
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioisomeric branched trisaccharides: a combined synthetic, biological, and computational approach to understand details of FimH-mediated bacterial adhesion†
Ingo
vom Sondern
 and
Thisbe K.
Lindhorst
*
and
Thisbe K.
Lindhorst
*
Otto Diels Institute of Organic Chemistry, Christiana Albertina University of Kiel, Otto-Hahn-Platz 3-4, 24118 Kiel, Germany. E-mail: tklind@oc.uni-kiel.de
First published on 2nd July 2025
Abstract
Carbohydrate-mediated interactions are crucial in many biological processes, including microbial adhesion and infections, where glycan recognition is often governed by the three-dimensional presentation of saccharide ligands. In this study, we systematically explored how regioisomeric structural variations of a Glc/Man pair, scaffolded on various monosaccharides, influence the mannose-specific adhesion of E. coli bacteria which is mediated by the bacterial lectin FimH. A targeted synthetic library of di- and trisaccharides featuring α-D-mannopyranoside/β-D-glucopyranoside (Man/Glc) and Man/Man motifs was synthesized, focusing on variations of regio- and stereochemistry. These glycans were functionalized with anomeric azido groups allowing for an eventual conjugation. Screening of all saccharides in bacterial adhesion-inhibition assays using GFP-tagged type 1 fimbriated live E. coli revealed pronounced differences in inhibitory potencies which could be correlated with the specific spatial arrangement of the saccharide units, underscoring the significance of glycan topology in lectin recognition. Complementary molecular modeling provided a structure-based rationale for the observed structure–function relationships. This work highlights the critical role of three-dimensional glycan architecture in heteromultivalent carbohydrate recognition events.
Introduction
Carbohydrate recognition plays a fundamental role in all organisms by enabling key biological processes including cell recognition, cell signalling, protein trafficking, microbial adhesion and infections.1–3 Many carbohydrate recognition events occur at the glycocalyx, which covers all cells with a dense layer of glycoconjugates at the outer site of the plasma membrane. The glycocalyx comprises, among other components, structurally diverse glycoproteins and glycolipids that serve as ligands in carbohydrate recognition.4 Intensive research has unravelled many structural key aspects of carbohydrate recognition;5 however, due to its inherent complexity, many mechanistic details of this biological process are still poorly understood.4 This underscores the need for innovative approaches to further foster our understanding of glycan interactions and eventually allow for their control and manipulation in a diagnostic and therapeutic context, for example to combat bacterial adhesion and biofilm formation.Carbohydrate–protein interactions are intrinsically weak but have a high specificity.6,7 Important aspects of specific glycan recognition involve the stereochemical features of glycoligands and furthermore, their multivalent presentation can enhance specificity of ligand recognition and lead to multivalency effects that increase avidity.8 Besides homomultivalency effects, recent research has shown that heteromultivalent effects also enhance avidity.9 The multitude of functional aspects in carbohydrate recognition can be deciphered by designing bespoke glycomimetics that allow to address specific features of glycoligand presentation.10 This is the focus of the present study.
We have recently investigated the biological effect of the differential three-dimensional presentation of carbohydrate ligands. Several heterobivalent and heterotrivalent glycoclusters were investigated, where an α-D-mannopyranoside and a β-D-glucopyranoside (Man/Glc) glycoligand pair were organized on non-carbohydrate scaffolds derived from enantiomeric serine derivatives11 or from TRIS (tris(hydroxymethyl)aminomethane), respectively.12 Additionally, regioisomeric heterobivalent Man/Glc glycoclusters were achieved based on a maltose scaffold.13 Biological testing was performed in inhibition-adhesion assays with type 1 fimbriated E. coli bacteria.14 Type 1 fimbriae are adhesive organelles expressed on the bacterial surface. Adhesion is mediated by the α-D-mannopyranoside-specific lectin FimH located at the tips of the fimbriae.15 Type 1 fimbriae are important virulence factors in bacterial infection processes such as in urinary tract infections and essential for biofilm formation.15
In this mannose-specific recognition process (specific E. coli adhesion to surfaces), we often observed differences in ligand affinity depending on the relative presentation of glycoligands on a scaffold, especially in the case of the heterobivalent Man/Glc glycoclusters.11,13 This was an intriguing finding as we were able to clearly demonstrate the importance of an appropriate three-dimensional presentation of glycoligands employing customized glycomimetics and corroborate previous evidence in the same direction.17–24
In this study, we aim to further substantiate the effect of differential three-dimensional glycoligand presentation through synthetic oligosaccharides according to the natural example structures. Notably, a complementary organization of a Man/Glc pair, as it was achieved earlier using enantiomeric serine derivatives (Fig. 1A) can be replicated by trisaccharides, where the Man/Glc pair is glycosidically linked to the 4- and 6-position of a carbohydrate scaffold, namely glucose or galactose (Fig. 1B). However, by employing a carbohydrate scaffold, an additional stereocenter is introduced and the conformational rigidity of the scaffold is increased.
 | ||
| Fig. 1 Analogous scaffolding of a Man/Glc glycoligand pair. Comparison of (A) D/L-serine-derived heterobivalent glycoclusters11 with (B) the structural motif of the investigated glycans of this work. A Newman projection illustrates the similarities between the two approaches. Both approaches share the same amount of carbon atoms between the two terminal glycoligands (highlighted in red) but differ in the scaffold moiety (serine versus glucose). (C) Saccharides of interest (SOI) in this work: glucose and galactose scaffolds were employed to gain branched trisaccharides with terminal α-D-mannopyranoside and β- or α-D-glucopyranoside units, respectively (a Man/Glc pair). Three trisaccharides comprising a Man/Man pair and four disaccharides were required as control compounds to evaluate the effect of a Glc residue in combination with a Man ligand in bacterial inhibition-adhesion assays. The anomeric position of all prepared oligosaccharides was functionalized with an azido (N3) or propargyl (Prop) group, respectively, to allow for an eventual conjugation via click chemistry. All glycans are represented according to the symbol nomenclature for glycans (SNFG) Glc = blue; Man = green; Gal = yellow circles.16 | ||
Based on these considerations on the structural similarity between glycomimetics and natural oligosaccharides, we have designed a targeted library of di- and trisaccharides in which the relative arrangement of a Man/Glc pair as well as the regiochemistry and the configuration of the anomeric linkages are systematically varied (Fig. 1C). To facilitate the eventual conjugation of these oligosaccharides via click chemistry, they were functionalized with an azido or alkyne functional group, respectively, at the anomeric center.
All synthesized glycans were investigated as inhibitors of mannose-specific (FimH-mediated) bacterial adhesion to study the effect of the isomeric orientation of the Man/Glc pair on a carbohydrate scaffold. The data obtained in biological testing were further evaluated by molecular modeling studies to allow for a structure-based interpretation of the measured effects.
Results and discussion
Synthesis
The synthesis of the glucose-centered regioisomeric trisaccharides requires the discrimination of the 4- and 6-position of the glucose scaffold. To this end, we combined three reaction steps starting with D-glucose (1) into a sequential one pot reaction to yield the benzylidene-protected glucosyl azide 2.25 This reaction can be carried out on a gram scale delivering 2 in 71% yield after a simple purification procedure (Scheme 1). For the introduction of the anomeric azido function, Shoda's reagent26 (2-chloro-1,3-dimethylimidazolinium chloride, DMC) was the method of choice. In the next reaction step, the benzylidene acetal was regioselectively cleaved to the 6-O-benzyl ether 3 by reductive ring opening with ethyldimethylsilane and copper(II) trifluoromethanesulfonate as a catalyst in 92% yield. The unprotected 4-position can subsequently be glycosylated with either a β-D-glucopyranosyl or an α-D-mannopyranosyl donor. Here, the respective acetyl-protected O-glycosyl-trichloroacetimidates Glc-TCA27 and Man-TCA28 were employed with boron trifluoride diethyl etherate as Lewis acid furnishing the disaccharides 4 and 5 in yields of 81% and 96%, respectively. In the next step, the 6-O-benzyl protecting group had to be removed to allow for the second glycosylation reaction. Here, cleavage of the benzyl ether under classical reductive conditions (H2/Pd) is incompatible with the anomeric azido group and therefore, a visible light-mediated oxidative debenzylation29 was employed and gave the 6-OH-free disaccharides 6 and 7 in yields of 88% and 85%, respectively. Then, the complementary glycosylation using Man-TCA28 and Glc-TCA,27 respectively, under the same conditions as applied in the first glycosylation step produced the regioisomeric αMan/βGlc trisaccharides 8 and 9 in 83% and 72% yield, respectively. Finally, the global deprotection of the O-acetyl esters under Zemplén conditions30 gave the unprotected target trisaccharides 10 and 11. | ||
| Scheme 1 Synthesis of the glucose-centered regioisomeric 4,6-branched αMan/βGlc trisaccharides 10 and 11. pTsOH: p-toluenesulfonic acid, DMC: 2-chloro-1,3-dimethylimidazolinium chloride; Me2EtSiH: ethyl(dimethyl)silane; Glc-TCA: O-(2,3,4,6-O-acetyl-α-D-glucopyranosyl)-trichloroacetimidate;27 Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate;28 DDQ: 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. | ||
Building on our synthetic strategy for the synthesis of the glucose-centered trisaccharides, we applied a similar approach for the preparation of the regioisomeric galactose-centered trisaccharides. Starting from the literature-known benzylidene-protected galactosyl azide 12,31 the benzylidene acetal was regioselectively cleaved by reductive ring opening with triethylsilane and triflic acid to furnish the 6-O-benzyl ether 13 in 65% yield (Scheme 2). Then, glycosylation of the free 4-OH group with either Glc-TCA27 or Man-TCA,28 respectively, under boron trifluoride diethyl etherate or trimethylsilyl trifluoromethanesulfonate catalysis led to the disaccharides 14 and 15 in 39% and 66% respective yield. The glycosylation yields are lower in comparison to the glycosylation of the glucosyl azide derivative 3, which is expected as the reactivity of the axial 4-hydroxy group of galactose is reduced compared to the equatorial 4-OH group in glucose.
 | ||
| Scheme 2 Synthesis of the galactose-centered regioisomeric 4,6-branched αMan/βGlc trisaccharides 20 and 21. Et3SiH: triethylsilane; TMSOTf: trimethylsilyl trifluoromethanesulfonate; Glc-TCA: O-(2,3,4,6-O-acetyl-α-D-glucopyranosyl)-trichloroacetimidate;27 Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate;28 DDQ: 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. | ||
The subsequent debenzylation under visible light-mediated oxidative conditions gave the 6-OH-free disaccharides 16 and 17 in yields of 77% and 87%, respectively. The final glycosylation step towards the galactose-centered αMan/βGlc trisaccharides was performed with the complementary glycosyl trichloroacetimidates and boron trifluoride diethyl etherate catalysis to furnish the regioisomeric trisaccharides 18 and 19 in yields of 89% and 69%, respectively. Finally, the global deprotection of the acetyl protecting groups under Zemplén conditions30 gave the unprotected target molecules 20 and 21.
In order to be able to compare the effect of an additional glucose moiety (Man/Glc pair, possible heteromultivalency effect) to a trisaccharide ligand comprising a Man/Man pair (possible multivalency effect), a glucose- as well as a galactose-centered trisaccharide were targeted carrying two terminal α-D-mannoside residues at the 4- and 6-positions of the sugar scaffold. In both cases, the synthesis started with the 4,6-benzylidene-protected glycosyl azide 2 and 12, respectively. The benzylidene group was cleaved under acidic conditions to yield the 4,6-unprotected glycosyl azides 2232 and 23 (Scheme 3). Subsequently, both hydroxy groups were glycosylated using the mannosyl trichloroacetimidate donor Man-TCA28 and boron trifluoride diethyl etherate as the promoter yielding the isomeric trisaccharides 24 and 25 in 44% and 20% respective yield. The final global deprotection of the acetyl protecting groups under Zemplén conditions30 gave the target Man/Man trisaccharides 26 and 27.
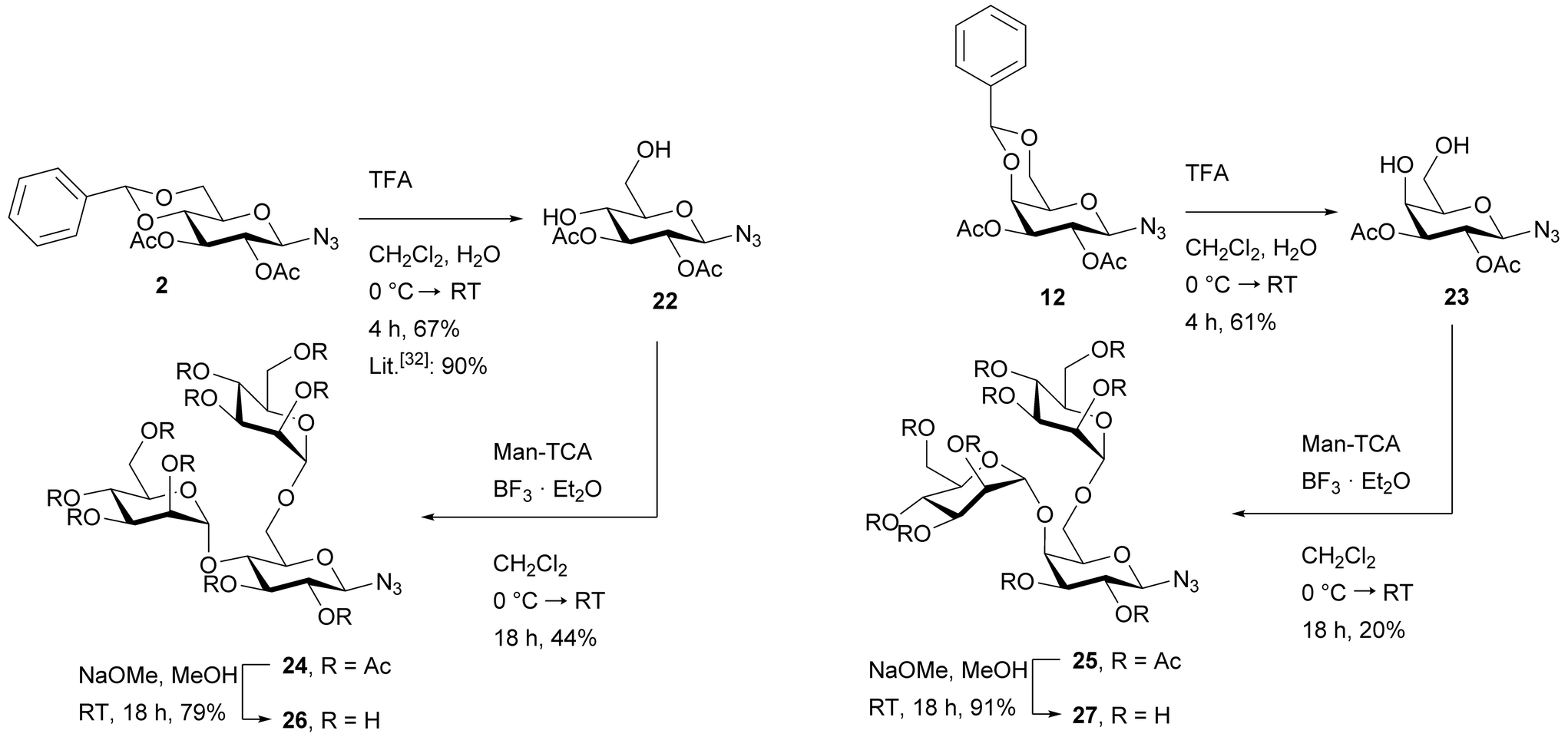 | ||
| Scheme 3 Synthesis of the glucose- and galactose-centered 4,6-branched αMan/αMan trisaccharides 26 and 27. The synthesis of 22 is literature-known and is shown for clarity.32 TFA: trifluoroacetic acid; Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate.28 | ||
In the next part of this work, we targeted an αGlcαMan-decorated trisaccharide as complementary saccharide of interest (SOI) to the βGlcαMan-bearing trisaccharide 10. In order to avoid a complex stereoselective α-glucosylation strategy or a tedious separation of anomers, we have built our synthetic strategy on a chiral pool compound, namely maltose, in which the Glc-α1,4-Glc linkage is already established. In analogy to the literature,13 maltose was first converted into the 1,6-anhydro derivative S9 (see ESI†) using Shoda's reagent (DMC), then globally benzoylated and subsequently the 1,6-anhydro ring was opened by acetolysis to give the maltose derivative 28 (Scheme 4, for experimental details see the ESI, chapters 2.44 and 2.26†). The anomeric azido group was then introduced with trimethylsilyl azide and tin(IV) chloride to give the glycosyl azide 29 in 80% yield. Subsequently, the 6-O-acetyl group was removed orthogonally to the benzoyl groups with acetyl chloride in a mixture of dichloromethane and methanol to give the 6-OH-free disaccharide 30 in a yield of 90%. Now, mannosylation with Man-TCA28 and boron trifluoride diethyl etherate gave the desired trisaccharide 31 in a yield of 81%, which displays the wanted αGlcαMan motif. Finally, the ester protecting groups were globally deprotected under Zemplén conditions30 to give the target trisaccharide 32.
 | ||
| Scheme 4 Synthesis of the glucose-centered 4,6-branched αMan/αGlc trisaccharide 32. TMSN3: trimethylsilyl azide; AcCl: acetyl chloride; Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate.28 | ||
Furthermore, building on the literature-known disaccharide 33,33 the 1,3-glycosidically linked regioisomer of 10 was obtained displaying a βGlcαMan motif (Scheme 5). 33 can be obtained by glucosylation of 2,4-di-O-benzoyl-1,6-anhydro-β-D-glucopyranose and subsequent acetolysis of the 1,6-anhydro ring. The anomeric azido group was introduced using trimethylsilyl azide and tin(IV) chloride to yield 34 in a yield of 71%. As before, the 6-O-acetyl group could be removed in the presence of the benzoyl groups leading to the 6-OH-free disaccharide 35 in 81% yield, which is now ready for a subsequent glycosylation step. Employing Man-TCA28 and boron trifluoride diethyl etherate, the desired trisaccharide 36 was obtained in a yield of 82% and its global deprotection under Zemplén conditions30 led to the target molecule 37.
 | ||
| Scheme 5 Synthesis of the glucose-centered 3,6-branched αMan/βGlc trisaccharide 37. TMSN3: trimethylsilyl azide; AcCl: acetyl chloride; Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate.28 | ||
The synthesis of β-trimannosyl azide 41 was inspired by the literature-known synthesis of its α-anomer.34,35 Accordingly, starting from β-D-mannopyranosyl azide 38,36 a 2,3:4,6-di-orthoester intermediate was formed with triethyl orthobenzoate and (+)-10-camphorsulfonic acid (CSA) and then hydrolysed under acidic conditions with a mixture of trifluoroacetic acid and water to yield 39 (Scheme 6). For the 2,3:4,6-di-orthoester of α-D-mannopyranosyl azide, Yockot et al. found a high regioselectivity for the opening of the 2,3-orthoester towards the axial benzoate, whereas opening of the 4,6-orthoester showed no pronounced regioselectivity.35 In contrast, for the 2,3:4,6-di-orthoester of β-D-mannopyranosyl azide, we observed a low regioselectivity for the opening of the 2,3-orthoester (2,4-benzoate 39 22%, 3,4-benzoate S4 39%) and a higher regioselectivity for opening of the 4,6-orthoester (only 14% of the 3,6-benzoate S5 were formed, see ESI†). All regioisomers formed upon orthoester opening were separable by column chromatography. Subsequently, a double glycosylation of 39 with Man-TCA as glycosyl donor and trimethylsilyl trifluoromethanesulfonate gave the desired trisaccharide 40 in a 63% yield, followed by global removal of the ester protecting groups under Zemplén conditions30 to give the target molecule 41.
 | ||
| Scheme 6 Synthesis of the mannose-centered 3,6-branched αMan/αMan trisaccharide 41. CSA: (+)-10-camphorsulfonic acid; TFA: trifluoroacetic acid; Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate;28 TMSOTf: trimethylsilyl trifluoromethanesulfonate. | ||
As the galactosyl azide 13 was already available (Scheme 2), it was employed for synthesis of the disaccharide 45 (Scheme 7). First, the hydroxy group of galactosyl azide 13 was acetylated to reach 42 in 97% yield and subsequently the benzyl ether was cleaved under visible light-mediated oxidative conditions to give the 6-OH-free galactosyl azide 43 in 84% yield. The following mannosylation with Man-TCA28 and boron trifluoride diethyl etherate gave the desired disaccharide 44 in a 85% yield and global deprotection of the ester protecting groups under Zemplén conditions30 delivered the target molecule 45. The disaccharides Man-α1,4-Glc-βN3 (S6) and Man-α1,4-Gal-βN3 (S7) were obtained by simple global deprotection of the disaccharides 7 and 17, respectively, under Zemplén conditions30 (see ESI, chapters 2.42–43† for details).
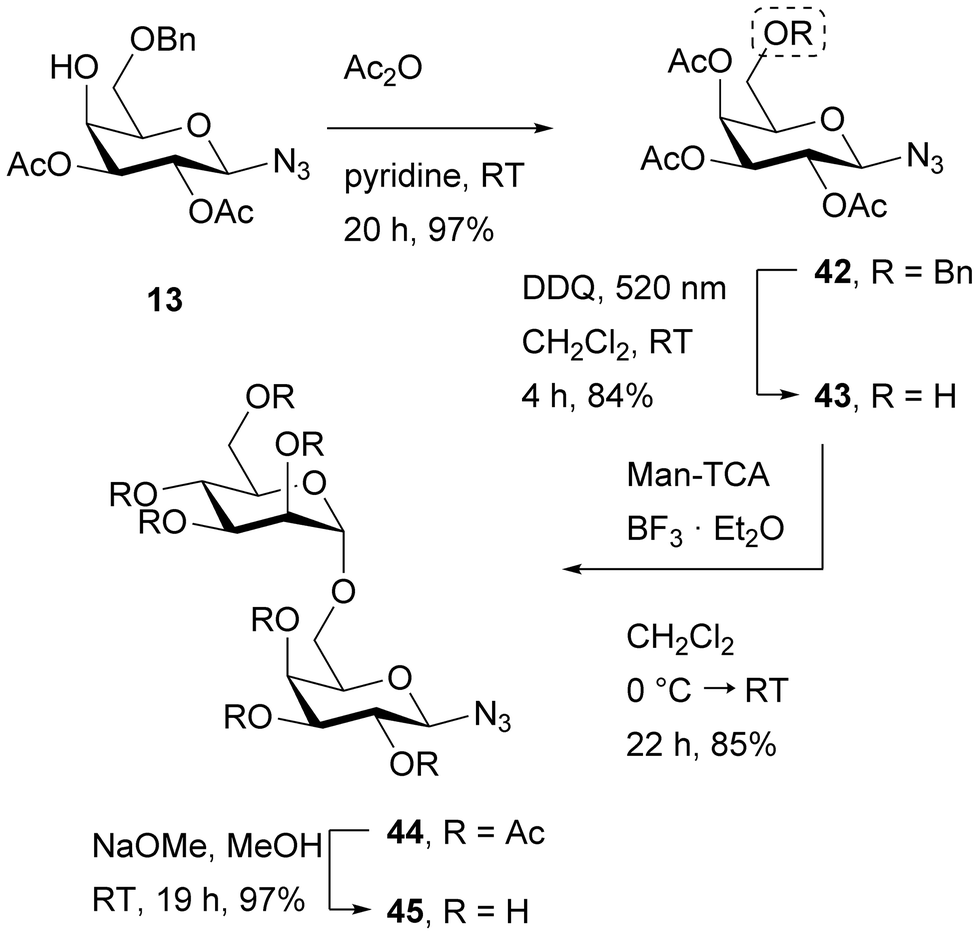 | ||
| Scheme 7 Synthesis of the disaccharide 45. DDQ: 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; Man-TCA: O-(2,3,4,6-O-acetyl-α-D-mannopyranosyl)-trichloroacetimidate.28 | ||
Overall, 12 novel glycosyl azides were synthesized to be tested in an adhesion-inhibition assay with focus on the relative orientation of the two terminal glycoligands Man and Glc. They were supplemented by three further glycans, the literature-known disaccharide Man-α1,6-Glc-βN3 (S1),37 and the known trisaccharides Man-α1,4-[Glc-β1,6]-Glc-βProp (S2)38 and Glc-β1,4-[Man-α1,6]-Glc-βProp (S3).38
Biological testing
The synthetic library of isomeric di- and trisaccharides (Fig. 1) were investigated as inhibitors of FimH-mediated α-D-mannopyranoside-specific bacterial adhesion (Fig. 2) to probe the effect of the different relative orientation of a Man/Glc ligand pair. A literature-known adhesion inhibition assay with live type 1 fimbriated E. coli bacteria, well established in our group, was used where the saccharides of interest (SOI) in solution compete with a mannan surface employed as an adhesive layer.14 Utilizing the GFP (green fluorescent protein)-containing strain PKL1162 allowed to correlate the fluorescence read-out of adhered bacteria left on the mannan surface after inhibition and washing with the inhibitory potency of the SOIs. The SOIs in solution were tested in serial dilutions and the obtained IC50 values were related to that of the standard inhibitor methyl α-D-mannopyranoside (MeMan) tested on the same microtiter plate, leading to relative inhibitory potencies (RIP values). MeMan has therefore by definition a RIP value of 1 and glycans with higher RIP values are more potent as inhibitors of bacterial adhesion to the mannan surface than MeMan. All obtained RIP values are summarized in Fig. 2 (see ESI† for details).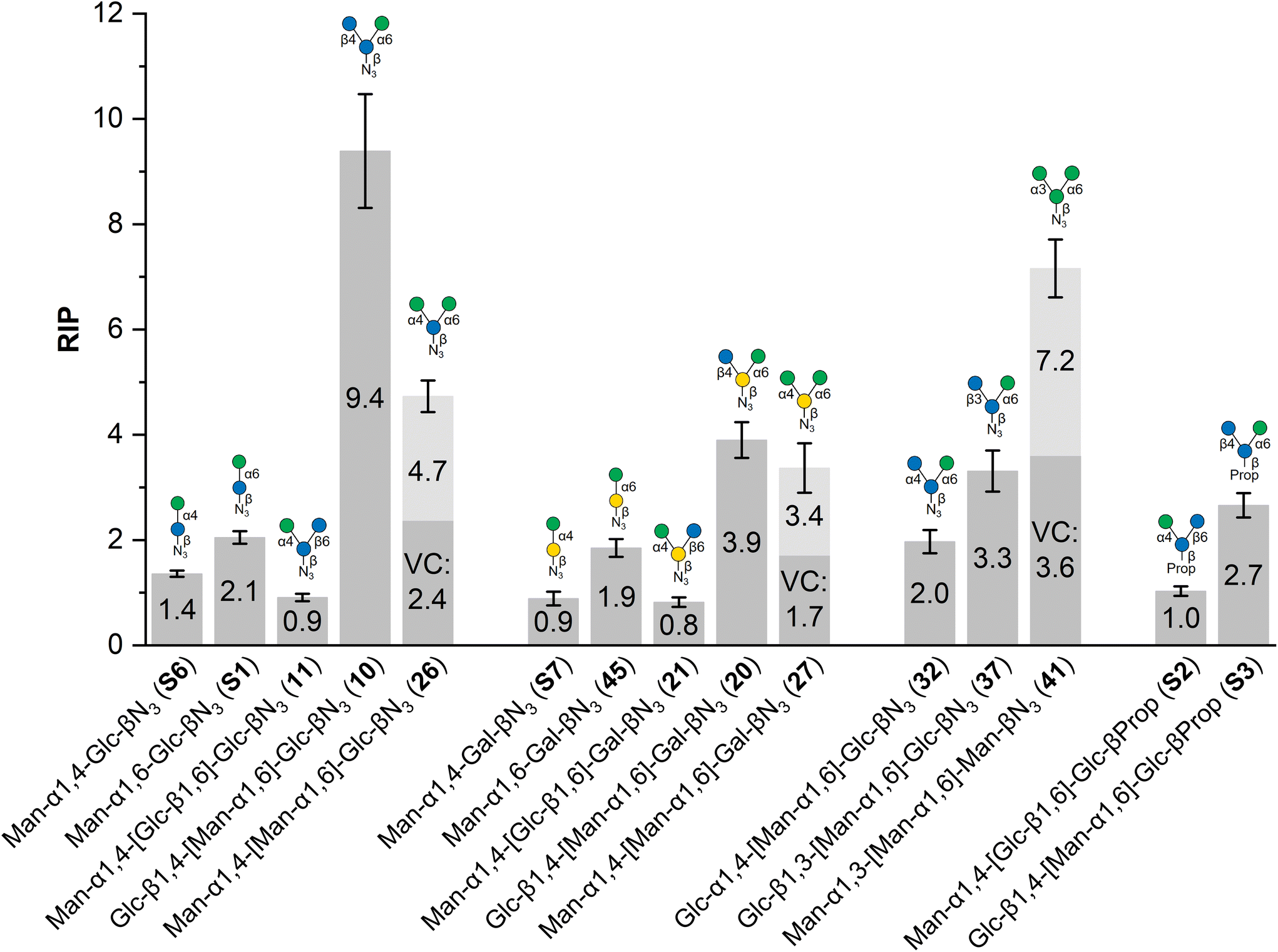 | ||
Fig. 2 Relative inhibitory potency (RIP) of the saccharides of interest (SOI) as inhibitors of bacterial adhesion relative to the standard inhibitor methyl α-D-mannopyranoside (MeMan, RIP![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 1) with indicated error bars deduced from error propagation (see ESI†). The structures of the tested oligosaccharides are abbreviated according to the symbol nomenclature for glycans (SNFG).25 For glycans with more than one terminal mannoside residue, a valency-corrected value (VC) is indicated in dark gray, whereas the measured RIP value is represented by the bar in light gray. All inhibition curves are provided in the ESI.† 1) with indicated error bars deduced from error propagation (see ESI†). The structures of the tested oligosaccharides are abbreviated according to the symbol nomenclature for glycans (SNFG).25 For glycans with more than one terminal mannoside residue, a valency-corrected value (VC) is indicated in dark gray, whereas the measured RIP value is represented by the bar in light gray. All inhibition curves are provided in the ESI.† | ||
The most striking difference in inhibitory potential can indeed be seen between the glucose-centered branched trisaccharides Man-α1,4-[Glc-β1,6]-Glc-βN3 (11) and Glc-β1,4-[Man-α1,6]-Glc-βN3 (10), where 11 shows approximately the inhibitory power of MeMan whereas 10 is about one order of magnitude (10.4-fold) stronger as inhibitor of type 1 fimbriated adhesion. The difference between the two isomers lies in the regiochemistry of an α-D-mannosyl and a β-D-glucosyl residue either attached to the 4- or to the 6-postion of the glucose scaffold. In other words, the trisaccharide in which the α-D-mannosyl residue is bound to the 6-position of the glucose scaffold is the far better ligand of FimH.
This unanticipated finding can not only be explained on the basis of the FimH affinities of the corresponding regioisomeric disaccharides Man-α1,4-Glc-βN3 (S6) and Man-α1,6-Glc-βN3 (S1) where the α1,6-linked disaccharide shows only a slightly stronger (1.5-fold) affinity for the bacterial lectin than the α1,4-linked regioisomer. Apparently, the additional β1,4-linked glucose residue in 10 plays an important role as “co-ligand” for the high affinity of this trisaccharide. This is especially evident since trisaccharide 26, which displays two α-D-Man residues at position 4 and 6 of the glucose scaffold, shows only half of the inhibitory potency of 10.
The same tendency can be recognized when looking at the galactose-centered branched saccharides 21 and 20, where the trisaccharide with the α1,6-linked Man residue (20) is 4.9-fold stronger as inhibitor of FimH-mediated bacterial adhesion than the regioisomeric trisaccharide with the α1,4-linked Man residue (21). Here, the ligand orientation effect is less pronounced than in the regioisomer pair 10 and 11 but also parallels with the corresponding disaccharides S7 and 45, where the α1,6-mannosylated 45 shows a 2.1-fold stronger FimH affinity than S7. In addition, just like in 26, also the trisaccharide 27, bearing two α-D-Man residues at positions 4 and 6, does not surpass the inhibitory power of 20, suggesting that the relative positioning of an α-D-Man ligand to a β-D-Glc ligand on a carbohydrate scaffold is more important for FimH binding than arbitrary divalency.
Consequently, we tested the effect of varied regiochemistry and also of the precise stereochemistry of the glycosidically bound Man/Glc pair on a glucose scaffold. We concentrated on the glucose scaffold, as the galactose-centered saccharides were less potent ligands. When the anomeric configuration of the 1,4-linked glucose residue was changed from β to α (32, Glc-α1,4-[Man-α1,6]-Glc-βN3) the orientation effect seen in 10/11 was almost completely abolished. Almost equally, when the regiochemistry of the β-glucosylation was changed from β1,4- to β1,3-linked (37, Glc-β1,3-[Man-α1,6]-Glc-βN3), the affinity of 10 cannot be reached, though there is a 1.6-fold affinity increase in comparison to the disaccharide S1.
To put these results into perspective, the mannotrioside 41 (Man-α1,3-[Man-α1,6]-Man-βN3), representing the branching unit of the core region in N-glycans, was investigated as well. In comparison to 26 and 27, here, the glucose scaffold is exchanged against a mannose central saccharide and the additional Man residue is α1,3-glycosidically bound instead of α1,4-linked. From all investigated glycans, 41 showed the second highest RIP value. It should be noted that the trisaccharides 26, 27, and 41, each bearing two αMan residues might exert a multivalency effect as both αMan termini can interact with the FimH binding site (vide infra). Therefore, also valency-corrected RIP values are displayed in Fig. 2 showing that the heterovalency effect provoked by an αMan/βGlc pair can surpass the effect of just “more” mannose residues.
Finally, to investigate the effect of the anomeric azido group, this was exchanged against a propargyl aglycon in the especially exciting regioisomeric trisaccharide pair 10/11. The corresponding branched trisaccharides Man-α1,4-[Glc-β1,6]-Glc-βProp (S2) and Glc-β1,4-[Man-α1,6]-Glc-βProp (S3) showed the same trend as it was found in 11/10, however, there is only a 2.7-fold difference between S2 and S3 and hence the regioisomeric effect is far less pronounced than between the glycosyl azides 11 and 10.
Previous studies on the FimH affinity of regioisomeric mannobiosides consistently reported a decreasing affinity in the order of α1-3 > α1-2 > α1-6.39,40 For α1-4-linked mannobiosides either a similar FimH affinity as for α1-6-linked mannobiosides was reported41 or an affinity stronger than that of the α1-2-linked regioisomer.42 In the herein investigated heterogeneous disaccharides Man-Glc (S1, S6) and Man-Gal (S7, 45), an α1-6 glycosidic linkage leads to higher FimH affinity than the α1-4 glycosidic linkage.
From the testing results, the following conclusions on FimH affinity of various saccharides can be drawn. (i) FimH affinity of an αMan-bearing saccharide can be increased by an appropriately oriented βGlc residue (“co-ligand”), as reported earlier.11,13 (ii) Scaffolding of ligand (αMan) and co-ligand (βGlc) is important for FimH affinity of a trisaccharide (compare a glucose (10) vs. a galactose scaffold (20)). (iii) The regiochemistry of the glycosidically bound Man/Glc pair is decisive for FimH affinity, however in interaction with other factors such as scaffolding (compare 10vs.11 and 20vs.21). (iv) The stereochemistry of the glycosidic bond of the co-ligand Glc plays a role (10vs.32); and (v) the anomeric azido group exerts an effect leading to increased FimH affinity (compare 11/10vs.S2/S3).
In order to better understand the measured FimH affinities, molecular modeling was performed, in particular to elucidate how an anomeric azido group in cooperation with secondary carbohydrate interactions of a βGlc co-ligand influences the conformation of the respective saccharide as well as receptor binding.
Molecular modeling
Docking studies were performed with all tested glycans using the Induced Fit Docking (IFD) protocol based on Glide and Prime.43 The interaction between the bacterial lectin FimH and a glycoligand is a dynamic process, where the so-called “tyrosine gate” at the entrance of the FimH carbohydrate binding pocket, formed by Tyr48 and Tyr137, can adopt different conformations. Crystal structures of the “open gate”, “half-open gate” and “closed gate” protein structure are known, where the tyrosine residues Tyr48 and Tyr137 are located at different distances from each other.44–47 To take these potential conformational differences into account in the modelling, the IFD protocol proved reliable in our previous work with the crystal structure in the “closed gate” conformation (pdb: 1UWF)45 as the starting point and was therefore also applied here.13 The obtained binding poses were then further ranked with binding pose metadynamics.48,49 In this additional refinement, the top five docked protein–ligand complexes were subjected to a multitude of 10 ns molecular dynamics simulations to determine the relative persistence of these poses by collective variables. This procedure allows for a more reliable validation of the computed binding pose than ranking by docking scores.49 In the next step, the most stable pose determined by binding pose metadynamics was selected and binding energies were computed with the MM-GBSA (molecular mechanics energies combined with generalized Born and surface area continuum solvation) method.50The evaluation of the computed binding poses shows that the α-D-mannosyl moiety is in all cases buried in the FimH binding pocket with the typical hydrogen bond network between the glycoligand and the FimH carbohydrate recognition domain (CRD). For glycans with two terminal mannoside residues (26, 41 and 27) either of the two Man residues can be complexed in the binding pocket as during IFD both possibilities were found. Further refinement of these protein-glycan complexes with binding pose metadynamics indicated that for 41 the 1,3-connected mannoside and for 26 the 1,4-connected mannoside unit is favorably complexed within the FimH CRD while for 27, complexation of the 1,6-glycosidically bound mannoside unit is favoured. These results confirm that a α-D-mannosyl moiety that is 1,3-glycosidically bound to a mannose scaffold (as in 41) contributes to high FimH affinity.39 When the complexation of 41 (Man-α1,3-[Man-α1,6]-Man-βN3) is compared with the literature-known crystal structure of FimH in complex with “oligomannose-3” (Man-α1,3-[Man-α1,6]-Man-α1,4-GlcNAc-β1,4-GlcNAc, pdb: 2VCO),46 it is noticeable that the Man-α1,3-Man core structure is positioned almost exactly the same in the binding pocket, while the orientation of the Man-α1,6-linked residue slightly differs between the two complexes (ESI Fig. S13†).
IFD produced glycan-protein complexes with open, half-open and closed gate conformations of the tyrosine gate. The open gate conformation was observed for the glycans Man-α1,3-[Man-α1,6]-Man-βN3 (41), Man-α1,4-[Man-α1,6]-Gal-βN3 (27) and Glc-β1,4-[Man-α1,6]-Glc-βProp (S3), where the central scaffold saccharide is “sandwiched” between the Tyr48 and Tyr137 residues of the tyrosine gate. The half-open gate conformation was observed for the glycans Man-α1,4-[Man-α1,6]-Glc-βN3 (26), Glc-α1,4-[Man-α1,6]-Glc-βN3 (32) and Glc-β1,3-[Man-α1,6]-Glc-βN3 (37), while in all other inspected glycan-protein complexes (10, 11, 20, 21, 45, S1, S2, S6, and S7), FimH adopts the closed gate conformation.
In almost every case, the second terminal saccharide, which is not buried in the binding pocket, forms secondary interactions with the polar region around Asn138 and Asp140 of FimH. A prototype of this binding pose is the trisaccharide 10, where the β1,4-connected glucoside unit forms hydrogen bonds with Asp140 (Fig. 3 and ESI, Fig. S11†). In 11, on the other hand, the β1,6-linked glucoside moiety is differently oriented, pointing towards Asp47 (Fig. 3), where is does not exert any favourable interactions. Hence, 10 is a much better FimH ligand as it can also be seen from the calculated binding energies (Table 1) showing −60.21 kcal mol−1 for 10 and −56.63 kcal mol−1 for 11.
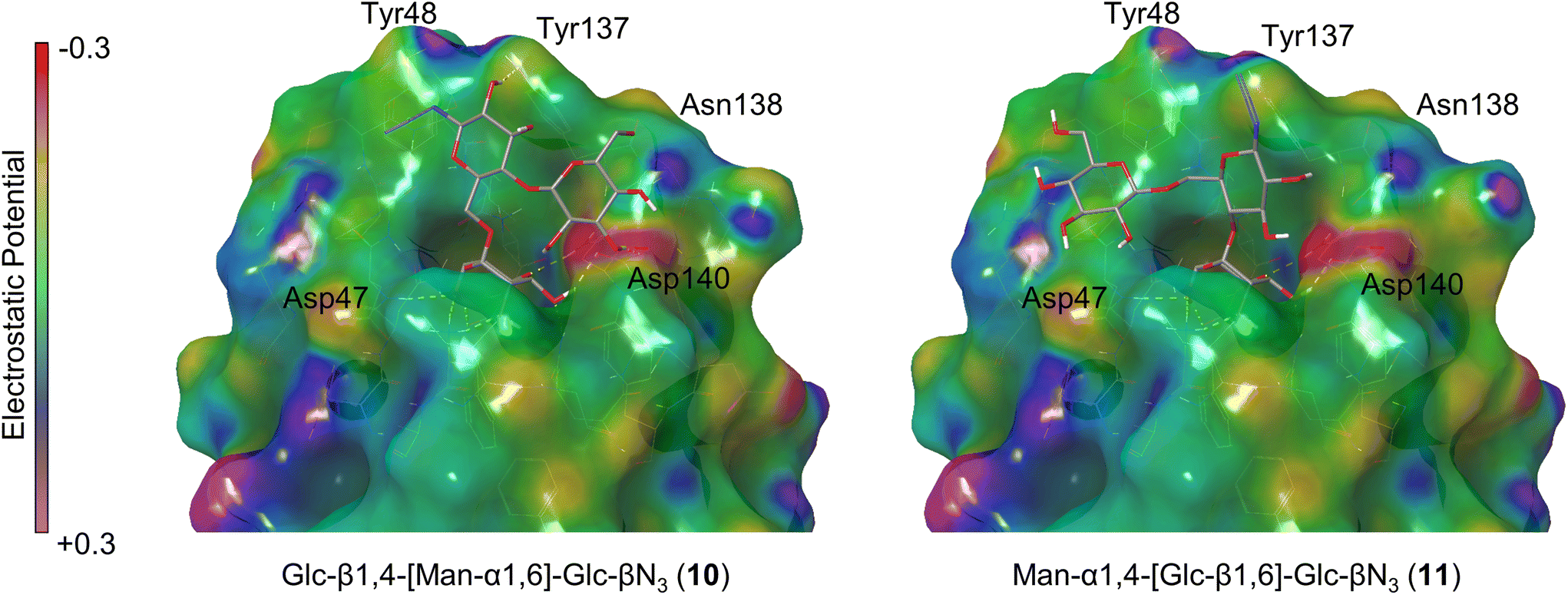 | ||
| Fig. 3 Two selected FimH-glycan complexes derived from induced fit docking (IFD). The protein surface is represented with a Connolly surface coloured according to the electrostatic potential of the surface (purple = positive, green = neutral, red = negative, cf. colour bar). Hydrogen bonds are indicated with yellow dashed lines. For all other computed FimH-glycan complexes see the ESI.† | ||
| Glycan | RIP | IFD score | Glide docking score | MM-GBSA binding energy [kcal mol−1] |
|---|---|---|---|---|
| Man-α1,4-Glc-βN3S6 | 1.4 | −341.27 | −9.938 | −55.69 |
| Man-α1,6-Glc-βN3S1 | 2.1 | −341.09 | −9.740 | −53.86 |
| Man-α1,4-[Glc-β1,6]-Glc-βN311 | 0.9 | −342.25 | −10.753 | −56.63 |
| Glc-β1,4-[Man-α1,6]-Glc-βN310 | 9.4 | −342.21 | −10.410 | −60.21 |
| Glc-α1,4-[Man-α1,6]-Glc-βN332 | 2.0 | −341.46 | −9.924 | −53.30 |
| Glc-β1,3-[Man-α1,6]-Glc-βN337 | 3.3 | −342.90 | −10.863 | −66.97 |
| Man-α1,4-[Man-α1,6]-Glc-βN326 | 4.7, VC 2.4 | −343.39 | −11.958 | −60.19 |
| Man-α1,3-[Man-α1,6]-Man-βN341 | 7.2, VC 3.6 | −342.59 | −11.580 | −45.53 |
| Man-α1,4-Gal-βN3S7 | 0.9 | −341.24 | −10.047 | −55.33 |
| Man-α1,6-Gal-βN345 | 1.9 | −340.52 | −9.207 | −57.47 |
| Man-α1,4-[Glc-β1,6]-Gal-βN321 | 0.8 | −343.13 | −12.078 | −53.87 |
| Glc-β1,4-[Man-α1,6]-Gal-βN320 | 3.9 | −342.35 | −10.314 | −59.09 |
| Man-α1,4-[Man-α1,6]-Gal-βN327 | 3.4, VC 1.7 | −341.83 | −10.811 | −48.41 |
| Man-α1,4-[Glc-β1,6]-Glc-βProp S2 | 1.0 | −343.06 | −11.200 | −48.38 |
| Glc-β1,4-[Man-α1,6]-Glc-βProp S3 | 2.7 | −343.55 | −11.484 | −53.13 |
The binding pose of the propargyl glycoside Man-α1,4-[Glc-β1,6]-Glc-βProp (S2) is very similar to that of 11. In Glc-β1,3-[Man-α1,6]-Glc-βN3 (37), on the other hand, the terminal glucoside unit is “sandwiched” between Tyr48 and Tyr137 and therefore can form secondary interactions with Thr51 (ESI, Fig. S11†). In all other investigated ligands, the Glc unit exerts more or less favourable secondary interactions in the FimH region around Asn138 and Asp140. Where secondary interactions with the polar region around Asn138 and Asp140 are observed, the anomeric azido group of the central scaffold saccharide is often positioned in close proximity to the tyrosine gate, where modelling suggests that favourable π-cation interactions with Tyr48 (for glycans 10, 20, 32, 41) or Tyr137 (for 21) are formed.
The computed binding energies correlate in most cases with the observed trends of measured RIP values for those trisaccharides containing one mannoside. For trisaccharides with two terminal mannoside residues, the computed binding energies are often lower than expected, most likely due to the potential complexity of multivalent binding events, which are not included in the docking studies. Overall, the molecular modeling provides a good basis for the structural classification of the biological test results.
Conclusion
We have synthesized and characterized a library of novel glycans as ligands of the bacterial lectin FimH with the aim to decipher the boundary conditions of the relative orientation of an αMan/βGlc ligand pair for FimH affinity. The stereochemistry and regiochemistry of the interglycosidic bonds were systematically altered using efficient synthesis routes. All glycans were comprehensively tested as inhibitors of FimH-mediated bacterial adhesion revealing that subtle structural changes within a trisaccharide can fine-tune FimH binding such that a low-affinity trisaccharide is turned into a high-affinity ligand. This effect can be allocated to the relative orientation of an αMan ligand versus a βGlc co-ligand and is particularly evident in the regioisomeric trisaccharide pair Man-α1,4-[Glc-β1,6]-Glc-βN3 (11) and Glc-β1,4-[Man-α1,6]-Glc-βN3 (10). Notably, the herein investigated branched azido trisaccharides with heterogeneous terminal residues (Glc/Man) exhibit markedly improved inhibitory potencies compared to their homogeneous counterparts (bearing a Man/Man pair), emphasizing the critical influence of co-ligands on binding affinity, an effect which can be explained by secondary interactions with the lectin and which highlights the relevance of the heterogeneous glycan architecture found on the cell surface.The bioassay suggested that the incorporation of an anomeric azido group can substantially enhance FimH affinity of a respective saccharide and this was confirmed and rationalised by molecular modeling studies. Indeed, the applied induced fit docking approach, combined with binding pose metadynamics and MM-GBSA calculations indicated an anomeric azido group in the tested saccharides exerts favourable interactions with the tyrosine gate at the entrance of the FimH carbohydrate binding site, hence increasing ligand affinity.
In summary, our results have not only provided valuable insights into the dynamic interactions between FimH and isomeric saccharide ligands but also advance our fundamental understanding of carbohydrate–protein recognition. Furthermore, the potential of tailored glycomimetics as inhibitors of bacterial adhesion and biofilm formation is also underscored. Future studies will aim to conjugate the herein investigated saccharide ligands to different scaffolds in order to probe further aspects of carbohydrate recognition on cell surfaces and various biological context.
Author contributions
Conceptualization: I. v. S. and T. K. L.; investigation: I. v. S.; methodology: I. v. S. and T. K. L.; supervision: T. K. L.; writing: I. v. S. and T. K. L.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We thank Christiana Albertina University for its fundamental support. We are grateful to Fabian Seidler, B.Sc. and Julius Bernhofen, M.Sc. for technical assistance.References
- A. Varki, Biological roles of glycans, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- N. Sharon and H. Lis, Lectins as Cell Recognition Molecules, Science, 1989, 246, 227–234 CrossRef CAS.
- R. D. Cummings and J. M. Pierce, The Challenge and Promise of Glycomics, Chem. Biol., 2014, 21, 1–15 CrossRef CAS PubMed.
- L. Möckl, The Emerging Role of the Mammalian Glycocalyx in Functional Membrane Organization and Immune System Regulation, Front. Cell Dev. Biol., 2020, 8, 253 Search PubMed.
- T. K. Dam, O. Hohman, L. Sheppard, C. F. Brewer and P. Bandyopadhyay, in Adv. Carbohydr. Chem. Biochem, ed. D. C. Baker, Academic Press, 2023, pp. 1–21 Search PubMed.
- V. Kumar and W. B. Turnbull, Targeted delivery of oligonucleotides using multivalent protein–carbohydrate interactions, Chem. Soc. Rev., 2023, 52, 1273–1287 RSC.
- M. Mammen, S.-K. Choi and G. M. Whitesides, Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- M. Hartweg, Y. Jiang, G. Yilmaz, C. M. Jarvis, H. V.-T. Nguyen, G. A. Primo, A. Monaco, V. P. Beyer, K. K. Chen, S. Mohapatra, S. Axelrod, R. Gómez-Bombarelli, L. L. Kiessling, C. R. Becer and J. A. Johnson, Synthetic Glycomacromolecules of Defined Valency, Absolute Configuration, and Topology Distinguish between Human Lectins, JACS Au, 2021, 1, 1621–1630 CrossRef CAS PubMed.
- M. González-Cuesta, C. O. Mellet and J. M. G. Fernández, Carbohydrate supramolecular chemistry: beyond the multivalent effect, Chem. Commun., 2020, 56, 5207–5222 RSC.
- A. Tamburrini, C. Colombo and A. Bernardi, Design and synthesis of glycomimetics: Recent advances, Med. Res. Rev., 2020, 40, 495–531 CrossRef CAS PubMed.
- J. Brekalo, G. Despras and T. K. Lindhorst, Pseudoenantiomeric glycoclusters: synthesis and testing of heterobivalency in carbohydrate–protein interactions, Org. Biomol. Chem., 2019, 17, 5929–5942 RSC.
- G. Despras, C. Spormann, F. Klockmann, D. Schollmeyer, A. Terfort and T. K. Lindhorst, Trivalent Heteroglycoclusters as Focal Point Pseudoenantiomers: Synthesis and Preliminary Biological Evaluation, Eur. J. Org. Chem., 2024, e202400809 CrossRef CAS.
- S. O. Jaeschke, I. vom Sondern and T. K. Lindhorst, Synthesis of regioisomeric maltose-based Man/Glc glycoclusters to control glycoligand presentation in 3D space, Org. Biomol. Chem., 2021, 19, 7013–7023 RSC.
- M. Hartmann, A. K. Horst, P. Klemm and T. K. Lindhorst, A kit for the investigation of live Escherichia coli cell adhesion to glycosylated surfaces, Chem. Commun., 2010, 46, 330–332 RSC.
- A. L. Flores-Mireles, J. N. Walker, M. Caparon and S. J. Hultgren, Urinary tract infections: epidemiology, mechanisms of infection and treatment options, Nat. Rev. Microbiol., 2015, 13, 269–284 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings, M. Aebi, N. H. Packer, P. H. Seeberger, J. D. Esko, P. Stanley, G. Hart, A. Darvill, T. Kinoshita, J. J. Prestegard, R. L. Schnaar, H. H. Freeze, J. D. Marth, C. R. Bertozzi, M. E. Etzler, M. Frank, J. F. Vliegenthart, T. Lütteke, S. Perez, E. Bolton, P. Rudd, J. Paulson, M. Kanehisa, P. Toukach, K. F. Aoki-Kinoshita, A. Dell, H. Narimatsu, W. York, N. Taniguchi and S. Kornfeld, Symbol Nomenclature for Graphical Representations of Glycans, Glycobiology, 2015, 25, 1323–1324 CrossRef CAS PubMed.
- Z. Wang, Z. S. Chinoy, S. G. Ambre, W. Peng, R. McBride, R. P. de Vries, J. Glushka, J. C. Paulson and G.-J. Boons, A General Strategy for the Chemoenzymatic Synthesis of Asymmetrically Branched N-Glycans, Science, 2013, 341, 379–383 CrossRef CAS PubMed.
- S. Hakomori, Carbohydrate-to-carbohydrate interaction in basic cell biology: a brief overview, Arch. Biochem. Biophys., 2004, 426, 173–181 CrossRef CAS PubMed.
- A. R. Todeschini and S. Hakomori, Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 421–433 CrossRef PubMed.
- N. Strömberg, P. G. Nyholm, I. Pascher and S. Normark, Saccharide orientation at the cell surface affects glycolipid receptor function., Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 9340–9344 CrossRef PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Multivalency as a Chemical Organization and Action Principle, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- R. Haag, Multivalency as a chemical organization and action principle, Beilstein J. Org. Chem., 2015, 11, 848–849 CrossRef CAS PubMed.
- A. K. Ciuk and T. K. Lindhorst, Synthesis of carbohydrate-scaffolded thymine glycoconjugates to organize multivalency, Beilstein J. Org. Chem., 2015, 11, 668–674 CrossRef CAS PubMed.
- C. Müller, G. Despras and T. K. Lindhorst, Organizing multivalency in carbohydrate recognition, Chem. Soc. Rev., 2016, 45, 3275–3302 RSC.
- M. Pintal, F. Charbonniere-Dumarcay, A. Marsura and S. Porwański, Synthesis of new saccharide azacrown cryptands, Carbohydr. Res., 2015, 414, 51–59 CrossRef CAS PubMed.
- A. J. Fairbanks, Applications of Shoda's reagent (DMC) and analogues for activation of the anomeric centre of unprotected carbohydrates, Carbohydr. Res., 2021, 499, 108197 CrossRef CAS PubMed.
- G. D'Adamio, M. Forcella, P. Fusi, P. Parenti, C. Matassini, X. Ferhati, C. Vanni and F. Cardona, Probing the Influence of Linker Length and Flexibility in the Design and Synthesis of New Trehalase Inhibitors, Molecules, 2018, 23, 436 CrossRef PubMed.
- M. Upreti, D. Ruhela and R. A. Vishwakarma, Synthesis of the Tetrasaccharide Cap Domain of the Antigenic Lipophosphoglycan of Leishmania donovani Parasite, Tetrahedron, 2000, 56, 6577–6584 CrossRef CAS.
- C. Cavedon, E. T. Sletten, A. Madani, O. Niemeyer, P. H. Seeberger and B. Pieber, Visible-Light-Mediated Oxidative Debenzylation Enables the Use of Benzyl Ethers as Temporary Protecting Groups, Org. Lett., 2021, 23, 514–518 Search PubMed.
- G. Zemplén and E. Pacsu, Über die Verseifung acetylierter Zucker und verwandter Substanzen, Ber. Dtsch. Chem. Ges. B, 1929, 62, 1613–1614 CrossRef.
- J. Kraft, D. Schmollinger, J. Maudrich and T. Ziegler, Synthesis of Sugar-Derived Triazole- and Pyridine-Based Metal Complex Ligands, Synthesis, 2015, 199–208 CAS.
- J. D’Onofrio, M. de Champdoré, L. De Napoli, D. Montesarchio and G. Di Fabio, Glycomimetics as Decorating Motifs for Oligonucleotides: Solid-Phase Synthesis, Stability, and Hybridization Properties of Carbopeptoid–Oligonucleotide Conjugates, Bioconjugate Chem., 2005, 16, 1299–1309 CrossRef PubMed.
- J. P. Lorentzen, B. Helpap and O. Lockhoff, Synthesis of an Elicitor-Active Heptaglucan Saccharide for Investigation of Defense Mechanisms of Plants, Angew. Chem., Int. Ed. Engl., 1991, 30, 1681–1682 CrossRef.
- N. Smiljanic, S. Halila, V. Moreau and F. Djedaïni-Pilard, Regioselective glycosylation of 3,6-unprotected mannoside derivatives: fast access to high-mannose type oligosaccharides, Tetrahedron Lett., 2003, 44, 8999–9002 Search PubMed.
- D. Yockot, V. Moreau, G. Demailly and F. Djedaïni-Pilard, Synthesis and characterization of mannosyl mimetic derivatives based on a β-cyclodextrin core, Org. Biomol. Chem., 2003, 1, 1810–1818 RSC.
- Z. Györgydeák and H. Paulsen, Synthese von β-D-Mannopyranosylaziden; Untersuchung der anomeren Strukturen, Justus Liebigs Ann. Chem., 1977, 1987–1991 CrossRef.
- L. Wang, H. Kong, M. Jin, X. Li, R. Stoika, H. Lin and K. Liu, Synthesis of disaccharide modified berberine derivatives and their anti-diabetic investigation in zebrafish using a fluorescence-based technology, Org. Biomol. Chem., 2020, 18, 3563–3574 RSC.
- C. M. Brunner, L. Pietsch, I. vom Sondern, M. Röhrl, C. Popov, M. F. W. Trollmann, R. W. Taylor, M. Blessing, C. Holler, K. Almahayni, S. O. Jaeschke, V. Sandoghdar, R. A. Böckmann, T. K. Lindhorst and L. Möckl, Bottom-up investigation of spatiotemporal glycocalyx dynamics with interferometric scattering microscopy, bioRxiv, 2025, preprint, DOI:10.1101/2025.05.09.653034.
- T. Dumych, C. Bridot, S. G. Gouin, M. F. Lensink, S. Paryzhak, S. Szunerits, R. Blossey, R. Bilyy, J. Bouckaert and E.-M. Krammer, A Novel Integrated Way for Deciphering the Glycan Code for the FimH Lectin, Molecules, 2018, 23, 2794 CrossRef PubMed.
- M. M. Sauer, R. P. Jakob, T. Luber, F. Canonica, G. Navarra, B. Ernst, C. Unverzagt, T. Maier and R. Glockshuber, Binding of the Bacterial Adhesin FimH to Its Natural, Multivalent High-Mannose Type Glycan Targets, J. Am. Chem. Soc., 2019, 141, 936–944 CrossRef CAS PubMed.
- J. Bouckaert, J. Mackenzie, J. L. D. Paz, B. Chipwaza, D. Choudhury, A. Zavialov, K. Mannerstedt, J. Anderson, D. Piérard, L. Wyns, P. H. Seeberger, S. Oscarson, H. D. Greve and S. D. Knight, The affinity of the FimH fimbrial adhesin is receptor-driven and quasi-independent of Escherichia coli pathotypes, Mol. Microbiol., 2006, 61, 1556–1568 CrossRef CAS PubMed.
- E.-M. Krammer, C. Bridot, S. Serna, B. Echeverria, S. Semwal, B. Roubinet, K. van Noort, R. H. P. Wilbers, G. Bourenkov, J. de Ruyck, L. Landemarre, N. Reichardt and J. Bouckaert, Structural insights into a cooperative switch between one and two FimH bacterial adhesins binding pauci- and high-mannose type N-glycan receptors, J. Biol. Chem., 2023, 299, 104627 CrossRef CAS PubMed.
- Schrödinger Release 2024–2: Induced Fit Docking protocol, Glide Schrödinger, LLC, New York, NY, 2024; Prime, Schrödinger, LLC, New York, NY, Induced Fit, 2024 Search PubMed.
- C.-S. Hung, J. Bouckaert, D. Hung, J. Pinkner, C. Widberg, A. DeFusco, C. G. Auguste, R. Strouse, S. Langermann, G. Waksman and S. J. Hultgren, Structural basis of tropism of Escherichia coli to the bladder during urinary tract infection, Mol. Microbiol., 2002, 44, 903–915 CrossRef CAS PubMed.
- J. Bouckaert, J. Berglund, M. Schembri, E. D. Genst, L. Cools, M. Wuhrer, C.-S. Hung, J. Pinkner, R. Slättegård, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. D. Greve, Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS PubMed.
- A. Wellens, C. Garofalo, H. Nguyen, N. V. Gerven, R. Slättegård, J.-P. Hernalsteens, L. Wyns, S. Oscarson, H. D. Greve, S. Hultgren and J. Bouckaert, Intervening with Urinary Tract Infections Using Anti-Adhesives Based on the Crystal Structure of the FimH–Oligomannose-3 Complex, PLoS One, 2008, 3, e2040 CrossRef PubMed.
- A. Wellens, M. Lahmann, M. Touaibia, J. Vaucher, S. Oscarson, R. Roy, H. Remaut and J. Bouckaert, The Tyrosine Gate as a Potential Entropic Lever in the Receptor-Binding Site of the Bacterial Adhesin FimH, Biochemistry, 2012, 51, 4790–4799 Search PubMed.
- Schrödinger Release 2024–2: Binding Pose Metadynamics protocol, Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2024. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, Binding Pose Metadynamics, 2024 Search PubMed.
- A. J. Clark, P. Tiwary, K. Borrelli, S. Feng, E. B. Miller, R. Abel, R. A. Friesner and B. J. Berne, Prediction of Protein–Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations, J. Chem. Theory Comput., 2016, 12, 2990–2998 Search PubMed.
- Schrödinger Release 2024–2: Prime, Schrödinger, LLC, New York, NY, Prime, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00927h |
| This journal is © The Royal Society of Chemistry 2025 |