
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible photocycloaddition of 8-pyrenylvinylguanine for photoreactive serinol nucleic acid (SNA)†
Keiji
Murayama
 *,
Ayaka
Ikeda
,
Fuminori
Sato
and
Hiroyuki
Asanuma
*
*,
Ayaka
Ikeda
,
Fuminori
Sato
and
Hiroyuki
Asanuma
*
Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: murayama@chembio.nagoya-u.ac.jp; asanuma@chembio.nagoya-u.ac.jp
First published on 5th July 2025
Abstract
Photoresponsive nanomachines are attractive components of functional nanodevices and nanosystems. To develop new photoresponsive nucleic acid-based nanomachines, we conjugated 8-pyrenylvinylguanine (PVG) to a serinol linker and incorporated it into an acyclic xeno nucleic acid, serinol nucleic acid (SNA). The two PVG residues incorporated into SNA underwent interstrand photocycloaddition upon 447 nm light irradiation in the duplex state, whereas previously reported 8-pyrenylvinyladenine (PVA) formed both intrastrand and interstrand photodimers. The PVG photodimer was converted to monomers by irradiation with 350 nm light. This photoreaction enabled reversible photoregulation of the formation and dissociation of the SNA/RNA duplex, although some byproducts were generated due to the slower photoreaction of PVGs than that of PVAs. In contrast, when a single PVG was incorporated into SNA, the interstrand photocycloaddition and cycloreversion were remarkably fast and effective in the single-stranded state. We utilized this to demonstrate a photocaging system that achieves one-way photoswitching of hybridization ability. The powerful photocycloaddition properties of PVG-SNA are expected to find applications in new photoresponsive nanodevices and nanosystems.
Introduction
DNA nanotechnology offers an attractive platform for realizing highly functional nanodevices and biological tools, such as DNA nanostructures,1 DNA computing systems,2 DNA-based nanomachines,3 and molecular robotics.4 Chemical modifications on nucleobases and additional incorporation of functional molecules into DNA strands facilitate the development of stimuli-responsive systems triggered by metal ions,5 pH,6 and photoirradiation,7 providing powerful systems for the design of novel nanomachines based on nucleic acids.To overcome the limitations of conventional DNA nanotechnology, it is essential to utilize xeno nucleic acids (XNAs)8 with modified backbone structures that exhibit high resistance to enzymatic and chemical degradation and unique hybridization properties distinct from those of natural nucleic acids. In fact, several examples of nanotechnology utilizing XNAs have been reported, demonstrating greater functionality than those utilizing DNA.9 We previously reported a reversible photoregulation of duplex formation and dissociation of serinol nucleic acid (SNA),10 an acyclic XNA, by introducing two 8-pyrenylvinyladenine (PVA) residues.11 When blue light is irradiated onto the duplex composed of SNA containing two PVA residues and complementary RNA, a [2 + 2] photocycloaddition reaction occurs between the two PVA residues, causing the duplex to dissociate into single strands (Fig. 1A). On the other hand, upon UV light irradiation, the PVA photodimer undergoes photocycloreversion to regenerate monomeric PVA, enabling the re-formation of the duplex. This system shows promise for the development of photon-driven nanomachines and photocontrollable biological tools that use XNAs as building blocks. However, these strategies are limited to photoresponsive modification of the adenine base, requiring two adjacent adenines to design a photocontrollable system, which limits the range of applicable sequences.
 | ||
| Fig. 1 Schematics of (A) previous work on the SNA strand modified with PVA residues and (B) this work on the SNA strand modified with PVG residues. | ||
We report on the photoregulation of SNA containing a photoresponsive nucleobase, 8-pyrenylvinylguanine (PVG) (Fig. 1B). The synthesis of PVG incorporated into deoxyribose has already been reported.12 Ogasawara demonstrated that PVG-modified DNA can be used as a photoswitch for G4 formation via the trans–cis isomerization of PVG, which was successfully applied to photoregulation of a transcription system.13 We hypothesized that two adjacent PVG residues conjugated with a flexible SNA strand are more likely to cause a photocycloaddition than cis-isomerization as in the case of PVA, which should lead to a photoregulation system different from the DNA-PVG system based on a trans–cis isomerization. Therefore, we synthesized a novel PVG-SNA phosphoramidite monomer and incorporated it into the SNA strand, evaluated the photoreactivity of the PVG residue, and compared it with the PVA modification.
Results and discussion
Synthetic procedures for the PVG monomer
First, we prepared a phosphoramidite monomer of PVG-SNA according to Fig. 2. The N-9 position of 2-amino-6-chloropurine was converted to the benzyl ester form of compound 1, and then the 6-position was substituted with oxygen to construct the guanine structure 3. The formamidine protection on the 2-amino group resulted in an unexpected ester exchange from benzyl ester to ethyl ester 4, but we continued the synthesis. After 8-bromination with NBS, the CuI/CsF-mediated Stille reaction14 was carried out to obtain the 8-vinylguanine skeleton 6. The Heck reaction with 1-bromopyrene followed by hydrolysis gave 2-protected-PVG 8. After coupling with DMTr-L-serinol, the desired phosphoramidite monomer 10 was successfully obtained using conventional phosphoramidite chemistry. The SNA strand with two PVG residues was synthesized using a DNA synthesizer, with each SNA monomer prepared as reported previously.10 | ||
| Fig. 2 Synthesis of a phosphoramidite monomer of PVG-SNA 10. (i) Benzyl bromoacetate, DIPEA, DMF, 0 °C to r.t., and o.n., (ii) K2CO3, DABCO, benzyl alcohol, 80 °C, and 16 h, (iii) TFA, DCM, r.t., and 1 h, (iv) N,N-dimethylformamide diethyl acetal, EtOH, 50 °C, and 16 h, (v) NBS, DMF, r.t., and 2 h, (vi) Pd(PPh3)4, tributyl(vinyl)tin, CuI, CsF, DMF, 55 °C, and 4 h, (vii) 1-bromopyrene, Pd(Ac)2, PPh3, NEt3, DMF, 110 °C, and 3 h, (viii) NaOH, 1,4-dioxane, MeOH, EtOH, H2O, r.t., and 20 min, and (ix) DMTr-L-serinol, DMT-MM, NEt3, DMF, and 0 °C for 20 min to r.t. for 1 h, and (x) 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, NEt3, CH2Cl2, 0 °C, and 30 min. | ||
Comparison of the photoreactivity between PVG and PVA introduced into the SNA strand
The melting temperature of the SNA-2PVG/RNA-2C duplex was 40.4 °C (Fig. 3A), which is slightly higher than that of SNA-2PVA/RNA-2U (31.0 °C, Fig. S1†). The stabilization of the duplex is probably caused by the strong stacking interaction of PVGs, as will be explained later. This result shows that SNA-2PVG and RNA-2C form a duplex at room temperature, suggesting that the PVG substitution does not suppress duplex formation. Subsequently, the photoreactivity of PVG in the SNA strand was evaluated (Fig. 3). Upon 447 nm light irradiation of the SNA-2PVG/RNA-2C duplex at 20 °C, the absorption band at around 400 nm decreased with the irradiation time, followed by the appearance of a new absorption band at around 330–360 nm, which was attributed to the pyrene moiety (Fig. 3B). The isosbestic points were 329 and 361 nm. This behavior is similar to the photocycloaddition of PVA residues in the SNA-2PVA/RNA-2U duplex, indicating that PVG residues also form a photodimer via photocycloaddition rather than cis-isomerization under the same conditions (Fig. S2†).12 When the photodimer of PVG was irradiated with 350 nm UV light, the absorption bands of the monomeric PVG were restored (Fig. 3C), indicating a cycloreversion reaction similar to that of PVA. We confirmed that pyrimidine dimers are not generated even when exposed to 350 nm light for 30 minutes under the present conditions (Fig. S12†). However, the kinetics of the photocycloaddition of SNA-2PVG/RNA-2C were rather slow compared to SNA-2PVA/RNA-2U: the half-life of the photodimerization of PVAs was about 10 s (Fig. 3D), while that of PVGs under the same conditions was over 10 min. Furthermore, the efficiency of cycloreversion also differed: 79% of the PVA photodimer was converted to monomers after 10 min of UV irradiation, whereas only 67% of the PVG photodimer was converted to monomers even after 30 min of UV irradiation (Fig. 3E). | ||
| Fig. 3 (A) Chemical structures of SNA units, sequences used in this study, and schematics of the irradiation experiment. (B) Absorption spectra of SNA-2PVG/RNA-2C before irradiation and at the indicated times of irradiation with 447 nm light. (C) Absorption spectra of SNA-2PVG/RNA-2C before irradiation, after 1 h of irradiation with 447 nm and after irradiation for the indicated times with 350 nm light. Irradiation was performed at 20 °C. (D) and (E) The ratio of monomeric PVG in SNA-2PVG/RNA-2C (purple circles) and that of monomeric PVA in SNA-2PVA/RNA-2U (blue triangles, Fig. S2† for UV spectra) as a function of irradiation time: (D) with 447 nm light and (E) with 350 nm light. Before irradiation with 350 nm light, PVG and PVA were crosslinked by irradiation with 447 nm light for 1 h and 10 min, respectively. The ratio of the monomers was calculated from the absorbance at 400 nm. | ||
To understand the reason for the different behavior of PVG and PVA, we performed denaturing PAGE analysis before and after light irradiation (Fig. 4A and B). To visualize the SNA strand, the entire gel was irradiated with 350 nm light after electrophoresis to restore the fluorescence of the monomeric PVG/PVA and detected using an imaging analyzer with a blue laser. Note that both PVG and PVA are emissive upon blue light excitation. In the case of SNA-2PVA/RNA-2U, irradiation with 447 nm light resulted in the appearance of two bands and the disappearance of the original band. The lower and the upper bands are assigned to the intrastrand and the interstrand crosslinking products, respectively (Fig. 4A). Under the present conditions employed, PVAs were able to crosslink not only intrastrand but also interstrand. This fact suggests that a dimer of the SNA-2PVA/RNA-2U duplexes is formed as an intermediate via association between PVAs in the duplexes. Subsequent irradiation with 350 nm light significantly reduced these bands and regenerated the original band of SNA-2PVA due to cycloreversion. In contrast, irradiation of SNA-2PVG/RNA-2C with 447 nm resulted in two additional bands in addition to the remaining unreacted SNA-2PVG strand (Fig. 4B, left). The upper high-intensity band is attributed to the product of interstrand crosslinking because it showed significantly reduced mobility compared to intrastrand-crosslinked SNA-2PVA. The lower band originated from unidentified by-products. Importantly, no band could be attributed to the intrastrand PVG-dimer, suggesting that intrastrand crosslinking of PVG was strongly suppressed. Upon irradiation with 350 nm light, the crosslinking product was converted to SNA-2PVG with PVG monomers, whereas the by-products remained. Unlike SNA-2PVA/RNA-2U, the SNA-2PVG/RNA-2C duplex may facilitate a static stacking interaction between the interstrand PVGs to form a dimer of the duplexes prior to photocrosslinking (Fig. 4C). In this case, it is difficult for PVG to adopt the necessary conformation for crosslinking, due to the rigid structure of the duplexes and the electrostatic repulsion between them.
 | ||
| Fig. 4 (A) and (B) Gel-shift assay with denaturing PAGE. (A) SNA-2PVA/RNA-2U duplex, (B, left) SNA-2PVG/RNA-2C duplex, and (B, right) SNA-2PVG single strand. The fluorescence of PVG and PVA was detected upon excitation using a 473 nm laser and a long-path filter (510 nm). (C) Illustration of different mechanisms of photocycloaddition between PVG and PVA. (D) The ratio of monomeric PVG in SNA-2PVG single strands upon irradiation with 447 nm (red circles) and 350 nm (blue squares) light. (E) Melting profiles of SNA-2PVG/RNA-2C before (black line) and after irradiation with 447 nm light (purple line) and 350 nm light (blue line). | ||
This hypothesis was supported by the results of the irradiation experiments in the absence of complementary RNA. Irradiation with 447 nm light for single-stranded SNA-2PVG showed significantly faster photocrosslinking than in the duplex state (Fig. 4D and Fig. S3†). The reaction half-life for photocycloaddition was approximately 1 min, which is about 10 times faster than that of the duplex state. The efficiency of the cycloreversion reaction for SNA-2PVG was also improved: approximately 80% of the crosslinked PVG was converted to the monomer by irradiation with 350 nm light for 20 min. Denaturing PAGE revealed that the main product of photoirradiation on single-stranded SNA-2PVG was also only the interstrand crosslinking product and that the formation of by-products was inhibited (Fig. 4B, right). The rapid photocycloaddition of single-stranded SNA-2PVG can be explained by the fact that the single-stranded state facilitates the interaction between PVGs on different strands due to the smaller electrostatic repulsion and that the flexibility of the single strand makes the dimerized structure of consecutive PVGs suitable for photocrosslinking reactions compared to the rigid duplex state. As a result, the efficiency of cycloreversion was also enhanced due to the relative suppression of the irreversible side reaction during irradiation with 447 nm light. The concentration dependence of the photocrosslinking kinetics provided further evidence for the interstrand reaction of PVGs. The crosslinking rate of 1 μM SNA-2PVG with 447 nm light was found to be slower than that of 5 μM SNA-2PVG (Fig. S3 and S4†). We conclude that two PVGs introduced into the SNA strand undergo photocycloaddition reactions in an interstrand manner.
Although the reaction mechanism was different from what we expected, we hypothesized that the photoreaction of PVGs would enable photoregulation of duplex formation. In SNA-2PVA/RNA-2U, which induces both intrastrand and interstrand crosslinks, clear photoregulation of duplex formation and dissociation was observed, which was reversible and consistent with our previous results (Fig. S5†).11 Irradiation of the SNA-2PVG/RNA-2C duplex with 447 nm light at 20 °C led to the complete disappearance of the sigmoidal melting curve, indicating effective dissociation of the duplex by interstrand photocycloaddition of PVGs (Fig. 4E, purple line). Upon irradiation of the photoadduct with 350 nm light, the sigmoidal curve partially recovered, supporting the re-formation of the duplex (Fig. 4E, blue line). However, PVG-SNA showed poor reversibility of the photoreaction (Fig. S6†), which is likely due to an unidentified by-product formed during the irradiation.
Although the precise mechanism remains unclear, it is postulated that PVG has a favorable orientation in photocycloaddition reactions, enabling it to photocrosslink solely via interstrand PVG dimerization (Fig. 4C). In addition, the strong stacking interaction between PVGs facilitates interstrand association during the photoreaction (Fig. S7†). The formation of by-products could be the reason for the low cycloreversion efficiency of the PVG system compared to the PVA system.
Single PVG incorporated into the SNA strand for effective interstrand photocrosslinking
By focusing on the strong stacking property and selective interstrand photocycloaddition between PVGs, we hypothesized that the SNA strand carrying a single PVG should undergo interstrand crosslinking and form a 4-arm branched SNA (Fig. 5A). We therefore prepared SNA-1PVG, which carried only a single PVG residue in the SNA strand, and recorded its absorption spectra after photoirradiation (Fig. 5B and C). Upon irradiation of single-stranded SNA-1PVG with 447 nm light, a slight blue shift of the absorption band was observed, which was attributed to the cis-isomer of PVG. It subsequently disappeared with the appearance of the pyrene band (Fig. 5B). The isosbestic point was not observed. Presumably, PVG in SNA-1PVG promotes rapid trans-to-cis isomerization of PVG, and cis-PVG is also excited and reverted to the trans form, causing photocrosslinking upon irradiation with 447 nm light. SNA-2PVG involving consecutive PVGs probably suppresses cis isomerization through strong stacking interaction between PVGs. The reaction rate of photocycloaddition for single stranded SNA-1PVG was as fast as that for single-stranded SNA-2PVG (Fig. 5D). Although cis-isomerization competed with photocrosslinking, the photocycloaddition reaction was remarkably fast. Fujimoto et al. reported that [2 + 2] photocycloaddition of 3-cyanovinylcarbazole (CNVK) occurred only with the trans isomer.15 In addition, the [2 + 2] photocycloaddition reaction is generally selective for trans-isomers.16 We therefore hypothesize that cisPVG may also need to revert to the trans form prior to photocycloaddition (Fig. S8†). However, since some stilbene derivatives have been reported to undergo photodimerization between trans- and cis-isomers,7h,17 it cannot be completely ruled out that the cis-PVG may undergo a [2 + 2] photocycloaddition reaction. Upon irradiation of the photocrosslinked SNA-1PVG with 350 nm light, the initial absorption band of PVG was rapidly restored, demonstrating that SNA-1PVG underwent highly effective cycloreversion compared to SNA-2PVG (Fig. 5C and D). Repeated irradiation experiments revealed that an effective photoreaction occurred for at least 5 cycles without severe photobleaching (Fig. S9†). Denaturing PAGE also confirmed the selective formation of the interstrand crosslinking product and the effective back reaction (Fig. 5E). The concentration dependence of the photoreaction confirmed an interstrand crosslinking reaction between PVGs: the difference in kinetics between 1 μM and 5 μM was approximately fivefold (Fig. S10 and S11†). | ||
| Fig. 5 (A) Schematic of the photoreaction of single-stranded SNA-1PVG under light irradiation. (B) Absorption spectra of single-stranded SNA-1PVG before and after irradiation for the indicated times with 447 nm light. (C) Absorption spectra of single-stranded SNA-1PVG before irradiation, after 1 h of irradiation with 447 nm light and after irradiation for the indicated times with 350 nm light. Irradiation was performed at 20 °C. (D) The ratio of monomeric PVG in SNA-1PVG single strands after irradiation at 447 nm (red circles) and 350 nm (blue squares). (E) Gel shift assay using denaturing PAGE before and after the light irradiation of the SNA-1PVG single strand. | ||
Photocaging strategy of PVG-tethered SNA for the photoactivation of hybridization with RNA
We demonstrated the one-way photoswitching of the hybridization ability of SNA-1PVG by photocycloreversion using the pre-dimerized strand (Fig. 6A). The single-stranded SNA-1PVG was irradiated with 447 nm light for 20 min to ensure complete photodimerization. Complementary RNA (RNA-2C) was then added, and the melting temperature was measured. The melting profile showed no transition, clearly indicating that the photocrosslinked SNA-1PVG had lost its hybridization ability (Fig. 6B). After irradiating this sample with 350 nm light for 10 min, a sigmoidal curve was observed due to duplex formation. Thus, we confirmed that the photocaging strategy with pre-crosslinked SNA-1PVG is feasible. We also confirmed that a single modification with PVA is applicable to the photocaging strategy (Fig. S13†).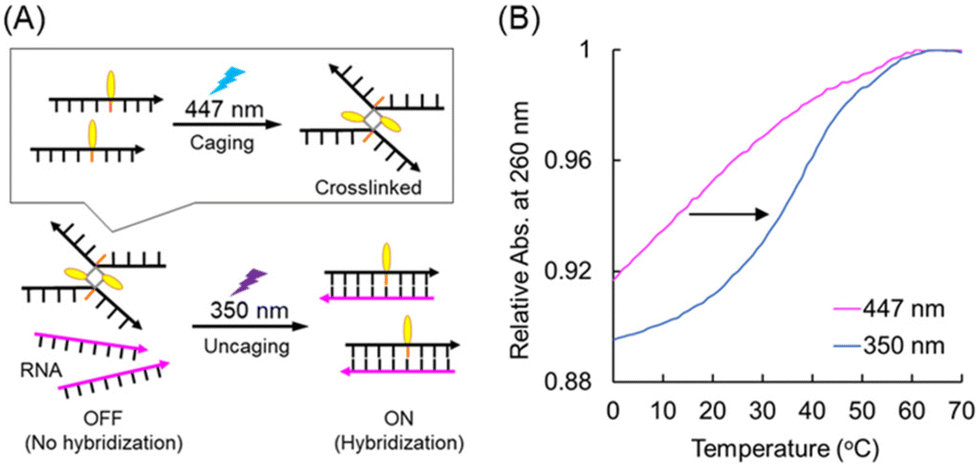 | ||
| Fig. 6 (A) Schematic illustration of the photocaging strategy using the pre-crosslinked SNA-1PVG dimer. (B) Melting profiles of the duplex of pre-crosslinked SNA-1PVG and RNA-2C (purple line) and after irradiation with 350 nm light (blue line). | ||
Experimental
Materials
Reagents for the synthesis of the PVG phosphoramidite monomer were purchased from Tokyo Kasei Co., Ltd, Wako Pure Chemical Industries, Ltd, and Aldrich. The reagents for oligomer synthesis and Poly-Pak II cartridges were purchased from Glen Research. The column for HPLC purification was purchased from Kanto Chemical Co., Ltd. RNA strands were purchased from Hokkaido System Science Co., Ltd.Photoirradiation
447 nm light-emitting diodes (LEDs; CCS) and a xenon light source (MAX-301, Asahi Spectra) equipped with interference filters centered at 350 nm was used for photoirradiation. The sample solution was added to a cuvette, and the temperature during the light irradiation was controlled using a programable temperature controller. All photoirradiation experiments were conducted at 20 °C.Spectroscopic measurements
UV/vis spectra were measured using a JASCO model V-730 spectrometer equipped with a programable temperature controller; 10 mm quartz cells were used. The sample solutions contained 100 mM NaCl and 10 mM phosphate buffer (pH 7.0). The duplex concentration was 5.0 μM, and the experiments were performed in a 10 mm quartz cell.Melting-temperature measurements
The melting curves of all nucleic acid duplexes were obtained using a JASCO model V-730 spectrometer equipped with a programable temperature controller by measuring the change in absorbance at 260 nm versus temperature. The melting curves for heating and cooling were recorded, and the average maximum value of their first derivatives was determined to be the melting temperature (Tm). The solution conditions were (unless otherwise noted) 100 mM NaCl, 10 mM phosphate buffer (pH 7.0), and 5.0 μM oligonucleotide.Denaturing PAGE
Samples were loaded into the wells of a denaturing gel (20% acrylamide, 8 M urea, and 1 × TBE). Electrophoresis was performed at 4 W and r.t. for 2 h. The gels were irradiated with 350 nm light after electrophoresis to recover the fluorescence of the monomeric PVG and PVA. The gels were analyzed using a Typhoon FLA 9500 with a 473 nm laser and an LPB filter (510 nm).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH2Cl2 = 1:1 solution and stirred for 1 h at room temperature. After brief evaporation, the residue was co-evaporated with acetonitrile three times. The residue was suspended in diethyl ether and then filtered to afford 3 as a solid (4.91 g, >100%). 1H-NMR [DMSO-d6, 500 MHz] δ = 10.88 (br, 1H), 7.88 (s, 1H), 7.40–7.32 (m, 5H), 6.72 (br, 1H), 5.19 (s, 1H), 4.97 (s, 1H). 13C-NMR [DMSO-d6, 125 MHz] δ = 167.8, 156.3, 154.3, 151.3, 137.9, 135.4, 128.6, 128.4, 128.1, 67.1, 44.5. HRMS (FAB) calcd for C14H13N5O3 (M + H+): 300.1096. Found 300.1094.
:5% Na2S2O4 aq. = 1:1) in an ice bath. After stirring for 5 min, the solution was extracted with chloroform and Na2S2O4, and the organic layer was collected. The water layer was washed with chloroform. The combined organic layer was evaporated. The residue was suspended in small amounts of chloroform and large amounts of diethyl ether. The precipitate was collected by filtration and dried in vacuo, yielding 5 (3.11 g, 92%).1H-NMR [DMSO-d6, 500 MHz] δ = 11.46 (br, 1H), 8.59 (s, 1H), 4.91 (s, 2H), 4.20 (q, 2H), 3.15 (s, 3H), 3.03 (s, 3H), 1.23 (t, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 167.6, 158.9, 158.2, 156.9, 151.9, 123.0, 119.6, 62.2, 44.7, 41.2, 35.2, 14.5. HRMS (FAB) calcd for C12H15BrN6O3 (M + H+): 371.0467. Found 371.0482.
:MeOH = 20:1), yielding 6 (280 mg, 47%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.33 (s, 1H), 8.57 (s, 1H), 6.71 (m, 1H), 6.17 (s, 1H), 5.47 (d, 1H), 5.06 (s, 2H), 4.17 (q, 2H), 3.14 (s, 1H), 3.03 (s, 1H), 1.21 (t, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 167.9, 158.2, 157.4, 157.2, 150.8, 145.3, 123.6, 119.5, 118.9, 61.5, 42.7, 40.7, 34.7, 14.0. HRMS (FAB) calcd for C14H18N6O3 (M + H+) 319.1518. Found 319.1534.
:MeOH = 20:1) to afford 7 (220 mg, 48%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.42 (br, 1H), 8.74 (d, 1H), 8.65–8.10 (m, 10 H), 7.58 (d, 1H), 5.29 (s, 1H), 4.21 (s, 2H), 3.17 (s, 3H), 3.05 (s, 3H), 1.24 (t, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 168.6, 158.6, 157.9, 157.6, 151.5, 146.6, 131.5, 131.4, 130.8, 130.5, 129.2, 128.7, 128.6, 128.2, 127.9, 127.0, 126.2, 125.9, 125.7, 124.8, 124.5, 124.3, 123.0, 120.1, 117.3, 61.9, 43.4, 41.2, 35.2, 14.5. HRMS (FAB) calcd for C30H26N6O3 (M + H+): 519.2144. Found 519.2173.
:EtOH = 1:1 solution was added to a stirred solution of compound 7 (220 mg, 0.42 mmol) in 1,4-dioxane:MeOH = 2:1 solution. The reaction mixture was stirred for 20 min at room temperature. After the addition of a large amount of H2O, the pH was adjusted to 2–3 using 1 N HCl aq. The solid was collected by filtration using diethyl ether to afford 8 (150 mg, 86%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.63 (br, 1H), 8.86 (d, 1H), 8.73–8.79 (m, 10H), 7.62 (d, 1H), 5.22 (s, 1H), 3.19 (s, 3H), 3.06 (s, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 169.1, 158.2, 157.5, 156.5, 150.5, 146.1, 131.4, 131.0, 130.3, 129.4, 128.5, 128.4, 127.4, 126.6, 125.9, 125.6, 125.3, 124.3, 123.9, 122.4, 118.1, 66.4, 43.2, 40.9. HRMS (FAB) calcd for C28H22N6O3 (M + H+): 491.1831. Found 491.1836.
:MeOH = 20:1, 1% triethylamine) to afford 9 (190 mg, 61%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.38 (br, 1H), 8.71–8.08 (m, 12H), 7.43–7.13 (m, 9H), 6.75 (m, 4H), 5.06 (dd, 2H), 4.81 (t, 1H), 4.07 (m, 1H), 3.642, 3.638 (s*2, 3H), 3.54 (m, 2H), 3.03 (m, 2H), 2.98 (s, 3H), 2.82 (s, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 166.4, 157.93, 157.90, 157.5, 157.0, 151.3, 146.3, 145.0, 135.8, 135.7, 131.0, 130.8, 130.4, 130.1, 129.7, 129.6, 128.4, 128.2, 128.1, 127.73, 127.66, 127.4, 126.5, 125.7, 125.4, 125.2, 124.3, 124.1, 123.6, 122.5, 119.7, 117.2, 113.0, 85.2, 62.4, 60.8, 55.0, 54.9, 51.3, 44.3, 34.6. HRMS (FAB) calcd for C52H47N7O6 (M + H+): 866.3666. Found 866.3697.
:acetone = 3:1 containing 3% triethylamine). The products were then dissolved in a small amount of dry CHCl3 and re-precipitated three times from hexane, yielding compound 10 (170 mg, 73%). Prior to synthesis of SNA using a DNA synthesizer, compound 10 was dried by co-evaporation with a mixture of dry acetonitrile and dry dichloromethane. 1H-NMR [DMSO-d6, 500 MHz] δ = 11.39 (br, 1H), 8.72–8.10 (m, 12H), 7.42–7.14 (m, 10H), 6.76 (m, 4H), 5.08 (d, 2H), 4.19 (m, 1H), 4.07 (m, 1H), 3.70–3.55 (m, 10H), 3.42 (m, 1H), 3.10 (m, 2H), 2.99, 2.98 (s*2, 3H), 2.83, 2.80 (s*2, 3H), 2.61 (m, 2H), 1.02 (m, 6H), 0.93 (m, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 166.4, 158.0, 157.9, 157.8, 157.5, 157.0, 151.2, 146.3, 144.9, 135.6, 135.5, 131.0, 130.8, 130.4, 130.1, 129.7, 129.6, 128.4, 128.2, 128.1, 127.7, 127.6, 127.4, 126.5, 125.7, 125.4, 125.2, 124.3, 124.0, 123.5, 122.5, 119.7, 118.8, 117.3, 113.0, 85.2, 62.0, 58.2, 58.1, 54.9, 44.2, 42.4, 42.3, 34.6, 32.1, 29.6, 24.3, 24.2, 24.1, 19.8, 19.7. 31P-NMR [DMSO-d6, 202 MHz] δ = 146.9, 146.6. HRMS (FAB) calcd for C61H64N9O7P (M + H+) 1066.4744. Found 1066.4730.
:CH2Cl2 = 1:1 solution and stirred for 1 h at room temperature. After brief evaporation, the residue was co-evaporated with acetonitrile three times. The residue was suspended in diethyl ether and then filtered to afford 3 as a solid (4.91 g, >100%). 1H-NMR [DMSO-d6, 500 MHz] δ = 10.88 (br, 1H), 7.88 (s, 1H), 7.40–7.32 (m, 5H), 6.72 (br, 1H), 5.19 (s, 1H), 4.97 (s, 1H). 13C-NMR [DMSO-d6, 125 MHz] δ = 167.8, 156.3, 154.3, 151.3, 137.9, 135.4, 128.6, 128.4, 128.1, 67.1, 44.5. HRMS (FAB) calcd for C14H13N5O3 (M + H+): 300.1096. Found 300.1094.
:5% Na2S2O4 aq. = 1:1) in an ice bath. After stirring for 5 min, the solution was extracted with chloroform and Na2S2O4, and the organic layer was collected. The water layer was washed with chloroform. The combined organic layer was evaporated. The residue was suspended in small amounts of chloroform and large amounts of diethyl ether. The precipitate was collected by filtration and dried in vacuo, yielding 5 (3.11 g, 92%).1H-NMR [DMSO-d6, 500 MHz] δ = 11.46 (br, 1H), 8.59 (s, 1H), 4.91 (s, 2H), 4.20 (q, 2H), 3.15 (s, 3H), 3.03 (s, 3H), 1.23 (t, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 167.6, 158.9, 158.2, 156.9, 151.9, 123.0, 119.6, 62.2, 44.7, 41.2, 35.2, 14.5. HRMS (FAB) calcd for C12H15BrN6O3 (M + H+): 371.0467. Found 371.0482.
:MeOH = 20:1), yielding 6 (280 mg, 47%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.33 (s, 1H), 8.57 (s, 1H), 6.71 (m, 1H), 6.17 (s, 1H), 5.47 (d, 1H), 5.06 (s, 2H), 4.17 (q, 2H), 3.14 (s, 1H), 3.03 (s, 1H), 1.21 (t, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 167.9, 158.2, 157.4, 157.2, 150.8, 145.3, 123.6, 119.5, 118.9, 61.5, 42.7, 40.7, 34.7, 14.0. HRMS (FAB) calcd for C14H18N6O3 (M + H+) 319.1518. Found 319.1534.
:MeOH = 20:1) to afford 7 (220 mg, 48%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.42 (br, 1H), 8.74 (d, 1H), 8.65–8.10 (m, 10 H), 7.58 (d, 1H), 5.29 (s, 1H), 4.21 (s, 2H), 3.17 (s, 3H), 3.05 (s, 3H), 1.24 (t, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 168.6, 158.6, 157.9, 157.6, 151.5, 146.6, 131.5, 131.4, 130.8, 130.5, 129.2, 128.7, 128.6, 128.2, 127.9, 127.0, 126.2, 125.9, 125.7, 124.8, 124.5, 124.3, 123.0, 120.1, 117.3, 61.9, 43.4, 41.2, 35.2, 14.5. HRMS (FAB) calcd for C30H26N6O3 (M + H+): 519.2144. Found 519.2173.
:EtOH = 1:1 solution was added to a stirred solution of compound 7 (220 mg, 0.42 mmol) in 1,4-dioxane:MeOH = 2:1 solution. The reaction mixture was stirred for 20 min at room temperature. After the addition of a large amount of H2O, the pH was adjusted to 2–3 using 1 N HCl aq. The solid was collected by filtration using diethyl ether to afford 8 (150 mg, 86%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.63 (br, 1H), 8.86 (d, 1H), 8.73–8.79 (m, 10H), 7.62 (d, 1H), 5.22 (s, 1H), 3.19 (s, 3H), 3.06 (s, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 169.1, 158.2, 157.5, 156.5, 150.5, 146.1, 131.4, 131.0, 130.3, 129.4, 128.5, 128.4, 127.4, 126.6, 125.9, 125.6, 125.3, 124.3, 123.9, 122.4, 118.1, 66.4, 43.2, 40.9. HRMS (FAB) calcd for C28H22N6O3 (M + H+): 491.1831. Found 491.1836.
:MeOH = 20:1, 1% triethylamine) to afford 9 (190 mg, 61%). 1H-NMR [DMSO-d6, 500 MHz] δ = 11.38 (br, 1H), 8.71–8.08 (m, 12H), 7.43–7.13 (m, 9H), 6.75 (m, 4H), 5.06 (dd, 2H), 4.81 (t, 1H), 4.07 (m, 1H), 3.642, 3.638 (s*2, 3H), 3.54 (m, 2H), 3.03 (m, 2H), 2.98 (s, 3H), 2.82 (s, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 166.4, 157.93, 157.90, 157.5, 157.0, 151.3, 146.3, 145.0, 135.8, 135.7, 131.0, 130.8, 130.4, 130.1, 129.7, 129.6, 128.4, 128.2, 128.1, 127.73, 127.66, 127.4, 126.5, 125.7, 125.4, 125.2, 124.3, 124.1, 123.6, 122.5, 119.7, 117.2, 113.0, 85.2, 62.4, 60.8, 55.0, 54.9, 51.3, 44.3, 34.6. HRMS (FAB) calcd for C52H47N7O6 (M + H+): 866.3666. Found 866.3697.
:acetone = 3:1 containing 3% triethylamine). The products were then dissolved in a small amount of dry CHCl3 and re-precipitated three times from hexane, yielding compound 10 (170 mg, 73%). Prior to synthesis of SNA using a DNA synthesizer, compound 10 was dried by co-evaporation with a mixture of dry acetonitrile and dry dichloromethane. 1H-NMR [DMSO-d6, 500 MHz] δ = 11.39 (br, 1H), 8.72–8.10 (m, 12H), 7.42–7.14 (m, 10H), 6.76 (m, 4H), 5.08 (d, 2H), 4.19 (m, 1H), 4.07 (m, 1H), 3.70–3.55 (m, 10H), 3.42 (m, 1H), 3.10 (m, 2H), 2.99, 2.98 (s*2, 3H), 2.83, 2.80 (s*2, 3H), 2.61 (m, 2H), 1.02 (m, 6H), 0.93 (m, 3H). 13C-NMR [DMSO-d6, 125 MHz] δ = 166.4, 158.0, 157.9, 157.8, 157.5, 157.0, 151.2, 146.3, 144.9, 135.6, 135.5, 131.0, 130.8, 130.4, 130.1, 129.7, 129.6, 128.4, 128.2, 128.1, 127.7, 127.6, 127.4, 126.5, 125.7, 125.4, 125.2, 124.3, 124.0, 123.5, 122.5, 119.7, 118.8, 117.3, 113.0, 85.2, 62.0, 58.2, 58.1, 54.9, 44.2, 42.4, 42.3, 34.6, 32.1, 29.6, 24.3, 24.2, 24.1, 19.8, 19.7. 31P-NMR [DMSO-d6, 202 MHz] δ = 146.9, 146.6. HRMS (FAB) calcd for C61H64N9O7P (M + H+) 1066.4744. Found 1066.4730.
Synthesis and purification of oligonucleotides
SNA phosphoramidite monomers involving T, G, A, and C were synthesized following reported procedures.9 SNA oligomers (SNA-2PVG and SNA-1PVG) were synthesized using an NTS M-6-MX_A12N DNA/RNA synthesizer (Nihon Techno Service Co., Ltd) using phosphoramidite chemistry. For the coupling of all SNA monomers, the incubation time was extended to 600 s. The concentrations of the SNA phosphoramidite monomers were adjusted to 0.1 M for 10 (PVG) and G, and 0.075 M for T, A, and C. 10 (PVG) was dissolved in a dry CH2Cl2:acetonitrile = 1:1 solution. The synthesized oligonucleotides were purified using Poly-Pak cartridges. The collected residue was further purified by reversed-phase HPLC (Kanto Chemical, Mightysil RP-18 GPII column). After purification, the synthesized oligonucleotides were characterized by MALDI-TOF MS. The preparation of SNA-2PVA was described in a previous report.11a
Conclusions
We synthesized a PVG-linked SNA as a photoresponsive system. A previous report showed that PVA-SNA favored the formation of intrastrand photodimers upon irradiation with blue light. In contrast, PVG selectively formed interstrand photodimers at a slower reaction rate, likely due to the different orientation favouring photocycloaddition. The interstrand crosslinking and cycloreversion of SNA-2PVG/RNA-2C enable the photoregulation of the dissociation and formation of duplexes upon light irradiation. The formation of by-products during irradiation of the SNA-2PVG/RNA-2C duplex may prevent the photoreaction from being reversible. In contrast, the interstrand photocycloaddition and cycloreversion of single-stranded SNA-1PVG were sufficiently fast and effective compared to the SNA-2PVG/RNA-2C duplex. Ogasawara et al. reported a photoregulation system using the trans–cis photoisomerization of PVG-DNA, but the photocrosslinking property of PVG was not described.12 The rigid cyclic structure of the DNA scaffold could suppress photocycloaddition, while the flexible acyclic SNA scaffold probably enables interstrand photocycloaddition with high efficiency. As we have demonstrated, effective and reversible interstrand crosslinking between PVGs is useful as a component of photoreactive materials such as photocaging systems. In particular, PVG-SNA is a suitable candidate as a photocrosslinker for various materials that respond to visible light, instead of other photocrosslinkers,18 because PVG-SNA facilitates effective intermolecular photocrosslinking even at μM concentrations due to strong stacking interactions. The powerful photocycloaddition properties of PVG-SNA promise applications in new photoresponsive nanodevices and advanced applications, such as chemical artificial intelligence.19Author contributions
K. M. and H. A. conceived and designed the experiments. A. I. performed all experiments and F. S. supervised the organic syntheses. All authors analyzed the data, discussed the results, and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the JST FOREST Program (JPMJFR2226, to K. M.); JSPS KAKENHI Grant Numbers JP20H05970, JP20H05968, JP23H00506, and JP25K00080 to K. M., and JP21H05025 to H. A.; and AMED (23am0401007 and 24ae0121056, to H. A.).References
- (a) N. C. Seeman, J. Theor. Biol., 1982, 99, 237 CrossRef CAS PubMed; (b) P. W. K. Rothemund, Nature, 2006, 440, 297 CrossRef CAS PubMed; (c) Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2021, 338, 1177 CrossRef PubMed.
- (a) L. M. Adleman, Science, 1994, 266, 1021 CrossRef CAS PubMed; (b) K. Sakamoto, H. Gouzu, K. Komiya, D. Kiga, S. Yokoyama, T. Yokomori and M. Hagiya, Science, 2000, 288, 1223 CrossRef CAS PubMed; (c) D. Y. Zhang and E. Winfree, J. Am. Chem. Soc., 2009, 131, 17303 CrossRef CAS PubMed.
- (a) C. Mao, W. Sun, Z. Shen and N. C. Seeman, Nature, 1999, 397, 144 CrossRef CAS PubMed; (b) B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605 CrossRef CAS PubMed; (c) E. S. Andersen, M. Dong, M. M. Nielsen, K. Jahn, R. Subramani, W. Mamdouh, M. M. Golas, B. Sander, H. Stark, C. L. P. Oliveira, J. S. Pedersen, V. Birkedal, F. Besenbacher, K. V. Gothelf and J. Kjems, Nature, 2009, 459, 73 CrossRef CAS PubMed.
- Y. Sato, Y. Hiratsuka, I. Kawamata, S. Murata and S. M. Nomura, Sci. Rob., 2017, 2, eaal3735 CrossRef PubMed.
- (a) A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300 CrossRef CAS PubMed; (b) K. Tanaka, G. H. Clever, Y. Takezawa, Y. Yamada, C. Kaul, M. Shionoya and T. Carell, Nat. Nanotechnol., 2006, 1, 190 CrossRef CAS PubMed; (c) J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46, 7587 CrossRef CAS PubMed; (d) S. Tanaka, K. Wakabayashi, K. Fukushima, S. Yukami, R. Maezawa, Y. Takeda, K. Tatsumi, Y. Ohya and A. Kuzuya, Chem. – Asian J., 2017, 12, 2388 CrossRef CAS PubMed; (e) Y. Takezawa, K. Mori, W.-E. Huang, K. Nishiyama, T. Xing, T. Nakama and M. Shionoya, Nat. Commun., 2023, 14, 4759 CrossRef CAS PubMed.
- (a) T. Wada, N. Minamimoto, Y. Inaki and Y. Inoue, J. Am. Chem. Soc., 2000, 122, 6900 CrossRef CAS; (b) D. Liu and S. Balasubramanian, Angew. Chem., Int. Ed., 2003, 42, 5734 CrossRef CAS PubMed; (c) T. Ohmichi, Y. Kawamoto, P. Wu, D. Miyoshi, H. Karimata and N. Sugimoto, Biochemistry, 2005, 44, 7125 CrossRef CAS PubMed; (d) T. Li and M. Famulok, J. Am. Chem. Soc., 2013, 135, 1593 CrossRef CAS PubMed; (e) A. Idili, A. Vallée-Bélisle and F. Ricci, J. Am. Chem. Soc., 2014, 136, 5836 CrossRef CAS PubMed; (f) Y. Hu, C.-H. Lu, W. Guo, M. A. Aleman-Garcia, J. Ren and I. Willner, Adv. Funct. Mater., 2015, 25, 6867 CrossRef CAS.
- (a) X. Liang, H. Nishioka, N. Takenaka and H. Asanuma, ChemBioChem, 2008, 9, 702 CrossRef CAS PubMed; (b) Y. Yoshimura and K. Fujimoto, Org. Lett., 2008, 10, 3227 CrossRef CAS PubMed; (c) S. Ogasawara and M. Maeda, Angew. Chem., Int. Ed., 2008, 47, 8839 CrossRef CAS PubMed; (d) X. Wamg and X. Liang, RSC Adv., 2016, 6, 93398 RSC; (e) S. Mori, K. Morihiro and S. Obika, Molecules, 2014, 19, 5109 CrossRef PubMed; (f) A. S. Lubbe, Q. Liu, S. J. Smith, J. W. Vries, J. C. M. Kistemaker, A. H. Vries, I. Faustino, Z. Meng, W. Szymanski, A. Herrmann and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 5069 CrossRef CAS PubMed; (g) M. W. Haydell, M. Centola, V. Adam, J. Valero and M. Famulok, J. Am. Chem. Soc., 2018, 140, 16868 CrossRef CAS PubMed; (h) A. M. Abdelhady, K. Onizuka, K. Ishida, S. Yajima, E. Mano and F. Nagatsugi, J. Org. Chem., 2022, 87, 2267 CrossRef CAS PubMed; (i) L. Rieger, B. Pfeuffer and H.-A. Wagenknecht, RSC Chem. Biol., 2023, 4, 1037 RSC; (j) T. Anderson, W. Wu, O. Sirbu, K. Tong, S. Winna, X. R. Liu, B. Kou, K. Murayama, H. Asanuma and Z. Wang, Angew. Chem., Int. Ed., 2024, 63, e202405250 CrossRef CAS PubMed; (k) T. Nakakuki, M. Toyonari, K. Aso, K. Murayama, H. Asanuma and T. de Greef, ACS Synth. Biol., 2024, 13, 521 CrossRef CAS PubMed; (l) L. Wohlrábová, M. Sahlbach, A. Heckel and T. Slanina, Chem. Commun., 2024, 60, 4366 RSC; (m) M. Wakano, M. Tsunoda, K. Murayama, J. Morimoto, R. Ueki, S. Aoyama-Ishiwatari, Y. Hirabayashi, H. Asanuma and S. Sando, J. Am. Chem. Soc., 2025, 147, 11477 CrossRef CAS PubMed.
- (a) C. J. Leumann, Bioorg. Med. Chem., 2002, 10, 841 CrossRef CAS PubMed; (b) J. C. Chaput and P. Herdewijn, Angew. Chem., Int. Ed., 2019, 58, 11570 CrossRef CAS PubMed; (c) K. Murayama and H. Asanuma, ChemBioChem, 2021, 22, 2507 CrossRef CAS PubMed.
- (a) U. Wenge and J. Wengel, Angew. Chem., Int. Ed., 2012, 51, 10026 CrossRef CAS PubMed; (b) R. S. Zhang, E. O. McCullum and J. C. Chaput, J. Am. Chem. Soc., 2008, 130, 5846 CrossRef CAS PubMed; (c) V. Kumar and V. Kesavan, RSC Adv., 2013, 3, 19330 RSC; (d) A. M. Kabza, B. E. Young and J. T. Sczepanski, J. Am. Chem. Soc., 2017, 139, 17715 CrossRef CAS PubMed; (e) W.-C. Hsieh, G. R. Martinez, A. Wang, S. F. Wu, R. Chamdia and D. H. Ly, Commun. Chem., 2018, 1, 89 CrossRef; (f) M. K. Skaanning, J. Bønnelykke, M. A. D. Nijenhuis, A. Samanta, J. M. Smidt and K. V. Gothelf, J. Am. Chem. Soc., 2024, 146, 20141 CrossRef CAS PubMed.
- H. Kashida, K. Murayama, T. Toda and H. Asanuma, Angew. Chem., Int. Ed., 2011, 50, 1285 CrossRef CAS PubMed.
- (a) K. Murayama, Y. Yamano and H. Asanuma, J. Am. Chem. Soc., 2019, 141, 9485 CrossRef CAS PubMed; (b) Y. Yamano, K. Murayama and H. Asanuma, Chem. – Eur. J., 2021, 27, 4599 CrossRef CAS PubMed.
- (a) S. Ogasawara and M. Maeda, Nucleic Acids Symp. Ser., 2008, 52, 369 CrossRef CAS PubMed; (b) Y. Saito, K. Matsumoto, Y. Takeuchi, S. S. Bag, S. Kodate, T. Morii and I. Saito, Tetrahedron Lett., 2009, 50, 1403 CrossRef CAS.
- S. Ogasawara, ACS Synth. Biol., 2018, 7, 2507 CrossRef CAS PubMed.
- S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed., 2004, 43, 1132 CrossRef CAS PubMed.
- K. Fujimoto, A. Yamada, Y. Yoshimura, T. Tsukaguchi and T. Sakamoto, J. Am. Chem. Soc., 2013, 135, 16161 CrossRef CAS PubMed.
- (a) M. S. Syamala and V. Ramamurthy, J. Org. Chem., 1986, 51, 3712 CrossRef CAS; (b) H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399 CrossRef; (c) F. D. Lewis, T. Wu, E. L. Burch, D. M. Bassani, J.-S. Yang, S. Schneider, W. Jager and R. L. Letsinger, J. Am. Chem. Soc., 1995, 117, 8785 CrossRef CAS; (d) W. Herrmann, S. Wehrle and G. Wenz, Chem. Commun., 1997, 1709 RSC; (e) B. Juskowiak and M. Chudak, Photochem. Photobiol., 2004, 79, 137 CrossRef CAS PubMed; (f) M. Schraub, H. Gray and N. Hampp, Macromolecules, 2011, 44, 8755 CrossRef CAS; (g) O. A. Fedorova, A. E. Saifutiarova, E. N. Gulakova, E. O. Guskova, T. M. Aliyeu, N. E. Shepel and Y. V. Fedorov, Photochem. Photobiol. Sci., 2019, 18, 2208 CrossRef CAS PubMed; (h) J. Seylar, D. Stasiouk, D. L. Simone, V. Varshney, J. E. Heckler and R. McKenzie, RSC Adv., 2021, 11, 6504 RSC; (i) T. P. Martyanov, A. P. Vorozhtsov, N. A. Aleksandrova, I. V. Sulimenkov, E. N. Ushakov and S. P. Gromov, ACS Omega, 2022, 7, 42370 CrossRef CAS PubMed.
- (a) D. M. Bassani, X. Sallenave, V. Darcos and J.-P. Desvergne, Chem. Commun., 2001, 1446 RSC; (b) H. Maeda, R. Hiranabe and K. Mizuno, Tetrahedron Lett., 2006, 47, 7865 CrossRef CAS; (c) C. T. Eckdahl, C. Ou, S. Padgaonkar, M. C. Hersam, E. A. Weiss and J. A. Kalow, Org. Biomol. Chem., 2022, 20, 6201 RSC; (d) V. X. Truong and C. Barner-Kowollik, Aust. J. Chem., 2022, 75, 899 CrossRef.
- (a) J. Zhang, S.-X. Tang, R. Fu, X.-D. Xu and S. Feng, J. Mater. Chem. C, 2019, 7, 13786 RSC; (b) K. Kalayci, H. Frisch, C. Barner-Kowollik and V. X. Truong, Adv. Funct. Mater., 2020, 30, 1908171 CrossRef CAS; (c) K. Kalayci, H. Frisch, V. X. Truong and C. Barner-Kowollik, Nat. Commun., 2020, 11, 4193 CrossRef PubMed; (d) J. Bai, Z. Shi, X. Ma, J. Yin and X. Jiang, Macromol. Rapid Commun., 2022, 43, 2200055 CrossRef CAS PubMed; (e) R. T. Michenfelder, L. Delafresnaye, V. X. Truong, C. Barner-Kowollik and H.-A. Wagenknecht, Chem. Commun., 2023, 59, 4012 RSC; (f) D. Hoenders, S. Ludwanowski, C. Barner-Kowollik and A. Walther, Angew. Chem., Int. Ed., 2024, 63, e202405582 CrossRef CAS PubMed; (g) C. Dohmen and H. Ihmels, ChemPhotoChem, 2024, 8(7), e202300318 CrossRef CAS.
- (a) S. Murata, T. Toyota, S. I. M. Nomura, T. Nakakuki and A. Kuzuya, Adv. Funct. Mater., 2022, 32, 2201866 CrossRef CAS; (b) A. Kuzuya, S. I. M. Nomura, T. Toyota, T. Nakakuki and S. Murata, IEEE Trans. Mol. Biol. Multi-Scale Commun., 2023, 9, 354 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, NMR charts, and MALDI-TOF MS charts. See DOI: https://doi.org/10.1039/d5ob00924c |
| This journal is © The Royal Society of Chemistry 2025 |