
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antiadhesive glycoconjugate metal complexes targeting pathogens Pseudomonas aeruginosa and Candida albicans†
Karolina
Wojtczak
 ab,
Emilie
Gillon
c,
Diana
Bura
d,
Karen
Richmond
e,
Megan
Joyce
e,
Emma
Caraher
e,
Keela
Kessie
f,
Trinidad
Velasco-Torrijos
f,
Cristina
Trujillo
gd,
Anne
Imberty
c,
Kevin
Kavanagh
h,
Gordon
Cooke
e and
Joseph P.
Byrne
*abij
ab,
Emilie
Gillon
c,
Diana
Bura
d,
Karen
Richmond
e,
Megan
Joyce
e,
Emma
Caraher
e,
Keela
Kessie
f,
Trinidad
Velasco-Torrijos
f,
Cristina
Trujillo
gd,
Anne
Imberty
c,
Kevin
Kavanagh
h,
Gordon
Cooke
e and
Joseph P.
Byrne
*abij
aSchool of Biological and Chemical Sciences, University of Galway, University Road, Galway, Ireland
bUCD School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: joseph.byrne@ucd.ie
cUniversité Grenoble Alpes, CNRS, CERMAV, Grenoble 38000, France
dSchool of Chemistry, Trinity Biomedical Sciences Institute, Trinity College Dublin, 152-160 Pearse St, Dublin 2, Ireland
eSchool of Chemical and BioPharmaceutical Sciences, TU Dublin, Ireland
fDepartment of Chemistry, Maynooth University, Maynooth, Co. Kildare, Ireland
gDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
hDepartment of Biology, Maynooth University, Maynooth, Co. Kildare, Ireland
iUCD Conway Institute for Biomedical and Biomolecular Research, Belfield, Dublin 4, Ireland
jUCD One Health Centre, University College Dublin, Belfield, Dublin 4, Ireland
First published on 25th June 2025
Abstract
Glycoconjugates are known to interact with carbohydrate-binding proteins involved in adhesion by pathogens, and offer opportunities to design antimicrobial agents. Metal complexes with Eu(III), Ni(II) and Zn(II) were prepared from glycoconjugate ligand 1Gal, which binds to P. aeruginosa's lectin LecA (Kd 9.6 ± 0.7 μM). In vitro anti-adhesive activity of these compounds was evaluated for both P. aeruginosa and C. albicans. Choice of metal ion played a crucial role in modulating anti-adhesive activity, with Eu(III) complexes most effective: [Eu·(1Gal)(H2O)6](CF3SO3)3 inhibits 60% biofilm formation by P. aeruginosa and [Eu·(1Gal)3](CF3SO3)3 inhibits 62% of C. albicans adhesion to buccal epithelial cells (both at 0.1 mM). The results presented demonstrate the potential for metal coordination to significantly enhance biological activity of glycoconjugates, surpassing the effect of the ligand's modest lectin-binding affinity alone.
Introduction
The traditional approach to tackling infectious diseases has been the discovery of molecules that can kill pathogenic bacteria or fungi, by leveraging the chemical weapons evolved for the natural warfare between microbial organisms, and using them (and derivatives) in treatment of infections on living tissues be it human, animal or plant. However, many pathogens developed resistance to clinically-approved antibiotics (AMR), creating critical risks to healthcare,1 and new antimicrobials are not being developed at an adequate rate.2 The WHO has identified Critical Priority pathogens, for which new treatments are urgently required. These pathogens of high concern include bacteria such as Pseudomonas aeruginosa and the yeast Candida albicans.3,4 AMR can be considered a glacial pandemic: slow moving, but vast, destructive and inexorable in its path, since multidrug-resistant strains have emerged in clinical settings, as pathogens continually evolve strategies to survive therapies. Progress in microbiology has made non-fatal strategies that instead target virulence factors, such as anti-adhesion, very attractive for development,5–9 to prevent or reverse infection without killing the pathogen. Targeting virulence factors, rather than pathogens as a whole, is only possible with a deeper understanding of a pathogen's biology and mechanism of infection. Targeting carbohydrate-binding proteins such as adhesins and lectins may disrupt pathogen binding to cell glycans or inhibit biofilm formation. This novel anti-virulence strategy has been reported for uropathogens Escherichia coli,10 for yeast C. albicans (CA)11 and also for P. aeruginosa (PA),6,12–16 and the fungus Aspergillus fumigatus,17 which are implicated in lung infections.5,7The core concept of antiadhesion strategies is that competition by soluble glycomimetics blocks pathogen adhesion to the tissue and therefore prevents the process of infection. Since the goal is to “unstick” a pathogen rather than eliminate it, as well as being non-toxic to the host, anti-adhesives should also be non-toxic to the pathogen and should not create selective pressure which would lead to the evolution of resistance; instead, mechanical expulsion is favoured through natural responses (coughing, urination etc.). Another advantage of this strategy is that it is effective against already resistant strains. Mathematical models predict anti-adhesion combined with antibiotic treatment is effective at removing resistant bacterial infections in synergy with each other;18 this has been shown in vitro with dendrimers targeting bacterial lectin LecB.19 Practical trials exist in hospital settings where combined administration of carbohydrate inhalations and antibiotics to people with Cystic Fibrosis accelerated the clearance of chronic PA infections in the airways when compared to antibiotic-only treatment.20,21 Often, the antiadhesive glycoconjugates can also prevent the formation of bacterial biofilms which are key to pathogenic behaviour and are known to decrease the susceptibility of bacteria to treatments.22
Progress has been made in the last two decades to develop glycoconjugates as high-affinity inhibitors for bacterium PA's two soluble lectins LecA and LecB,8 which are selective for galactosides and fucosides/mannosides, respectively. Nanomolar affinities are achieved by multivalent presentation of carbohydrates on scaffolds as diverse as peptide dendrimers, calixarenes and nanoparticles,13,14,19 as well as simpler carefully-designed lower valency druglike molecules.16,23,24 Affinity is not directly predictive for antiadhesive activity, but a range of successful antiadhesive compounds were recently reviewed by Titz and co-workers,6 while Vidal and co-workers reported glycoconjugate treatment providing protection against PA lung infection in an in vivo mouse model.14
While only 27 treatments are in the clinical pipeline for WHO priority bacterial pathogens,2 in the case of antimycotic agents the situation is also limited:4 three new antifungals are currently in the pipeline, two of them first-in-class.25 Much innovation in the field is centred on novel formulations of Amphotericin-B such as its incorporation into nanoparticles26 or on monoclonal antibody therapies.25–27 Fungi represent a smaller proportion of infections however they continue to accumulate resistance to current treatments; an anti-adhesive strategy presents an attractive opportunity for development as anti-mycotics if suitable targets are identified. Velasco-Torrijos and co-workers reported a library of molecules, including 3 and 4 (Fig. 1), showing that suitably presented divalent galactosides have anti-adhesive effects against CA.11,28,29 Bis-triazole 3 was the first glycoconjugate known to inhibit CA's adhesion to human Buccal Epithelial Cells (BECs) (45% vs. controls). Structure–activity relationships indicate a mechanism that is driven by the identity of the carbohydrate motif, and while the precise molecular target is not yet identified, it is likely a cell wall adhesin.11 A follow-up study varied the central scaffold to lock a “closed” conformation of the sugars, and norbornene-based derivative 4 had comparable activity albeit requiring much higher concentrations.28 A multivalent peptide-based dendrimer presenting four copies of motif 3 was constructed, leveraging the glycoside cluster effect, and resulted in an increase in activity at a much lower concentration than its predecessors.29
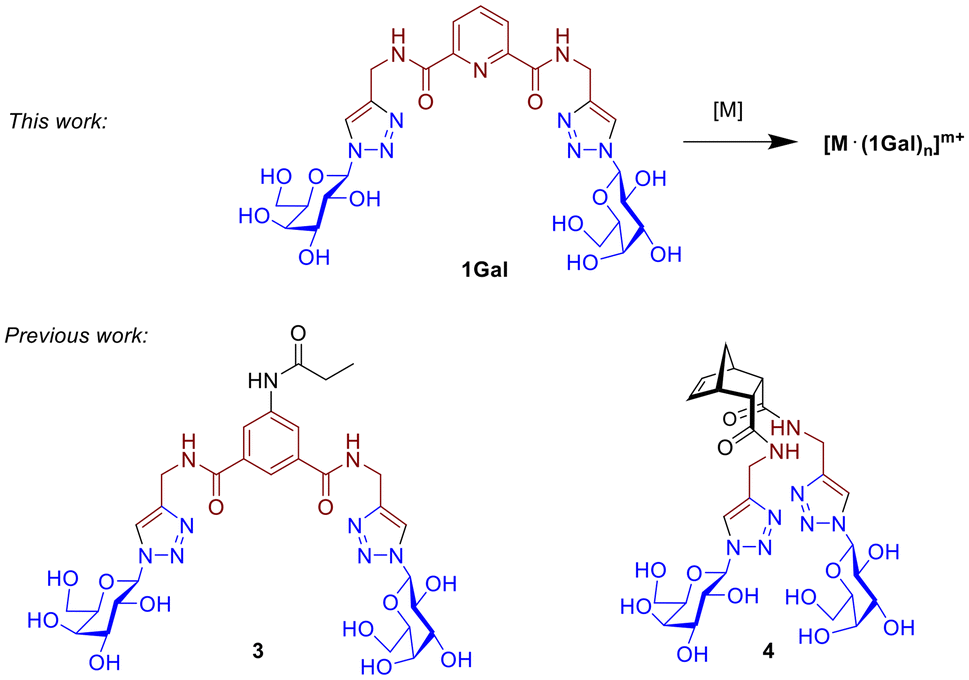 | ||
| Fig. 1 Structure of ligand 1Gal and its metal complexes, compared with other previously-reported digalactoside compounds 3 and 4. | ||
Metallodrugs such as Salvarsan and Pepto-Bismol were among the very first antimicrobial agents used to consciously treat infections in modern medicine.30 After the discovery of penicillin and the advent of small-molecule antibiotics, metallodrugs were relegated to a secondary league, and only viewed as viable candidates for development after the success of Cisplatin in the treatment of cancer since the 1970s.31 Metallodrugs continue to be an underexplored and underdeveloped class of molecules with immense potential; a recent assessment of the proportion of inorganic complexes submitted to the CO-ADD initiative showed 27% of complexes had antimicrobial activity, while only 2% of the purely organic molecules submitted demonstrated any antimicrobial effects.32,33 However, no metallodrugs are currently in clinical trials as antimicrobials, despite approval of silver sulfadiazine and bismuth tribromophenate.30,32,34,35 Schiff base transition metal complexes are reported to kill CA, with a Ni(II) complex particularly effective.36 Ruthenium complexes are best represented in the antibacterial metallodrug research, generally being more active against Gram-positive bacteria (e.g. MRSA)37–39 than Gram-negative species like PA.37,40–42 Machine learning is also being used to predict antimicrobial metal complexes, as a potential method of metallodrug discovery.43
Glycoconjugate metallodrugs have been considered as targeted anti-cancer agents,44 however targeted antiadhesive strategies with metal complexes based on glycoconjugates has not be widely explored. Au(I)-based drug Auranofin has shown activity against S. aureus,45 and glucose-containing Ir(III) complexes have been reported to inhibit PA biofilm at 16 μg mL−1 with a MIC of 4 μg mL−1,46 however it is unclear in either case if the mechanism of action of these involves carbohydrate recognition by lectins/adhesins. Previous work published by the Byrne Group reported the first example of a metal ion used to template glycoconjugate ligands, which inhibited biofilm formation by targeting the galactophilic lectin LecA expressed by PA;47 it was found that templating the glycoconjugate ligands into a multivalent topology was required for activity not seen for ligands alone, while the Ru(II) played only a structural role. The work presented here investigates activity of related glycoconjugate derivatives 1 (Fig. 1), based on the dpa (dipicolinic acid) motif, a class of ligand widely used to coordinate metals with diverse properties and geometries including lanthanide and transition metals.48,49 The combination of glycoconjugate ligands with targeting epitopes curated to interfere with lectins or adhesins, and the presentation topologies metal complexes presents a family of new potential antiadhesive agents.
Results and discussion
Affinity of 1Gal for LecA
The synthesis of digalactoside 1Gal was previously described in a study of luminescent lectin-sensing by Tb(III)-glycoconjugate complexes.50 The complex of 1Gal was not an effective sensor for LecA, but analogues with flexible spacers between the carbohydrate motif and the triazole function as ‘switch-on’ sensors for unlabelled bacterial lectin LecA.50 We have since further investigated affinity of 1Gal by isothermal calorimetry (ITC) and surface plasmon resonance (SPR) to establish that this compound has surprisingly high affinity for PA's galactophilic lectin, considering the short spacing between galactose epitopes, precluding chelate binding.24 In ITC experiments (Fig. 2), binding data for 1Gal indicated this digalactoside binds to LecA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry, only availing of one galactose epitope at a time, which is unsurprising, due to the relatively wide spacing of neighbouring receptor sites in the LecA tetramer (ca. 29 Å).51 An average Kd of 9.6 ± 0.7 μM for LecA was determined. The interaction had a typical enthalpy for a single carbohydrate recognition event with LecA of about −30 kJ mol−1. Moreover, SPR data, determined by immobilising LecA on a chip to investigate kinetic interactions also gave a low micromolar affinity of 6.6 ± 0.5 μM, using a steady-state fitting model and 5.9 ± 0.1 μM by kinetics fitting model. A more flexible related ligand 5 (Fig. S21, ESI†), with a triethyleneglycol linker50 had marginally better affinity by ITC (6.5 ± 0.1 μM) and comparable Kd by SPR (6.0 ± 1 μM) indicating that for this motif, the added length and flexibility conferred by the linker does not affect affinity to the lectin to a major extent, while lanthanide-coordination by either ligand has minimal impact on Kd.52
:1 stoichiometry, only availing of one galactose epitope at a time, which is unsurprising, due to the relatively wide spacing of neighbouring receptor sites in the LecA tetramer (ca. 29 Å).51 An average Kd of 9.6 ± 0.7 μM for LecA was determined. The interaction had a typical enthalpy for a single carbohydrate recognition event with LecA of about −30 kJ mol−1. Moreover, SPR data, determined by immobilising LecA on a chip to investigate kinetic interactions also gave a low micromolar affinity of 6.6 ± 0.5 μM, using a steady-state fitting model and 5.9 ± 0.1 μM by kinetics fitting model. A more flexible related ligand 5 (Fig. S21, ESI†), with a triethyleneglycol linker50 had marginally better affinity by ITC (6.5 ± 0.1 μM) and comparable Kd by SPR (6.0 ± 1 μM) indicating that for this motif, the added length and flexibility conferred by the linker does not affect affinity to the lectin to a major extent, while lanthanide-coordination by either ligand has minimal impact on Kd.52
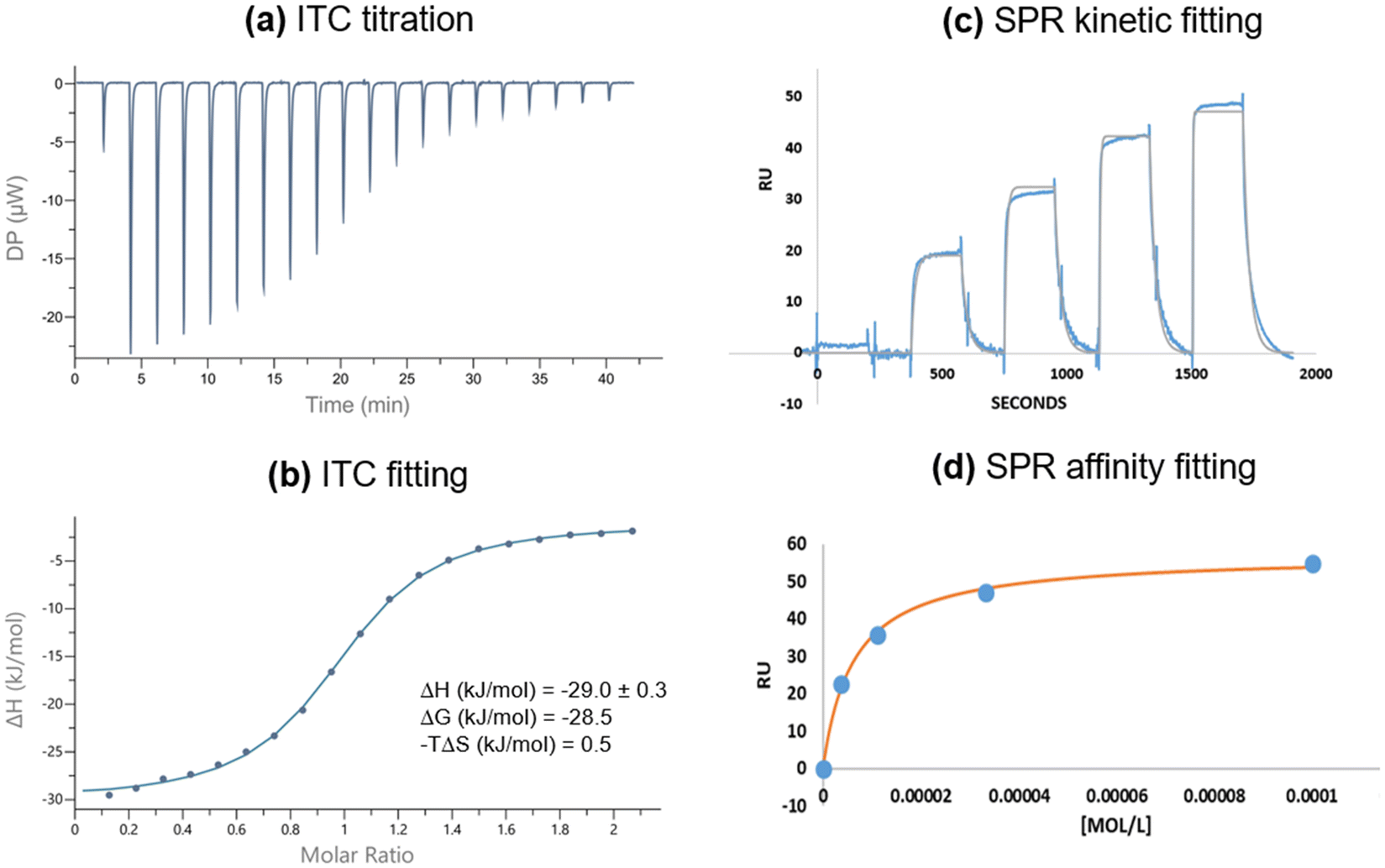 | ||
| Fig. 2 Biophysical analysis of interactions between 1Gal and LecA. (a and b) Isothermal calorimetry titration and fitting in 10 mM Tris/HCl buffer (pH 7.2, 100 mM NaCl, 10 μM CaCl2), [LecA] = 300 μM; (c and d) surface plasmon resonance kinetic and affinity fitting, carried out in PBS buffer (10 mM phosphate buffer pH 7.4, 2.7 mM KCl, 137 mM NaCl, 100 μM CaCl2, 0.05% Tween 20) with [1Gal] = 0–100 μM. | ||
These experiments establish that 1Gal has lectin-targeting motifs that bind to LecA (and likely other galactose-binding proteins), thus opening possibilities to test this ligand as part of inorganic complexes for bacteriostatic or antimicrobial effects. Its low micromolar affinities, achieved through single-site binding, are modest compared to several carefully designed mono- and di-valent examples, of varying degrees of synthetic complexity, but review of the field has established that affinity is not directly predictive of antiadhesive or other biological effects.8,15,23 Moreover, this high-yielding and straightforward preparation allows efficient modular ligand synthesis without sacrificing much affinity, encouraging us to develop derivatives with other carbohydrate moieties by leveraging the efficiency of click reactions, and complex these to a variety of metals as potential anti-biofilm agents targeting PA's lectins.
Synthesis of glycoconjugate ligands
The dipicolinic amide motif (dpa) is well known tridentate coordinating motif for lanthanides and transition metals.48,49 Ligand 1Gal was prepared as described previously,50 while for this study glucose and lactose analogues (1Glc, 1Lac) were also synthesised (Scheme 1). In brief, fully acetylated saccharide azide derivatives were prepared conveniently from acetylated α-bromoacetylglycopyranosides and NaN3. From these azides, protected triazoles 1OAc were prepared through Cu(I)-catalysed azide–alkyne ‘click’ reactions (CuAAC) with alkyne 2. Acetylated triazoles were quite polar and could be easily purified by flash chromatography in ethyl acetate, with good yields (60–70%).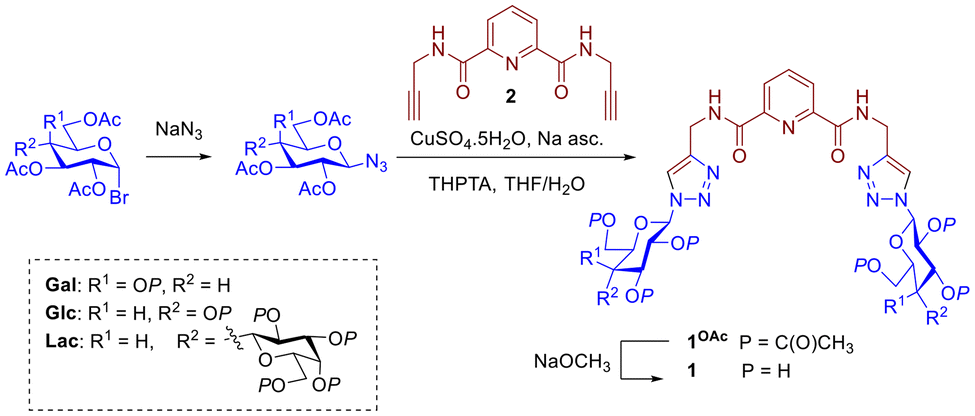 | ||
| Scheme 1 Synthesis of dpa glycoconjugate ligands 1Gal, 1Glc and 1Lac. | ||
The triazoles were then deprotected under Zemplén conditions to obtain pure ligands 1 as white solids in high yields (>95%) at a scale of hundreds of milligrams; facile isolation makes them convenient synthetic targets. Disappearance of a characteristic ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band in IR spectrum of 1Gal compared to 1GalOAc confirmed deprotection (see ESI†), alongside absence of acetyl CH3 resonances in 1H NMR. Each glycoconjugate ligand's 1H NMR spectrum shows a single set of peaks, corresponding to formation of single anomers without epimerisation, evidenced by coupling constants of the anomeric proton resonances at of 9.2 Hz for 1Gal and 1Glc, and 9.2 Hz (lactoside glucose) and 7.8 Hz (lactoside terminal galactose) for 1Lac respectively. The lactoside is linked to the triazole via the glucoside anomeric position thus making it more de-shielded than the galactoside moiety. High resolution mass spectrometry (ESI+, Q-TOF) was used to further characterise the ligands and in all cases the mass found agreed with the calculated m/z of the ion. For instance, for 1Glc, calculated for C25H33N9O12+ [M + H]+m/z = 652.2326; found m/z = 652.2320.
O band in IR spectrum of 1Gal compared to 1GalOAc confirmed deprotection (see ESI†), alongside absence of acetyl CH3 resonances in 1H NMR. Each glycoconjugate ligand's 1H NMR spectrum shows a single set of peaks, corresponding to formation of single anomers without epimerisation, evidenced by coupling constants of the anomeric proton resonances at of 9.2 Hz for 1Gal and 1Glc, and 9.2 Hz (lactoside glucose) and 7.8 Hz (lactoside terminal galactose) for 1Lac respectively. The lactoside is linked to the triazole via the glucoside anomeric position thus making it more de-shielded than the galactoside moiety. High resolution mass spectrometry (ESI+, Q-TOF) was used to further characterise the ligands and in all cases the mass found agreed with the calculated m/z of the ion. For instance, for 1Glc, calculated for C25H33N9O12+ [M + H]+m/z = 652.2326; found m/z = 652.2320.
Preparation of metal complexes of 1
Complexes with transition metals were assembled by reflux in MeOH solution of 1Gal with NiCl2 or Zn(OAc)2, in a 2:1 molar ratio. Complexes formed readily and could be isolated by simple evaporation of the solvent or centrifugation followed by drying, with no evidence of unreacted ligand. All complexes had distinct 1H NMR spectra featuring characteristic broadening of peaks, particularly those corresponding to the dpa motif and anomeric protons. IR spectra showed slight shifts in characteristic peaks corresponding to the amide CO stretch, and the fingerprint region to varying degrees (Fig. S20, ESI†). UV-Vis spectra for both complexes, Fig. 3, show a structured absorbance feature with λmax = 274 nm, with stronger absorbance than the ligand at the same concentration, and a band at λmax = 224 nm, features which are typical for dpa derivatives.
 | ||
| Fig. 3 UV-Vis absorbance spectra of transition metal complexes of 1Gal. | ||
To establish the self-assembly behaviour of 1Gal with lanthanide Eu(III) and its ability to form complexes, a self-assembly titration was performed by adding aliquots of Eu(CF3SO3)3 to a 2 × 10−5 M aqueous solution of 1Gal and monitoring changes in the UV-Vis absorbance and Eu(III)-centred emission spectra (Fig. S24, ESI†). Changes in absorbance spectra were modest, with hyperchromic shift in the spectrum around the maximum at 223 nm and higher energies, and only slight increase in absorbance in the peak with λmax = 274 nm. Upon addition of Eu(III), the non-phosphorescent ligand sensitised metal-centred emission with characteristic line-like bands at 591, 614, 625 and 695 nm which correspond to 5D0 → 7F1–4 transitions, which increased in intensity with lanthanide ion concentration. Global stability constants were estimated by fitting spectroscopic data by non-linear regression (ReactLab Equilibria, Jplus Consulting Pty Ltd) to a model including the formation of Eu: 1Galn (n = 1,3) self-assemblies in solution. When 1:2 assembly was included in the model, the fit did not converge. The ground state data indicated logβ1:1 = 6.4 and logβ1:3 = 16.1 ± 0.2, while fitting data from the emission of the excited state broadly agreed, with logβ1:1 = 5.5 and logβ1:3 = 14.9 ± 0.2. These stability constants with Eu(III) agree closely with those obtained with Tb(III) for related ligand structures by us and others.50,53,54
Based on these self-assembly studies, complexes with Eu(III) were prepared with 1:1 and 1:3 stoichiometries by reflux of Eu(CF3SO3)3 and 1Gal in the appropriate ratios, followed by evaporation to give white solids which luminesce red-orange under UV irradiation (Fig. S26, ESI†), namely [Eu·(1Gal)(H2O)6](CF3SO3)3 and [Eu·(1Gal)3](CF3SO3)3, each demonstrating luminescent signals whose decay curves fit to a single exponential curve.55 Attempts to prepare 1:2 complexes failed, giving instead a mixture of the 1:1 and 1:3 complexes. Analogous complexes with 1Glc and 1Lac were also prepared. Due to the paramagnetic nature of the lanthanide ion, 1H NMR spectra (500 MHz, D2O) of these complexes were broadened and shifted compared to those of the corresponding ligands (Fig. S11 and S12, ESI†). Emission spectra of the isolated complexes demonstrate characteristic Eu(III)-centred emission as seen for the self-assembly studies. Quantum yields (Φ) of these complexes were determined by a relative method, using Cs3[Eu·(dpa)3] as a secondary standard,56 and it was found that the 1:1 complexes were more emissive than trileptic complexes with Φ = 4% for the 1:1 complex of 1Gal, approximately double that for the 1:3 complex (Table 1).
| Complex | Φ (%) |
|---|---|
| [Eu·(1Gal)(H2O)6](CF3SO3)3 | 3.8 |
| [Eu·(1Glc) (H2O)6](CF3SO3)3 | 6.2 |
| [Eu·(1Lac) (H2O)6](CF3SO3)3 | 4.4 |
| [Eu·(1Gal)3](CF3SO3)3 | 1.8 |
| [Eu·(1Glc)3](CF3SO3)3 | 3.2 |
| [Eu·(1Lac)3](CF3SO3)3 | 0.4 |
Biological activity against Pseudomonas aeruginosa
Based on the affinity of ligand 1Gal for LecA, it was anticipated that this series of glycoconjugate complexes with varying topologies and stoichiometries might interact with PA with antimicrobial or anti-virulence effects. Initially, bactericidal behaviour was assessed by testing the ability of galactosides incubated with bacteria at a range of concentrations from 1 μM to 10 mM to inhibit growth of PAO1 strain of the bacterium, as compared with bacteria-only control and a positive control (where 1 μg mL−1 of commercial antibiotic ciprofloxacin was added). No biologically-significant toxicity was seen for 1Gal, or any of its complexes, even at concentrations as high as 10 mM, see Fig. 4a. The zinc and nickel complexes of 1Gal were both found to be completely innocuous to the bacteria, even at high concentrations (Fig. S27, ESI†). Interestingly, while each metal salt precursor (Eu(CF3SO3)3, NiCl2 and Zn(OAc)2) showed toxicity at 10 mM, this was not replicated by their complexes or by salts at lower concentrations for Eu(III) and Zn(II), illustrating that coordination chemistry modulates the biological effects of the metal ion, and complexes are sufficiently stable to dissociation to demonstrate their own biological activity profile. | ||
| Fig. 4 (a) Bactericidal activity to PA for ligand 1Gal, its Eu(III) complexes and the precursor metal salts (at 10 mM and 1 mM) compared to positive and negative control; (b) effect of Eu(III) complexes on biofilm formation (±SD, compared to control experiments with PA alone), with percentage inhibition indicated in text and statistical significance indicated by asterisks (One-way ANOVA). Biofilm formation measured by crystal violet assay (N = 5, 3 replicates per experiment). | ||
Since these complexes have negligible toxicity, potential anti-virulence activity was also probed by in vitro assay. Biofilm inhibition was investigated for all galactoside compounds by crystal violet assay,57 and percentage inhibition of biofilm measured as optical density (OD) at 590 nm, normalised to the mean of a bacteria only control (all concentrations shown Fig. S28, ESI†). Ligand 1Gal did not show any biofilm inhibition, even at higher concentrations, nor did the Ni(II) complex, while alternative ligands with glucosyl or lactosyl epitopes were also ineffective at biofilm inhibition. Non-toxic complex [Zn·(1Gal)2](OAc)2 only showed significant inhibition at 1 mM (45%, p < 0.01) and not at 10 mM, this counterintuitive and non-linear drop in inhibitory effect may be explained by zinc being an endogenous metal for organisms, which upon higher concentration could be abstracted from the complex by the bacteria and used as a micronutrient.58
The biofilm inhibition activity of monoleptic complex [Eu·(1Gal)(H2O)6](CF3SO3)3 was of most note, as it persisted with significance to lower concentrations: it showed a remarkable 60% biofilm inhibition at 0.1 mM, and 48% even as dilute as 0.01 mM (p < 0.0001), Fig. 4b. Control compounds of the same complex structure with 1Glc or 1Lac did not demonstrate decrease in biofilm formation (Fig. S28, ESI†), suggesting that this activity is facilitated by the micromolar interactions of this galactoside with LecA established by ITC above, since LecA is known to play a role in biofilm formation.6,59 While the complexes with 1:1 stoichiometry demonstrate strong effects, the Eu(III) complex with 1:3 stoichiometry also had more modest effects with 39% biofilm inhibition when bacteria were incubated with 0.1 mM of the complex, despite presenting additional galactoside epitopes. This suggests that Eu(III) plays an important role in the mechanism of inhibition but it is unlikely that the biofilm inhibition is caused simply by chelation between carbohydrate-binding active sites by the ligands, as the 1:3 complex does not benefit from the ‘glycocluster effect’. It is worth highlighting that even though the affinity of the ligand for LecA is moderate compared to some other known examples, not all potent LecA ligands disrupt biofilm behaviour, and this complex's biofilm inhibition effect emerged upon complexation to a particular metal ion, and so is not reliant solely on the potency of lectin inhibition.
These results point to an interaction with PAO1 bacteria in vitro for this range of complexes that demonstrate low toxicity, but nonetheless in the case of Eu(III) complexes in particular, metal coordination gives rise to a carbohydrate-targeted biofilm inhibition effect that is noteworthy, particularly in the case of complexes with a 1:1 stoichiometry. The largest effect measured at subinhibitory concentration, at approximately 50% decrease in biofilm formation vs. control compares favourably with Ru(II) and Ag(I) complexes reported previously.41,47,60
Given that antiadhesive activity here relies on the identity of the carbohydrate epitope as well as the metal ion, it is likely that some of these complexes might also be effective for antiadhesive therapeutic effects against other pathogens with relevant receptors for galactosides.
Molecular modelling of ligand 1Gal and its complexes
Carbohydrate-presentation plays an important role in glycoconjugate–protein interactions, so to gain insight into the structure of these compounds, and how complexation might influence galactoside presentation, DFT calculations were carried out to predict the lowest energy conformations in water (SMD method). Noting structural similarity of ligand 1Gal to previously published compounds possessing a bis-galactoside motif, such as 3 (Fig. 1), we hypothesised that 1Gal and its complexes, in addition to the PA biofilm inhibition already described, also have potential for antiadhesive activity against fungus CA.11,28,29 The most stable conformation of 1Gal was found to be a closed ‘basket’ configuration (Fig. 5), which is analogous to that of the previously-reported molecules 3 and 4 with known CA antivirulence activity.28 Calculated energies of the conformers and the inter-anomeric carbon distances in each correspond closely, being 5.90 Å for the most stable conformer (Fig. S30, ESI†). This suggested that the dpa derivative and its metal complexes have potential for anti-adhesive activity against CA. Complexation to metals gives access to topological presentation patterns inaccessible or difficult to access with purely organic molecules.13,19,29 The most stable conformer of the Eu(III) complex shows similar overall configuration and inter-anomeric carbon distances (6.14 Å) to the ligand, whereas in calculated Ni(II) and Zn(II) complex conformers, this distance (within the same ligand) is extended by ca. 1 Å. This variation, along with the change in multivalency may interplay with the varying biological activity observed for these systems.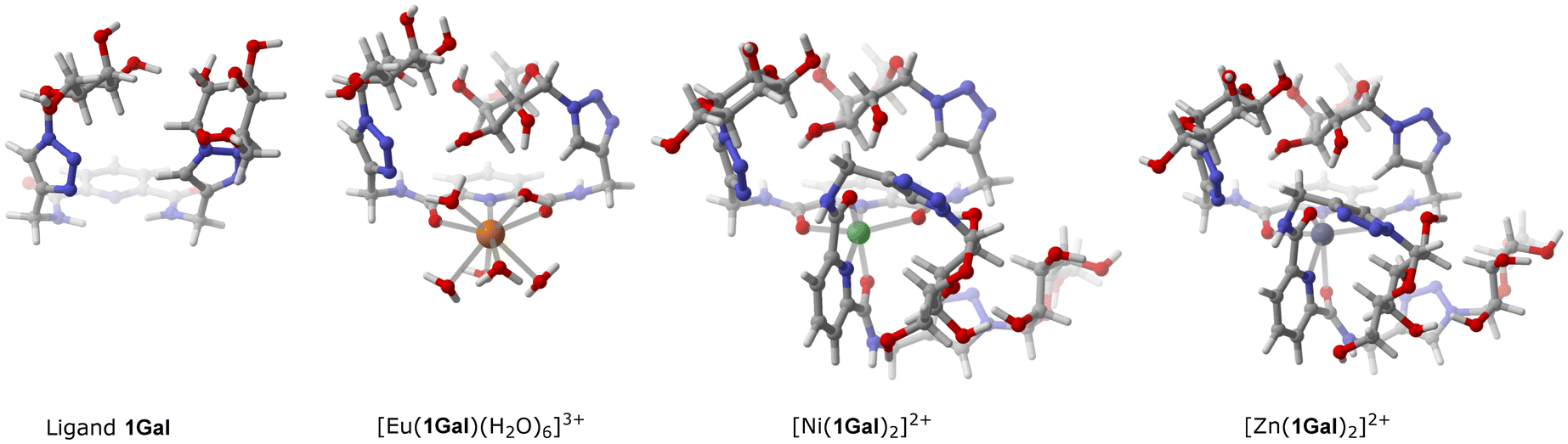 | ||
| Fig. 5 Most stable conformer structures calculated by DFT for 1Gal and its complexes. Details of parameters given in the experimental section; additional illustrations in ESI.† | ||
Adhesins belonging to yeasts of the Candida genus are known and lectin-like interactions are recognised as one of the three types of adhesive interactions along with integrin mediated protein–protein interactions and as of yet undefined interactions.61,62 Lectin-like interactions CA's surface are often attributed to mannoprotein and fucoside recognition, with some examples also citing GlcNAc as epithelial cell receptors,63,64 but to date the exact protein responsible for galactoside recognition in CA is not known, therefore the characterisation of interactions at a molecular level (e.g. ITC) is not yet possible. Nevertheless, several studies, including the data presented below, demonstrate adhesion to human buccal epithelial cells (BECs) can be significantly inhibited by glycoconjugates with galactose epitopes (particularly divalent galactosides), providing potential medicinal value at early stages of infection.
Biological activity against Candida albicans
To probe the interactions of 1Gal with CA, two types of assays were carried out: a pre-treatment assay, and a competition assay. These experiments allowed insight into the effects of digalactosides on protecting human BECs from adhesion and for competing with adherent fungi, respectively. These data are directly comparable to existing examples in the literature.11,28,29The pre-treatment exclusion assay, Fig. 6a and b, 0.1 mM 1Gal, was incubated with CA before exposing BECs to the yeast; this assay showed decrease in CA cells attached to the human cells: 35% less than the control. While in a competitive assay, where BECs were co-incubated with yeast cells and the glycoconjugate, 1Gal achieved a 47% decrease in adhesion versus control (p < 0.0001), Fig. 6c. This ligand compared very well with previously reported activity of 3.11 Compound 3 gave ca. 35% reduction in both assays, while displacement assays with both 1Gal and 3 also showed comparable results (ESI†).
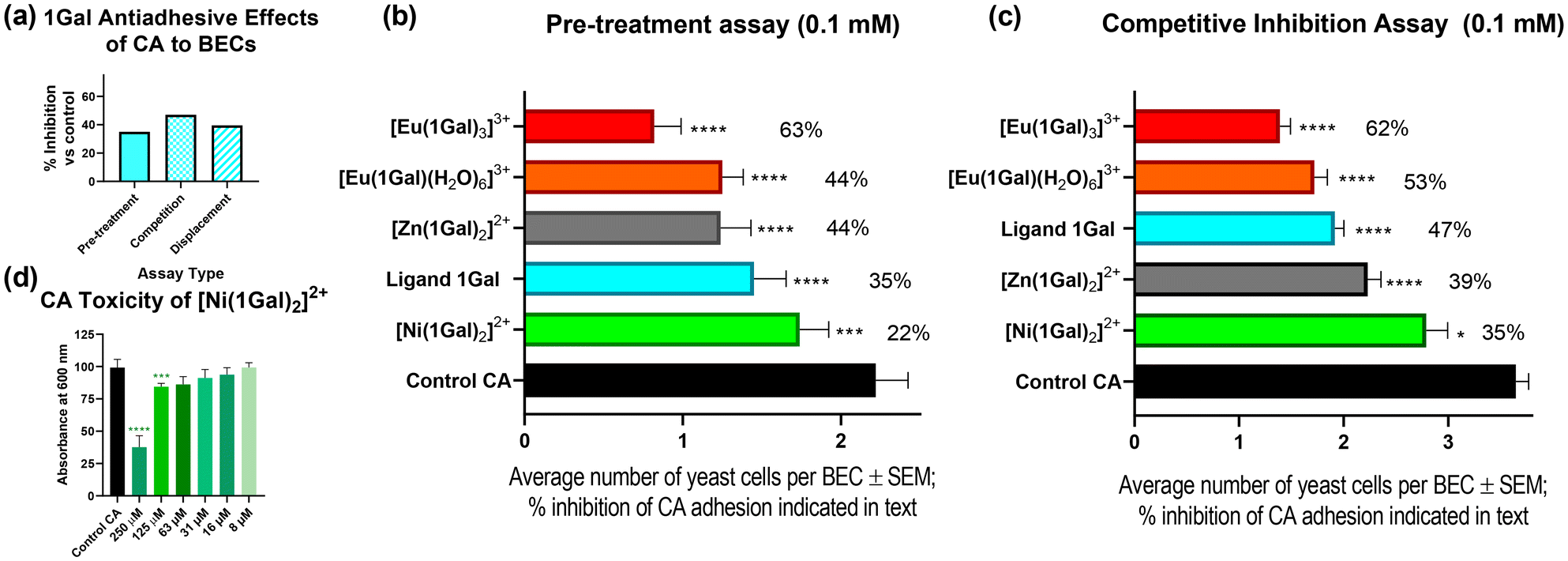 | ||
| Fig. 6 Biological assays of 1Gal and its complexes against C. albicans (CA). (a) Inhibition of adhesion of CA to BECs by 1Gal measured by three different assay types; (b) inhibition of CA in a pre-treatment exclusion assay; (c) inhibition of adhesion of CA in a competition assay, compared to control experiments. (d) Toxicity of Ni(II) complex towards CA at low concentrations. | ||
Having confirmed the effectiveness of the dpa-based ligand scaffold, we examined the impact complexation with a variety of metals had on CA yeast cells and their antiadhesive activity. Since toxicity is a concern in medicinal chemistry when proposing metallodrug candidates,30 particularly as antiadhesives, toxicity assays were first performed to identify any fungicidal potential for this series of complexes (Fig. S33, ESI†). Upon incubating CA culture with various concentrations of 1Gal and its complexes, the nickel(II) complex was the only one to inhibit growth with any statistical significance, doing so even at concentrations as low as 63 μM (Fig. 6d). One-way ANOVA analysis was performed at each concentration to determine statistical significance of results (p < 0.1). Europium(III) complex [Eu·(1Gal)3](CF3SO3)3 exhibited no toxicity, a very encouraging result given its effectiveness against PA above, and likely impact on CA adhesion.
In the pre-treatment exclusion assay, Fig. 6b, where each complex was tested at 0.1 mM, the Ni(II) complex was less effective than the ligand alone (22% inhibition), and it was observed that the yeast cells were particularly small compared to other experiments, a result that may be explained by this complex's toxicity, Fig. 7a and b. Indeed, under the same assay conditions, NiCl2 alone also gave rise to 18% inhibition (ESI†), suggesting that biological activity here is likely metal-driven.
 | ||
| Fig. 7 Optical microscope images (100×) of human buccal epithelial cells (BECs) in the presence of C. albicans (a) without any inhibitor (control sample); (b) in competition assay with the Ni(II) complex, demonstrating smaller fungal cells; (c) in competition assay with 1:3 Eu(III) complex, demonstrating of clumping of C. albicans cells (indicated with arrow) and human BECs with minimal fungal adhesion. | ||
On the other hand, non-toxic complexes [Eu·(1Gal)(H2O)6](CF3SO3)3 and [Zn·(1Gal)2]Cl2 both outperformed the ligand (44%), without any unusual morphology of the fungal cells. The most effective antiadhesive was trileptic complex [Eu·(1Gal)3](CF3SO3)3, which inhibited 63% of adherence of CA cells to the BECs. This result at 0.1 mM was comparable with a tetravalent peptide-based dendrimer derivative of 3 which inhibits 64% of adhesion at 0.212 mM, but with much simplified synthetic procedure in the case of this work, merely requiring the assembly of lower-valency ligands in the presence of a metal ion, and slightly lower concentration.29 Importantly, control experiments with the metal salt precursors showed negligible antiadhesive properties with Eu(CF3SO3)3 alone (ESI†), indicating that increased activity arises from enhancing the capacity of the glycoconjugate to interact with CA.
Competition assays with the same metal complexes, Fig. 6c, demonstrated very similar trends to those seen above. As in the pre-treatment assay, the Eu(III) complexes, when added to BECs at the same time as the yeast compete most effectively to prevent adherence of CA. The 1:3 complex inhibits 62% of adhesion and the 1:1 complex 53%. The Zn(II) complex also had an inhibitory effect of ca. 40%. In the presence of the 1:3 Eu(III) complex, clumping of fungal cells was observed under microscope, Fig. 7c. This had also been observed in the pre-treatment assay. It may point towards a multivalent-glycocluster mechanism of action for this compound relying on presentation of multivalent digalactoside epitopes in three-dimensions, bringing together several pathogen cells, thereby preventing their adhesion to the human cells. This clumping, not observed for the slightly less antiadhesive 1:1 complexes, might present challenges in vivo, and thus must be considered carefully in further exploration of these systems. We are currently developing analogues of these ligands to include more conjugated chromophores which might allow for excitation of probes with blue light for potential fungal imaging applications; this may give further insight into the precise protein target of this class of anti-adhesive.
Conclusions
We have demonstrated that metal coordination chemistry is able to assemble glycoconjugate ligands in such a way as to enhance antiadhesive effects from binding to pathogens’ carbohydrate-binding proteins, in the case of P. aeruginosa and C. albicans (both WHO Critical Priority pathogens). Galactoside 1Gal has moderate micromolar binding affinity for PA's lectin LecA, but while the ligand alone doesn't demonstrate biofilm inhibition for this bacterium in vitro, the Eu(III) complexes do so at subinhibitory concentrations (up to 60% for the 1:1 complex). DFT modelling of these compounds suggested that the ligand and complexes shared structural properties with previously-reported anti-CA agents, and indeed 1Gal and its complexes with Ni(II), Zn(II) and Eu(III) also display antiadhesive effects on the fungus’ interactions with human BECs in vitro, with the most effective agent again being the 1:3 Eu(III) complex. This lead complex inhibits or outcompetes >60% of yeast adhesion, and demonstrates clumping of the yeast cells. These results indicate that while carbohydrate epitopes target pathogens’ carbohydrate-binding proteins, this alone may not directly translate into antiadhesive effects – this is pathogen specific, and antiadhesive activity may be unlocked or modulated through choice of coordinated metal ions. Eu(III) complexes, in particular, show promise from this study and we are seeking to optimise these structures’ photophysical profile and lectin-affinity to allow for future use of these luminescent compounds for imaging in more detailed models of lectin-mediated pathogenesis (e.g. biofilm flow cell confocal microscopy, study of epithelial cell adhesion/toxicity), and to probe whether this activity is shared across the range of lanthanide(III) complexes, or if Eu(III) plays a unique role in the mode of action of these complexes. Moreover, optimised complexes may sensitise these and other pathogens to adjunct therapy approaches with traditional antibiotics. These promising results for glycoconjugate coordination compounds open up opportunities to develop further antiadhesive metal complexes, with variation in topology, ligand design and metal-centred activity, such as redox activity.
Experimental
General experimental details
NMR spectra were obtained on a Varian VNMRS 500 MHz 54 mm AR Spectrometer (UoG) operating under VnmrJ software, JEOL 400 MHz NMR ECX-400 Spectrometer (UoG), and Varian VnmrS 400 MHz spectrometer (UCD). Chemical shifts (δ in ppm, J in Hz) were referenced to residual solvent resonances and are reported downfield from SiMe4. Deuterated solvents CDCl3, D2O, DMSO-d6 were obtained commercially and used without further purification. Spectra were visualised and analysed using MestReNova software. Infrared spectra were obtained on a PerkinElmer Spectrum 400 FTIR/FT-FIR spectrometer equipped with a universal ATR accessory. High resolution mass spectroscopy was performed on an Agilent 6530 Accurate Mass Q-TOF LC/MS coupled to an Agilent 1290 Infinity UPLC system (UoG), Agilent 6546 Quadrupole Time-Of-Flight MS System coupled with an Agilent 1260 Infinity Prime II LC system (UCD) and on a Bruker ‘autoflex maX’ MALDI TOF/TOF system equipped with a HTX Technologies TM-Sprayer (using Super-DHB matrix). TLC experiments were performed using aluminium sheets pre-coated with silica gel 60 (HF254, E. Merck, Darmstadt, Germany). Chromatography was performed with silica gel 60 mesh (Sigma Aldrich, Wicklow, Ireland). Reactions performed under microwave irradiation were carried out in a CEM Discover-SP Microwave Reactor, 2012 model, in the appropriate bespoke vessels (UoG). All starting materials were obtained from commercial sources and used without further purification, unless otherwise stated. Dialkyne 2,65 glycopyranose azides,66 and dpa-based compounds (1GalOAc, 1Gal and 5)50 were prepared as previously reported.Expression and purification of recombinant LecA were performed as previously described67 and the protein was lyophylized. ITC experiments were performed on an MicroCal ITC200 instrument by Malvern Panalytical and all data analysis was performed using MicroCal PEAQ-ITC analysis software (Malvern Instruments). SPR measurements were performed on a Biacore™ X100 instrument using Biacore CM5 gold chips and data analysed with the analysis software (Biacore). All stock solutions were made from lyophilised material prior to use.
Synthetic procedures
:H2O (2:1) and filtered to obtain white crystals. Yield 0.516 g, 0.52 mmol (59%). HRMS (QTOF ESI+) calcd for [M + H]+ C41H49N9O20+m/z = 988.3167, found m/z = 988.3171 and [M + Na]+ C41H49N9O20Na+m/z = 1010.2986, found m/z = 1010.2993; 1H NMR (500 MHz, DMSO-d6): δ = 9.94 (t, J = 6.3 Hz, 2H, NH), 8.29 (s, 2H, triazole), 8.24–8.18 (m, 3H, pyr-CH), 6.30 (d, J = 9.2 Hz, 2H, Glc H-1), 5.66 (t, J = 9.4 Hz, 2H, Glc H-2), 5.51 (t, J = 9.5, 9.5 Hz, 2H, Glc H-3), 5.15 (t, J = 9.8 Hz, 2H, Glc H-4), 4.62 (m, 4H, Glc H-6,6′), 4.32 (m, 2H, Glc H-5), 4.07 (m, 4H, CH2), 2.01 (s, 6H, OCH3), 1.97 (s, 6H, OCH3), 1.94 (s, 6H, OCH3), 1.77 (s, 6H, OCH3); 13C NMR (126 MHz. DMSO-d6): δ = 170.5, 170.0, 169.8, 168.9 (4 × OAc CO), 163.7 (amide CO), 148.9 (qt), 146.1 (qt), 140.0 (Pyr CH), 125.0 (Pyr CH), 122.5 (Tz CH), 84.3 (Glc C-1), 73.7 (Glc C-5), 72.7 (Glc C-3), 70.5 (Glc C-2), 68.0 (Glc C-4), 62.3 (Glc C-6), 34.9 (CH2), 20.9, 20.8, 20.7, 20.4 (4 × OAc CH3). IR (ATR, cm−1): 1741, 1673, 1535, 1432, 1367, 1211, 1136, 1039, 953, 899, 842, 753.
O), 163.7 (amide CO), 148.9 (Tz-C-1), 146.0(Pyr C-2,6), 140.0 (Pyr C-4), 124.9 (Pyr C-3,5), 122.6 (Tz C-2), 100.5 (Gal C-1), 84.1 (Glc C-1), 76.2, 74.8, 72.9, 70.8, 70.2, 69.3, 67.5 (all Lac CH), 62.7 (Lac C-6), 61.3 (Lac C-6′), 34.9 (CH2), 21.0, 20.9, 20.8, 20.8, 20.7, 20.4 (4 × OAc CH3); FT-IR (ATR, cm−1): 1741, 1673, 1535, 1432, 1367, 1211, 1136, 1039, 953, 899, 842, 753.
O), 147.3 (qt), 144.6 (qt), 139.7 (Pyr CH), 124.8 (Pyr CH), 123.2 (Tz CH), 87.4 (Glc C-1), 78.8 (Glc CH), 75.8 (Glc CH), 72.2 (Glc C-2), 68.9 (Glc C-4), 60.4 (Glc CH2-6), 34.2 (CH2); FT-IR (ATR, cm−1): 3303, 2922, 2459, 1655, 1425, 1341, 1237, 1093, 1042, 898, 842, 751, 696.
O), 147.4 (Tz C), 144.7 (Pyr C), 139.8 (Pyr CH), 124.9 (Pyr CH), 123.2 (Tz CH), 102.9 (Gal C-1), 87.2 (Glc C-1), 77.6, 77.3, 75.3, 74.5, 72.5, 71.9, 70.9, 68.5 (all Lac CH), 61.0 (Lac CH2), 59.7 (Lac CH2), 34.2 (CH2); FT-IR (ATR, cm−1): 3317, 2886, 1661, 1538, 1447, 1404, 1239, 1040, 1000, 895, 843, 783, 750, 697, 670.
:3) and heated, in the ratios indicated below. Eu(III) complexes were heated in microwave reactor at 90 °C for 30 minutes, while Ni(II) and Zn(II) complexes were formed by reflux overnight. Solvent was removed in vacuo and solids dried in oven at 60 °C.
:1 molar ratio, according to the general method described above, yielding a white solid (luminescing red under UV irradiation). Yield 0.1160 g (93%). HRMS (MALDI+) calcd for [Eu(1Gal·)(CF3SO3)2 + Na]+ C27H33N9O18EuCF6S2Na+m/z = 1125.0396, found m/z = 1125.0325. 1H NMR (500 MHz, D2O): δ = 8.24 (br s, Tz), 8.15 (br app. s, Pyr), 7.91 (br app s, Pyr), 5.62–5.56 (br d, Gal H-1), 5.35, 4.25 (br s, CH2), 4.13–4.09 (br m, Gal CH), 3.97–3.95 (br m, Gal CH), 3.89–3.88 (br m, Gal CH), 3.78–3.74, 3.67–3.66 (br d, Gal Gal CH2–6,6′); IR (ATR, cm−1): 3317, 1635, 1566, 1462, 1436, 1399, 1223, 1171, 1089, 1025, 892, 811, 762, 697.
:1 molar ratio, according to the general method described above, yielding a hygroscopic white solid (luminescing red under UV irradiation). Yield 0.0827 g (97%). 1H NMR (500 MHz, D2O): δ = 8.25 (br s, Tz), 8.11–7.96 (br m, Pyr), 5.62–5.54 (split d, Gal H-1), 4.60 (br s, CH2), 4.26 (br s), 4.14–4.09 (br m, Gal CH), 3.98–3.95 (br m, Gal CH), 3.91–3.86 (br m, Gal CH), 3.78–3.74 (br m, Gal CH), 3.67–3.65 (br m, Gal CH2–6,6′); 13C NMR (126 MHz, D2O): δ = 144.97, 122.90, 87.99, 78.24, 72.86, 69.69, 68.51, 60.82, 34.43, 16.71. IR (ATR, cm−1): 3317, 2906, 1635, 1596, 1565, 1459, 1432, 1355, 1241, 1225, 1167, 1125, 1088, 1051, 1026, 890, 822, 757, 723, 699.
:1 molar ratio, according to the general method described above, yielding a lime green solid, fine powder. Yield 0.0682 g (95%). HRMS (QTOF ESI+) calcd for [M − 2Cl]2+ NiC50H64N18O24+m/z = 679.1842, found m/z = 679.1915; 1H NMR (500 MHz, D2O): δ = 8.07 (br s, 4H, Tz), 7.97–7.90 (br m, 6H, Pyr), 5.54–5.52 (br d, 4H, Gal H-1), 4.54 (br s, 8H, CH2), 4.08–4.04 (br t, 4H, Gal CH), 3.95–3.94 (br d, 4H, Gal CH), 3.87–3.84 (br t, 4H, Gal CH), 3.75–3.72 (br dd, 4H, Gal CH), 3.65–3.64 (br app. d, 8H, Gal CH2–6,6′), residual EtOH. 13C NMR (126 MHz, D2O): δ = 165.6, 147.6, 144.8, 139.8, 125.0, 122.9, 88.0, 78.2, 72.9, 69.7, 68.5, 60.8, 57.4, 34.3, 16.7. IR (ATR, cm−1): 3262, 2892, 1635, 1542, 1447, 1346, 1237, 1049, 881, 820, 746, 698.
:1 molar ratio, according to the general method described above, yielding a fine white hygroscopic solid (weakly fluorescent under UV irradiation). Yield 0.0569 g (92%). 1H NMR (500 MHz, D2O): δ = 8.07 (s, 4H, Tz), 7.99 (d, J = 7.0 Hz, 4H, Pyr), 7.94 (dd, J = 8.9, 6.5 Hz, 2H, Pyr), 5.53 (d, J = 9.2 Hz, 4H, Gal H-1), 4.55 (d, J = 2.0 Hz, 8H, CH2), 4.07 (t, J = 9.5 Hz, 4H, Gal H-2), 3.95 (dd, J = 3.4, 1.1 Hz, 4H, Gal H-4), 3.86 (td, J = 6.1, 1.1 Hz, 4H, Gal H-5), 3.74 (dd, J = 9.8, 3.3 Hz, 4H, Gal H-3), 3.65 (d, J = 6.0 Hz, 8H, Gal CH2–6,6′), 1.78 (d, J = 1.0 Hz, 3H). 13C NMR (126 MHz, D2O): δ = 165.6, 147.6, 144.8, 139.8, 125.0, 122.9, 88.0, 78.2, 72.9, 69.7, 68.5, 60.8, 34.4, 23.1. IR (ATR, cm−1): 3304, 2925, 1657, 1653, 1539, 1447, 1398, 1339, 1237, 1090, 1051, 1017, 889, 843, 819, 747.
:1 molar ratio, according to the general method described above, yielding a hygroscopic white solid (luminescing red under UV irradiation). Yield 0.0287 g (98%). 1H NMR (500 MHz, D2O): δ = 8.35 (br s, Tz), 8.23 (br s, Pyr), 5.79 (m, Gal H-1), 4.36 (br s, CH2), 4.04–3.60 (br m, Gal CH and CH2). IR (ATR, cm−1): 3316, 2919, 2852, 1635, 1567, 1433, 1383, 1243, 1226, 1168, 1094, 1046, 1025, 897, 802, 761, 726, 696.
:1 molar ratio, according to the general method described above, yielding a hygroscopic white solid (luminescing red under UV irradiation). Yield 0.0260 g (quantitative). 1H NMR (500 MHz, D2O): δ = 8.20 (br, s, Tz), 8.10–8.07 (br, d, pyr), 5.67–5.60 (m, Gal H-1), 4.20 (br, s, CH2), 3.89–3.48 (br, m's, Gal CH and CH2).
:1 molar ratio, according to the general method described above, yielding a white solid (luminescing red under UV irradiation). Yield 0.0439 g (78%). 1H NMR (400 MHz, D2O): δ = 8.36–7.69 (br m, 5H, Tz Pyr), 5.85–5.56 (br split m, 2H, Glc H-1), 4.40 (d, J = 7.5 Hz, 2H, Gal H-1), 4.27 (s, 1H), 4.09–3.37 (m, 29H, Lac CH and CH2), 2.45 (s).
:1 molar ratio, according to the general method described above, yielding a white solid (luminescing orange under UV irradiation). Yield 0.0290 g (69%). 1H NMR (400 MHz, D2O): δ = 8.16–7.78 (br m, 15H, Tz and Pyr), 5.63 (br d, J = 9.1 Hz, 6H, Glc H-1), 4.56 (s, 12H, CH2), 4.37 (d, J = 7.7 Hz, 6H, Gal H-1), 4.07– 3.32 (m, 75H, Lac CH and CH2), 0.57 (s).
Isothermal calorimetry measurements with LecA
ITC measurements were carried out on a MicroCal ITC200 (Malvern Panalytical) and the data was analyzed using the MicroCal PEAQ-ITC analysis software. The experiments were carried out at 25 °C. Ligands, i.e.1Gal and 5, and LecA were dissolved in the same buffer, i.e., 10 mM Tris/HCl buffer (pH 7.2, 100 mM NaCl, 10 μM CaCl2). The lectin concentration in the microcalorimeter cell (0.2 mL) was 300 μM. A total of 20 injections of 2 μL of 3 mM ligand solutions were added at intervals of 120 s while stirring at 750 rpm. Titrations were carried out in duplicate and Kd reported as an average.Surface plasmon resonance measurements with LecA
Surface plasmon resonance experiments were performed on a BIACORE X100 instrument (GE Healthcare) at 25 °C. For LecA immobilization, the system was pre-equilibrated with PBS buffer (10 mM phosphate buffer pH 7.4, 2.7 mM KCl, 137 mM NaCl, 100 μM CaCl2, 0.05% Tween 20), followed by an activation of the CM5 chip surface by 3 injections of 1:1 N-hydroxysuccinimide (NHS)/1-ethyl-3(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) mixture on channel 1 and 2 (contact time of 540 s, flow rate 10 μL min−1) until the binding response was above 800 RU. LecA was dissolved in 10 mM sodium acetate pH 4.5 (100 μg mL−1) and injected over the activated chip surface on channel 2 (contact time of 540 s, flow rate 10 μL min−1) until the response of 1625 RU was reached. Excess free NHS-ester groups were capped with an injection of 1 M ethanolamine (contact time 540 s, flow rate 10 μL min−1). 2.5 μM ligand stocks (i.e.1Gal and 5) were diluted to required concentrations in a running buffer (10 mM phosphate buffer pH 7.4, 2.7 mM KCl, 137 mM NaCl, 100 μM CaCl2, 0.05% Tween 20) then subjected to single-cycle kinetics analyses (contact time of 120 s and dissociation time of 120 s, flow rate 30 μL min−1) consisting of injections of the analytes at 0, 3.7, 11.1, 33.3, and 100 μM over the immobilized-LecA. The chip surface was regenerated by 5× injections of 5 mM galactose followed by 5× injections of the running buffer (contact time of 120 s, flow rate 30 μL min−1). Kd determination was performed using BIACORE X100 evaluation software (version 2.0) by applying the 1:1 binding model to fit the experimental data.
Antimicrobial assays with P. aeruginosa
Ligand 1Gal and complexes were tested for their ability to inhibit growth of P. aeruginosa (PAO1) at various concentrations. In brief: PAO1 was seeded into wells at 106 CFU mL−1 in Mueller Hinton broth. Serial dilutions of each of the compounds was then added to the wells and a set of control wells with no compound was also set up. Each biological replicate experiment was performed in three technical replicates. Plates were incubated for 24 h at 37 °C before absorption readings at 590 nm were taken to determine the extent of inhibition of in vitro growth of the microorganism versus control.Anti-biofilm assays with P. aeruginosa
Biofilm formation assays were based on methodology from previously published work.57 In brief, starting from an overnight liquid culture in Tryptone Soya Broth, a dilution containing approximately 108 CFU mL−1 was made of PAO1 strain (kind donation from Prof. Seamas Donnelly, School of Medicine, Trinity College Dublin). For each biofilm experiment (each biological replicate), 3 wells of a round-bottomed polypropylene 96-well micro plate (Corning Costar, Sigma) were inoculated with 100 μL of this dilution, 3 wells were inoculated with the dilution and treated with the test compounds (in concentrations from 0 to 10 mM) and 3 control wells were filled with sterile medium (bacteria alone). Following 4 hours of adhesion, the supernatant, containing non-adhered cells, was removed from each well and plates rinsed using PBS solution. Following this 100 μL of fresh media was added to the control wells and fresh media with each concentration of ligand 1 or its complexes was added to the appropriate wells, the plate was then incubated for a further 24 h. After 24 h biofilm formation, the supernatants were again removed, and the wells rinsed with PBS again. Once the wells were washed, 100 μL of a 0.5% crystal violet (CV) solution was added to all wells. After 20 min, the excess CV was removed by washing the plates under running tap water. Finally, bound CV was released by adding 150 μL of 33% acetic acid (Sigma). Absorbance was measured at 590 nm.DFT calculations for ligand and complexes
Five different conformer orientations were designed as the starting structures for the conformational analysis of the 1Gal compound. Once the conformers were generated, they were optimised by DFT methodology using the Gaussian16 program.68 Geometry optimisations and vibrational frequency calculation of the 1Gal conformers were carried out at the B3LYP-D3/6-31+g(d) computational level.69–73 The Europium complex geometry optimisations and frequency calculations were performed using B3LYP with the Stuttgart-Dresden (SDD) effective core potential (ECP) on Eu and 6-31G(d,p) for light atoms (C, H, N, O), with GD3BJ dispersion correction.74,75 For the Nickel and Zinc complexes, geometry optimisations were conducted at the B3LYP/6-31+G(d) level with GD3BJ dispersion correction. The Eu(III) complex was modelled as a septet,76 the Ni(II) complex as a doublet,77 and the Zn(II) complex as a singlet.78 All the calculations were performed in water as the solvent (SMD method) and at a temperature of 298.15 K.79 The vibrational frequencies were used to ensure that the structures were fully optimised and represented a minimum on the potential energy surface.Adherence assays with C. albicans
Adherence assays were based on methodology from previously published work and will be reiterated in brief.11 Buccal epithelial cells (BECs): cells were harvested from healthy volunteers by gently scraping the inside of the cheek with a sterile tongue depressor. Cells were washed with PBS and resuspended at a density of 1.5 × 105 mL−1.:1 in a final volume of 2 mL and incubated at 37 °C for 100 minutes. The BEC/yeast cell mixture was harvested by passing through a polycarbonate membrane containing 30 μm pores which trapped the BECs but allowed unattached yeast cells to pass through. This was washed × 2 with 10 mL PBS and cells remaining on the membrane were collected and spread on glass microscope slides which were left to slowly air dry overnight. Cells were heat fixed and stained with 0.1% (w/v) crystal violet solution, and left to air dry for 30 minutes. For each slide, the CA cells attached to 100 BECs were counted and the average number of adhered yeast cells averaged and expressed as a percentage inhibition compared to untreated controls.
Author contributions
The work was conceptualised by KW, JPB, TVT and KKa. TVT and KKa's methodologies were utilised in this article. Funding from Taighde Éireann - Research Ireland (formerly SFI and IRC) was acquired by JPB, and additional funding from French Embassy in Ireland by KW. Primary investigation was carried out by KW (synthesis, characterisation, ITC/SPR analysis, CA assays), EG (SPR), DB (DFT), KR and MJ (PA assays), KKe (synthesis, displacement assay and additional CA assays). Formal analysis of PA assay data by KR, MJ, GC, EC and JPB. Formal analysis of computational data by DB. Formal analysis of CA data by KW, KKa, KKe and JPB. Visualisation of data by KW, GC, DB, JPB. Supervision and resources were provided by JPB, GC, EC, KKa, TVT, AI and CT. Project administration by JPB. The original draft was written by KW and JPB, with reviewing and editing by GC, KKa, TVT, AI and CT.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was primarily funded by Taighde Éireann - Research Ireland under SFI Starting Investigator Research Grant (18/SIRG/5501; JPB, KW) and ITC/SPR experiments facilitated by a French Research Residency award by the French embassy in Ireland to KW to visit Grenoble (AI). We thank Irish Research Council for financial support (IRCLA/2022/3703). Support of GlycoNanoProbes and InnoGly COST Actions (CA18132 and CA18103) is also acknowledged. AI further acknowledges Glyco@Alps (ANR-15-IDEX-0002) and Labex Arcane/CBH-EUR-GS (ANR-17-EURE-0003). We thank Prof Paul Murphy for mentorship and support at University of Galway and Dr Jimmy Muldoon for mass spectroscopy support and insight, as well as technical staff in all institutions.References
- C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. D. Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- WHO, 2021 Antibacterial agents in clinical and preclinical development: an overview and analysis, World Health Organization, Geneva, 2022 Search PubMed.
- E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. A. Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. G. Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M. P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. van der Meer, N. V. Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318–327 CrossRef PubMed.
- WHO, WHO fungal priority pathogens list to guide research, development and public health action, World Health Organization, 2022 Search PubMed.
- S. Sattin and A. Bernardi, Trends Biotechnol., 2016, 34, 483–495 CrossRef CAS PubMed.
- M. B. Calvert, V. R. Jumde and A. Titz, Beilstein J. Org. Chem., 2018, 14, 2607–2617 CrossRef CAS PubMed.
- S. Behren and U. Westerlind, Molecules, 2019, 24, 1004 CrossRef CAS.
- K. Wojtczak and J. P. Byrne, ChemMedChem, 2022, 17, e202200081 CrossRef CAS.
- U. Theuretzbacher and L. J. V. Piddock, Cell Host Microbe, 2019, 26, 61–72 CrossRef CAS.
- S. Kleeb, L. Pang, K. Mayer, D. Eris, A. Sigl, R. C. Preston, P. Zihlmann, T. Sharpe, R. P. Jakob, D. Abgottspon, A. S. Hutter, M. Scharenberg, X. Jiang, G. Navarra, S. Rabbani, M. Smiesko, N. Lüdin, J. Bezençon, O. Schwardt, T. Maier and B. Ernst, J. Med. Chem., 2015, 58, 2221–2239 CrossRef CAS PubMed.
- H. Martin, M. M. Govern, L. Abbey, A. Gilroy, S. Mullins, S. Howell, K. Kavanagh and T. Velasco-Torrijos, Eur. J. Med. Chem., 2018, 160, 82–93 CrossRef CAS PubMed.
- J. L. Reymond, M. Bergmann and T. Darbrea, Chem. Soc. Rev., 2013, 42, 4814–4822 RSC.
- M. Bergmann, G. Michaud, R. Visini, X. Jin, E. Gillon, A. Stocker, A. Imberty, T. Darbre and J. L. Reymond, Org. Biomol. Chem., 2015, 14, 138–148 RSC.
- A. M. Boukerb, A. Rousset, N. Galanos, J.-B. Méar, M. Thépaut, T. Grandjean, E. Gillon, S. Cecioni, C. Abderrahmen, K. Faure, D. Redelberger, E. Kipnis, R. Dessein, S. Havet, B. Darblade, S. E. Matthews, S. de Bentzmann, B. Guéry, B. Cournoyer, A. Imberty and S. Vidal, J. Med. Chem., 2014, 57, 10275–10289 CrossRef CAS PubMed.
- E. Zahorska, S. Kuhaudomlarp, S. Minervini, S. Yousaf, M. Lepsik, T. Kinsinger, A. K. H. Hirsch, A. Imberty and A. Titz, Chem. Commun., 2020, 56, 8822–8825 RSC.
- R. Sommer, D. Hauck, A. Varrot, S. Wagner, A. Audfray, A. Prestel, H. M. Möller, A. Imberty and A. Titz, ChemistryOpen, 2015, 4, 756–767 CrossRef CAS PubMed.
- M. Duca, D. Haksar, J. van Neer, D. M. E. Thies-Weesie, D. Martínez-Alarcón, H. de Cock, A. Varrot and R. J. Pieters, ACS Chem. Biol., 2022, 17, 3515–3526 CrossRef CAS PubMed.
- P. A. Roberts, R. M. Huebinger, E. Keen, A.-M. Krachler and S. Jabbari, PLoS Comput. Biol., 2019, 15, e1007211 CrossRef PubMed.
- G. Michaud, R. Visini, M. Bergmann, G. Salerno, R. Bosco, E. Gillon, B. Richichi, C. Nativi, A. Imberty, A. Stocker, T. Darbre and J. L. Reymond, Chem. Sci., 2015, 7, 166–182 RSC.
- H.-P. Hauber, M. Schulz, A. Pforte, D. Mack, P. Zabel, U. Schumacher, P. D. Med and H.-P. Hauber, Int. J. Med. Sci., 2008, 5, 371–376 CrossRef CAS PubMed.
- I. Bucior, J. Abbott, Y. Song, M. A. Matthay and J. N. Engel, Am. J. Physiol., 2013, 305, L352 CAS.
- D. Sharma, L. Misba and A. U. Khan, Antimicrob. Resist. Infect. Control, 2019, 8, 76 CrossRef PubMed.
- F. Pertici and R. J. Pieters, Chem. Commun., 2012, 48, 4008–4010 RSC.
- F. Pertici, N. J. D. Mol, J. Kemmink and R. J. Pieters, Chem. – Eur. J., 2013, 19, 16923–16927 CrossRef CAS PubMed.
- A. Vitiello, F. Ferrara, M. Boccellino, A. Ponzo, C. Cimmino, E. Comberiati, A. Zovi, S. Clemente and M. Sabbatucci, Biomedicines, 2023, 11, 1063 CrossRef CAS PubMed.
- S. Fairuz, R. S. Nair and N. Billa, Pharmaceutics, 2022, 14, 1823 CrossRef CAS PubMed.
- R. Kovács and S. Mahmoudi, Front. Cell. Infect. Microbiol., 2023, 13, 1184922 CrossRef PubMed.
- H. Martin, T. Somers, M. Dwyer, R. Robson, F. M. Pfeffer, R. Bjornsson, T. Krämer, K. Kavanagh and T. Velasco-Torrijos, RSC Med. Chem., 2020, 11, 1386–1401 RSC.
- H. Martin, D. Goyard, A. Margalit, K. Doherty, O. Renaudet, K. Kavanagh and T. Velasco-Torrijos, Bioconjugate Chem., 2021, 32, 971–982 CrossRef CAS PubMed.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- A. Frei, A. G. Elliott, A. Kan, H. Dinh, S. Bräse, A. E. Bruce, M. R. Bruce, F. Chen, D. Humaidy, N. Jung, A. P. King, P. G. Lye, H. K. Maliszewska, A. M. Mansour, D. Matiadis, M. P. Muñoz, T.-Y. Pai, S. Pokhrel, P. J. Sadler, M. Sagnou, M. Taylor, J. J. Wilson, D. Woods, J. Zuegg, W. Meyer, A. K. Cain, M. A. Cooper and M. A. T. Blaskovich, JACS Au, 2022, 2, 2277–2294 CrossRef CAS PubMed.
- D. J. Barillo, A. R. Barillo, S. Korn, K. Lam and P. S. Attar, Burns, 2017, 43, 1189–1194 CrossRef PubMed.
- A. Evans and K. A. Kavanagh, J. Med. Microbiol., 2021, 70, 001363 CrossRef CAS PubMed.
- M. A. Malik, S. A. Lone, M. Y. Wani, Md. I. A. Talukdar, O. A. Dar, A. Ahmad and A. A. Hashmi, Bioorg. Chem., 2020, 98, 103771 CrossRef CAS PubMed.
- X. Li, K. Heimann, F. Li, J. M. Warner, F. R. Keene and J. G. Collins, Dalton Trans., 2016, 45, 4017–4029 RSC.
- F. Li, J. G. Collins and F. R. Keene, Chem. Soc. Rev., 2015, 44, 2529–2542 RSC.
- S. V. Kumar, S. O. Scottwell, E. Waugh, C. J. McAdam, L. R. Hanton, H. J. L. Brooks and J. D. Crowley, Inorg. Chem., 2016, 55, 9767–9777 CrossRef CAS PubMed.
- S. Li, C. Wu, X. Tang, S. Gao, X. Zhao, H. Yan and X. Wang, Sci. China: Chem., 2013, 56, 595–603 CrossRef CAS.
- P. Rogala, G. Czerwonka, S. Michałkiewicz, M. Hodorowicz, B. Barszcz and A. Jabłońska-Wawrzycka, Chem. Biodivers., 2019, 16, e1900403 CrossRef CAS PubMed.
- G. Czerwonka, D. Gmiter, A. Guzy, P. Rogala, A. Jabłońska-Wawrzycka, A. Borkowski, T. Cłapa, D. Narożna, P. Kowalczyk, M. Syczewski, M. Drabik, M. Dańczuk and W. Kaca, Biofouling, 2019, 35, 59–74 CrossRef CAS PubMed.
- M. Orsi, B. S. Loh, C. Weng, W. H. Ang and A. Frei, Angew. Chem., Int. Ed., 2024, 63, e202317901 CrossRef CAS PubMed.
- D. Iacopini, J. Vančo, S. Di Pietro, V. Bordoni, S. Zacchini, F. Marchetti, Z. Dvořák, T. Malina, L. Biancalana, Z. Trávníček and V. Di Bussolo, Bioorg. Chem., 2022, 126, 105901 CrossRef CAS PubMed.
- S. Thangamani, H. Mohammad, M. F. N. Abushahba, T. J. P. Sobreira, V. E. Hedrick, L. N. Paul and M. N. Seleem, Sci. Rep., 2016, 6, 22571 CrossRef CAS PubMed.
- E. Sauvageot, M. Elie, S. Gaillard, R. Daniellou, P. Fechter, I. J. Schalk, V. Gasser, J.-L. Renaud and G. L. A. Mislin, Metallomics, 2017, 9, 1820–1827 CrossRef CAS PubMed.
- C. O'Reilly, S. Blasco, B. Parekh, H. Collins, G. Cooke, T. Gunnlaugsson and J. P. Byrne, RSC Adv., 2021, 11, 16318–16325 RSC.
- M. J. Celestine, J. L. Bullock, S. Boodram, V. H. Rambaran and A. A. Holder, Rev. Inorg. Chem., 2015, 35, 57–67 CAS.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 RSC.
- K. Wojtczak, E. Zahorska, I. J. Murphy, F. Koppel, G. Cooke, A. Titz and J. P. Byrne, Chem. Commun., 2023, 59, 8384–8387 RSC.
- A. Imberty, M. Wimmerová, E. P. Mitchell and N. Gilboa-Garber, Microbes Infect., 2004, 6, 221–228 CrossRef CAS PubMed.
- For Tb(III) complexes of 1Gal and 5, Kd with LecA was determined by ITC as 13.6 and 11.1, respectively.
- O. Kotova, J. A. Kitchen, C. Lincheneau, R. D. Peacock and T. Gunnlaugsson, Chem. – Eur. J., 2013, 19, 16181–16186 CrossRef CAS PubMed.
- A. T. O'Neil, N. Zhang, J. A. Harrison, S. M. Goldup and J. A. Kitchen, Supramol. Chem., 2021, 33, 160–173 CrossRef.
- Hydration number (q), determined by measuring luminescent lifetime in D2O (3.00 ms) and H2O (0.180). Applying modified Horrock's equation, q=5.5 ± 0.5. Six coordinated water molecules is analogous to our previously-reported Tb(III) complexes, ref. 50. See also: T. Ala-Kleme, et al., Lanthanide Luminescence Photophysical, Analytical and Biological Aspects, Springer Verlag, Heidelberg, 2011 RSC; A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- A. Chauvin, F. Gumy, D. Imbert and J. G. Bünzli, Spectrosc. Lett., 2004, 37, 517–532 CrossRef CAS.
- E. Peeters, H. J. Nelis and T. Coenye, J. Microbiol. Methods, 2008, 72, 157–165 CrossRef CAS PubMed.
- D. A. Capdevila, J. Wang and D. P. Giedroc, J. Biol. Chem., 2016, 291, 20858–20868 CrossRef CAS PubMed.
- C. Chemani, A. Imberty, S. D. Bentzmann, M. Pierre, M. Wimmerová, B. P. Guery and K. Faure, Infect. Immun., 2009, 77, 2065–2075 CrossRef CAS PubMed.
- B. Đ. Glišić, L. Senerovic, P. Comba, H. Wadepohl, A. Veselinovic, D. R. Milivojevic, M. I. Djuran and J. Nikodinovic-Runic, J. Inorg. Biochem., 2016, 155, 115–128 CrossRef PubMed.
- M. K. Hostetter, Clin. Microbiol. Rev., 1994, 7, 29–42 CrossRef CAS PubMed.
- B. Timmermans, A. De Las Peñas, I. Castaño and P. Van Dijck, J. Fungi, 2018, 4, 60 CrossRef PubMed.
- G. Cotter and K. Kavanagh, Br. J. Biomed. Sci., 2000, 57, 241–249 CAS.
- Y. Fukazawa and K. Kagaya, J. Med. Vet. Mycol., 1997, 35, 87–99 CrossRef CAS PubMed.
- T. Ziegler and C. Hermann, Tetrahedron Lett., 2008, 49, 2166–2169 CrossRef CAS.
- F. M. Ibatullin and K. A. Shabalin, Synth. Commun., 2000, 30, 2819–2823 CrossRef CAS.
- S. Kuhaudomlarp, E. Gillon, A. Varrot and A. Imberty, in Lectin Purification and Analysis: Methods and Protocols, ed. J. Hirabayashi, Springer US, New York, NY, 2020, pp. 257–266 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS.
- X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2002, 581, 139–147 CrossRef CAS.
- S. Carlotto, L. Babetto, M. Bortolus, A. Carlotto, M. Rancan, G. Bottaro, L. Armelao, D. Carbonera and M. Casarin, Inorg. Chem., 2021, 60, 15141–15150 CrossRef CAS PubMed.
- J. L. Mancuso, C. A. Gaggioli, L. Gagliardi and C. H. Hendon, J. Phys. Chem. C, 2021, 125, 22036–22043 CrossRef CAS.
- J. G. O. Neto, J. R. Viana, J. B. O. Lopes, A. D. S. G. Lima, M. L. Sousa, M. R. Lage, S. R. Stoyanov, R. Lang and A. O. Santos, J. Mol. Model., 2022, 28, 341 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR, UV/Vis absorbance and emission spectra, additional ITC/SPR, DFT calculation details, and full biological assay data. See DOI: https://doi.org/10.1039/d5ob00970g |
| This journal is © The Royal Society of Chemistry 2025 |