of i-PrBpin to i-BuBpin with no formation of 4b detected by NMR or mass spectrometry (Fig. S2). This experiment indicates that the lack of homologation observed with 4a cannot be attributed to inhibition of the lithium–halogen exchange reaction between chloroiodomethane and n-BuLi.
 | ||
| Scheme 4 (a) Successful homologation of i-PrBpin with standard conditions (2.5 equiv. ClCH2I, 2.1 equiv. n-BuLi, 0.5 equiv. ZnCl2, 0.6 M THF, −78 °C to rt, 16 h). (b) N-Benzyl protected 4a does not homologate under standard conditions. (c) Competition experiment: an equimolar combination of i-PrBpin and 4a results in only homologation of the pinacol boronic ester and no recovery of 4b, demonstrating that lithium–halogen exchange of n-BuLi and ICH2Cl to form chloromethyllithium does proceed in the presence of 4a. | ||
Having established that chloromethyllithium was formed under the reaction conditions, we next considered if failure of nucleophilic addition to the boron atom of 4a might account for the lack of homologation. We began by understanding what reactions 4a does undergo with alkyllithium reagents generally. Previous research from our laboratory has shown that vinyl BN naphthalene reacts with n-BuLi in a substitution reaction, resulting in the formation of n-butyl BN naphthalene 4d (Table 1, entry 1) and ethylene, which was hypothesized to arise from protonation of vinyllithium.9 Because the formation of a more stable C(sp2) carbanion from C(sp3) n-BuLi was thought to contribute to a favorable exchange reaction, elimination of 2-propyllithium was not initially considered to be a concern for 4a. However, treatment of 4a with n-BuLi yielded measurable conversion to 4d, as estimated by 1H NMR, and high conversion to 4d could be observed with excess n-BuLi (Table 1, entries 2, 3 and Fig. S3, S4). We found that the more sterically hindered tert-butyllithium (t-BuLi) did not result in substitution (Table 1, entry 4 and Fig. S5). These data suggest that direct nucleophilic attack by LiR2 on 4a could yield an azaboronate complex such as II, which can either regenerate starting materials or yield the exchange product. The lack of substitution with t-BuLi is consistent with slow nucleophilic attack to form azaboronate II due to the greater steric hindrance of the t-Bu group.
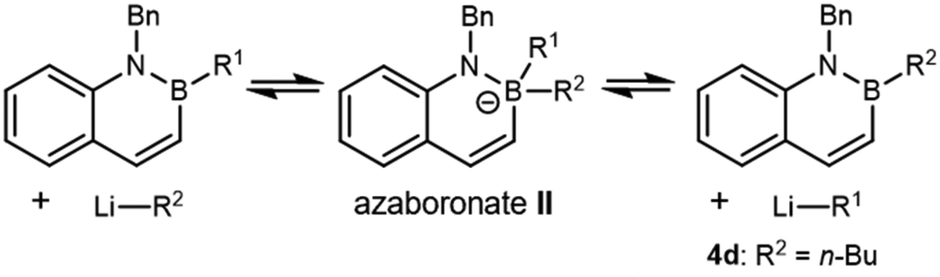
These results suggest that the reaction between 4a and ICH2Cl/t-BuLi should cleanly generate complex I as the only boronate in solution (Fig. S4), a key step towards homologation. However, the ultimate product distribution would depend on the relative rates of i-Pr migration and exchange, leading to products 4b or 4c, respectively (Fig. 2a). Prior work has shown that Lewis acids can accelerate 1,2-migration.44–46 Therefore, we hypothesized that addition of ZnCl2 to our synthetic protocol could accelerate i-Pr migration over exchange.
 | ||
| Fig. 2 (a) Azaboronate complex I as a common intermediate in migration or exchange pathways. (b) (left) Cropped 1H NMR (400 MHz, CDCl3) spectra of the unpurified reaction mixture compared to independently synthesized 4a and 4b as well as purified samples of 5a and 5b (right) dentification of 5a and 5b as the major products of azaboronate rearrangement of 4a. | ||
The reaction of 4a and LiCH2Cl under this revised protocol was reinvestigated. Independent synthesis of 4b and 4c (Scheme 3a) provided authentic samples for comparison. However, analysis of the 1H NMR spectrum of the unpurified reaction mixture suggested no evidence of either the homologation product 4b or the substitution product 4c (Fig. 2b). Instead, we identified the products as 5a and 5b (Fig. 2c), which could be isolated after column chromatography in 58% combined yield (5a:5b, ca. 1:2). The isomeric products were challenging to separate, but after a second purification by preparatory thin layer chromatography, small quantities of pure samples of both (Z)-5a and 5b could be obtained, which, combined with mass spectrometry, confirmed the structural assignment. (E)-5a is known47,48 and our spectroscopic data match, with exception of the coupling constants for the vinyl protons ((Z)-5a, J = 11.2 Hz; (E)-5a, J = 15.4 Hz).49 Allylbenzene 5b is a known compound and our spectroscopic data match the reported characterization data.50,51
Formation of 5a and 5b from 4a under a variety of conditions (Table 2, entries 2–4) is consistent with an unprecedented alternative decomposition of azaboronate I. Rather than exchange (dominant in the reaction of 4a and n-BuLi, see Table 2, entry 1) or migration of the iso-propyl group (dominant in the reactions of i-PrBpin and LiCH2Cl, see Scheme 4a), we hypothesize that migration of the endocyclic alkenyl group results in ring-expansion product 6 (not isolated, Fig. 3). Intermediate boracycle 6 was unable to be isolated, and instead (Z)-alkene 5a and allylbenzene 5b, which are related by isomerization, are observed. Without the stabilizing effect of aromaticity, which is expected to be around 20 kcal mol−1,52 we suggest that 6 readily undergoes deborylation prior to or upon quenching to yield the shown products. This is in line with the mechanistic understanding of deborylation reactions53–56 and with established lability of the B–N bond under the given conditions.57–61
 | ||
| Fig. 3 Overview of the three potential decomposition pathways for azaboronate complex I, formed by reaction of 4a with chloromethyllithium or 4c with isopropylmagnesium chloride. Ring expansion product 6 is not isolable, but 5a and 5b can be recovered. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Starting material | Carbanion | ZnCl2 | Migration | Exchange | Ring expansion |
| 1 | 4a (R1 = i-Pr) | n-BuLi | — | n/a | 72–96% | 0 |
| 2 | 4a (R1 = i-Pr) | LiCH2Cl | — | 0 | 0 | 20% |
| 3 | 4a (R1 = i-Pr) | LiCH2Cl | 0.5 equiv. | 0 | 0 | 58% |
| 4 | 4a (R1 = i-Pr) | LiCH2Br | 0.5 equiv. | 0 | 0 | 33% |
| 5 | 4c (R1 = CH2Cl) | i-PrMgCl | 0.5 equiv. | 0 | 0 | >99% |
| 6 | 4c (R1 = CH2Cl) | n-BuLi | 0.5 equiv. | 0 | 0 | >99% |
We appreciated that if azaboronate I is the key intermediate leading to 5a and 5b, then azaboronate I should also be accessible from the reaction between 4c and iso-propylmagnesium chloride (i-PrMgCl, Fig. 3). Indeed, the reaction of 4c and i-PrMgCl exclusively yielded ring-opened products 5a and 5b (Table 2, entry 5). Neither the starting material 4c, the exchange product 4a, nor the homologated product 4b were observable by either NMR or mass spectrometry. 5a–b were also observed as the major products in the reaction of 4c with n-BuLi (Table 2, entry 6). These data strongly support ring-expansion of azaboronate I as the pathway leading to 5a and 5b.
Ring-expansion via O-migration in pinacol-derived boronates has been long known to compete with exocyclic migration. For various reasons, including conformational demands, it is typically a minor or unobserved product.41,62 Observation of exclusively an endocyclic migration product and dearomatization prompted further investigation.
To support ring expansion and compound 6 as a plausible intermediate towards the formation of the isolable 5a and 5b, we performed computational analysis at the B3LYP-D3(BJ)/def2-TZVP//B3LYP-D3(BJ)/def2-SVP (SMD, THF) level of theory. Complex I is shown coordinated to ZnCl2 (I·ZnCl2), which was present in the reaction conditions with the highest yields of 5a and 5b. Of the two possible diastereoisomers, the favored one has the carbenoid and ZnCl2 in a syn-relationship, which facilitates halide abstraction, as previously reported in related studies on the Matteson homologation.63 The starting material 1,2-azaborine does not strongly bind ZnCl2, but addition of a nucleophile to form azaboronate complex I breaks aromaticity by preventing interaction of the N lone pair with B. This means that the N in complex I is much more Lewis basic than in azaborine 4a.
We observed a low-energy rotational conformation of I·ZnCl2 in which the Cl–C–B–C(sp2) dihedral angle is 179.99°. This orients the internal carbon perfectly for ring-expansion via 1,2-metallate migration, which occurs in an antiperiplanar fashion (Fig. 4). The barrier to the subsequent migration is low (5.4 kcal mol−1). The exocyclic i-Pr group is not antiperiplanar (torsion angle = 56.03°) suggesting that migration of the exocyclic substituent is less favorable. Low but observable formation of 5a and 5b in the absence of ZnCl2 could be due to coordination to the lithium counterion in a similar fashion.64 While further experimentation and calculations would be necessary to thoroughly compare the possible decomposition pathways of azaboronate complex I, these data support ring-expansion as a kinetically plausible pathway under experimentally-relevant conditions.
 | ||
| Fig. 4 Calculated relative free energies (ΔG, kcal mol−1) for the ring-expansion migration pathway of I·ZnCl2 to form 6·ZnCl3 at the B3LYP-D3(BJ)/def2-TZVP//B3LYP-D3(BJ)/def2-SVP (SMD, THF) level of theory. | ||
Conclusions
Here we describe the observed behavior of alkyl-substituted azaborines towards the formation and decomposition of a tetracoordinate azaboronate complex via addition of a carbon-based nucleophile. We found that the starting azaborine was readily deprotonated upon addition of butyllithium, so an N-benzyl variant was synthesized. However, this structure readily underwent nucleophilic addition and exchange with n-BuLi, which was not desired but nonetheless demonstrated susceptibility of the boron to nucleophilic addition. Upon treatment with conditions which generated chloromethyllithium as the sole nucleophile in solution, we isolated products which indicated that the azaboronate complex was decomposed by an unexpected C(sp2)-migration. This hypothesis was further substantiated by forming the same azaboronate by a different method, which yielded similar results, and by computational analysis which identified a stable conformation of the intermediate which was geometrically aligned to perform the observed ring-expansion.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ob01286d.CCDC 2471064–2471066 contain the supplementary crystallographic data for this paper.65a–c
Acknowledgements
This material is based upon work supported by the National Science Foundation under grant CHE-2304952. The NIH (1S10OD030352 to M. A. S.) is gratefully acknowledged for financial support.References
- H. C. Brown and B. C. S. Rao, A new technique for the conversion of olefins into organoboranes and related alcohols, J. Am. Chem. Soc., 1956, 78(21), 5694–5695, DOI:10.1021/ja01602a063
.
- H. C. Brown and G. Zweifel, Convenient new procedures for the hydroboration of olefins, J. Am. Chem. Soc., 1959, 81(15), 4106–4107, DOI:10.1021/ja01524a071
- S. N. Mlynarski, A. S. Karns and J. P. Morken, Direct Stereospecific Amination of Alkyl and Aryl Pinacol Boronates, J. Am. Chem. Soc., 2012, 134(40), 16449–16451, DOI:10.1021/ja305448w
- E. Edelstein, A. Grote, M. Palkowitz and J. Morken, A Protocol for Direct Stereospecific Amination of Primary, Secondary, and Tertiary Alkylboronic Esters, Synlett, 2018,(13), 1749–1752, DOI:10.1055/s-0037-1610172
- N. Miyaura and A. Suzuki, Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds, Chem. Rev., 1995, 95(7), 2457–2483, DOI:10.1021/cr00039a007
- G. Zhou, Z. Guo, S. Liu and X. Shen, Divergent Synthesis of Fluoroalkyl Ketones through Controlling the Reactivity of Organoboronate Complexes, J. Am. Chem. Soc., 2024, 146(6), 4026–4035, DOI:10.1021/jacs.3c12150
- Y. Lin, Y. Guo, S. Mu, G. Lin, W. Kong, X. Lei, J. Xu and Q. Song, Radical 1,2-Boron Shift-Enabled Synthesis of β-Thioalkylboronic Esters from β-Boryl NHPI Esters, Org. Lett., 2025, 27(35), 9825–9830, DOI:10.1021/acs.orglett.5c03295
- H.-C. Shen and V. K. Aggarwal, Merging Organocatalysis with 1,2-Boronate Rearrangement: A Lewis Base-Catalyzed Asymmetric Multicomponent Reaction, J. Am. Chem. Soc., 2024, 146(40), 27305–27311, DOI:10.1021/jacs.4c11113
- W.-J. Pang, X.-C. Wang, J. Sun, X.-S. Li and M.-D. Zhou, Zn-Mediated Fragmentation of β-Boryl NHPI Esters Enabling the Radical 1,2-Boron Shift for the Synthesis of Gem -Difluoroalkenyl Boronates, Org. Lett., 2025, 27(20), 5270–5274, DOI:10.1021/acs.orglett.5c01486
- M. Chen and G. Dong, Atom-by-Atom Iterative Synthetic Logic: Laying the Foundation for Programmable Automated Construction of Small Organic Molecules, ACS Cent. Sci., 2025, 11(6), 843–854, DOI:10.1021/acscentsci.5c00526
- S. R. Angle, H. A. Sharma, C. K. Choi, K. E. Carlson, Y. Hou, J. C. Nwachukwu, S. H. Kim, B. S. Katzenellenbogen, K. W. Nettles, J. A. Katzenellenbogen and E. N. Jacobsen, Iterative Catalyst-Controlled Diastereoselective Matteson Homologations Enable the Selective Synthesis of Benzestrol Isomers, J. Am. Chem. Soc., 2024, 146(45), 30771–30777, DOI:10.1021/jacs.4c12857
- D. J. Blair, S. Chitti, M. Trobe, D. M. Kostyra, H. M. S. Haley, R. L. Hansen, S. G. Ballmer, T. J. Woods, W. Wang, V. Mubayi, M. J. Schmidt, R. W. Pipal, G. F. Morehouse, A. M. E. Palazzolo Ray, D. L. Gray, A. L. Gill and M. D. Burke, Automated Iterative Csp3−C Bond Formation, Nature, 2022, 604(7904), 92–97, DOI:10.1038/s41586-022-04491-w
- Q. Xie, R. Zhang and G. Dong, Programmable Amine Synthesis via Iterative Boron Homologation, Angew. Chem., Int. Ed., 2023, 62(35), e202307118, DOI:10.1002/anie.202307118
- F. Valerio, R. Mykura, J. Fordham, J. Rogers, B. Banecki, A. Noble and V. Agaarwal, Automated Stereocontrolled Assembly-Line Synthesis of Organic Molecules, Nat. Synth., 2002, 1, 902–907, DOI:10.1038/s44160-022-00158-6
- F. Sun, M. Huang, Z. Zhou and X. Fang, 4a,8a-Azaboranaphthalene-4-Yl Phosphine Ligands: Synthesis and Electronic Modulation in Suzuki–Miyaura Coupling Reactions, RSC Adv., 2015, 5(92), 75607–75611, 10.1039/C5RA15929F
- H. L. van de Wouw, J. Y. Lee, E. C. Awuyah and R. S. Klausen, A BN Aromatic Ring Strategy for Tunable Hydroxy Content in Polystyrene, Angew. Chem., Int. Ed., 2018, 57(6), 1673–1677, DOI:10.1002/anie.201711650
- Y. Ji and R. S. Klausen, Chain Transfer to Solvent in BN 2−vinylnaphthalene Radical Polymerization, J. Polym. Sci., 2021, 59(21), 2521–2529, DOI:10.1002/pol.20210362
- Y. Ji, T. Zhou, H. L. Van De Wouw and R. S. Klausen, Organoborane Strategy for Polymers Bearing Lactone, Ester, and Alcohol Functionality, Macromolecules, 2020, 53(1), 249–255, DOI:10.1021/acs.macromol.9b02201
- H. L. Van De Wouw, E. C. Awuyah, J. I. Baris and R. S. Klausen, An Organoborane Vinyl Monomer with Styrene-like Radical Reactivity: Reactivity Ratios and Role of Aromaticity, Macromolecules, 2018, 51(16), 6359–6368, DOI:10.1021/acs.macromol.8b01368
- H. L. Van De Wouw, J. Y. Lee and R. S. Klausen, Gram-Scale Free Radical Polymerization of an Azaborine Vinyl Monomer, Chem. Commun., 2017, 53(53), 7262–7265, 10.1039/C7CC02300F
- J. Huang, Y. Jiang, Z. Zhang, S. Li and D. Cui, Stereoselective Polymerization of an Aromatic Vinyl Monomer to Access Highly Syndiotactic Poly(Vinyl Alcohol), Macromol. Rapid Commun., 2020, 41(10), 2000038, DOI:10.1002/marc.202000038
- S. N. Mendis, T. Zhou and R. S. Klausen, Syndioselective Polymerization of a BN Aromatic Vinyl Monomer, Macromolecules, 2018, 51(17), 6859–6864, DOI:10.1021/acs.macromol.8b01707
- S. J. Melvin, B. A. Mediavilla, E. G. Ambrosius, Q. Jiang, F. Fang, Y. Ji, T. Mukhopadhyaya, H. E. Katz and R. S. Klausen, RAFT Polymerization of an Aromatic Organoborane for Block Copolymer Synthesis, Polym. Chem., 2023, 14(38), 4454–4464, 10.1039/D3PY00706E
- H. Wakefield Iv, Q. Jiang and R. S. Klausen, Azaborine Benzylic Ion Stability and Reactivity in Ionic Polymerization, Org. Biomol. Chem., 2022, 20(7), 1407–1414, 10.1039/D1OB02467A
- M. J. S. Dewar and R. Dietz, 546. New Heteroaromatic Compounds. Part III. 2,1-Borazaro-Naphthalene (1,2-Dihydro-1-Aza-2-Boranaphthalene), J. Chem. Soc. Resumed, 1959, 2728, 10.1039/jr9590002728
- P. Paetzold, C. Stanescu, J. R. Stubenrauch, M. Bienmüller and U. Englert, 1−Azonia–2−boratanaphthalenes, Z. Anorg. Allg. Chem., 2004, 630(15), 2632–2640, DOI:10.1002/zaac.200400333
- M. R. Sánchez Casado, M. Ciordia Jiménez, M. Ariza Bueno, M. Barriol, J. E. Leenaerts, C. Pagliuca, C. Martínez Lamenca, A. I. De Lucas, A. García, A. A. Trabanco and F. J. R. Rombouts, Synthesis of 2,1−Borazaroquinolines and 2,1-Borazaroisoquinolines from Vinyl-aminopyridines and Potassium Organotrifluoroborates by Microwave–Assisted Heating, Eur. J. Org. Chem., 2015,(23), 5221–5229, DOI:10.1002/ejoc.201500622
- P. I. Paetzold, G. Stohr, H. Maisch, H. Lenz and I. V. Borimide, Die, Reaktion von Borimiden Mit Phenylacetylen, Chem. Ber., 1968, 101(8), 2881–2888, DOI:10.1002/cber.19681010837
- S. R. Wisniewski, C. L. Guenther, O. A. Argintaru and G. A. Molander, A Convergent, Modular Approach to Functionalized 2,1-Borazaronaphthalenes from 2-Aminostyrenes and Potassium Organotrifluoroborates, J. Org. Chem., 2014, 79(1), 365–378, DOI:10.1021/jo402616w
- G. H. M. Davies, Z.-Z. Zhou, M. Jouffroy and G. A. Molander, Method for Accessing Nitrogen-Containing, B -Heteroaryl-Substituted 2,1-Borazaronaphthalenes, J. Org. Chem., 2017, 82(1), 549–555, DOI:10.1021/acs.joc.6b02574
- F.-D. Zhuang, J.-M. Han, S. Tang, J.-H. Yang, Q.-R. Chen, J.-Y. Wang and J. Pei, Efficient Modular Synthesis of Substituted Borazaronaphthalene, Organometallics, 2017, 36(14), 2479–2482, DOI:10.1021/acs.organomet.6b00811
- S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Aromatic Metamorphosis of Indoles into 1,2-Benzazaborins, Org. Lett., 2019, 21(10), 3855–3860, DOI:10.1021/acs.orglett.9b01353
- R. Soundararajan, G. Li and H. C. Brown, Homologation of Representative Boronic Esters Using in Situ Generated (Halomethyl)Lithiums: A Comparative Study, Tetrahedron Lett., 1994, 35(48), 8957–8960, DOI:10.1016/0040-4039(94)88399-8
- M. C. Elliott and K. Smith, Migratory Aptitudes of Alkyl Groups on Boron: A Computational Study of Halomethyllithium-Induced Migration Reactions, Organometallics, 2013, 32(17), 4878–4881, DOI:10.1021/om4006164
- M. C. Elliott, K. Smith, D. H. Jones, A. Hussain and B. A. Saleh, Factors Affecting Migration of Tertiary Alkyl Groups in Reactions of Alkylboronic Esters with Bromomethyllithium, J. Org. Chem., 2013, 78(7), 3057–3064, DOI:10.1021/jo4000459
- A. Bottoni, M. Lombardo, A. Neri and C. Trombini, Migratory Aptitudes of Simple Alkyl Groups in the Anionotropic Rearrangement of Quaternary Chloromethyl Borate Species: A Combined Experimental and Theoretical Investigation, J. Org. Chem., 2003, 68(9), 3397–3405, DOI:10.1021/jo026733e
- R. Davenport, M. Silvi, A. Noble, Z. Hosni, N. Fey and V. K. Aggarwal, Visible–Light–Driven Strain–Increase Ring Contraction Allows the Synthesis of Cyclobutyl Boronic Esters, Angew. Chem., Int. Ed., 2020, 59(16), 6525–6528, DOI:10.1002/anie.201915409
- M. E. Fairchild, A. Noble and V. K. Aggarwal, Diastereodivergent Synthesis of Cyclopentyl Boronic Esters Bearing Contiguous Fully Substituted Stereocenters, Angew. Chem., Int. Ed., 2022, 61(35), e202205816, DOI:10.1002/anie.202205816
- C. J. Cope, M. Zanini, M. R. P. George, D. Otero, J. Sendra, A. Noble, J. Lefranc, R. Robiette and V. K. Aggarwal, Intramolecular Lithiation–Borylation for the Stereoselective Synthesis of Cyclopentyl and Cyclobutyl Bis–Boronic Esters, Angew. Chem., Int. Ed., 2025, e202512896, DOI:10.1002/anie.202512896
- K. Yamaguchi, T. Shimizu, A. Miura, K. Ishida and H. Kusama, Photoinduced Intramolecular Cyclization of Acylsilanes Bearing a Boronate Moiety: Construction of a Highly Strained Trans- Fused Bicyclo[3.3.0]Octane Skeleton, Org. Lett., 2022, 24(31), 5807–5811, DOI:10.1021/acs.orglett.2c02337
- R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Enantioselective Construction of Quaternary Stereogenic Centers from Tertiary Boronic Esters: Methodology and Applications, Angew. Chem., Int. Ed., 2011, 50(16), 3760–3763, DOI:10.1002/anie.201008067
- D. P. Hari, J. C. Abell, V. Fasano and V. K. Aggarwal, Ring-Expansion Induced 1,2-Metalate Rearrangements: Highly Diastereoselective Synthesis of Cyclobutyl Boronic Esters, J. Am. Chem. Soc., 2020, 142(12), 5515–5520, DOI:10.1021/jacs.0c00813
- S. J. Dolman, R. R. Schrock and A. H. Hoveyda, Enantioselective Synthesis of Cyclic Secondary Amines through Mo-Catalyzed Asymmetric Ring-Closing Metathesis (ARCM), Org. Lett., 2003, 5(25), 4899–4902, DOI:10.1021/ol036026n
- D. S. Matteson, Asymmetric Synthesis with Boronic Esters, Acc. Chem. Res., 1988, 21(8), 294–300, DOI:10.1021/ar00152a002
- D. S. Matteson, Alpha.-Halo Boronic Esters: Intermediates for Stereodirected Synthesis, Chem. Rev., 1989, 89(7), 1535–1551, DOI:10.1021/cr00097a009
- D. S. Matteson and G. D. Hurst, Asymmetric Synthesis of Tertiary Alcohols from A–halo Boronic Esters, Heteroat. Chem., 1990, 1(1), 65–74, DOI:10.1002/hc.520010110
- A. D. J. Calow, D. Dailler and J. F. Bower, Carbonylative N-Heterocyclization via Nitrogen-Directed C–C
Bond Activation of Nonactivated Cyclopropanes, J. Am. Chem. Soc., 2022, 144(25), 11069–11074, DOI:10.1021/jacs.2c02921
- A.-M. L. Hogan and D. F. O'Shea, Enantioselective Carbolithiation Initiated Cascade Reactions, J. Am. Chem. Soc., 2006, 128(32), 10360–10361, DOI:10.1021/ja060076g
-
R. Silverstein, F. Webster and D. Kiemle, Spectromeric Identification of Organic Compounds, John Wiley & Sons, Inc., 7th edn, 2005 Search PubMed
- G. Tafurt, J. R. Martínez, E. E. Stashenko, S. L. Gomez and A. Palma, Chemical reactivity in the intramolecular Friedel-Crafts alkylation of N-benzyl substituted orthoa-allylanilines, Rev. Colomb. Quím., 2009, 38(3), 409–423 CAS
- A. Palma, N. Galeano and A. Bahsas, A Simple and Practical Approach to the Dibenzo[c,f]Thiazolo[3,2-a]Azepines: A Novel Fused Tetracyclic Azepine System, Synthesis, 2010,(08), 1291–1302, DOI:10.1055/s-0029-1218674
- M. H. Matus, S.-Y. Liu and D. A. Dixon, Dehydrogenation Reactions of Cyclic C2 B2 N2 H12 and C4 BNH12 Isomers, J. Phys. Chem. A, 2010, 114(7), 2644–2654, DOI:10.1021/jp9102838
- H. L. D. Hayes, R. Wei, M. Assante, K. J. Geogheghan, N. Jin, S. Tomasi, G. Noonan, A. G. Leach and G. C. Lloyd-Jones, Protodeboronation of (Hetero)Arylboronic Esters: Direct versus Prehydrolytic Pathways and Self-/Auto-Catalysis, J. Am. Chem. Soc., 2021, 143(36), 14814–14826, DOI:10.1021/jacs.1c06863
- P. A. Cox, A. G. Leach, A. D. Campbell and G. C. Lloyd-Jones, Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: pH–Rate Profiles, Autocatalysis, and Disproportionation, J. Am. Chem. Soc., 2016, 138(29), 9145–9157, DOI:10.1021/jacs.6b03283
- P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, Base-Catalyzed Aryl-B(OH)2 Protodeboronation Revisited: From Concerted Proton Transfer to Liberation of a Transient Aryl Anion, J. Am. Chem. Soc., 2017, 139(37), 13156–13165, DOI:10.1021/jacs.7b07444
- G. Wang, H. T. Mekonnen, A. Shaw, X. Liu, C. Villa, A. Raghuraman, V. Bernales, S. Arora, S. Bhargava, J. M. Notestein and L. J. Broadbelt, Modeling of Protodeborylation of Tris(Pentafluorophenyl)Borane (BCF) under Conditions Relevant to Epoxide Ring-Opening Reactions, Chem. Eng. J., 2025, 506, 159851, DOI:10.1016/j.cej.2025.159851
- Y. Lee and C. Cheon, Synthesis of Benzimidazole–Substituted Arylboronic Acids via Aerobic Oxidation of 1,2−Arylenediamines and Formyl–Substituted Aryl MIDA Boronates Using Potassium Iodide as a Catalyst, Adv. Synth. Catal., 2015, 357(13), 2951–2956, DOI:10.1002/adsc.201500302
- M. Lauer and G. Wulff, Arylboronic Acids with Intramolecular B-N Interaction: Convenient Synthesis through Ortho-Lithiation of Substituted Benzylamines, J. Organomet. Chem., 1983, 256, 1–9, DOI:10.1016/S0022-328X(00)99290-8
- M. Afandiyeva, X. Wu, W. Brennessel, A. Kadam and C. R. Kennedy, Secondary-Sphere Preorganization by an NHC-Pyridonate Ligand Enables Nickel-Catalyzed Hydroboration of Nitriles, Chem. Comm., 2023, 59, 13450–13453, 10.1039/D3CC04229D
- A. G. Karatjas and E. Vedejs, Formation of Pinacol Boronate Esters via Pyridine Iodoborane Hydroboration, J. Org. Chem., 2008, 73(23), 9508–9510, DOI:10.1021/jo8020049
- W. D. English, A. L. McCloskey and H. Steinberg, Transamination of Boron-Nitrogen Compounds, J. Am. Chem. Soc., 1961, 83(9), 2122–2124, DOI:10.1021/ja01470a018
- S. Nave, R. P. Sonawane, T. G. Elford and V. K. Aggarwal, Protodeboronation of Tertiary Boronic Esters: Asymmetric Synthesis of Tertiary Alkyl Stereogenic Centers, J. Am. Chem. Soc., 2010, 132(48), 17096–17098, DOI:10.1021/ja1084207
- V. Fasano and V. K. Aggarwal, Origin of Stereocontrol in the Matteson Reaction: Importance of Attractive Electrostatic Interactions, Tetrahedron, 2021, 78, 131810, DOI:10.1016/j.tet.2020.131810
- S. P. Thomas, R. M. French, V. Jheengut and V. K. Aggarwal, Homologation and Alkylation of Boronic Esters and Boranes by 1,2−metallate Rearrangement of Boron Ate Complexes, Chem. Rec., 2009, 9(1), 24–39, DOI:10.1002/tcr.20168
-
(a)
CCDC 2471064: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nybrq
| This journal is © The Royal Society of Chemistry 2025 |