
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence detection of DNA with a single-base mismatch by a Tm-independent peptide nucleic acid (PNA) twin probe†
Koki
Ishii
a,
Hajime
Shigeto
 b,
Shohei
Yamamura
b,
Yoshitane
Imai
a,
Takashi
Ohtsuki
c and
Mizuki
Kitamatsu
*a
b,
Shohei
Yamamura
b,
Yoshitane
Imai
a,
Takashi
Ohtsuki
c and
Mizuki
Kitamatsu
*a
aDepartment of Applied Chemistry, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
bHealth and Medical Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 2217-14 Hayashi-cho, Takamatsu, Kagawa 761-0395, Japan
cDepartment of Interdisciplinary Science and Engineering in Health Systems, Okayama University, 3-1-1 Tsushimanaka, Okayama 700-8530, Japan
First published on 13th June 2025
Abstract
There is a need to develop efficient methods for detecting target nucleic acids to enable the rapid diagnosis and early treatment of diseases. We previously demonstrated that a peptide nucleic acid (PNA) twin probe, consisting of two PNAs each containing a fluorescent dye, with pyrene at one end, detects target DNA sequence-specifically through pyrene excimer emission. In this study, to advance the development of this probe system, we further investigated the fluorescence properties of the PNA twin probe P1 and P2, and found that the excimer fluorescence was significantly reduced when a mismatched base in the DNA sequence was present at the site of P1 closest to the pyrene. In other words, this probe was found to detect single-base mismatches without taking into account the thermal stability of the PNA/DNA hybrid. The detection limit of this PNA twin probe for the single-base-mismatched DNA was 2.7 nM. In the future, this probe should lead to a method to detect point mutations in endogenous nucleic acids within cells.
Introduction
Peptide nucleic acid (PNA) is a nucleic acid analog that contains a peptide as the main chain and nucleic acids as the side chains.1 PNA forms sequence-specific hybrids with DNA and RNA. These hybrids have higher thermal stability, higher resistance to proteases and nucleases, and superior sequence specificity relative to hybrids formed from DNA or RNA themselves. In particular, the superior sequence specificity of PNA2,3 makes it promising for use as a probe for single-nucleotide polymorphisms (SNPs).PNAs are expected to be used as probes for detecting nucleic acids with specific base sequences. Among these probes, those combining PNA and fluorescent dyes have been widely studied4–7 because they provide visual output of PNA recognizing nucleic acids. We have also investigated a PNA twin probe, which consists of two PNAs each modified with the fluorescent dye pyrene (Pyr).8 When the two PNAs of this probe are present together with DNA containing perfectly complementary base sequences, the two pyrenes of this probe assemble on the DNA via hybrids. As a result, the pyrene monomer fluorescence is converted to excimer fluorescence, allowing the detection of DNA (upper and middle panels in Fig. 1).
 | ||
| Fig. 1 (Upper panel) Chemical structures of PNA twin probe P1 and P2 and their sequences. The red and blue colors of P1 and P2 in the structures indicate PNA, respectively. The N-terminus of each peptide is a free amino (H2N–) group, and the C-terminus is a primary amide (–CONH2). The solid underlined red and dotted underlined blue italic letters in the sequences indicate the base sequences of PNA in P1 and P2, respectively. Pyr indicates pyrenylalanine, and C6 indicates an alkyl linker [–NH–(CH2)6–CO–]. (Middle panel) Schematic diagram of base sequence-specific pyrene excimer emission of the PNA twin probe against a target DNA. When the DNA contains base sequences complementary to P1 and P2, each of these forms hybrids with the DNA. At this time, the two Pyr at the ends of P1 and P2 are designed to face each other and be close to each other on the DNA template. As a result, the Pyr are appropriately positioned to emit excimers. (Lower panel) The base sequences of DNA D1–D18 used in this study. The solid underlined red and dotted underlined blue letters in the sequences indicate the DNA base sequences that are antiparallel and complementary to P1 and P2, respectively. | ||
The fluorescent detection technique using two PNAs is expected to be able to distinguish nucleic acids with longer sequences with higher selectivity than a single PNA probe, and to be able to detect them in a ratiometric manner, unlike detection by differences in fluorescence intensity. Such techniques have been well developed for DNA probes,9–11 but are rarely seen for PNA probes.4 Further efforts should thus be made to aid the development of PNA twin probes. Several studies have demonstrated this approach using various dye combinations, for example, fluorescein (Fam)/Cy5 by Taylor's group,12,13 thiazole orange (TO)/AlexaFluor-594 by Peteanu's group,14 Cy3/Cy5 by Artero's group,15 Fam/tetramethylrhodamine16 and TO/oxazolopyridine analog/NIR66417 by Seitz's group, and Pyr/Pyr8 and boronic acid/its ligand18 by our group.
As described in this report, we investigated the fluorescence properties of the PNA twin probe for DNA with a single-base mismatch as further development of this probe.
Results and discussion
Design of PNA twin probe P1 and P2
We used the PNA twin probe P1 and P2 for this investigation (Fig. 1; upper panel). P1 is the same as P1C68 used in a previous study, and a C6 linker [–NH–(CH2)6–CO–] is introduced between the pyrenylalanine with Pyr in the side chain and the N-terminus of the PNA. The presence of the alkyl linker has a significant effect on the excimer fluorescence derived from the PNA twin probe. We previously reported that the excimer formation via two Pyr-PNAs on a DNA template depends on the length of the linker, and the introduction of the C6 linker most enhanced the excimer fluorescence of the probe.8 Therefore, P1 was used in this study. P2 is also the same peptide as used in the previous study.8 Both PNAs are 10-mers. In previous studies,8P1/P2 showed the highest excimer/monomer ratio (E/M value) for the target DNA D1, which has sequences complementary to each of them and has two adenine bases inserted between these sequences. Both P1 and P2 contain tetra-arginine at one end, which is expected to improve the water solubility and to facilitate intracellular delivery as a cell-penetrating peptide (CPP)19 in the future. We have shown that tetra-arginine plays these roles in another report on a PNA twin probe modified with a boronic acid and its ligand, instead of Pyr.18We used DNAs D1–D18 in this study (Fig. 1; lower panel). Among them, D1 contains a sequence perfectly complementary to both P1 and P2. D9–D13 have one mismatched base at positions 9, 7, 5, 3, and 1 from the N-terminus of P1, respectively. D14–D18 have one mismatched base at positions 1, 3, 5, 7, and 9 from the C-terminus of P2. D2 is a scrambled sequence of D1.
Response of P1 and P2 to DNA with a single-base mismatch
First, we measured UV melting curves of P1 with D3, which is complementary to P1, or D4–D8, which have one mismatched base, to confirm the thermal stability of the PNA/DNA hybrid before investigating the fluorescence response of the PNA twin probe to DNA (Fig. 2A).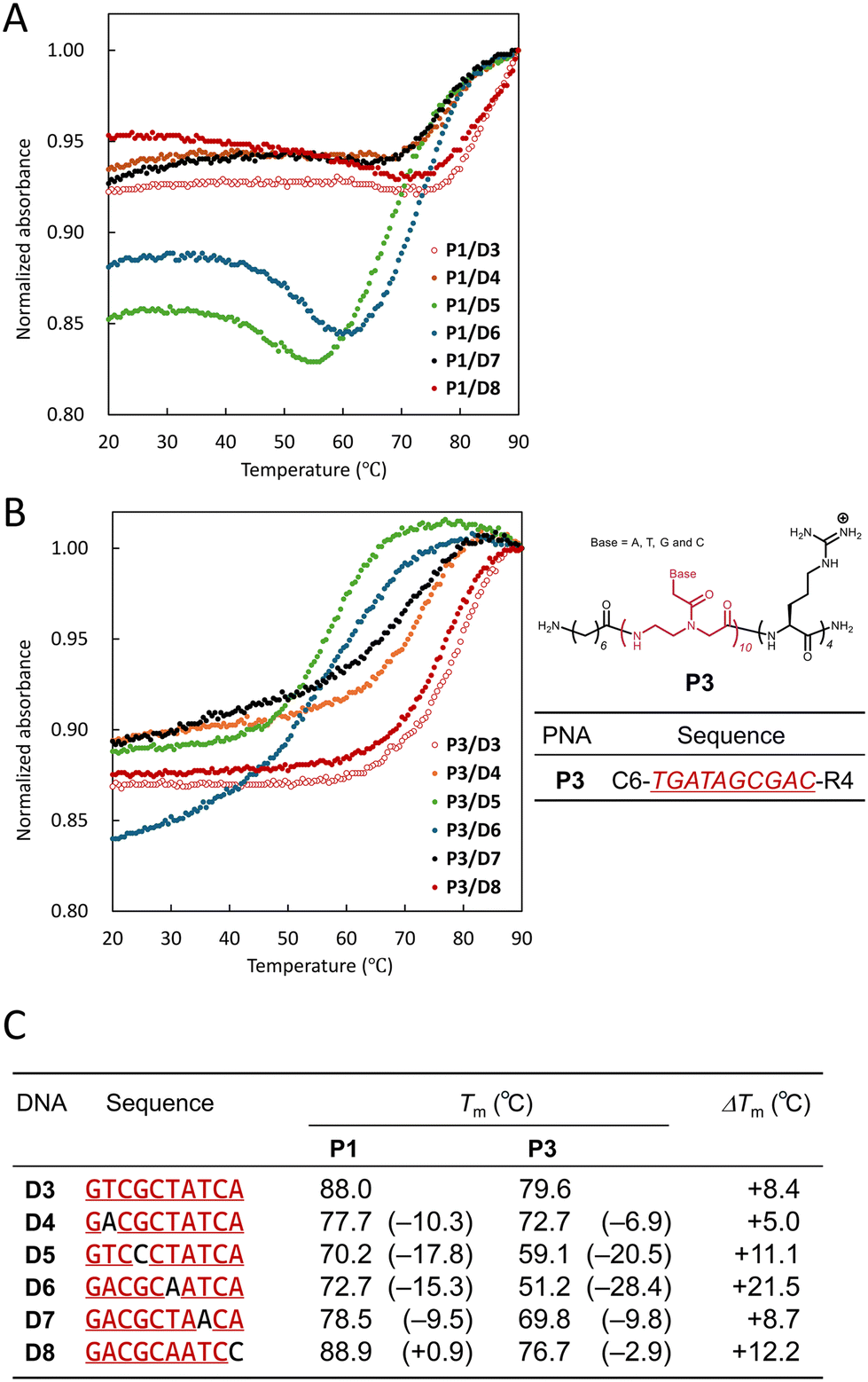 | ||
| Fig. 2 UV melting curves of equimolar mixtures of P1 (A) and P3 (B) with DNA D3–D8 and their Tm values (C). The values in brackets in C are the Tm values of D4–D8 subtracted from that of D3, and ΔTm is the Tm value of P1/DNA subtracted from that of P3/DNA. The concentrations of PNA and DNA were 5 μM each, dissolved in an aqueous solution (10 mM PBS, pH 7.0). The absorption wavelength was measured at 260 nm. The measurement range was 20–90 °C. | ||
The UV melting curve of an equimolar mixture of P1/D3 (open red circles) showed a hypochromic effect above 80 °C, and a sigmoidal curve was observed. The melting temperature (Tm) was determined to be 88.0 °C. P1/D8, which has one mismatched base at the first position from the N-terminus (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Pyr side) of P1 (filled red circles), showed a profile similar to that of P1/D3, and Tm was 88.9 °C, slightly higher than that of P1/D3. In the case of P1/D7 and P1/D4, which have one mismatched base at the 3rd and 9th positions from the N-terminus of P1 (black and orange circles, respectively), the sigmoidal curves were shifted toward lower temperatures than that of P1/D3. Their Tms were 78.5 °C and 77.7 °C, respectively. P1/D6 and P1/D5, which have one mismatched base at the 5th and 7th positions from the N-terminus of P1 (blue and green circles, respectively), showed a large hypochromic effect in the range of 60–90 °C. Their Tms were 72.7 °C and 70.2 °C, respectively. In addition, the absorbance of P1/D6 and P1/D5 increased in the range of 40–60 °C and remained almost constant in the range of 20–40 °C. Although there is no clear reason for this change in absorbance, it is speculated to be due to the influence of Pyr in P1.
Pyr side) of P1 (filled red circles), showed a profile similar to that of P1/D3, and Tm was 88.9 °C, slightly higher than that of P1/D3. In the case of P1/D7 and P1/D4, which have one mismatched base at the 3rd and 9th positions from the N-terminus of P1 (black and orange circles, respectively), the sigmoidal curves were shifted toward lower temperatures than that of P1/D3. Their Tms were 78.5 °C and 77.7 °C, respectively. P1/D6 and P1/D5, which have one mismatched base at the 5th and 7th positions from the N-terminus of P1 (blue and green circles, respectively), showed a large hypochromic effect in the range of 60–90 °C. Their Tms were 72.7 °C and 70.2 °C, respectively. In addition, the absorbance of P1/D6 and P1/D5 increased in the range of 40–60 °C and remained almost constant in the range of 20–40 °C. Although there is no clear reason for this change in absorbance, it is speculated to be due to the influence of Pyr in P1.
Following these results, we also measured the UV melting curves of DNA with P3, a Pyr-free P1 (Fig. 2B). All of the UV melting curves of P3/D3–D8 showed clear sigmoidal curves, and the change in absorbance at low temperature observed for P1/D6 and P1/D5 was not observed.
We summarize the Tm of P1/D3–D8 and P3/D3–D8 in Fig. 2C. For P1, D8, which has one mismatched base at the first position, has higher thermal stability (+0.9 °C) than D3, which is a full match. This suggests that Pyr enhances the hybrid stability between PNA and DNA. In addition, the hybrid stability of P1/DNA with a mismatched base in the center (D5: −17.8 °C and D6: −15.3 °C) is lower than that of DNA with a mismatched base at the end (D4: −10.3 °C and D7: −9.5 °C). This tendency was also observed for P3/DNA. It is reasonable that the presence of a mismatched base in the center of PNA has a greater effect on the PNA/DNA hybrid stability. Comparing the thermal stability between P1/D3–D8 and P3/D3–D8 hybrids, it is found that P1/DNA is more stable than P3/DNA in all cases (ΔTm = +5.0–+21.5 °C). Furthermore, the differences in Tm between P1/D4–D8 and P1/D3 ranged from +0.9 °C to −17.8 °C, whereas those between P3/D4–D8 and P3/D3 ranged from −2.9 °C to −28.4 °C. In other words, the presence of Pyr in PNA reduces the differences in hybrid stability due to the presence of a mismatched base and the position of the mismatched base. These findings also indicate that the presence of Pyr enhances the hybrid stability of PNA/DNA, as mentioned above.
These results indicate that PNAs can discriminate a single mismatch in DNA by temperature tuning, but that the tuning must be strictly controlled, and that Pyr-modified PNAs have more difficulty discriminating DNA mismatches by temperature control.
Based on the results of Fig. 2, we measured fluorescence spectra of an equimolar mixture of P1/P2 and DNA with one mismatch at ambient temperature to investigate the fluorescence properties of the PNA twin probe for DNA in detail (Fig. 3). The fluorescence spectra of P1/P2/D9–D13 with one mismatch at the P1 site are shown in the left panel of Fig. 3A. In the case of D2 (a blue dashed line), a scrambled sequence where P1/P2 are not expected to form a hybrid, only the fluorescence from Pyr monomer was observed at 380 nm (3090) and 400 nm (2980). On the other hand, in the case of D13 (a red solid line), which has one mismatch at the first base from the N-terminus of P1, only the fluorescence from the monomer was observed, as in D2, but the fluorescence intensity was significantly reduced (1000 and 970, respectively). The decrease in the monomer fluorescence suggests that P1 is in the vicinity of DNA; that is, Pyr is quenched by the nucleobase in DNA upon hybrid formation of P1 with D13, as also reported for the twin probe based on two Pyr-DNA by Kool et al.20 In other words, under the conditions applied here, even DNA with a single mismatch can form a hybrid with the PNA twin probe, which is supported by the results shown in Fig. 2.
 | ||
| Fig. 3 (A) Fluorescence spectra of equimolar mixtures of P1/P2 and DNA (D1, D2, and D9–D13, left panel; and D1, D2, and D14–D18, right panel) in aqueous solution (10 mM PBS, pH 7.0) at 25 °C. Each concentration of P1, P2, and DNA was 500 nM. Excitation wavelength was 350 nm. (B) E/M values of P1/P2/DNA. Each bar represents the mean of three measurements. (C) Plausible illustrations of a P1/P2/D13 hybrid (top) and a P1/P2/D14 hybrid (bottom). | ||
Next, in the case of D1 (a red dashed line), which is perfectly complementary to P1/P2, fluorescence from the Pyr monomer was observed at 380 nm (1030) and 400 nm (1230), as well as excimer fluorescence at 480 nm (1230). This indicates that P1 and P2 are assembled on D1 through their hybridization, and efficient excimer formation occurs between the two Pyr. This is consistent with our previous report.8D9–D12 (orange, green, blue, and black solid lines, respectively), which have one mismatch at the 9th, 7th, 5th, and 3rd bases on the N-terminus of P1, showed a profile similar to that of D1. In addition, the fluorescence intensity from the monomers in all cases was significantly reduced compared with that of D2 or was almost the same as that of D1, which is presumably due to the conversion from monomer to excimer fluorescence and/or the quenching of Pyr fluorescence by hybridization of PNA with DNA. Notably, among D9–D13, which have one-base mismatches at various positions relative to P1, only D13, which contains a mismatch at the position closest to the N-terminus (Pyr side) of P1, almost eliminated excimer fluorescence (see below).
We also measured the fluorescence spectra of mixtures of P1/P2/D14–D18 with one mismatch at the P2 site (right panel of Fig. 3A). D14 and D15 (red and black solid lines, respectively), which have one mismatch at the first and third positions from the C-terminus of P2 (i.e., the mismatch is located close to Pyr), showed monomer fluorescence at 380 and 400 nm and excimer fluorescence at 480 nm, but the excimer fluorescence intensity was lower than that of D1. The monomer fluorescence was also significantly reduced, similar to that of D13. Meanwhile, D16–D18 (blue, green, and orange solid lines, respectively), which contain one mismatch at the 5th, 7th, and 9th positions from the C-terminus of P2 (i.e., the mismatch is located far from Pyr), showed almost the same profile as D1.
Based on the results in Fig. 3A, we summarized the ratio of excimer emission intensity at 480 nm to monomer emission intensity at 380 nm (E/M value) for each DNA (Fig. 3B). The E/M value of P1/P2/D1 was 1.05, and that of P1/P2/D2 was 0.07. These results indicate that the PNA twin probe clearly distinguishes the target DNA. The E/M values of P1/P2/D9–D12 were in the range of 0.66–0.94 (D9: 0.65, D10: 0.94, D11: 0.84, and D12: 0.69). These values are almost the same as that of D1, suggesting that, even if a single-base mismatch exists in the DNA, hybrids are formed under the present conditions and do not affect the excimer formation between Pyr. Meanwhile, the E/M value of P1/P2/D13 was 0.16. This is almost the same as that of D2, suggesting that the hybrids between P1/P2 and D13 are formed, but the excimer formation is significantly inhibited. We also assessed the E/M values of other mismatch bases (T/T and G/T mismatches; D13T and D13G, respectively) other than the first C/T mismatch (D13) from the N-terminus of P1 (Fig. S1†). The results showed E/M values of 0.13 and 0.12, respectively, and the presence of the mismatch bases showed a significant decrease in excimer fluorescence compared to the full match D1. Next, the E/M values of P1/P2/D16–D18 were in the range of 0.89–0.99 (D16: 0.89, D17: 0.98, and D18: 0.99). These values were also the same as that of D1. However, the E/M values of P1/P2/D14 and P1/P2/D15 were 0.40 and 0.34, respectively. These values were smaller than that of D1, and although not as large as that of D13, they suggested that excimer formation was inhibited.
These results indicate that the presence of a mismatched base near the Pyr of P1 or P2 inhibits excimer formation (Fig. 3C). Presumably, in the hybridization of each PNA on the DNA template, the binding between the nucleobases of the PNA and DNA, especially near the Pyr, suppresses a swing (positional instability) of the Pyr and strongly fixes it in place, thereby providing stable excimer formation between Pyr. In other words, these results indicate that, if the fixation of Pyr through the PNA on the DNA template is insufficient, the swing of Pyr cannot be suppressed, inhibiting excimer formation. It is speculated that D13 caused a large decrease in the E/M value compared with D14 because of the effect of the C6 linker. In P1, where the linker is introduced between Pyr and PNA, the swing is amplified by the linker when the bond between the nucleobases closest to Pyr is lost. It is reasonable to consider that this instability significantly hinders the excimer formation of Pyr, resulting in a larger decrease in the E/M value than the loss of the bond between the nucleobases at the P2 site.
We investigated the concentration dependence of fluorescence detection of DNA with one mismatch by the PNA twin probe (Fig. 4). Within the measured concentration range (500 pM–500 nM), almost no excimer emission was observed for P1/P2/D2 and P1/P2/D13, and the E/M values of P1/P2/D2 and P1/P2/D13 were almost constant, in the ranges of 0.06–0.12 and 0.12–0.16, respectively. On the other hand, P1/P2/D1 showed high E/M values in the range of 27.5–500 nM (27.5 nM: 0.85, 50 nM: 0.89, 275 nM: 0.91, and 500 nM: 0.96). Below 10 nM, the E/M value was lower, but even at 2.8 nM it was 0.37. Using the results in the low concentration range, we calculated the limit of detection (LoD) to be 2.7 nM.
 | ||
| Fig. 4 E/M values of equimolar mixtures of P1/P2/D1 (red circles), P1/P2/D2 (blue circles), and P1/P2/D13 (black circles) at various concentration. | ||
In the study of SNPs, the LoD of various PNA systems has been reported. Zhao's group used a bare 12-mer PNA in combination with S1 nuclease/single-walled carbon nanotube/hemin/tetramethylbenzidine21 and S1 nuclease/gold nanoparticles,22 while Tamiya's group used an 11-mer PNA modified on a gold electrode surface in combination with S1 nuclease/ferrocene (Fc)-conjugated chitosan nanoparticles23 to successfully distinguish DNA with a single mismatch colorimetrically and electrochemically, respectively. Their LoDs were 0.11 nM, 6.1 nM, and 1 fM, respectively. Zhao's group also succeeded in electrochemically identifying RNAs with one mismatch (LoD: 0.15 fM) using a system combining two hairpin DNAs modified with Fc on a gold electrode surface.24 Our group has successfully identified RNAs with one mismatch by fluorescence emission (LoD: 4.7 nM) using a system combining a 10-mer PNA modified with a fluorescein and quencher-modified DNA.6 Meanwhile, Seitz's group has successfully identified DNAs with one mismatch by mass using chemical ligation on a DNA template using a system with two 11–12-mer PNAs (sister probes) modified with cysteine or thioester at one end (LoD: 29 pM).25 Among these, our PNA twin-probe system is relatively simple and is considered to show reasonable results for detection in solution.
Conclusions
In this report, we first discussed the fluorescence response of PNA twin probes to DNA with a single mismatch. In the fluorescence spectra of an equimolar mixture of the probe and DNA in aqueous solution, the appearance and disappearance of excimer fluorescence through Pyr was observed depending on the position of the mismatch in DNA. This suggested that, if we design the PNA twin probe so that a single mismatch exists closest to the pyrene, the mismatched DNA can be detected. Most fluorescent PNA probes, except for systems using enzymes such as S1 nuclease,21–23 distinguish and detect their targets by utilizing the difference in thermal stabilities of hybrids with nucleic acids.4 In other words, it is necessary to consider the thermal stability of the hybrid when designing the PNA probe, and precise control of the temperature will be required, although the PNA has the superior sequence specificity for nucleic acids. In contrast, the PNA twin probe will be able to detect nucleic acids with a single mismatch without worrying about the thermal stability, as long as it is set at the temperature (usually room temperature) at which it hybridizes with nucleic acids, in addition to the superior sequence specificity.While various methods for intracellular delivery of PNA have been reported so far,26 the use of PNA as tools in live cells is expected. The Tm-independent fluorescence detection of DNA using the PNA twin probe shown in this study is expected to be applicable to the fluorescence detection of endogenous nucleic acids containing a single-base mismatch in live cells. Although there are some points that need to be considered, such as the incubation concentration of the probe to prevent the excimer signal from being masked by the monomer signal, the hybridization of the probe to RNA, and versatility of the probe to various base sequences, we are currently conducting research on the fluorescence detection of nucleic acids in live cells using this probe, taking these points into consideration.
Experimental
Materials
9-Fluorenylmethyloxycarbonyl group (Fmoc)-derivatized amino acids, Fmoc-derivatized super acid labile-poly(ethylene)glycol (Fmoc-NH-SAL-PEG) resin, Fmoc-Ala(Pyn)-OH (Pyr), Fmoc-Ahp(7)-OH (C6 linker), Fmoc-Arg(Pbf)-OH, piperidine, O-(1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU), N-methylmorpholine (NMM), trifluoroacetic acid (TFA), and triisopropylsilane (TIPS) were purchased from Watanabe Chemicals (Hiroshima, Japan). Fmoc-derived PNA monomers [Fmoc-A(Bhoc)-OH, Fmoc-T-OH, Fmoc-G(Bhoc)-OH, and Fmoc-C(Bhoc)-OH] were purchased from Panagene (Daejeon, South Korea). N,N′-Dimethylformamide (DMF), N-methyl-2-pyrrolidinone (NMP), diethyl ether, acetonitrile, and dichloromethane (DCM) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Phosphate-buffered saline (PBS, 100 mM, pH 7.0) was purchased from Nacalai Tesque (Kyoto, Japan). DNA oligomers were purchased from Thermo Fisher Scientific (Waltham, MA, USA).Peptide synthesis
As the PNA twin probe, P1 and P2, and P3 (Fig. 1) were prepared by Fmoc-based solid-phase peptide synthesis, in accordance with a previous report.8 Fmoc-NH-SAL-PEG resin containing 7.2 μmol Fmoc on its surface was used as a solid-phase support resin for the peptide synthesis after swelling with DMF for 1 day. An Fmoc deprotection and coupling process of the peptide extending on the resin surface was carried out at room temperature without capping. For the deprotection process, 20% piperidine in DMF was added to the resin and stirred for 7 min, and for the coupling process, 4 parts of Fmoc-derived PNA monomer, Fmoc-Ala(Pyn)-OH, Fmoc-Ahp(7)-OH, or Fmoc-Arg(Pbf)-OH, 3.6 parts of HBTU, and 11.5 parts of NMM in DMF or DMF/NMP were added to the resin and stirred for 50 min. After the final deprotection step, the resin was washed with DCM, dried, and stirred with 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 (v/v) TFA/TIPS/water at room temperature for 90 min to globally deprotect the side chain protecting groups of a crude peptide on the resin and cleave the peptide from the resin. The crude peptide in the cleavage cocktail was added to cold diethyl ether to precipitate the peptide, which was then repeatedly washed with diethyl ether until neutral pH was reached. The solid peptide was air-dried and then dissolved in 0.1% aqueous TFA/acetonitrile. The peptide solution was purified by reversed-phase high-pressure liquid chromatography (HPLC; Shimadzu, Kyoto, Japan) equipped with a C18 preparative column (Cadenza 5CD-C18; Imtakt, Kyoto, Japan). The purification was performed using a linear gradient of 0.1% aqueous TFA and acetonitrile at a detection wavelength of 340 nm and a flow rate of 10.0 mL min−1. The fractionated solutions were analysed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (AXIMA Confidence; Shimadzu) (Fig. S2, S4, and S6†) and HPLC on a C18 analytical column (Cadenza CD-C18; Imtakt) (Fig. S3, S5, and S7†) to obtain the final products P1, P2 and P3.
:2.5:2.5 (v/v) TFA/TIPS/water at room temperature for 90 min to globally deprotect the side chain protecting groups of a crude peptide on the resin and cleave the peptide from the resin. The crude peptide in the cleavage cocktail was added to cold diethyl ether to precipitate the peptide, which was then repeatedly washed with diethyl ether until neutral pH was reached. The solid peptide was air-dried and then dissolved in 0.1% aqueous TFA/acetonitrile. The peptide solution was purified by reversed-phase high-pressure liquid chromatography (HPLC; Shimadzu, Kyoto, Japan) equipped with a C18 preparative column (Cadenza 5CD-C18; Imtakt, Kyoto, Japan). The purification was performed using a linear gradient of 0.1% aqueous TFA and acetonitrile at a detection wavelength of 340 nm and a flow rate of 10.0 mL min−1. The fractionated solutions were analysed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (AXIMA Confidence; Shimadzu) (Fig. S2, S4, and S6†) and HPLC on a C18 analytical column (Cadenza CD-C18; Imtakt) (Fig. S3, S5, and S7†) to obtain the final products P1, P2 and P3.
UV melting curve measurement
Molar concentrations of P1, P3 and D3–D8 were estimated from the absorbance at 260 nm and were measured using a UV-vis spectrometer (JASCO V-560) with molar extinction coefficients of the nucleobases. The UV melting curves of the equimolar mixtures of P1 or P3 with D3–D8 in aqueous buffer (100 mM PBS; pH 7.0), were measured using a Shimadzu TMSPC-8 Tm Analysis System that was equipped with a UV-visible spectrometer (UV-1700) and an eight-position Peltier temperature controller. Each concentration of the PNA and DNA was 5.0 μM, and the quartz cell length was 1 cm. The melting curves were recorded with cooling of the solution by 0.5 °C/0.5 min, from 90 to 20 °C, while measuring the absorbance at 260 nm. The observed absorbance was normalized to that at 90 °C. The melting temperature at which 50% of the strands remained hybridized (Tm) was obtained using a TMSPC-8 with Tm analysis software.Fluorescence measurement
All fluorescence measurements were conducted with a JASCO FP-8200 fluorescence spectrometer and a 1 cm quartz cell at an excitation wavelength of 350 nm in aqueous buffer (10 mM PBS, pH 7.0) at 25 °C. Emission wavelengths were measured between 370 and 580 nm. The final concentrations of each of P1, P2, and DNA were adjusted to 500 nM. To determine the limit of detection of the PNA twin probe, the final concentrations of each of P1, P2, and DNA (D1, D2, and D13) were further adjusted to 0.5, 2.75, 5, 27.5, 50, and 275 nM. The detection limit of the probe to the target DNA was determined using the following equation: detection limit = [3 × (standard deviation at 0 nM)]/(slope). In this case, the standard deviation of 0 nM was substituted with the standard deviation when P1 and P2 were adjusted to 0.5 nM, respectively.Author contributions
K. Ishii: data curation, formal analysis, investigation, validation, visualization, writing – original draft. H. Shigeto: funding acquisition, investigation, resources and writing – review & editing. S. Yamamura: funding acquisition, resources and writing – review & editing. Y. Imai: conceptualization and writing – review & editing. T. Ohtsuki: conceptualization, formal analysis and writing – review & editing. M. Kitamatsu: conceptualization, data curation, funding acquisition, methodology, resources, supervision, visualization and writing – original draft. All authors approved the final version of the manuscript for submission.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
We thank Sakura Tsuchitani and Miyu Toyama (Kindai University) for providing technical support. This work was supported by 2023 Kindai University Research Enhancement Grant (IP009).References
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497, DOI:10.1126/science.1962210
.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Norden and P. E. Nielsen, Nature, 1993, 365, 566, DOI:10.1038/365566a0
- J. Wang, E. Palecek, P. E. Nielsen, G. Rivas, X. Cai, H. Shiraishi, N. Dontha, D. Luo and P. A. M. Farias, J. Am. Chem. Soc., 1996, 118, 7667, DOI:10.1021/ja9608050
- T. Vilaivan, Beilstein J. Org. Chem., 2018, 14, 253, DOI:10.3762/bjoc.14.17
- H. Shigeto, T. Kishi, K. Ishii, T. Ohtsuki, S. Yamamura and M. Kitamatsu, Processes, 2022, 10, 722, DOI:10.3390/pr10040722
- K. Tabara, K. Watanabe, H. Shigeto, S. Yamamura, T. Kishi, M. Kitamatsu and T. Ohtsuki, Bioorg. Med. Chem. Lett., 2021, 51, 128359, DOI:10.1016/j.bmcl.2021.128359
- H. Shigeto, E. Yamada, M. Kitamatsu, T. Ohtsuki, A. Iizuka, Y. Akiyama and S. Yamamura, Micromachines, 2020, 11, 628, DOI:10.3390/mi11070628
- K. Ishii, S. Tsuchitani, M. Toyama, H. Shigeto, S. Yamamura, T. Ohtsuki, Y. Imai and M. Kitamatsu, Bioorg. Med. Chem. Lett., 2022, 71, 128838, DOI:10.1016/j.bmcl.2022.128838
- J. Chen, C. Shi, X. Kang, X. Shen, X. Lao and H. Zheng, Anal. Methods, 2020, 12, 884, 10.1039/c9ay02332a
- A. S. Boutorine, D. S. Novopashina, O. A. Krasheninina, K. Nozeret and A. G. Venyaminova, Molecules, 2013, 18, 15357, DOI:10.3390/molecules181215357
- D. M. Kolpashchikov, Chem. Rev., 2010, 110, 4709, DOI:10.1021/cr900323b
- Z. Wang, K. Zhang, Y. Shen, J. Smith, S. Bloch, S. Achilefu, K. L. Wooley and J. Taylor, Org. Biomol. Chem., 2013, 11, 3159, 10.1039/c3ob26923j
- B. Y. Oquare and J. Taylor, Bioconjugate Chem., 2008, 19, 2196, DOI:10.1021/bc800284x
- K. L. Robertson, L. Yu, B. A. Armitage, A. J. Lopez and L. A. Peteanu, Biochemistry, 2006, 45, 6066, DOI:10.1021/bi052050s
- A. M. Blanco, L. Rausell, B. Aguado, M. Perez-Alonso and R. Artero, Nucleic Acids Res., 2009, 37, e116, DOI:10.1093/nar/gkp551
- C. Dose, S. Ficht and O. Seitz, Angew. Chem., Int. Ed., 2006, 45, 5369, DOI:10.1002/anie.200600464
- G. Fang, J. Chamiolo, S. Kankowski, F. Hövelmann, D. Friedrich, A. Löwer, J. C. Meier and O. Seitz, Chem. Sci., 2018, 9, 4794, 10.1039/c8sc00457a
- K. Ishii, Y. Hattori, H. Shigeto, S. Yamamura and M. Kitamatsu, Chem. Lett., 2024, 53, upae169, DOI:10.1093/chemle/upae169
- S. Futaki, Adv. Drug Delivery Rev., 2005, 57, 547, DOI:10.1016/j.addr.2004.10.009
- P. L. Paris, J. M. Langenhan and E. T. Kool, Nucleic Acids Res., 1998, 26, 3789, DOI:10.1093/nar/26.16.3789
- M. Xu, S. Xing, Y. Zhao and C. Zhao, Talanta, 2021, 232, 122420, DOI:10.1016/j.talanta.2021.122420
- S. Xing, X. Xu, P. Fu, M. Xu, T. Gao, X. Zhang and C. Zhao, Colloids Surf., B, 2019, 181, 333, DOI:10.1016/j.colsurfb.2019.05.069
- K. Kerman, M. Saito and E. Tamiya, Anal. Bioanal. Chem., 2008, 391, 2759, DOI:10.1007/s00216-008-2204-8
- X. Yu, S. Ding, Y. Zhao, M. Xu, Z. Wu and C. Zhao, Talanta, 2024, 266, 125020, DOI:10.1016/j.talanta.2023.125020
- P. L. Lareo, M. W. Linscheid and O. Seitz, J. Mass Spectrom., 2019, 54, 676, DOI:10.1002/jms.4382
- C. Avitabile, M. T. Cerasa, A. D'Aniello, M. Saviano and M. Moccia, Chem. – Eur. J., 2025, e202500469, DOI:10.1002/chem.202500469
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00873e |
| This journal is © The Royal Society of Chemistry 2025 |