Selective monophosphorylation of cyclic diols and polyols via hemiboronic acid catalysis†
Received
16th May 2025
, Accepted 9th June 2025
First published on 13th June 2025
Abstract
The phosphorylation of organic molecules is a biologically essential chemical transformation. Consequently, there is high demand for methods that allow for the direct, selective, and catalytic monophosphorylation of diols and complex polyols. Due to their ability to form reversible covalent bonds with hydroxy (–OH) groups, hemiboronic acids present the unique capacity to catalytically activate diols in a nucleophilic manner. Herein, we disclose a hemiboronic acid-catalyzed monophosphorylation protocol, amenable to a variety of acyclic and cyclic diols, along with the site-selective functionalization of polyols including saccharides. Mechanistic analyses comprising of kinetic experiments and computational investigation were performed to probe for the origin of the observed site-selectivities. We propose that the observed site-selectivity originates from a difference in calculated nucleophilicity between the diol oxygens in the lower energy epimer of the reactive complex, which also exhibits a kinetically stabilizing hydrogen bonding effect with the approaching electrophile.
Introduction
Coined as one of Nature's favourite modifications, the phosphorylation of organic molecules is a fundamental transformation central to numerous cellular signalling pathways, energy transduction, protein synthesis, and maintenance of genetic material.1–3 When considering that modern chemical and biological methods often aim to mimic natural processes, it is unsurprising that the synthetic manipulation of phosphorylation processes is of significant interest and remains one of the great challenges in organic chemistry.3 In the pharmaceutical industry, phosphorylation is a commonly used prodrug tactic, affording improved bioavailability of therapeutics by bestowing high membrane permeability and aqueous solubility.4,5 In this approach, upon administration of the phosphate ester-bearing prodrug, the active pharmaceutical agent is released by endogenous phosphatases.4,6,7 This strategy has been exemplified in countless marketed polyol therapeutics such as fosamprenavir, fludarabine phosphate, and etoposide phosphate (Fig. 1a).8–10 Synthetic methods for the generation of phosphate monoesters are based on the facile nucleophilic attack of alcohols onto electrophilic phosphate sources. Despite this apparent simplicity, catalytic activation and/or prerequisite substrate derivatization are often necessary to increase hydroxy group reactivity and achieve site-selectivity in substrates bearing multiple reactive sites.11–15 Using kinase enzymes, Nature phosphorylates complex biomolecules with high selectivity, circumventing the need for protecting groups. Paradoxically, the adoption of biocatalytic or pseudo-biocatalytic strategies for the preparation of therapeutic and agrochemical compounds has been limited due to active site specificity requiring substrates similar to the native ligand.16 Despite this challenge, a pioneering report by Miller and coworkers exemplified this strategy using a peptide-based kinase mimic for the synthesis of D-myo-inositol-1-phosphate in excellent yield and enantioselectivity (Fig. 1b).17 Yet, current biomimetic methods pale in comparison to kinases as they require impractical conditions, achieve suboptimal yields, and present poor tolerance in substrate variability. The synthetic phosphorylation of alcohols is typically performed using P(v) reagents such as chlorophosphates or pyrophosphates but can also be achieved using P(III) reagents such as phosphoramidites, phosphites or phosphorochloridites, followed by an oxidation step that is subject to chemoselectivity issues with complex substrates.11,18 Amongst these strategies, phosphate transfer using P(V) reagents is prevalent, using either Lewis acid catalysis for hydroxy group activation, or nucleophilic catalysts for phosphosphorylating agent activation. Numerous procedures have achieved the catalytic phosphorylation of alcohols using catalysts such as pyridine-N-oxide derivatives,2,18 tetrabutylammonium hydrogen sulfate,19 Ti(IV) complexes,20–22 Cu(II) salts,11 Zn(II) salts,23 Mo(VI) salts,24 and dimethylaminopyridine (DMAP) derivatives.25 Conversely, strategies achieving diol monophosphorylation have been seldomly reported, with key reports using catalytic Ti(OiPr)4, attaining moderate success when extended to enantioselective diol desymmetrization (Fig. 1b).26,27 Recently, Dong and coworkers reported on the Fe(II)-catalyzed site-selective monophosphinoylation of carbohydrate derivatives, however monophosphorylation is not readily amenable to this system (Fig. 1b).28 Finally, Dong and coworkers recently devised a method employing catalytic SnCl2 for the phosphorylation of alcohols, diols, and protected carbohydrates.29 Under the reported conditions, the monophosphorylation of carbohydrate derivatives bearing three unprotected hydroxy groups was unsuccessful; monophosphinoylation was instead achieved. It remains clear that the selective monophosphorylation of unprotected diols and polyols using mild catalytic conditions is of significant interest, and there is considerable demand for innovative methods.
 |
| | Fig. 1 Introduction. (a) Phosphate prodrug pharmaceutical agents. (b) Previous work in catalytic monophosphorylation. (c) This work: hemiboronic acid catalyzed monophosphorylation of acyclic and cyclic diols, and polyols. | |
Boron based catalysis (boronic acid, hemiboronic acid, and borinic acid) has transformed the field of selective diol functionalization. It operates through the ability to dynamically exchange with hydroxy groups and form an anionic tetrahedral boron adduct, temporarily increasing the nucleophilicity of the diol oxygens (Fig. 1c).30 Borinic acids readily form the desired tetrahedral adduct through the single exchangeable site at boron, however they suffer from poor oxidative stability, requiring special precautions such as employing the more robust ethanolamine adduct as a pre-catalyst.31 In recent years, the synthetic applicability of these species in catalytic diol and polyol activation for selective carbohydrate monoacylation, sulfonylation, and alkylation has been exemplified by Taylor and coworkers.31,32 Hemiboronic acids present the unique ability of having both a single exchangeable site and superior oxidative stability, with tunable Lewis acidity tailored to the desired catalytic application. Despite having first been synthesized in the 1950s by Snyder and Dewar, benzoxazaborine and boroxarophenanthrene have only recently been demonstrated as competent catalysts for the nucleophilic activation of 1,2-diols and 1,3-diols, respectively.33–36 Using benzoxazaborine (BAC 1a), we previously reported the selective monophosphorylation of a small subset of acyclic 1,2-diols, but the wider potential of this system for the site-selective monophosphorylation of biologically relevant diols and polyols remained to be explored (Fig. 1b).33
Herein, the successful implementation of hemiboronic acid catalysis to the selective monophosphorylation of a variety of acyclic and cyclic 1,2-diols, and unprotected polyols is disclosed. In addition, the determination of preferred catalyst electronics, and computational insight into the origin of the observed site-selectivity with carbohydrates are described (Fig. 1c).
Results and discussion
Optimization
We began our investigation by subjecting cis–1,2-cyclohexanediol to our previously reported conditions, which resulted in the formation of monophosphorylated product 2a in an 81% yield, in addition to pyrophosphate 3 as an undesired by-product in a 20% yield (Table 1, entry 2).33 This unreactive species resulted in onerous purification, as it was difficult to separate from the desired monophosphorylated product. We hypothesized that formation of 3 stemmed from the presence of residual water, which was corroborated by the observed increase in by-product formation with the stoichiometric addition of water (entry 3). The incorporation of 3 Å molecular sieves suppressed the formation of 3 (entry 1). Numerous cyclic hemiboronic acid catalysts of varying Lewis acidity were screened as potential catalysts, yet benzoxazaborine (BAC 1a) was found to be the most efficient at catalyzing this transformation (entries 4–11).33,34,37 A variety of non-nucleophilic bases were tested both in the presence and absence of BAC 1a to monitor background reactivity, unveiling N,N-diisopropylethylamine (DIPEA) as the ideal base for this system (entries 12–17). Chlorophosphates are the most commonly employed P(V) reagents as they are generally commercially available and are synthetically straightforward to use.20,22,38 Within this subclass, diethyl-, dimethyl-, diphenyl-, and dibenzylchlorophosphate were assessed as phosphorylating agents (entries 18–20). Both dimethyl- and diphenylchlorophosphate were not competent phosphorylating agents due to poor yields with the former, and product cyclization in the latter as was also observed by Dong and coworkers with certain substrates (see ESI for details†).29 As diethylchlorophosphate is commercially available and led to slightly increased yields in comparison to dibenzylchlorophosphate, it was selected as the primary phosphorylating agent for this study. Finally, tetrabenzylpyrophosphate, and 2-cyanoethyl N,N-diisopropylchlorophosphoramidite, popular in oligonucleotide synthesis,39 were not reactive in this system (entries 21 & 22).
Table 1 Reaction optimization
|

|
| Entry |
Deviation |
Yield 2aa (%) |
|
All reactions performed on a 0.1 mmol scale, with yields determined by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard.
Yield of cyclized phosphate product obtained (see ESI for details†). Yields in parentheses denote background product formation in the absence of boronic acid catalyst. Yields in brackets denote byproduct 3 formation.
|
| 1 |
None |
82% [7%] (<1%) |
| 2 |
No 3 Å MS |
81% [20%] |
| 3 |
No 3 Å MS, 1 equiv. H2O |
35% [27%] |
| 4 |
BAC 2
|
0% |
| 5 |
BAC 3
|
6% |
| 6 |
BAC 4
|
0% |
| 7 |
BAC 5
|
22% |
| 8 |
BAC 6
|
0% |
| 9 |
BAC 7
|
0% |
| 10 |
BAC 8
|
40% |
| 11 |
BAC 9
|
42% |
| 12 |
2,6-lutidine |
12% (0%) |
| 13 |
2,6-di-tert-butylpyridine |
0% (0%) |
| 14 |
DBU |
13% (0%) |
| 15 |
pyridine |
20% (21%) |
| 16 |
K2CO3 |
62% (0%) |
| 17 |
Et3N |
70% (0%) |
| 18 |
ClP(O)(OMe)2 |
10% |
| 19 |
ClP(O)(OPh)2 |
45%b |
| 20 |
ClP(O)(OBn)2 |
70% |
| 21 |
(BnO)2P(O)OP(O)(OBn)2 |
0% |
| 22 |
(iPr)2NP(Cl)OCH2CH2CN |
0% |
Scope
With optimized conditions in hand, we turned our attention to the scope of diols and polyols. In all cases, scaling the reaction up from 0.1 mmol to 0.3 mmol resulted in a higher isolated yield in the latter than what was observed by 1H NMR yield during optimization. Monophosphorylated products of cyclic cis-1,2-diols were obtained in near quantitative yields for 5- (2b), 6- (2a), and 8-membered rings (2c), and in moderate yield for 12-membered ring (2d) and anhydroerythritol (2f) (Fig. 2). Despite cis-1,2-diols being the preferred motif for boronic acid complexation,30,40 monophosphorylated trans-1,2-cyclohexanediol (2e) was isolated in a 98% yield. Acyclic diols were also re-examined under these optimal conditions, focusing on 1,2-disubstituted-1,2-diols and 1,3-diols that were not studied in our prior work.33 Both 2R,3R-2,3-butanediol (2j) and 1R,2R-1,2-diphenylethanediol (2g) lead to moderate isolated yields. Furthermore, 4-CF3 (2h) and 2-Me (2i) derivatives of 1R,2R-1,2-diphenylethanediol, were successfully monophosphorylated in a 51% and 74% yield, respectively, demonstrating tolerance of electron poor diols, and increased steric hindrance. In addition to acyclic 1,2-diols, the products originating from 2-phenyl-1,3-propanediol (2k) and chloramphenicol (2l) were isolated in moderate to poor yields, with selective functionalization of the primary alcohol in the latter. Remarkably, the site-selective monophosphorylation of six pyranose derivatives (2m–r), including the lincosamide antibiotic clindamycin (2r), proceeded with excellent to near quantitative yields and complete site-selectivity. In all cases (2m–r), monophosphorylation occurred at the C3 position, as has previously been observed in catalytic nucleophilic functionalization of this substrate subclass.28,29,31,32 Site-selectivity assignments were performed using 2D gCOSY NMR to first assign the 1H signals around the sugar ring. In conjunction with this analysis, 31P decoupled 1H NMR data was recorded. The spectrum exhibited a simplification in the coupling pattern for the 1H resonance on the carbon bound to the phosphorylated oxygen through the removal of the observed 3J1H–31P (see ESI for details†). Unfortunately, this synthetic methodology was not conducive to the site-selective monophosphorylation of furanose derivatives, as it resulted in an equal mixture of constitutional isomers (see ESI for details†). Likewise, complex polyols bearing more than three unprotected hydroxy groups or possessing unprotected phenol moieties afforded poor site-selectivity (see ESI for details†).
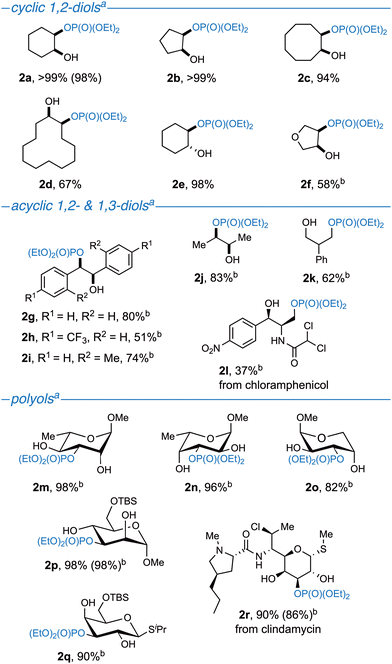 |
| | Fig. 2 Reaction scope. aReactions performed on a 0.3 mmol scale, with diethylchlorophosphate (1.1 equiv.), DIPEA (1.1 equiv.), BAC 1a (10 mol%), in MeCN (0.1 M) with 3 Å MS, at room temperature, for 1.5 h. Isolated yields reported. Yields in parenthesis run on 1.0 mmol scale. bReaction run for 3 h. | |
Phosphate ester deprotection
As phosphate prodrugs are almost exclusively found in their free phosphoric acid form, it is important to demonstrate the applicability of this method to the synthesis of these medicinally valuable compounds. Since current methods to access polyol-based phosphate drugs require multiple additional steps for protecting group installation and cleavage, our catalytic system provides a notable improvement.41a–c Despite the commercial availability of diethylchlorophosphate, the harsh deprotection of phosphorylated polyols using conventional methods, such as the McKenna deprotection, was unsuccessful in our hands.42a–c We then sought to identify a different phosphate ester that is deprotectable under milder conditions. As dibenzylchlorophosphate is both easily prepared from commercially available dibenzylphosphite, and the deprotection of benzyl esters is well established, it was selected as an alternative phosphorylating agent.43,44 Thus, it was applied to the monophosphorylation of four previous substrates, in low to moderate yields (Fig. 3, examples 4a, 4m, 4p, 4r). Subsequently, the corresponding benzyl phosphate products were subjected to standard hydrogenation conditions, yielding the corresponding phosphoric acid products in near quantitative yields, circumventing column chromatography purification (Fig. 3, examples 5a, 5m, 5p, 5r).43,44 In the case of 5p, the global deprotection of 4p was achieved, with concurrent removal of the silyl protecting group.45
 |
| | Fig. 3 Product deprotection. aReactions performed on a 1.0 mmol scale, isolated yields reported. bReactions performed on a 0.3 mmol scale, isolated yields reported. cReaction run with MeOH (0.1 M). *Denotes hydroxy group that is phosphorylated. | |
Mechanistic investigation
Kinetic studies
It is well established that the pKa of a boronic acid catalyst has a significant impact on its catalytic efficacy.30,33 Catalysts possessing a moderate pKa are typically sought after for nucleophilic activation, as they allow a compromise between efficient diol complexation and sufficient build-up of negative charge within the complex, while also enabling catalyst turnover. Previous work by our group alluded to a three-step mechanism for boronic acid catalyzed diol monophosphorylation (Fig. 4a): initial catalyst complexation (benefitting from a low pKa catalyst), nucleophilic attack on the phosphorylating agent (benefitting from a higher pKa catalyst), and catalyst turnover via product dissociation.33 We hypothesized that by synthesizing electronic variants of benzoxazaborine catalyst (BAC 1a) possessing a range of pKa values, we could investigate the impact catalyst pKa has on the kinetic profile of the reaction. We successfully synthesized four new variants with pKa values ranging from 5.5–7.7 (Fig. 4b, BAC 1a–1e), with substitution para to boron, maximizing the electronic effects at the boron centre, while minimizing possible steric interactions with the incoming diol or phosphorylating agent. The relative ordering of determined pKa values agrees with the substituents’ established σp values.46 The selected substrate for this experiment was 1R,2R-1,2-diphenylethanediol, as phosphorylation occurred on an appropriate time scale for monitoring by 1H NMR. In order to minimize possible manifestations of product inhibition, kinetics were only examined for the first 30 minutes of reaction time (Fig. 4b). The lowest pKa catalyst (BAC 1b) had the slowest onset of reactivity, and nearly inexistent catalytic activity. Catalyst BAC 1d had the quickest onset of reactivity and is the highest yielding throughout the experiment. Interestingly, catalysts BAC 1a, 1d, 1e, exhibit the beginning of a plateau in yield, while the highest pKa catalyst (BAC 1c) shows a slow but steady increase in yield throughout the experiment. These observations are in line with either the phosphorylation or catalyst turnover steps being rate limiting, as higher pKa catalysts are superior. To assess the possible correlation between the initial rate of the reaction and catalyst Lewis acidity, pKa values were plotted against the initial slopes of product formation for each catalyst, showing good correlation (R2 = 0.9670) between the two parameters for BAC 1a, 1b, 1c, and 1e (Fig. 4b). However, catalyst BAC 1d did not fall within this trend, which is not entirely unexpected given previous studies showing that a weaker correlation between the Hammett parameters and pKa values of 4-halo substituted boron heterocycles appears to exist.47
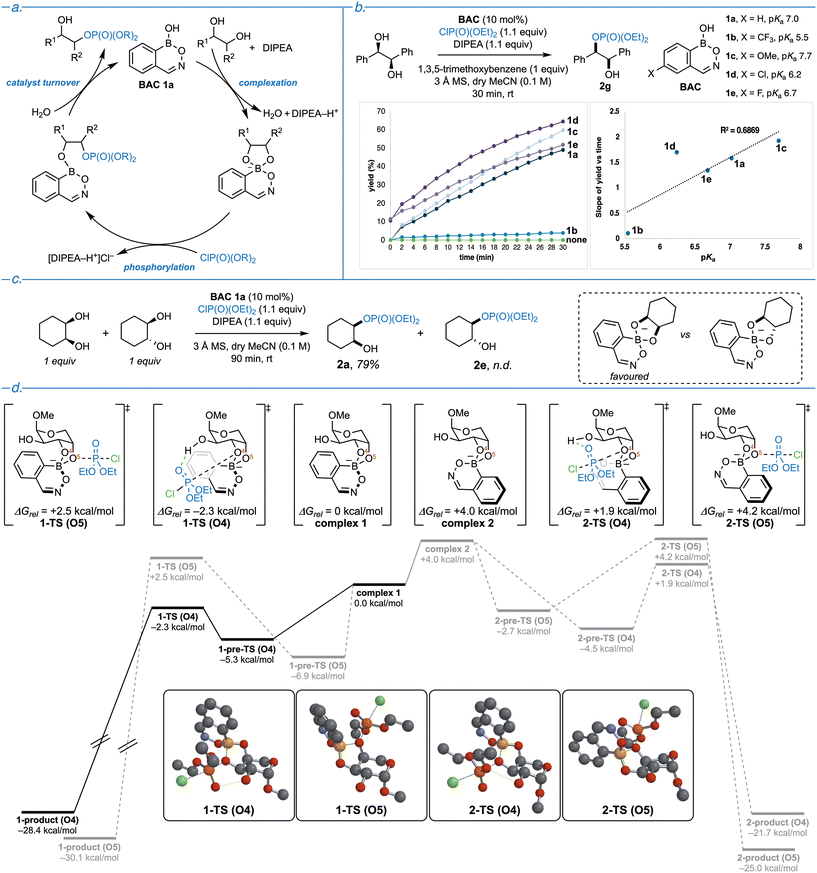 |
| | Fig. 4 Mechanistic investigations. (a) Proposed catalytic cycle. (b) Initial rates kinetics study studying the impact of hemiboronic acid catalyst pKa. Reactions performed on a 0.1 mmol scale, with 1R,2R-1,2-diphenylethanediol (1.0 equiv.), diethylchlorophosphate (1.1 equiv.), DIPEA (1.1 equiv.), BAC (10 mol%), 1,3,5-trimethoxybenzene (1.0 equiv.) in MeCN (0.1 M) with 3 Å MS, for 30 min, at room temperature in an NMR tube. NMR yields reported, using 1,3,5-trimethoxybenzene as the internal standard. (c) cis- & trans-1,2-cyclohexanediol competition study. Reactions performed on a 0.3 mmol scale. Isolated yield reported. (d) Transition state analysis via DFT computations (SMD(MeCN)/ωB97X-V(6-311+G(2df,2p))//ωB97X-D/6-31G(d)). | |
Origin of selectivity
Given the well-established preference for cyclic hemiboronic acids to bind to cis-diols, and the observed site-selectivity with polyol substrates, the nearly quantitative yield obtained with product 2e was surprising, as reaction conditions were identical to cis-diol examples. 11B NMR complexation experiments confirmed that when subjected to the reaction conditions, both cis- and trans-1,2-cyclohexanediol form a tetrahedral boronate complex, with characteristic signals at 6.9 and 7.0 ppm, respectively (see ESI for details†). Thus, to better understand the site-selectivity observed with polyol examples 2m–2r, a competition experiment with equimolar cis- and trans-1,2-cyclohexanediol was performed. Remarkably, only the cis-diol product 2a was monophosphorylated and isolated, albeit in a slightly diminished yield (Fig. 4c). This outcome demonstrates that when both cis- and trans-diols are present, this system is fully selective for the former, while if only the latter is present, it will still react.
Computational studies
To gain insight into the origin of the observed site-selectivity with monosaccharides, Fukui indices (fk–) were determined for example 2n, assessing O4 and O5 in both complex 1 and complex 2 (Fig. 4d). This metric is a means of evaluating the relative nucleophilicity of various atoms in a molecular structure, which is calculated by comparing the Mulliken charges of each nucleophilic atom at ground state and upon the removal of one electron from the system.48 This method was successfully employed by Taylor and coworkers to rationalize their observed site-selectivity in the borinic acid-catalyzed acylation of the same class of carbohydrate derivatives.32 Unfortunately, we observed that this parameter is not suitable in the current system owing to the electronic differences between hemiboronic acid catalyst BAC 1a with the previously reported borinic acid catalyst (see ESI for details†). Using density functional theory calculations (SMD(MeCN)/ωB97X-V(6-311+G(2df,2p))//ωB97X-D/6-31G(d)), transition state analysis was conducted using example 2n to better understand the observed site-selectivity (Fig. 4d). As two possible tetracoordinate boron epimers can form upon condensation of BAC 1a onto 2n, both geometries were optimized (Fig. 4d, complex 1 and complex 2), revealing a 4.0 kcal mol−1 energy difference in favour of complex 1. Condensation of BAC 1a onto 2n activates two oxygen atoms (O4 and O5) that can undergo nucleophilic attack onto the incoming diethylchlorophosphate. As such, the transition states were calculated for each oxygen atom of both epimers of the BAC 1a condensate. Starting from the more energetically favourable complex 1, pre-transition state and transition state energies were obtained for the phosphorylation at O4 (observed product) and O5. Despite 1-pre-TS (O4) being higher in energy than 1-pre-TS (O5), the activation barrier from 1-pre-TS (O4) to 1-TS (O4) is only 3.0 kcal mol−1, while it is 9.4 kcal mol−1 from 1-pre-TS (O5) to 1-TS (O5). Similarly, for complex 2, nucleophilic attack from O4 is more kinetically accessible over O5. Interestingly, a hydrogen bonding interaction was observed between the diethyl chlorophosphate and (H) O3 within 1-TS (O4) and 2-TS (O4). It is plausible that this interaction is key to the exquisite site-selectivity by stabilizing the transition state of the nucleophilic attack at O4 over O5.
Conclusion
We have disclosed a practical methodology leveraging the nucleophilic mode of cyclic hemiboronic acid catalysis for the site-selective monophosphorylation of a variety of cyclic diols and polyols, as well as acyclic diols. This method is amenable to the preparation of phosphate ester and free phosphate containing polyol drugs under ambient conditions. Mechanistic studies have uncovered a plausible explanation for the observed site-selectivity of the transformation, and a study of initial-rates kinetic study revealed that this system benefits from a more electron-rich boron catalyst. Future work will be driven towards developing a catalyst capable of achieving enantioselective monophosphorylation of symmetrical, prochiral substrates, in addition to extending the tolerance of this system towards polyols of increased complexity.
Author contributions
D. G. H. obtained funding. D. G. H. and G. F. O. conceptualized the study. G. F. O. led investigation and methodology development, and supervised D. L. A. D. J. K. carried out computational studies. G. F. O. wrote the original manuscript, with review and editing in collaboration with D. G. H. and D. J. K.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
Data for this article, including experimental details, analytical data, and spectral reproductions for all new compounds, and details of all mechanistic and computational data have been included as part of the ESI.†
Acknowledgements
This work was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery grant RGPIN-2023-04436 to D. G. H., CGS-D scholarship to G. F. O. and D. J. K.). The authors thank Mark Miskolzie for assistance with NMR spectroscopy experimentation, Dr Jason P. G. Rygus for insightful discussions, and Dr Angie Morales-Izquierdo for help with mass spectrometry.
References
- S. C. L. Kamerlin, O. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1 CrossRef CAS PubMed.
- J. I. Murray, R. Woscholski and A. C. Spivey, Chem. Commun., 2014, 50, 13608 RSC.
- H. J. Jessen, N. Ahmed and A. Hofer, Org. Biomol. Chem., 2014, 12, 3526 RSC.
- S. Kleeb, X. Jiang, P. Frei, A. Sigl, J. Bezençon, K. Bamberger, O. Schwardt and B. Ernst, J. Med. Chem., 2016, 59, 3163 CrossRef CAS PubMed.
- C. Schultz, Bioorg. Med. Chem., 2003, 11, 885 CrossRef CAS PubMed.
- J. B. Zawilska, J. Wjcieszak and A. B. Olejniczak, Pharmacol. Rep., 2013, 65, 1 CrossRef CAS PubMed.
- C. R. Strader, C. J. Pearce and N. H. Oberlies, J. Nat. Prod., 2011, 74, 900 CrossRef CAS PubMed.
- M. B. Wire, M. J. Shelton and S. Studenberg, Clin. Pharmacokinet., 2006, 45, 137 CrossRef CAS PubMed.
- J. Rautio, H. Kumpulainen, T. Heimbach, R. Oliyai, D. Oh, T. Järvinen and J. Savolainen, Nat. Rev. Drug Discovery, 2008, 7, 255 CrossRef CAS PubMed.
- A. Fulmali and S. S. Bharate, Drug Dev. Res., 2022, 83, 1059 CrossRef CAS PubMed.
- S. Jones and C. Smanmoo, Org. Lett., 2005, 7, 3271 CrossRef CAS PubMed.
- T. Aiba, M. Sato, D. Umegaki, T. Iwasaki, N. Kambe, K. Fukase and Y. Fujimoto, Org. Biomol. Chem., 2016, 14, 6672 RSC.
- D. J. Trader and E. E. Carlson, Mol. BioSyst., 2012, 8, 2484 RSC.
- P. A. Jordan, K. J. Kayser-Bricker and S. J. Miller, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20620 CrossRef CAS PubMed.
- B. R. Sculimbrene, A. J. Morgan and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 11653 CrossRef CAS PubMed.
- J. P. Adams, M. J. B. Brown, A. Diaz-Rodriguez, R. C. Lloyd and G.-D. Roiban, Adv. Synth. Catal., 2019, 361, 2421 CrossRef CAS.
- B. R. Sculimbrene and S. J. Miller, J. Am. Chem. Soc., 2001, 123, 10125 CrossRef CAS PubMed.
- J. I. Murray, R. Woscholski and A. C. Spivey, Synlett, 2015, 985 CAS.
- K. Domon, M. Puripat, K. Fujiyoshi, M. Hatanaka, S. A. Kawashima, K. Yamatsugu and M. Kanai, ACS Cent. Sci., 2020, 6, 283 CrossRef CAS PubMed.
- S. Jones, D. Selitsianos, K. J. Thompson and S. M. Toms, J. Org. Chem., 2003, 68, 5211 CrossRef CAS PubMed.
- S. Jones and D. Selitsianos, Org. Lett., 2002, 4, 3671 CrossRef CAS PubMed.
- O. S. Fenton, E. E. Allen, K. P. Pedretty, S. D. Till, J. E. Todaro and B. R. Sculimbrene, Tetrahedron, 2012, 68, 9023 CrossRef CAS.
- Y. Saito, S. M. Cho, L. A. Danieli and S. Kobayashi, Org. Lett., 2020, 22, 3171 CrossRef CAS PubMed.
- C.-Y. Liu, V. D. Pawar, J.-Q. Kao and C.-T. Chen, Adv. Synth. Catal., 2010, 352, 188 CrossRef CAS.
- A. Fallek, M. Weiss-Shtofman, M. Kramer, R. Dobrovetsky and M. Portnoy, Org. Lett., 2020, 22, 3722 CrossRef CAS PubMed.
- K. A. Coppola, J. W. Testa, E. E. Allen and B. R. Sculimbrene, Tetrahedron Lett., 2014, 55, 4203 CrossRef CAS.
- E. T. Ouellette, M. G. Lougee, A. R. Bucknam, P. J. Endres, J. Y. Kim, E. J. Lynch, E. J. Sisko and B. R. Sculimbrene, J. Org. Chem., 2021, 86, 7450 CrossRef CAS PubMed.
- J. Zhao, X. Zhang, Q. Yang, T. Bi, B. Du, J. Lv, S.-X. Gu and H. Dong, Org. Lett., 2025, 27, 1555 CrossRef CAS PubMed.
- J. Lv, C.-Y. Liu, X. Zhang, S.-X. Gu and H. Dong, Eur. J. Org. Chem., 2025, e202500209 Search PubMed.
- D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475 RSC.
- D. Lee, C. L. Williamson, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260 CrossRef CAS PubMed.
- D. Lee and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 3724 CrossRef CAS PubMed.
- J. P. G. Rygus and D. G. Hall, Nat. Commun., 2023, 14, 2563 CrossRef CAS PubMed.
- C. D. Estrada, H.-T. Ang, K.-M. Vetter, A. A. Ponich and D. G. Hall, J. Am. Chem. Soc., 2021, 143, 4162 CrossRef CAS PubMed.
- H. R. Snyder, A. J. Reedy and W. J. Lennarz, J. Am. Chem. Soc., 1958, 80, 835 CrossRef CAS.
- M. J. S. Dewar; and R. Dietz, Tetrahedron Lett., 1959, 14, 21 CrossRef.
- J. J. Blackner, O. M. Schneider, W. O. Wong and D. G. Hall, J. Am. Chem. Soc., 2024, 146, 19499 CrossRef CAS PubMed.
- P. B. Brady, E. M. Morris, O. S. Fenton and B. R. Sculimbrene, Tetrahedron Lett., 2009, 50, 975 CrossRef CAS.
- N. D. Sinha, J. Biernat and H. Köster, Tetrahedron Lett., 1983, 24, 5843 CrossRef CAS.
- C. Lü, H. Li, H. Wang and Z. Liu, Anal. Chem., 2013, 85, 2361 CrossRef PubMed.
-
(a)
W. L. Matier, C. Woo and Y.-C. Lee, US Pat, 4849515, 1989 Search PubMed;
(b)
G. Yiping, Q. Li, X. Lingyue, C. Renqiang, Z. Xin and C. Yangyi, CN Pat, CN108794550A, 2018 Search PubMed;
(c)
W. Xuewen, D. Hongxu, S. Ling, P. Qi, W. Zongda, J. Xinming, S. Xiaolan and X. Yuli, CN Pat, CN113121617A, 2021 Search PubMed.
-
(a) C. E. McKenna, M. T. Higa, N. H. Cheung and M.-C. McKenna, Tetrahedron Lett., 1977, 18, 155 CrossRef;
(b) C. E. McKenna and J. Schmidhuser, J. Chem. Soc., Chem. Commun., 1979, 739 RSC;
(c) K. Justyna, J. Małolepsza, D. Kusy, W. Maniukiewicz and K. M. Błażewska, Beilstein J. Org. Chem., 2020, 16, 1436 CrossRef CAS PubMed.
- M. Xu, R. Galindo-Murillo, T. E. Cheatham III. and R. M. Franzini, Org. Biomol. Chem., 2017, 15, 9855 RSC.
- D. W. Koh, D. L. Coyle, N. Mehta, S. Ramsinghani, H. Kim, J. T. Slama and M. K. Jacobson, J. Med. Chem., 2003, 46, 4322 CrossRef CAS PubMed.
- S. Kim, S. M. Jacobo, C.-T. Chang, S. Bellone, W. S. Powell and J. Rokach, Tetrahedron Lett., 2004, 45, 1973 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- J. W. Tomsho, A. Pal, D. G. Hall and S. J. Benkovic, ACS Med. Chem. Lett., 2012, 3, 48 CrossRef CAS PubMed.
- W. Yang and W. J. Mortier, J. Am. Chem. Soc., 1986, 108, 5708 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Dawson J.
Konowalchuk
,
Dawson J.
Konowalchuk




