
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral diketopyrrolo[3,4-c]pyrrole dyes with different substitution symmetry: impact of adamantyl groups on the photo-physical properties in solution and thin films†
Martin
Cigánek
 a,
Claudio
Vatteroni
b,
Francesca Romana
Lauro
b,
Francesco
Zinna
b,
Gennaro
Pescitelli
b,
Jozef
Krajčovič
*a and
Gianluigi
Albano
*bc
a,
Claudio
Vatteroni
b,
Francesca Romana
Lauro
b,
Francesco
Zinna
b,
Gennaro
Pescitelli
b,
Jozef
Krajčovič
*a and
Gianluigi
Albano
*bc
aFaculty of Chemistry, Brno University of Technology, Purkyňova 118, 612 00, Brno, Czech Republic. E-mail: krajcovic@fch.vut.cz
bDipartimento di Chimica e Chimica Industriale, Università di Pisa, Via Giuseppe Moruzzi 13, 56124 Pisa, Italy
cDipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università degli Studi di Parma, Parco Area delle Scienze, 17/A, 43124 Parma, Italy. E-mail: gianluigi.albano@unipr.it
First published on 12th June 2025
Abstract
Here we propose a comparative study on four chiral 1,4-diketo-3,6-dithienylpyrrolo[3,4-c]pyrrole (DPP) dyes with different substitution symmetry on the lactam moieties: (i) symmetrically N-substituted dyes 1–2, bearing two (S)-β-citronellyl (DPP-CT) or (S)-2-methylbutyl (DPP-MT) chains; (ii) non-symmetrically N-substituted dyes 3–4, where an adamantylmethyl appendage replaced one of the (S)-β-citronellyl (DPP-ACT) or (S)-2-methylbutyl (DPP-AMT) chains. The four molecules were synthesized and subjected to detailed investigation of their photo-physical properties in solution (absorbance, fluorescence, cyclic voltammetry) and thin films (absorbance, electronic circular dichroism, optical microscopy), evaluating the impact of different chiral moieties (citronellyl vs. 2-methylbutyl chains) and of different substitution patterns exerted by the adamantane moiety (symmetrical vs. non-symmetrical DPPs). Special emphasis was given to the chiroptical study of thin films, discussing the origin of different supramolecular arrangements in the solid state. Putting together experimental and computational results, we rationalize the chiroptical behavior of these systems in comparison with that of other classes of DPP dyes recently investigated by our group.
Introduction
In the last few decades, organic π-conjugated systems have gained extraordinary development as semiconducting materials in optoelectronics,1 spintronics2 and nanotechnology.3 Compared to traditional inorganic semiconductors, they provide all the advantages of plastic-like materials, including ease of processing, mechanical flexibility and easy disposal.4 However, the most appealing aspect of these systems is that all the properties required for the technological applications (molar extinction coefficients, fluorescence quantum yield, HOMO–LUMO band gap, magnetism, etc.) can be finely tuned through modifications in the chemical structure,5 for example tweaking the π-conjugation length of the chromophore or incorporating in topical positions electron-donating or electron-withdrawing functional groups or other specific moieties.6 All these structural modifications at molecular level then allow for a fine tuning of the solid-state arrangement at all hierarchical levels, nanoscopic (first-order supramolecular aggregates, 1–10 nm),7 mesoscopic (larger scale morphologies, 10–1000 nm)8 and microscopic (local domains, >1 μm),9 which in turn influence all the above-mentioned functional properties. Therefore, a thorough understanding of the structure–property relationships in thin films is crucial for optimizing the performance of these materials as active layers in devices for technological applications.To achieve precise control over the solid-state organization of organic π-conjugated materials in thin films, a combination of chemical design strategies and processing techniques is required. On the one hand, the extraordinary progress in the development of highly efficient transition metal-promoted synthetic methodologies for carbon–carbon bond formation has greatly expanded the structural diversity of these systems: traditional cross-coupling reactions between organic halides and organometallic reagents,10 more sustainable direct C–H activation strategies11 and emerging cross-dehydrogenative coupling (double C–H bond activation) methods.12 On the other hand, many external factors such as deposition techniques (drop casting vs. spin coating), post-deposition treatments (solvent vs. thermal annealing), and other environmental conditions can strongly influence the solid-state arrangement at all hierarchical levels,13 complicating a correct rationalization of the intimate structure–property relationships.
Thin films of organic π-conjugated materials are often characterized by a marked polymorphism: multiple aggregation pathways (e.g., kinetic vs. thermodynamic states),14 each of them exhibiting different structural features and physico-chemical properties, take place under different working conditions (in terms of deposition technique and post-deposition treatments) or, in some cases, also co-exist in the same sample segregated in local domains.13 The relative weight and distribution of these polymorphs are typically very dependent on external stimuli, which may lead to even dramatic changes in the functional properties of interest,14 and are therefore very difficult to rationalize without a precise knowledge of the intimate structure. Given the complexity of these materials, a thorough characterization of thin films at different hierarchical scales is essential for elucidating their organizational patterns – including local polymorphisms – and correlating them with functional properties. In this regards, optical microscopy (OM) and polarized optical microscopy (POM) are used for the characterization of thin films at the micrometric scale,15 while electron microscopy (TEM and SEM) and atomic force microscopy (AFM) allow for investigating samples at the mesoscopic scale.16 However, none of them can provide insight into the supramolecular organization at the nanoscopic scale, thus necessitating the integration of advanced spectroscopic and diffraction methods to achieve a comprehensive structural understanding.
In recent years, the development of chiral organic π-conjugated systems has provided new lifeblood to this research field.17 The possibility to combine all the above-mentioned features of π-conjugated materials with the properties of chirality has found three main advantages. First, the functionalization of these compounds with chiral moieties could represent a valid tool for controlling their solid-state organization at different hierarchical levels. The introduction of enantiopure appendages (such as terpene, α-amino acid or monosaccharide units) as side chains in topical positions of the π-conjugated backbone prevents undesirable disordered structures of chromophores and promotes their organization at all hierarchical levels under the effect of intermolecular forces. At nanoscopic scale, they could be arranged into first-order supramolecular three-dimensional (3D) chiral architectures, showing tilted stacks of the component molecules under clockwise or counterclockwise orientation.18 By preventing a perfect co-facial stacking between adjacent π-conjugated units, this could reasonably ensure longer exciton lifetime19 and lower fluorescence aggregation-caused quenching (ACQ).20 These first-order supramolecular helices can be then further organized at mesoscopic scale into larger 3D chiral structures, like helical fibers or twisted ribbons.21 However, in some cases, chiral π-conjugated molecules can also self-assemble at microscopic scale to give locally anisotropic domains with two-dimensional (2D) chirality.22 It is worth emphasizing that a perfect structural control is not trivial here: competition between several weak non-covalent interactions can lead to many local minimum energy supramolecular architectures, resulting in multiple aggregation modes in dynamic transitions from one another, depending on experimental procedures for thin films fabrication.
A second relevant advantage associated with the development of chiral organic π-conjugated materials is the birth of a variety of cutting-edge technological applications, only thanks to their unique properties. One of them is the chirality-induced spin selectivity (CISS) effect,23 which can be applied in spintronics or in spin-controlled electrochemical reactions, such as oxygen evolution reaction (OER) or oxygen reduction reaction (ORR).24 Other very relevant applications in organic optoelectronics are strongly related to their chiroptical activity: this is the case for circularly polarized (CP) organic light-emitting diodes,25 organic field-effect transistors26 and organic photodetectors.27 In addition to these, enantiopure chiral magnets,28 chiral electrochemical sensors29 and stimuli-responsive materials in optical information storage and processing30 must also be added. Studies about potential optoelectronic applications of thin films of chiral organic π-conjugated materials are still emerging, with plenty of room for improvement.31
However, the most relevant advantage related to the study of chiral organic π-conjugated materials as thin films is the possibility of characterizing them by means of chiroptical spectroscopies, particularly electronic circular dichroism (ECD). In fact, ECD spectroscopy is very helpful in providing information on the solid-state organization at different hierarchical levels. In fact, it is sensitive to the arrangement of adjacent chromophores at the nanoscopic scale, discriminating and recognizing 3D-chiral supramolecular structures which would appear indistinguishable to non-polarized light investigation.32 At the same time, ECD may provide some clues to higher hierarchical levels of organization, detecting the presence of microscopic local structures exhibiting 2D chirality.33 For the sake of completeness, it is also worth mentioning the existence of spatially resolved ECD techniques: localized chiroptical spectroscopy (LCS) and circularly polarized microscopy (CPM), revealing polymorphs which would be indistinguishable by means of conventional OM.34
For thin films of chiral π-conjugated compounds, the measurement of ECD spectra is much more complex than solution, due to the potential presence of macroscopic anisotropies, linear dichroism (LD) and linear birefringence (LB), often responsible for significant interference contributions to the emergent ECD signals. According to the Mueller matrix theory, a mathematical model for understanding the physical meaning of emerging chiroptical signals in polarization spectroscopy,35 the experimental ECD in thin films can be expressed as follows:
| ECD ≈ CDiso + ½ (LD′·LB − LD·LB′) +… | (1) |
CDiso36 is the intrinsic component of the experimental ECD signal, invariant upon sample rotation or flipping with respect to the instrument optical axis. This term is proportional to the rotational strength of electronic transitions (the same characterizing ECD signals in solution), and for this reason it is also called “true CD”. It is an expression of 3D-chiral structures at molecular and/or first-order supramolecular level (nanoscopic scale).
The second term of eqn (1), typically named LDLB36 or even “apparent CD”,37 “pseudo CD”38 and “polarity reversal of ellipticity”,39 is the non-reciprocal component of the experimental ECD signal. It is invariant upon sample rotation around the instrument optical axis but inverts its sign by sample flipping. LDLB does not arise from the combined effect of electric and magnetic dipole transition moments at nanoscopic scale (as for CDiso), but rather from the coupling between LD and LB in locally anisotropic domains at microscopic scale, when their principal axes are non-parallel and non-orthogonal to each other. These domains are 2D-chiral, each of them responsible for local chiroptical signals, whose sign depends on the skew angle β between the two linear terms: positive if 0° < β < 45°, negative if −45° < β < 0°. Such domains may be randomly oriented in the plane, thus making macroscopic linear anisotropies negligible when averaged on the whole sample. LDLB then arises from the prevalence of domains with a well-defined enantiomeric face adhering onto the glass surface during thin film preparation.40 The origin, as well as possible applications, of non-reciprocal chiroptical properties of thin films has been recently theoretically and experimentally investigated by several research groups.41 In this context, a semiclassical theory combining Mueller matrix analysis and a Lorentz oscillator model was recently developed by Tempelaar et al., offering insight into the supramolecular arrangement of π-conjugated dyes able to generate LDLB.42
According to the Mueller matrix theory,35 if other terms leading to optical artifacts are negligible, CDiso and LDLB could be extracted by taking, respectively, the semi-sum and the semi-difference of ECD spectra recorded for the front (organic layer towards light source) and the back face (organic layer towards detector):43
| CDiso = ½ × (ECDfront + ECDback) | (2) |
| LDLB = ½ × (ECDfront − ECDback) | (3) |
To quantitatively evaluate their relative weights, it is also possible to define the ρ ratio between the integrated areas of the absolute values of semi-difference  and semi-sum
and semi-sum  . These very simple mathematical operations allow one to assess both intrinsic and non-reciprocal terms by using a benchtop spectropolarimeter, avoiding the need for dedicated and costly equipment, such as artifact-free spectropolarimeters44 or Mueller Matrix polarimetry (MMP)45 technique.
. These very simple mathematical operations allow one to assess both intrinsic and non-reciprocal terms by using a benchtop spectropolarimeter, avoiding the need for dedicated and costly equipment, such as artifact-free spectropolarimeters44 or Mueller Matrix polarimetry (MMP)45 technique.
1,4-Diketo-3,6-di(hetero)arylpyrrolo[3,4-c]pyrroles (DPPs) are a class of organic π-conjugated compounds46 that have been extensively used as high-performance industrial pigments in coatings, varnishes and plastics,47 mainly thanks to their excellent thermal and photostability.48 More recently, they have attracted significant attention as building blocks of functional and smart materials, with applications in a variety of solution-processed organic electronic devices. Such interest stems from the electronic deficient nature of the DPP core, which facilitates the design of donor–acceptor materials with narrow band gaps.49 DPP-based π-conjugated materials exhibited excellent charge-carrier mobilities in organic field-effect transistors (OFETs)50 and high power conversion efficiencies in organic solar cells (OSCs).51 At the same time, outstanding optical properties such as strong oscillator strength, tuneable absorption and high photoluminescence quantum yields make them good candidates as fluorescent probes in sensing and bio-imaging,52 as well as two-photon-absorbing materials.53
Structurally, the DPP unit is a planar bicyclic scaffold incorporating two strong electron-withdrawing lactam moieties, which can be easily functionalized by Pd-catalyzed cross-coupling reactions on the (hetero)aryl groups at positions 3 and 6 or by nucleophilic substitution reactions with the nitrogen atoms under basic conditions. The N-substitution of lactam moieties of DPPs with groups of different steric hindrance and polarity may have a large impact on their solubility, as well as on their photophysical properties in solution and thin films.54 In particular, the DPP core is typically functionalized with two identical moieties, thus resulting in chromophores of C2h symmetry group. However, recent research has increasingly focused on non-symmetrically N-substituted DPP dyes, where distinct appendages are introduced at each lactam moiety. Notably, in 2018 Zhang et al. have reported a remarkable charge mobility enhancement for π-conjugated DPP-based copolymers by simply replacing one branched alkyl chain with a linear one at each DPP unit.55 More recently, Bronstein et al. also highlighted substantial changes in the optical properties of DPP dyes with different substitution symmetry.56
Despite the large interest in DPP-based compounds, the development of chiral DPP dyes remains surprisingly underexplored. Some DPP derivatives incorporating helicene,57 binaphthyl,58 pyrido-fused myrtanyl,59 ethyl lactate60 or β-citronellyl61 groups at the terminal positions of the π-conjugated backbone were investigated in solution and/or thin films by ECD and other related spectroscopies. However, chiroptical studies on DPPs modified with chiral substituents on the lactam moieties are still in their early stages. For instance, in recent studies by Meskers et al.62 and Würthner et al.,63 DPP dyes N-substituted with chiral appendages were synthesized and characterized in solution and thin films by several techniques, yet no chiroptical spectroscopies were used. Only in the past few years have the first chiroptical studies emerged for this class of chiral DPPs.64 However, all these investigations have so far been limited to systems where identical chiral groups are attached to both lactam moieties. To the best of our knowledge, no examples of non-symmetrically N-substituted chiral DPP dyes have been reported to date.
In this work we propose a comparative study on four chiral 1,4-diketo-3,6-dithienylpyrrolo[3,4-c]pyrroles dyes 1–4 with different substitution symmetry on the lactam moieties: (i) the symmetrically N-substituted DPPs 1–2, functionalized with two enantiopure (S)-β-citronellyl (DPP-CT, 1) or (S)-2-methylbutyl (DPP-MT, 2) chains; (ii) the non-symmetrically N-substituted DPPs 3–4, where an adamantylmethyl appendage replaced one of the two (S)-β-citronellyl (DPP-ACT, 3) or (S)-2-methylbutyl (DPP-AMT, 4) chains on the lactam moieties (see Fig. 1). The choice of such chiral appendages is due to multiple reasons. First, they can be easily introduced on the π-conjugated chromophore starting from (S)-(−)-β-citronellol and (S)-(−)-2-methylbutanol, which are in turn available in Nature as a component of, respectively, rose oils and fusel oil.65 Second, both moieties can transfer their molecular asymmetry (stereogenic centre at the C2/C3 of the branched chain) to supramolecular or larger scale mainly through dispersion forces and steric effects. Furthermore, we aim to investigate how the position of the stereogenic center on branched alkyl chains may impact on the solid-state arrangement of DPP dyes, and thus on their photophysical properties. For non-symmetrically N-substituted DPP dyes 3–4, the bulky and compact methyladamantane appendage was selected as it could have a significant impact on the solid state arrangement, thus resulting in a highly ordered molecular packing of DPPs responsible for an exceptional ambipolar charge transport.66
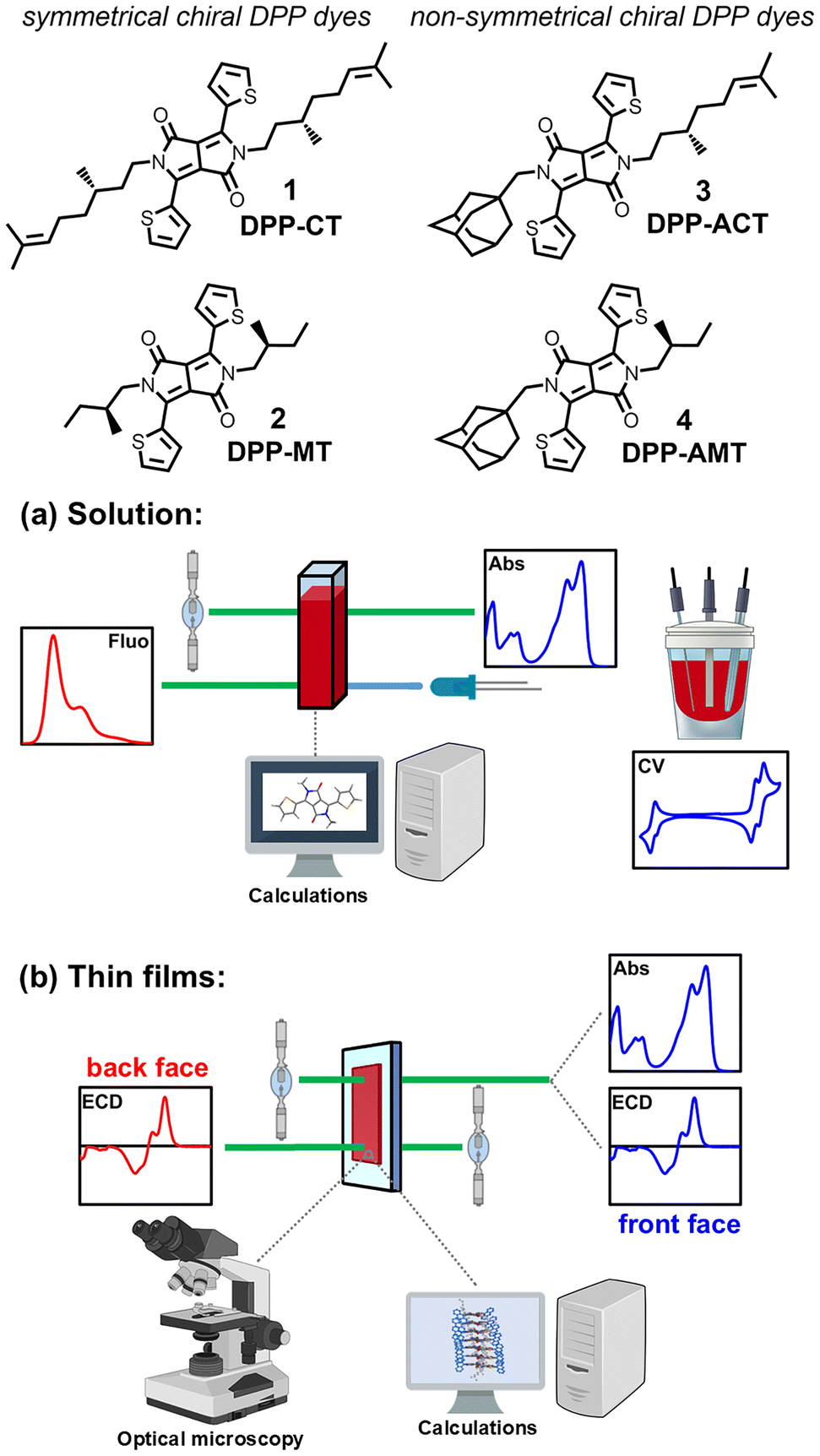 | ||
| Fig. 1 Aim of the work. Four chiral DPP dyes with different substitution symmetry on the lactam moieties were investigated: symmetrically N-substituted 1–2, functionalized with two enantiopure (S)-β-citronellyl (DPP-CT, 1) or (S)-2-methylbutyl (DPP-MT, 2) chains; non-symmetrically N-substituted 3–4, where an adamantylmethyl appendage replaced one of the two (S)-β-citronellyl (DPP-ACT, 3) or (S)-2-methylbutyl (DPP-AMT, 4) chains. The compounds were characterized: (a) in solution, by means of absorbance, fluorescence and cyclic voltammetry (CV); (b) in thin films, by means of absorbance, ECD, OM and POM. The impact of different chiral moieties and different substitution symmetry exerted by the adamantane moiety was unravelled also thanks to computational analysis (TD-DFT calculations). | ||
The four target molecules 1–4 were synthesized and subjected to a detailed investigation of their photophysical properties in solution (absorbance, fluorescence, cyclic voltammetry (CV)) and thin films (absorbance, ECD, OM, POM), evaluating the impact of different chiral moieties (citronellyl vs. 2-methylbutyl chains) and of different substitution pattern due to the combination of chiral and adamantane moieties (symmetrical vs. non-symmetrical DPPs). Special emphasis was given to the chiroptical study of thin films, discussing the origin of different supramolecular arrangements in the solid state, and therefore of the emerging ECD spectra, also with the support of time-dependent density functional theory (TD-DFT) calculations (Fig. 1).
Results and discussion
The synthetic pathway for obtaining the four final chiral DPP dyes products 1–4 is depicted in Scheme 1. It involves a bimolecular nucleophilic substitution (SN2) reaction, where a DPP pigment molecule with monosubstituted amide nitrogen (8 or 9) is converted using basic conditions (potassium carbonate) to the corresponding N-anion, which then acts as a nucleophile to attack the electrophilic centre in an alkyl halide (5) or an alkyl tosylate (6 and 7). | ||
| Scheme 1 Synthetic pathway for the preparation of four chiral DPP dyes 1–4 investigated in the present work. | ||
The preparation of these electrophilic alkylating agents started from readily commercially available alcohols (R-OH) in the form of (adamantan-1-yl)methanol, (S)-(−)-β-citronellol and (S)-(−)-2-methylbutanol. While in the case of the preparation of 1-(bromomethyl)adamantane (5) the transformation of the alcohol to the alkyl bromide using 48% hydrobromic acid at reflux was used, chiral alcohols were converted to the corresponding (S)-β-citronellyl and (S)-2-methylbutyl tosylates 6–7 using milder conditions.
The preparation of chiral DPP products started from 3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8) as the basic DPP pigment, prepared by the well-known succinic method using thiophene-2-carbonitrile and diisopropyl succinate as starting materials under strongly basic conditions.46a Symmetrical DPPs 1 and 2 were prepared by direct alkylation of 8 using a 5-molar excess of K2CO3 base and a 3.5-molar excess of alkyl tosylates 6 and 7, respectively, in DMF at 125 °C. Although tosylates are generally better leaving groups than halides and the substitution should therefore proceed more efficiently, the yields of products 1 and 2 were relatively low (18% and 12%, respectively). This is due to the ambident nature of the nucleophilic DPP anion, where the negative charge delocalizes between the nitrogen and oxygen atoms and the reaction produces undesirable by-products, i.e. N,O- and O,O-alkylated derivatives, as previously reported.66b The preparation of non-symmetrical DPPs 3 and 4 started with the synthesis of mono-N-alkylated derivative 9, for which adamantylmethyl bromide 5 was used. Due to its neopentyl character, which increases steric hindrance and thus makes the course of the SN2 reaction more difficult, it allows the preparation of the monosubstituted product 9 in sufficient yield (15%). In particular, a 3-molar excess of base and a 1.5-molar excess of alkyl bromide 5 were used to carry out the reaction, in DMF at 140 °C (that is, a temperature 15 °C higher than the reaction with alkyl tosylates). Finally, the mono-N-alkylated DPP 9 was treated with alkyl tosylates 6 and 7 under the same reaction conditions used for the preparation of symmetrical derivatives, and the desired non-symmetrical DPPs 3 and 4 were obtained in 9% and 31% yields, respectively.
As for other similar 1,4-diketo-3,6-dithienylpyrrolo[3,4-c]pyrroles previously reported in the literature,64 the four chiral compounds 1–4 appear in the form of dark purple powders. All of them showed quite good solubility in chlorinated solvents, whereas in highly polar solvents (such as MeOH and H2O) their solubility was low.
We started our comparative study evaluating the optical properties of four chiral DPP dyes 1–4 in solution: the UV-Vis absorption and photoluminescence emission spectra in CHCl3 are depicted in Fig. 2. At first glance, UV-Vis absorption spectra of four dyes appear roughly similar, although it is possible to appreciate minor variations between systems with different substitution pattern. In particular, all the compounds showed the most prominent broad band between 400 and 600 nm, with maximum absorbance at 549 nm for symmetrical DPPs 1–2 and 540 nm for non-symmetrical DPPs 3–4. In analogy to other dyes of similar structure previously reported in the literature, this band could be attributed to a π → π* transition localized on the DPP moiety,67 rather than to an intramolecular charge-transfer (CT) transition from thiophene rings to the DPP.68 Quite interestingly, the vibrational structure associated with this band was also found to be different depending on the nature of the substituent, with the N-methyladamantane group causing less resolved vibrational structure. At higher energy we found two smaller bands, possibly associated to other electronic transitions involving the π-conjugated backbone: a broad band between 300 and 400 nm, with maximum at 340 nm (for symmetrical systems 1–2) or 341 nm (for non-symmetrical systems 3–4), and a sharp peak centred at 292 nm. A computational analysis was run on the N,N-dimethyl model of 1–4, here indicated as DPP-Me (see Fig. S1 in ESI†). As expected, the lowest energy structure has coplanar aromatic rings with a s-trans orientation, i.e., with S atoms directed towards the closest N atoms. The major UV band is calculated at 520 nm at B3LYP/def2-TZVP level and corresponds to a π–π* transition delocalized on the π-conjugated skeleton; the transition electric dipole moment is oriented approximately along the C3–C6 direction of the DPP ring. Other higher energy transitions are also predicted, with different orientation, including a transverse one (i.e., directed along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/CO direction) for the band <300 nm.
O/CO direction) for the band <300 nm.
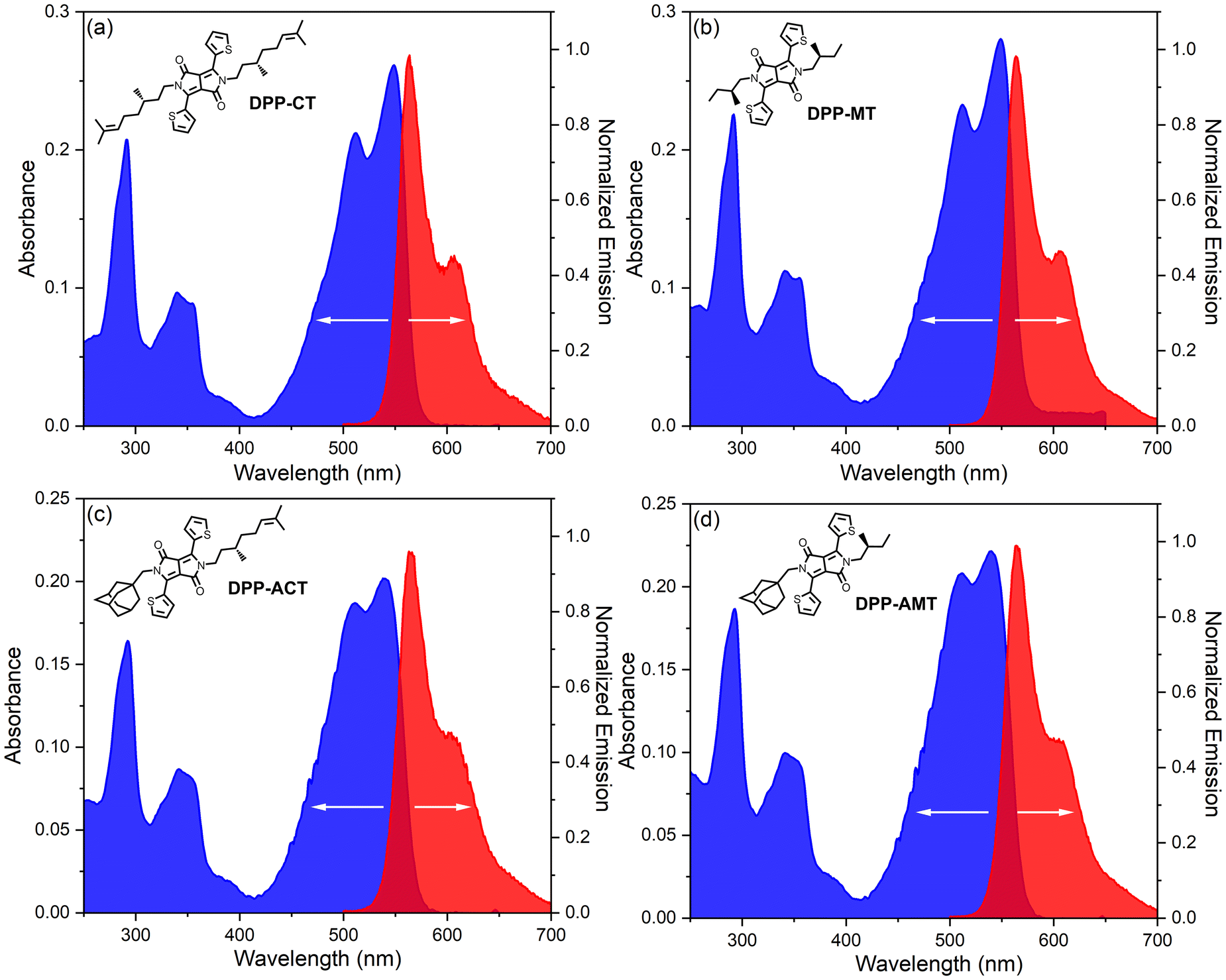 | ||
| Fig. 2 Optical investigation of chiral DPP dyes 1–4 in CHCl3 solution. Absorbance (blue) and photoluminescence (red) spectra of: (a) DPP-CT; (b) DPP-MT; (c) DPP-ACT; (d) DPP-AMT. For absorbance measurements: cell length 1 cm; sample concentration 10−5 M. For photoluminescence measurements: sample concentration 10−5 M, excitation wavelength 467 nm. | ||
Moving on to photoluminescence spectra, in all cases we found a strong emission band between 500 and 700 nm, exhibiting a quite specular profile to the low energy UV-Vis absorption band (thus slightly more resolved for symmetrical DPPs), with maximum centred at 564 nm (for symmetrical systems 1–2) or 563 nm (for non-symmetrical systems 3–4). As a result, the four chiral dyes also showed modest Stokes shift values: 15 nm for 1–2, 23 nm for 3–4. Remarkable differences were also found in the fluorescence quantum yields Φ: symmetrically N-substituted DPP-CT and DPP-MT showed higher values (69% and 76%, respectively) than non-symmetrically N-substituted DPP-ACT and DPP-AMT (57% and 39%, respectively). A more efficient nonradiative decay for DPP-ACT and DPP-AMT is in line with larger broadening observed in the absorption spectra. On the contrary, both the nature of chiral groups and the molecular symmetry did not impact the values of optical band gaps (Eoptg), estimated from the absorption onset (λonset) in the UV-Vis spectra (obtained from the intercept of the red-side slope of low energy absorption band with the wavelength axis): for all the compounds, it was determined as about 2.18 eV.
All the most relevant optical properties of chiral DPP dyes 1–4 measured in CHCl3 solution are summarized in Table 1, while all the plots for the determination of molar extinction coefficients ε (Fig. S2–S5†), fluorescence quantum yields Φ (Fig. S6–S9†) and UV-Vis absorption onset λonset (Fig. S10†) can be found in ESI.†
| Chiral DPP dye |
λ
maxabs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (nm) a (nm) |
ε
maxb (104 M−1 cm−1) |
λ
maxemc (nm) |
Φ (%) | Stokes shifte (nm eV−1) |
λ
onsetf (nm) |
E
optgg (eV) |
|---|---|---|---|---|---|---|---|
| a Wavelength of the absorbance maximum. b Molar extinction coefficient corresponding to λmaxabs. c Wavelength of the emission maximum (excitation wavelength: 467 nm). d Fluorescence quantum yield determined by the relative method using rhodamine B in absolute ethanol as the reference. e Stokes shift defined as λmaxem − λmaxabs. f Onset wavelength determined from the intercept of the red-side slope of the absorbance main band with the wavelength axis. g Optical band gap evaluated as Eoptg = 1240/λonset. | |||||||
| DPP-CT | 549 | 2.57 | 564 | 69 | 15/0.06 | 569 | 2.18 |
| 340 | 0.95 | ||||||
| 292 | 2.04 | ||||||
| DPP-MT | 549 | 2.87 | 564 | 76 | 15/0.06 | 570 | 2.18 |
| 340 | 1.14 | ||||||
| 292 | 2.31 | ||||||
| DPP-ACT | 540 | 2.02 | 563 | 57 | 23/0.09 | 569 | 2.18 |
| 341 | 0.87 | ||||||
| 292 | 1.64 | ||||||
| DPP-AMT | 540 | 2.18 | 563 | 39 | 23/0.09 | 569 | 2.18 |
| 341 | 0.98 | ||||||
| 292 | 1.84 | ||||||
To gain more insight into the impact of different chiral alkyl chains, as well as of the adamantyl moiety on the HOMO and LUMO energy levels of DPP dyes, their electrochemical properties in solution were investigated by means of CV (Fig. 3). All the compounds showed four electrochemical processes: two in oxidation, two in reduction. However, quite interestingly their reversibility was found to be dependent on the nature of chiral alkyl group, rather than on the substitution symmetry of DPP moiety. For DPP-CT and DPP-ACT, bearing the (S)-β-cytronellyl chain, both the first oxidation and the first reduction were electrochemically and chemically reversible, while the other two processes were irreversible (Fig. S11 and S12 of ESI†). More details about the observed electrochemical processes were obtained by repeating CV experiments of DPP-CT at different scanning rates (Fig. S13 of ESI†): according to the Randles–Sevcik equation,69 the linearity between the peak current and the square root of scanning rate testified a diffusive nature for first oxidation and first reduction processes. Furthermore, linear sweep voltammetry (LSV) experiments carried out on both DPP-CT and DPP-ACT (Fig. S14 and S15 of ESI†) revealed comparable current intensities for the oxidation processes, indicating that the same number of electrons is involved. It can thus be reasonably assumed that both the oxidation processes are monoelectronic. Moving on DPP-MT and DPP-AMT, functionalized with (S)-2-methylbutyl chain, not only first oxidation and first reduction, but also the second oxidation are electrochemically and chemically reversible processes; only the second reduction was found irreversible (Fig. S16 and S17 of ESI†). This quite unexpected result suggested that the second oxidation in DPP-CT and DPP-ACT actually involved the double bond of the (S)-β-cytronellyl chain, which is not present in DPP-MT and DPP-AMT. However, CVs at different scanning rates revealed also for DPP-MT (Fig. S18 of ESI†) a diffusive nature for first oxidation and first reduction processes.
 | ||
| Fig. 3 Electrochemical investigation of chiral DPP dyes 1–4 in solution. Cyclic voltammograms of: (a) DPP-CT; (b) DPP-MT; (c) DPP-ACT; (d) DPP-AMT, recorded on a Pt disk (surface: 0.07 cm2) working electrode in CH2Cl2/n-Bu4NPF6 0.2 M solution. Sample concentration 10−3 M; scanning rate 100 mV s−1. | ||
Beyond these significant differences between (S)-β-cytronellyl- and (S)-2-methylbutyl-functionalized DPPs concerning the nature of the electrochemical processes, only minimal variations were found in the values of oxidation and reduction potentials, and consequently also in the HOMO and LUMO energy levels (see Table 2 for details). In particular, symmetrical DPP dyes 1–2 exhibited electrochemical band gap Eelg values slightly lower (1.99 eV for DPP-CT, 2.01 eV for DPP-MT) than non-symmetrical DPP dyes 3–4 (2.05 eV for DPP-ACT, 2.04 eV for DPP-AMT). In this regard, for the model compound DPP-Me used in the computational analysis we also calculated a HOMO–LUMO energy gap (Ecompg) of 2.46 eV in vacuo (see Fig. S19 of ESI†), slightly higher than those evaluated experimentally.
| Chiral DPP dye |
E
onoxa (V) |
HOMOb (eV) |
E
onredc (V) |
LUMOd (eV) |
E
elge (eV) |
|---|---|---|---|---|---|
| a Oxidation potential determined from the onset of the oxidation wave of the DPP dye vs. the ferrocene/ferrocenium (Fc/Fc+) reference. b HOMO energy level estimated by the empirical equation HOMO = −e(Eonox + 5.1 V). c Reduction potential determined from the onset of the reduction wave of the DPP dye vs. the ferrocene/ferrocenium (Fc/Fc+) reference. d LUMO energy level estimated by the empirical equation LUMO = −e(Eonred + 5.1 V). e Electrochemical band gap evaluated as Eelg = e(Eonox − Eonred). | |||||
| DPP-CT | 0.43 | −5.53 | −1.56 | −3.54 | 1.99 |
| DPP-MT | 0.44 | −5.54 | −1.57 | −3.53 | 2.01 |
| DPP-ACT | 0.46 | −5.56 | −1.59 | −3.51 | 2.05 |
| DPP-AMT | 0.44 | −5.54 | −1.59 | −3.51 | 2.04 |
To conclude the characterization of the four chiral dyes in solution, we also studied their optical and chiroptical behaviour in conditions of potential solution aggregation, induced by the use of solvent mixtures consisting of a good (CHCl3) and a poor solvent (MeOH) in different ratios. In general, spectral profiles recorded with the progressive addition of a polar protic solvent could provide a step-by-step picture of the self-assembly processes occurring in solution. Unfortunately, all the UV-Vis absorption and ECD spectra of 1–4 (see Fig. S20–S23 of ESI†) showed only negligible differences with those recorded in pure chloroform: the four DPPs, both symmetrical and non-symmetrical ones, would not undergo aggregation in CHCl3/MeOH mixtures. This result is not completely surprising: similarly to that previously reported in the literature for other symmetrically N-substituted chiral 3,6-dithienyl-DPPs of similar structure,62,63 compounds 1–4 exhibited a very small region of π-electron delocalization, especially if compared to the length of the branched chiral alkyl chains and to the high steric hindrance of the adamantylmethyl appendage. This is responsible for only very modest π-interactions, easily overwhelmed by conformational disorder and entropy effects associated with the chiral alkyl chains and/or the bulky adamantane moiety.
If in solution we found that both the chiral alkyl chain and the substitution symmetry may have a significant impact on the photophysical properties, this was even more evidently in thin films, where our comparative study was mostly focused. All the samples were prepared by spin-coating technique, depositing about 100 μL of a 2.0 × 10−2 M CHCl3 solution of each dye on square glass slides, followed by fast evaporation of the solvent while spinning the plates (time 30 s, rate 1000 rpm, acceleration 1000 rpm s−1). All the obtained films, appearing macroscopically as dark purple semi-transparent layers, were then subjected to a detailed optical and chiroptical investigation. Since solid-state organization in thin films may be strongly dependent not only on the intimate chemical structure of the material, but also on post-deposition operations, for each chiral DPP dye we compared the chiroptical properties of samples before (as-cast) and after prolonged solvent or thermal annealing. Under each condition, ECD spectra of thin films were recorded for the front face (organic layer towards light source) and the back side (organic layer towards detector), in order to evaluate the contribution of the above mentioned intrinsic (CDiso) and non-reciprocal (LDLB) terms.
ECD spectra and chiroptical properties of thin films of 1–4 as-cast and under the best experimental conditions of annealing are depicted in Fig. 4 and Table 3; all the UV-Vis absorption, ECD and dissymmetry factor gabs spectra of samples under all the investigated conditions can be found in Fig. S24–S39 of ESI.† For some of these samples, OM images in bright field and under cross-polarized filters were also acquired, depicted in Fig. 5 and Fig. S40–S46 of ESI.†
 | ||
| Fig. 4 Chiroptical investigation for thin films of chiral DPP dyes 1–4 prepared by spin coating technique under the most relevant experimental conditions. ECD spectra recorded for: (a) DPP-CT, before (continuous lines) and after (dashed lines) annealing (CHCl3, 40 min); (b) DPP-MT, before (continuous lines) and after (dashed lines) annealing (CHCl3, 10 min); (c) DPP-ACT, before (continuous line) and after (dashed lines) annealing (110 °C, 30 min); (d) DPP-AMT with no annealing. For panels (a)–(c): blue lines are spectra recorded for the front face (organic layer towards light source), red lines are spectra recorded for the back face (organic layer towards detector). | ||
 | ||
| Fig. 5 Microscopy investigation for thin films of chiral DPP dyes 1–4 prepared by spin coating technique under the most relevant experimental conditions. OM images of: (a) DPP-CT, before (up) and after (down) annealing (CHCl3, 40 min); (b) DPP-MT, before (up) and after (down) annealing (CHCl3, 10 min); (c) DPP-ACT, before (up) and after (down) annealing (110 °C, 30 min); (d) DPP-AMT, with no annealing. All images were recorded in bright field; only for DPP-ACT after annealing (panel c, down), the image under cross-polarized filters is also shown. | ||
| Chiral DPP dye | Annealinga |
λ
maxECDb (nm) |
g
maxabsc (10−2) |
ρ |
|---|---|---|---|---|
a Experimental details of solvent or thermal annealing (if any) performed before absorbance and ECD measurements.
b Wavelength of the maximum ECD intensity.
c Maximum dissymmetry factor value, defined as gabs = (Ellipticity (mdeg)/32980)/Absorbance, calculated at λmaxECD.
d Ratio of the integrated areas of the absolute value of semi-difference  and semi-sum and semi-sum  . .
|
||||
| DPP-CT | No | Front: 299 | 0.51 | 0.20 |
| Back: 299 | 0.38 | |||
| CHCl3, 40 min | Front: 293 | −0.32 | 0.32 | |
| Back: 293 | −0.65 | |||
| DPP-MT | No | Front: 518 | 0.84 | 0.27 |
| Back: 518 | 0.54 | |||
| CHCl3, 10 min | Front: 460 | −0.27 | 0.33 | |
| Back: 460 | −0.27 | |||
| DPP-ACT | No | n.a. | n.a. | n.a. |
| 110 °C, 30 min | Front: 530 | −0.15 | 1.52 | |
| Back: 489 | 0.22 | |||
| DPP-AMT | No | n.a. | n.a. | n.a. |
Starting our discussion from the symmetrically N-substituted DPP-CT dye, the UV-Vis absorption spectrum of the as-cast sample (Fig. S24 of ESI†) exhibited three main bands, similarly to that found in solution. However, a clear difference from the solution spectrum concerns the lowest energy band, which is red shifted by 30–40 nm and characterized by a shoulder between 600 and 650 nm. This is presumably an “aggregation band”,70 suggesting the presence of strong intermolecular interactions between adjacent π-conjugated chains. This was then confirmed by ECD characterization (Fig. 4a, continuous lines), where we found an asymmetric, positive exciton couplet between 420 and 650 nm, corresponding to the lowest energy absorption band of the π → π* transition on the DPP moiety, and two further positive bands at higher energy, associated to the other electronic transitions. Moreover, the spectral profiles recorded for the two faces of the film were very similar, which suggested an almost pure CDiso contribution associated to 3D chiral structures at nanoscopic level. In particular, the positive ECD couplet could indicate the occurrence of a right-handed supramolecular arrangement between adjacent DPP moieties. A positive couplet might also be seen around 300 nm for the transverse-polarized transition. We recall that for a pack of helically stacked chromophores with rectangular shape, the exciton chirality defined by longitudinal-polarized transitions coincide with that defined by transverse-polarized transitions. To study the impact of post-deposition treatments, thin films of DPP-CT were then subjected to solvent annealing, by keeping the sample in a close chamber saturated with CHCl3 vapours. Quite interestingly, if annealing for 10 minutes did not have relevant impact on the chiroptical features (Fig. S25 of ESI†), after a longer time more significant variations in the ECD spectra were appreciated (Fig. S26 and S27 of ESI†). In particular, after 40 minutes (Fig. 4a, dashed lines), the two positive bands in the high energy region (between 280 and 375 nm) reversed their sign; at the same time, the positive ECD couplet at lower energy evolved into a single broad positive band. These variations did not significantly impact the ρ ratio (which increased from 0.20 to 0.32), thus confirming a predominant intrinsic CDiso term (due to 3D chiral structures). Therefore, annealing is responsible of a significant change in the solid-state organization, where a supramolecular helical arrangement has been presumably replaced, or accompanied, by structures involving a chiral intramolecular twist of the π-conjugated moiety. In general, in closely packed supramolecular arrangements of chiral π-conjugated moieties, a combination of intra-molecular (intra-chain) twist and supramolecular (inter-chain) twist should always be expected.36,71 It is worth emphasizing that such differences in the solid-state arrangement were appreciated only thanks to the superior capability of ECD measurements in providing information about the first level of supramolecular organization, which may be very difficult to identify with other spectroscopy or microscopy technique. Indeed, no appreciable differences were found comparing OM images of DPP-CT before and after annealing (Fig. 5a and Fig. S40, S41 of ESI†). Similar result was observed when thin films of DPP-CT were subjected to thermal annealing at 100 °C, although slightly longer times (1–2 h) were needed (Fig. S28 and S29 of ESI†). In contrast, in the ECD spectra of samples after 1 month of ageing (Fig. S30 of ESI†) only very small variations compared to as-cast thin films were observed. This proved that, even in freshly prepared samples, the DPP-CT molecules were arranged in an energetically stable aggregation state, although the molecular mobility provided by annealing facilitated an evolution towards the most thermodynamically stable organization.
Moving on to DPP-MT, a simple change in the position of the stereogenic center on chiral alkyl chains, by keeping the same substitution symmetry on the DPP moiety, resulted in significant variations of the solid-state organization in thin films. Also in this case, the absorption spectrum of as-cast sample (Fig. S31 of ESI†) showed major differences with respect to solution, especially on the lowest energy band: although it also fell between 450 and 600 nm, the shape was here quite different, with a maximum at 514 nm and a further peak at 560 nm (the latter potentially identifiable as “aggregation band”). Interestingly, this band also appeared very dissimilar from that of as-cast thin films of DPP-CT, thus suggesting the possible occurrence of different solid-state aggregates. Indeed, this was further confirmed by the chiroptical investigation. The ECD spectra of as-cast DPP-MT (Fig. 4b, continuous lines), characterized by an almost pure intrinsic CDiso (ρ = 0.27, with minimal changes between front and back faces likely due to reproducibility issues related to a sample off-set), revealed a rather complex profile. Besides a large asymmetric positive ECD couplet between 450 and 600 nm, corresponding to the above-mentioned π → π* transition band in the absorption spectrum, a further negative band was found above 600 nm, not present for DPP-CT, likely associated with scattering. Other differences with respect to freshly prepared spin-coated samples of DPP-CT were observed in the higher energy region, with a second positive ECD couplet between 300 and 400 nm (in correspondence with the absorption band centred at around 350 nm), and a sharp negative ECD signal at 289 nm. If the positive sign of the two asymmetric ECD couplet could suggest the existence, also for as-cast samples of DPP-MT, of 3D chiral supramolecular structures with a right-handed helicity, we can still imagine important structural differences compared to DPP-CT films under the same conditions. The higher intensity and more pronounced vibronic pattern seen for DPP-MT indicates a higher degree of supramolecular order, possibly associated with the shorter and less flexible chain. Another relevant difference of DPP-MT with respect to DPP-CT concerns the impact of post-deposition operations (Fig. S32–S34 of ESI†). In particular, after only 10 minutes of solvent annealing in a closed chamber saturated with CHCl3 vapours, in the UV-Vis absorption spectrum (Fig. S32 of ESI†) we found a strong decrease of the “aggregation band” at 560 nm, suggesting a partial dissolution of solid-state aggregates, yielding a situation where DPP molecules are more distant and held together by weaker intermolecular interactions. In fact, even in the corresponding ECD spectrum (Fig. 4b, dashed lines) we observed a strong decrease in the chiroptical signals, confirming a partial loss of aggregation. At the same time, we also found some changes in the spectral shape with respect to the as-cast sample: if the positive couplet between 400 and 550 nm is still present, a disappearance of the negative band above 600 nm and a sign reversal of bands at higher energy was observed. Longer time of solvent annealing (Fig. S33 of ESI†) simply resulted in a further decrease of chiroptical signals. As in the case of DPP-CT, OM images of thin film samples of DPP-MT acquired before and after solvent annealing (Fig. 5b and Fig. S42, S43 of ESI†) did not exhibit any remarkable change in the organic layer texture. Similarly, 1 h of thermal annealing at 170 °C (Fig. S34 of ESI†) also resulted in an almost complete disappearance of all ECD bands, except for a very weak couplet between 450 and 550 nm. In contrast, no changes were appreciated after 1 month of ageing (Fig. S35 of ESI†). This aspect is in common with DPP-CT: for as-cast thin films of DPP-MT, molecules were arranged in a thermodynamically stable aggregation state, which can be disturbed only by increasing the molecular motion in the presence of solvent vapours or at high temperatures.
If the different nature of the enantiopure branched chain already had a significant impact on the optical and chiroptical properties of symmetrical DPP dyes 1–2 in thin films, a change in the substitution symmetry by replacing one of these chiral chains with an adamantylmethyl appendage resulted in a totally different (chiro)optical behaviour, indicating the occurrence of a different solid-state organization. Starting with the study of DPP-ACT, differences with its symmetrical analogue DPP-CT already emerged in the UV-Vis absorption spectrum of the as-cast film (Fig. S36 of ESI†). Surprisingly, the spectral profile was almost entirely coincident with that recorded in CHCl3 solution: no appreciable bathochromic shift was observed, nor the “aggregation band” previously found at low energy for both symmetrical dyes. This would indicate the presence of DPP-ACT molecules in the isolated state, or sufficiently distant from each other to result in a quasi-isolated form despite the thin film state. Such hypothesis was confirmed by the ECD spectrum of the same sample (Fig. 4c, continuous blue line), exhibiting no chiroptical signal. Although the formation of neither achiral aggregates nor a racemate of right- and left-handed helical supramolecular structures cannot be excluded, both the lower molecular symmetry and the steric hindrance of the adamantane appendage may have favoured totally disordered solid-state structures, or alternatively ordered ones with an intermolecular distance much greater than that of symmetric systems. A further puzzling aspect came from thermal annealing of the same sample: by keeping it in an oven at 110 °C for 30 min, we observed the appearance of some weak dichroic signals in the ECD spectrum (Fig. 4c, dashed lines and Fig. S37 of ESI†), exhibiting a predominant non-reciprocal LDLB contribution, as testified by the ρ ratio value (1.52). The increased molecular mobility acquired at high temperatures allows DPP-ACT molecules to organize into 2D chiral microscopic domains, although the weak dichroic signals could suggest only a very slight prevalence of domains leaning on the glass substrate with a well-defined enantiomeric face. In this specific case, even OM measurements were useful: if the as-cast sample (Fig. 5c, up and Fig. S44 of ESI†) showed a very homogeneous texture, without any notable LD and LB, after thermal annealing we observed the appearance of small grains, few μm sized, exhibiting moderate LB under cross-polarized filters (Fig. 5c, down and Fig. S45 of ESI†). These grains could represent the above mentioned 2D chiral microscopic domains, characterized by interacting local LD and LB, responsible for the predominant non-reciprocal chiroptical signal.
Even in the case of non-symmetrical DPP-AMT dye, the UV-Vis absorption spectrum of the as-cast thin film (Fig. S38 of ESI†) was very similar to that one recorded in CHCl3 solution, and no dichroic signal was detected in the corresponding ECD spectrum (Fig. 4d). We hypothesize here the same behaviour reported above for DPP-ACT, with a solid-state organization involving molecules sufficiently distant from each other to result in a quasi-isolated form. Behaviour similar to DPP-ACT also occurred in the OM investigation (Fig. 5d and Fig. S46 of ESI†), where no structure was observed. However, a difference between the two non-symmetrically substituted dyes occurred after thermal annealing: keeping the DPP-AMT samples for 1 h at 120 °C, no difference was observed in both absorption and ECD spectra (Fig. S39 of ESI†). Therefore, even more than for DPP-ACT, in thin films of DPP-AMT the quasi-isolated solid state is such energetically stable, that it did not undergo any modification even by thermally increasing the molecular motions.
The present optical and chiroptical study of the four DPP dyes revealed that the nature of chiral alkyl chain, both in terms of length and position of the stereogenic center, can lead to important variations in the solid-state organization (DPP-CTvs.DPP-MT, as well as DPP-ACTvs.DPP-AMT). However, the most substantial differences came from comparing dyes of different substitution pattern: from 3D chiral structures of the symmetrically substituted DPP-CT and DPP-MT, to disordered/isolated or, possibly, 2D chiral structures of the non-symmetrically substituted DPP-ACT and DPP-AMT. While a simulation of (chiro)optical properties of large and disordered supramolecular assemblies remains a formidable task,36 we substantiated the observation that bulky adamantyl groups disrupt the tendency to aggregation by running metadynamics simulations by CREST (conformer-rotamer sampling tool),72 coupled with semi-empirical tight binding calculations at GFN2-xTB level.73 We built small oligomers (dimers and trimers) of DPP-MT (2) and DPP-AMT (4) included in an ellipsoid wall potential designed to model non-covalent interactions (NCI). The results for the trimers are analysed in Fig. S47 of ESI† in terms of plane-to-plane distance and inclination between proximate DPP moieties (1–2 and 2–3 units), while representative snapshots are shown in Fig. 6. It emerges that DPP-MT is able to reach more compact and ordered aggregates, with DPP moieties at stacking distance (<3.8 Å) and almost parallel to each other. The simulations also highlight that, in the stacks, the DPP moieties are tilted toward alternating opposite directions, rather than aligned with each other. This situation prevents a simple interpretation of exciton-coupled ECD spectra in terms of staircase-like helical stacks.
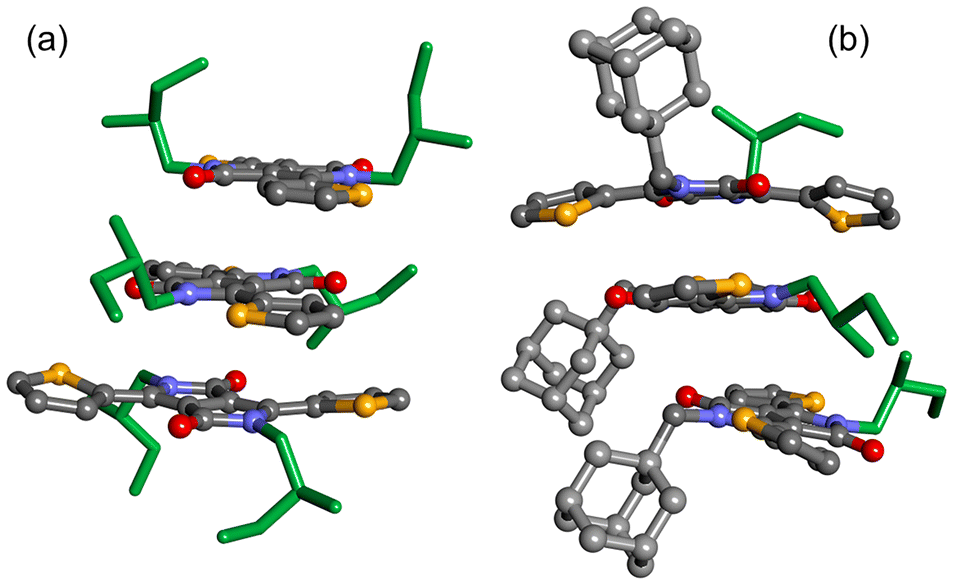 | ||
| Fig. 6 Representative snapshots from the CREST simulations of: (a) DPP-MT; (b) DPP-AMT. | ||
These results are in line with the different chiroptical behaviour observed for the present and other classes of DPP dyes recently investigated by our research group,40b,67 as summarised in Fig. 7. Th2-DPP-Th2 derivatives, where the central DPP core is connected to two lateral 2,2′-bithienyl units, tend to adopt a staircase-like aggregate structure (Fig. 7a), thus favouring exciton coupling between first neighbours which dominates the CDiso.67 In the case of Anth-Th-DPP-Th-Anth derivatives, where two lateral (9-anthracenyl)thienyl moieties are present, exciton-coupled ECD is hampered by an almost perfect alignment between DPP units (Fig. 7b), which instead strengthens the LDLB terms.40b Finally, for the present Th-DPP-Th derivatives, the alternating tilted orientation between DPP moieties (Fig. 7c) downplays exciton coupling with respect to Th2-DPP-Th2, yielding ECD spectra with less evident couplets or showing contribution from intra-chain mechanism.
 | ||
| Fig. 7 Pictorial representation of the aggregation modes observed for three different classes of DPP dyes recently investigated by our group: (a) Th2-DPP-Th2 derivatives, (b) Anth-Th-DPP-Th-Anth derivatives, (c) Th-DPP-Th derivatives. | ||
Conclusions
In conclusion, here we have carried out a comparative study on four chiral 1,4-diketo-3,6-dithienylpyrrolo[3,4-c]pyrrole (DPP) dyes, focusing on how the nature and symmetry of N-substitution affect their photo-physical (and, in particular, chiroptical) properties. Our results clearly demonstrate that both types of chiral side chains (β-citronellyl vs. 2-methylbutyl) and the substitution pattern (symmetrical vs. non-symmetrical) play crucial roles, particularly in dictating the solid-state organization at different hierarchical levels of these dyes in thin films.On the one hand, symmetrically substituted derivatives (DPP-CT and DPP-MT) exhibit the propensity to form more ordered, 3D chiral aggregates. On the other, the introduction of a bulky adamantyl group in the non-symmetrical derivatives (DPP-ACT and DPP-AMT) appears to disrupt such organization, leading instead to more disordered or possibly 2D chiral arrangements.
This interpretation is further supported by metadynamics simulations: while the two symmetrically substituted DPPs can form compact, tilted, yet organized aggregates, the non-symmetrically substituted systems display only a reduced tendency to stack efficiently. More importantly, the observed alternating tilt between DPP units within the aggregates prevents straightforward interpretation of their ECD signatures in terms of conventional helical stacking, indicating a more complex excitonic landscape.
When compared to other previously investigated DPP dyes having different lateral π-conjugated units, the Th-DPP-Th framework investigated here reveals a distinct chiroptical behaviour. Unlike Th2-DPP-Th2 derivatives, where 3D-chiral staircase-like assemblies, promoting strong exciton coupling, are responsible for intrinsic CDiso terms, or Anth-Th-DPP-Th-Anth systems, where tight alignment favours the occurrence of 2D-chiral structures responsible for strong non-reciprocal LDLB contributions, the alternating orientation observed in Th-DPP-Th compounds reduces the strength of exciton coupling, thus generating weaker intrinsic CDiso contributions in the ECD spectra, characterized by less evident couplets and contributions due to intra-chain mechanism.
Overall, this work underscores the delicate interplay between molecular design, supramolecular organization and photo-physical properties, offering valuable insights for the future development of functional organic materials with the desired chiroptical properties in thin films.
Experimental section
Synthetic procedures
1H-NMR and 13C-NMR spectra were recorded from solutions in deuterated CDCl3 solvent on Benchtop NMR Spinsolve 80 MHz or FT-NMR spectrometer Bruker AVANCE™ III 500 MHz, and chemical shifts (δ) are given in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal reference. Melting points were determined on a Kofler apparatus (type KB T300 equipped with an optical system with a 40× magnification) and the temperature was not calibrated. Elemental analysis of pure products was performed on a Elementar Vario Micro Cube CHNS-analyzer.
1-(Bromomethyl)adamantane (5). (Adamantan-1-yl)methanol (6.7 g, 40.30 mmol, 1.0 eq.) was stirred in 48% hydrobromic acid (37 mL, 8.0 eq.) at reflux for 18 h. The biphasic reaction mixture was then cooled to RT and washed with chloroform (3 × 30 mL). The collected organic phases were then washed with 96% sulphuric acid (2 × 30 mL), followed by saturated NaHCO3 solution (2 × 150 mL), then with distilled water (2 × 200 mL), finally with brine (120 mL) and then dried over anhydrous Na2SO4 and filtered through filter aid (Celite). After removal of the solvent under vacuum, the obtained crude mixture was purified by crystallisation in methanol (50 mL) to get pure product 5 as a white crystal material (4.42 g, yield 48%). Analytical data are in agreement with those previously reported in the literature.75 Melting point 42 °C (lit. 41–45 °C).751H-NMR (80 MHz, CDCl3, ppm): δ = 3.15 (s, 2H), 1.99 (m, 3H), 1.67 (m, 6H), 1.56 (m, 6H).
(S)-3,7-Dimethyloct-6-en-1-yl 4-methylbenzenesulfonate (6). (S)-(−)-β-Citronellol (0.49 g, 3.15 mmol, 1.00 eq.) was stirred in DCM (7 mL) and Et3N (0.38 g, 3.76 mmol, 1.20 eq.) under argon atmosphere. After that, TsCl (0.67 g, 3.53 mmol, 1.12 eq.) was added to the reaction mixture and it was stirred at room temperature for 5.5 hours. The reaction was then quenched with saturated aqueous NaHCO3 solution (4 mL) and extracted with DCM (3 × 5 mL). The collected organic phases were then dried over anhydrous Na2SO4 and filtered through Celite. After removal of the solvent under vacuum, the obtained crude mixture was purified by flash chromatography (eluent petroleum ether/ethyl acetate 20
:1, Rf = 0.38) to get pure product 6 as a colourless liquid (0.84 g, yield 86%). Analytical data are in agreement with those previously reported in the literature.761H-NMR (80 MHz, CDCl3, ppm): δ = 7.78 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.2 Hz, 2H), 5.12–4.89 (m, 1H), 4.05 (t, J = 6.4 Hz, 2H), 2.43 (s, 3H), 1.95–1.75 (m, 2H), 1.66–1.60 (m, 4H), 1.58–1.35 (m, 5H), 1.30–1.23 (m, 1H), 1.17–1.07 (m, 1H), 0.80 (d, J = 5.8 Hz, 3H).
(S)-2-Methylbutyl 4-methylbenzenesulfonate (7). (S)-(−)-2-Methylbutanol (1.07 g, 12.10 mmol, 1.00 eq.) was stirred in pyridine (7 mL) under argon atmosphere and the reaction mixture was cooled to 0 °C. After that, TsCl (2.76 g, 14.49 mmol, 1.20 eq.) was added to the reaction mixture and it was stirred at 0 °C for 20 hours. The reaction was then quenched with ice-water (80 mL) and extracted with diethyl ether (3 × 15 mL). The collected organic phases were then washed with distilled water (2 × 45 mL), followed by 2 M HCl solution (1 × 45 mL), then with saturated aqueous NaHCO3 solution (1 × 45 mL), finally with brine (45 mL) and then dried over anhydrous Na2SO4 and filtered through Celite. After removal of the solvent under vacuum, the obtained crude mixture was purified by flash chromatography (eluent petroleum ether/ethyl acetate 15
:1, Rf = 0.47) to get pure product 7 as a colourless liquid (2.64 g, yield 90%). Analytical data are in agreement with those previously reported in the literature.771H-NMR (80 MHz, CDCl3, ppm): δ = 7.81 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 3.87 (d, J = 5.9 Hz, 2H), 2.47 (s, 3H), 1.91–1.00 (m, 3H), 0.94 (s, 3H), 0.87–0.72 (m, 3H).
2-(Adamantan-1-yl)methyl-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (9). 3,6-Di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8) (0.85 g, 2.83 mmol, 1.0 eq.) and anhydrous K2CO3 (1.17 g, 8.49 mmol, 3.0 eq.) were stirred at 60 °C for 1 h in anhydrous DMF (60 mL) and under argon atmosphere. Then, reagent 5 (0.97 g, 4.25 mmol, 1.5 eq.) was added in one portion to the reaction mixture, and it was stirred at 140 °C for 20 h. After that, the reaction mixture was poured into distilled water (350 mL), and the formed precipitate was filtered off and washed first with distilled water (3 × 150 mL) to remove unreacted K2CO3 and residual DMF. The filter cake was then washed with hot chloroform (3 × 180 mL) to dissolve the product and separate it from the unreacted starting material 8. The chloroform filtrate was then concentrated to give the crude product, which was further purified twice by flash chromatography (eluent DCM/n-heptane 4
:1, Rf = 0.39, →1:1) to get pure product 9 as a dark red solid (0.21 g, yield 15%). Melting point >340 °C. 1H-NMR (500 MHz, CDCl3, ppm): δ = 8.54 (dt, J = 3.9; 1.0 Hz, 1H), 8.37 (dt, J = 3.9; 1.0 Hz, 1H), 8.25 (s, 1H), 7.61 (ddd, J = 4.9; 3.9; 1.0 Hz, 2H), 7.26–7.23 (m, 2H), 3.93 (s, 2H), 1.89 (s, 2H), 1.63 (s, 1H), 1.57–1.47 (m, 10H), 1.25 (s, 4H), 0.90–0.83 (m, 1H).
2,5-Bis((S)-3,7-dimethyloct-6-en-1-yl)-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (DPP-CT, 1). 3,6-Di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8) (0.16 g, 0.53 mmol, 1.0 eq.) and anhydrous K2CO3 (0.37 g, 2.65 mmol, 5.0 eq.) were stirred at 60 °C for 35 min in anhydrous DMF (7.5 mL) and under argon atmosphere. Then, reagent 6 (0.57 g, 1.84 mmol, 3.5 eq.) was added dropwise to the reaction mixture, and it was stirred at 125 °C for 17 h. After that, the reaction mixture was reduced by vacuum distillation, then suspended in methanol (20 mL), filtered and the obtained filter cake was washed with water (3 × 15 mL), methanol (3 × 15 mL) and toluene (3 × 25 mL). The toluene filtrate was then washed in a separator funnel with distilled water (3 × 80 mL), brine (80 mL) and then dried over anhydrous Na2SO4 and filtered through Celite. After removal of the solvent under vacuum, the obtained crude mixture was purified by flash chromatography (eluent toluene, Rf = 0.55) followed by recrystallization in n-heptane/DCM (2.5
:0.5 mL) to get pure DPP-CT product 1 as a dark violet-red crystal material (54.5 mg, yield 18%). Melting point 121 °C. 1H-NMR (500 MHz, CDCl3) δ = 8.90 (dd, J = 3.9, 1.2 Hz, 2H), 7.63 (dd, J = 5.0, 1.2 Hz, 2H), 7.28 (dd, J = 5.0, 3.9 Hz, 2H), 5.08 (tdt, J = 5.8, 2.9, 1.5 Hz, 2H), 4.19–4.02 (m, 4H), 2.07–1.90 (m, 4H), 1.81–1.71 (m, 2H), 1.67 (d, J = 1.4 Hz, 6H), 1.64–1.53 (m, 10H), 1.41 (dddd, J = 13.4, 9.5, 6.4, 5.2 Hz, 2H), 1.24 (dddd, J = 13.4, 9.4, 7.5, 5.9 Hz, 2H), 1.02 (d, J = 6.3 Hz, 6H). 13C-NMR (126 MHz, CDCl3) δ = 161.50, 140.19, 135.33, 131.51, 130.76, 129.88, 128.76, 124.72, 107.98, 40.94, 37.10, 36.85, 31.04, 25.85, 25.60, 19.55, 17.82. Anal. calcd for C34H44N2O2S2: C, 70.79; H, 7.69; N, 4.86; S, 11.12; found: C, 70.73; H, 7.74; N, 4.85; S, 11.11.
2,5-Bis((S)-2-methylbutyl)-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (DPP-MT, 2). 3,6-Di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8) (0.21 g, 0.70 mmol, 1.0 eq.) and anhydrous K2CO3 (0.48 g, 3.50 mmol, 5.0 eq.) were stirred at 60 °C for 35 min in anhydrous DMF (10 mL) and under argon atmosphere. Then, reagent 7 (0.59 g, 2.45 mmol, 3.5 eq.) was added dropwise to the reaction mixture, and it was stirred at 125 °C for 23 h. After that, the reaction mixture was reduced by vacuum distillation, then suspended in methanol (25 mL), filtered and the obtained filter cake was washed with water (3 × 15 mL), methanol (3 × 15 mL) and toluene (3 × 25 mL). The toluene filtrate was then washed in a separator funnel with distilled water (3 × 100 mL), brine (80 mL) and then dried over anhydrous Na2SO4 and filtered through Celite. After removal of the solvent under vacuum, the obtained crude mixture was purified by flash chromatography (eluent toluene, Rf = 0.14) followed by recrystallization in n-heptane/DCM (2.5
:0.5 mL) to get pure DPP-MT product 2 as a deep violet crystal material (37.3 mg, yield 12%). Melting point 190 °C. 1H-NMR (500 MHz, CDCl3) δ = 8.96 (dd, J = 3.9, 1.2 Hz, 2H), 7.62 (dd, J = 5.0, 1.2 Hz, 2H), 7.27 (dd, J = 5.0, 3.9 Hz, 2H), 4.05–3.92 (m, 4H), 1.93 (dddd, J = 15.4, 8.3, 6.9, 4.6 Hz, 2H), 1.50 (dqd, J = 13.5, 7.5, 4.5 Hz, 2H), 1.29–1.16 (m, 2H), 0.95–0.88 (m, 12H). 13C-NMR (126 MHz, CDCl3) δ = 161.88, 140.50, 135.65, 130.73, 130.06, 128.63, 107.97, 47.75, 35.56, 27.12, 16.74, 11.38. Anal. calcd for C24H28N2O2S2: C, 65.42; H, 6.41; N, 6.36; S, 14.55; found: C, 65.47; H, 6.44; N, 6.36; S, 14.54.
2-((Adamantan-1-yl)methyl)-5-((S)-3,7-dimethyloct-6-en-1-yl)-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (DPP-ACT, 3). 2-(Adamantan-1-yl)methyl-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (9) (0.16 g, 0.36 mmol, 1.0 eq.) and anhydrous K2CO3 (0.15 g, 1.08 mmol, 3.0 eq.) were stirred at 60 °C for 35 min in anhydrous DMF (5.5 mL) and under argon atmosphere. Then, reagent 6 (0.17 g, 0.54 mmol, 1.5 eq.) was added dropwise to the reaction mixture, and it was stirred at 125 °C for 21 h. After that, the reaction mixture was reduced by vacuum distillation, then suspended in methanol (25 mL), filtered and the obtained filter cake was washed with water (3 × 15 mL), methanol (3 × 15 mL) and toluene (3 × 25 mL). The toluene filtrate was then washed in a separator funnel with distilled water (3 × 100 mL), brine (80 mL) and then dried over anhydrous Na2SO4 and filtered through Celite. After removal of the solvent under vacuum, the obtained crude mixture was purified by flash chromatography (eluent toluene, Rf = 0.21) followed by recrystallization in n-heptane/DCM (1.0
:0.4 mL) to get pure DPP-ACT product 3 as a bright red crystal material (19.4 mg, yield 9%). Melting point 144 °C. 1H-NMR (500 MHz, CDCl3) δ = 8.92 (dd, J = 3.9, 1.2 Hz, 1H), 8.57 (dd, J = 3.8, 1.2 Hz, 1H), 7.63 (dd, J = 5.0, 1.1 Hz, 1H), 7.58 (dd, J = 5.0, 1.2 Hz, 1H), 7.26 (ddd, J = 19.9, 5.0, 3.8 Hz, 2H), 5.12–5.05 (m, 1H), 4.09 (dddd, J = 37.0, 14.4, 10.7, 5.2 Hz, 2H), 3.92 (s, 2H), 1.99 (ddp, J = 21.7, 14.2, 6.9 Hz, 2H), 1.88 (q, J = 3.1 Hz, 3H), 1.77 (ddt, J = 16.4, 9.2, 4.6 Hz, 1H), 1.67 (d, J = 1.4 Hz, 3H), 1.64–1.52 (m, 12H), 1.49 (d, J = 2.9 Hz, 5H), 1.45–1.36 (m, 1H), 1.24 (dddd, J = 13.4, 9.4, 7.4, 5.9 Hz, 1H), 1.01 (d, J = 6.4 Hz, 3H). 13C-NMR (126 MHz, CDCl3) δ = 162.40, 161.56, 141.41, 140.53, 135.48, 134.68, 131.51, 130.79, 130.52, 129.84, 128.79, 128.19, 124.72, 108.85, 107.70, 52.81, 41.03, 40.93, 37.11, 36.86, 36.84, 36.80, 31.06, 28.51, 25.86, 25.60, 19.55, 17.83. Anal. calcd for C35H42N2O2S2: C, 71.63; H, 7.21; N, 4.77; S, 10.93; found: C, 71.68; H, 7.16; N, 4.79; S, 10.92.
2-((Adamantan-1-yl)methyl)-5-((S)-2-methylbutyl)-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (DPP-AMT, 4). 2-(Adamantan-1-yl)methyl-3,6-di(thiophen-2-yl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (9) (0.21 g, 0.47 mmol, 1.0 eq.) and anhydrous K2CO3 (0.19 g, 1.39 mmol, 3.0 eq.) were stirred at 60 °C for 40 min in anhydrous DMF (6.8 mL) and under argon atmosphere. Then, reagent 7 (0.17 g, 0.70 mmol, 1.5 eq.) was added dropwise to the reaction mixture, and it was stirred at 125 °C for 21 h. After that, the reaction mixture was reduced by vacuum distillation, then suspended in methanol (25 mL), filtered and the obtained filter cake was washed with water (3 × 15 mL), methanol (3 × 15 mL) and toluene (3 × 25 mL). The toluene filtrate was then washed in a separator funnel with distilled water (3 × 100 mL), brine (100 mL) and then dried over anhydrous Na2SO4 and filtered through Celite. After removal of the solvent under vacuum, the obtained crude mixture was purified by flash chromatography (eluent toluene, Rf = 0.20) followed by recrystallization in n-heptane/DCM (2.5
:1.0 mL) to get pure DPP-AMT product 4 as a dark red crystal material (74.8 mg, yield 31%). Melting point 181 °C. 1H-NMR (500 MHz, CDCl3) δ = 8.96 (dd, J = 3.9, 1.2 Hz, 1H), 8.58 (dd, J = 3.8, 1.2 Hz, 1H), 7.62 (dd, J = 5.0, 1.2 Hz, 1H), 7.58 (dd, J = 5.0, 1.2 Hz, 1H), 7.30–7.21 (m, 2H), 4.03–3.89 (m, 2H), 3.93 (s, 2H), 1.97–1.86 (m, 2H), 1.62 (dt, J = 12.4, 2.9 Hz, 4H), 1.58–1.50 (m, 5H), 1.50 (s, 3H), 1.49 (s, 3H), 1.27–1.22 (m, 1H), 0.95–0.85 (m, 6H). 13C-NMR (126 MHz, CDCl3) δ = 162.37, 161.94, 141.41, 140.83, 135.71, 134.78, 130.77, 130.47, 130.03, 129.81, 128.63, 128.20, 108.67, 107.85, 53.55, 52.82, 47.71, 41.02, 36.85, 36.80, 35.55, 32.03, 28.51, 27.12, 22.83, 16.76, 14.25, 11.38. Anal. calcd for C30H34N2O2S2: C, 69.46; H, 6.61; N, 5.40; S, 12.36; found: C, 69.50; H, 6.57; N, 5.41; S, 12.35.
Characterization
Fluorescence emission measurements in solution were performed at room temperature using a Horiba Jobin Yvon FluoroLog®-3 fluorescence spectrophotometer, with temperature control to within ±0.1 °C, at excitation wavelength of 467 nm. Stock solutions of each chiral DPP dye in CHCl3 were 10−2 M, while working solutions were 10−5 M. Fluorescence quantum yields Φ, determined by the relative method using rhodamine B in absolute ethanol as the reference, were calculated by using five working solutions of the chiral DPP dye in CHCl3 at different concentrations, from 1.0 × 10−6 M to 5.0 × 10−6 M.
Cyclic voltammetry (CV) measurements were performed at room temperature with a PalmSens4 potentiostat instrument interfaced with a laptop equipped with the PSTrace5 electrochemical software, using a three-electrode home-built electrochemical cell. A Pt disk (surface: 0.07 cm2) was used as working electrode (WE), a Pt wire (BASi) as counter electrode (CE) and a leak-free Ag/AgCl/KCl (3.4 mol L−1) (eDAQ) as reference electrode (RE). The experiments were carried out under Ar using a 0.2 M solution of n-Bu4NPF6 in anhydrous CH2Cl2 as the supporting electrolyte. A proper amount of chiral DPP dyes 1–4 was dissolved in the supporting electrolyte solution, in order to achieve a final sample concentration of 1.0 × 10−3 M. CV measurements were carried out with a scanning rate of 100 mV s−1. Only for symmetrical DPP dyes 1 and 2, CV experiments were also repeated at other different scanning rates (ranging from 20 mV s−1 to 1000 mV s−1). The measured potentials were subsequently referenced to the Fc/Fc+ (ferrocene/ferricenium) redox couple, which was added as an internal standard. Under the present experimental conditions, the one-electron oxidation of ferrocene occurred at  vs. Ag/AgCl/KCl (3.4 mol L−1). Linear sweep voltammetry (LSV) measurements were acquired in a similar way, with a Pt rotating disk electrode as WE, a Pt wire as CE and a leak-free Ag/AgCl/KCl (3.4 mol L−1) as RE, using a scanning rate of 20 mV s−1.
vs. Ag/AgCl/KCl (3.4 mol L−1). Linear sweep voltammetry (LSV) measurements were acquired in a similar way, with a Pt rotating disk electrode as WE, a Pt wire as CE and a leak-free Ag/AgCl/KCl (3.4 mol L−1) as RE, using a scanning rate of 20 mV s−1.
Electronic circular dichroism (ECD) measurements under solution aggregation were performed at room temperature with a Jasco J-1500 spectropolarimeter. For each chiral DPP dye, solution aggregation was induced using solvent mixtures of CHCl3 as good solvent and THF as poor solvent in different ratio. Stock solutions of each dye in the good solvent (CHCl3) were 10−2 M, while solution aggregation samples in CHCl3/MeOH mixtures were 1.0 × 10−5 M.
UV-Vis absorbance spectra in thin films were recorded at room temperature using a Jasco V-650 spectrophotometer.
ECD spectra in thin films were recorded at room temperature using a Jasco J-1500 spectropolarimeter. In all cases, at least 3 independent thin film samples were prepared and subjected to full ECD analysis. Invariance of ECD signals upon sample rotation was first testified, by rotating thin film sample by 90°, 180° and 270° around the optical axis: in no cases were found significant variations. Then, for each sample two different measurements were recorded: one with the organic layer facing the light source (front) and one with the organic layer facing the detector (back). The semi-sum and semi-difference of front and back ECD spectra were calculated in order to obtain intrinsic CDiso and non-reciprocal LDLB contributions, according to eqn (2) and (3). For each thin film, the dissymmetry factor gabs spectrum was calculated as “ECD spectrum/UV-Vis absorbance spectrum”, i.e., as gabs = (Ellipticity (mdeg)/32980)/Absorbance.
Optical microscopy (OM) images of thin films were obtained at room temperature using a ZEISS SteREO Discovery V8 microscope equipped with a Canon PowerShot A640 camera.
Computational details
Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including NMR data (FID) of final products 1–4, optical, chiroptical and electrochemical raw data in solution, solution aggregation and thin films, microscopy images of thin films and XYZ coordinates generated by CREST calculations, are available in the Zenodo repository at https://doi.org/10.5281/zenodo.15668949.Acknowledgements
This work has benefited from the equipment and framework of the COMP-R Initiative, funded by the ‘Departments of Excellence’ program of the Italian Ministry for University and Research (MUR, 2023–2027). MC and JK would like to thank MEYS for financial support in the scope of the grant mobility CZ-AT 8J24AT 022. JK thanks project AKTION no. 95p3. The authors thank Dr Tiziana Funaioli for helpful discussions on electrochemical measurements.References
- (a) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed; (c) P. Friederich, A. Fediai, S. Kaiser, M. Konrad, N. Jung and W. Wenzel, Adv. Mater., 2019, 31, 1808256 CrossRef PubMed; (d) M. Cigánek, J. Richtár, M. Weiter and J. Krajčovič, Isr. J. Chem., 2022, 62, e202100061 CrossRef.
- Y. Zhang, L. Guo, X. Zhu and X. Sun, Front. Chem., 2020, 8, 589207 CrossRef CAS PubMed.
- G. P. Neupane, W. Ma, T. Yildirim, Y. Tang, L. Zhang and Y. Lu, Nano Mater. Sci., 2019, 1, 246–259 CrossRef.
- (a) S. Logothetidis, Mater. Sci. Eng., B, 2008, 152, 96–104 CrossRef CAS; (b) S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed; (c) K.-J. Baeg and J. Lee, Adv. Mater. Technol., 2020, 5, 2000071 CrossRef CAS.
- (a) J. Cornil, D. Beljonne, J.-P. Calbert and J.-L. Brédas, Adv. Mater., 2001, 13, 1053–1067 CrossRef CAS; (b) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (c) Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed; (d) T. L. Andrew and T. M. Swager, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 476–498 CrossRef CAS.
- (a) J. Gierschner, J. Cornil and H.-J. Egelhaaf, Adv. Mater., 2007, 19, 173–191 CrossRef CAS; (b) O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- (a) P. Leclère, M. Surin, P. Brocorens, M. Cavallini, F. Biscarini and R. Lazzaroni, Mater. Sci. Eng., R, 2006, 55, 1–56 CrossRef; (b) B. J. Rancatore, C. E. Mauldin, S.-H. Tung, C. Wang, A. Hexemer, J. Strzalka, J. M. J. Fréchet and T. Xu, ACS Nano, 2010, 4, 2721–2729 CrossRef CAS PubMed; (c) K. Ariga, T. Mori, M. Akamatsu and J. P. Hill, Thin Solid Films, 2014, 554, 32–40 CrossRef CAS.
- (a) P. Gemünden, C. Poelking, K. Kremer, K. Daoulas and D. Andrienko, Macromol. Rapid Commun., 2015, 36, 1047–1053 CrossRef PubMed; (b) V. Y. Rudyak, A. A. Gavrilov, D. V. Guseva, S.-H. Tung and P. V. Komarov, Mol. Syst. Des. Eng., 2020, 5, 1137–1146 RSC; (c) C. Kim, H. Joung, H. J. Kim, K. Paeng, L. J. Kaufman and J. Yang, NPG Asia Mater., 2023, 15, 32 CrossRef CAS.
- (a) R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed; (b) Y. Yao, H. Dong and W. Hu, Polym. Chem., 2013, 4, 5197–5205 RSC; (c) G. E. J. Hicks, S. Li, N. K. Obhi, C. N. Jarrett-Wilkins and D. S. Seferos, Adv. Mater., 2021, 33, 2006287 CrossRef CAS PubMed.
- (a) J. K. Stille, Pure Appl. Chem., 1985, 57, 1771–1780 CrossRef CAS; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (c) B. Betzemeier and P. Knochel, Angew. Chem., Int. Ed. Engl., 1997, 36, 2623–2624 CrossRef CAS.
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (c) G. Albano, A. Punzi, M. A. M. Capozzi and G. M. Farinola, Green Chem., 2022, 24, 1809–1894 RSC.
- (a) Y. Wu, J. Wang, F. Mao and F. Y. Kwong, Chem. – Asian J., 2014, 9, 26–47 CrossRef CAS PubMed; (b) G. Albano, Org. Chem. Front., 2024, 11, 1495–1622 RSC.
- (a) Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed; (b) Y. Diao, L. Shaw, Z. Bao and S. C. B. Mannsfeld, Energy Environ. Sci., 2014, 7, 2145–2159 RSC; (c) A. M. Hiszpanski, R. M. Baur, B. Kim, N. J. Tremblay, C. Nuckolls, A. R. Woll and Y.-L. Loo, J. Am. Chem. Soc., 2014, 136, 15749–15756 CrossRef CAS PubMed; (d) M. Chang, G. T. Lim, B. Park and E. Reichmanis, Polymers, 2017, 9, 212 CrossRef PubMed.
- (a) P. A. Korevaar, T. F. A. de Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef CAS; (b) Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem. – Eur. J., 2019, 25, 5848–5864 CrossRef CAS PubMed; (c) Z. Xu, K. S. Park and Y. Diao, Front. Chem., 2020, 8, 583521 CrossRef CAS PubMed; (d) L. Ding, Z.-Y. Wang, Z.-F. Yao, N.-F. Liu, X.-Y. Wang, Y.-Y. Zhou, L. Luo, Z. Shen, J.-Y. Wang and J. Pei, Macromolecules, 2021, 54, 5815–5824 CrossRef CAS.
- M. J. Booth, Light: Sci. Appl., 2014, 3, e165–e165 CrossRef.
- (a) F. J. Giessibl, Rev. Mod. Phys., 2003, 75, 949–983 CrossRef CAS; (b) J. Cazaux, J. Microsc., 2005, 217, 16–35 CrossRef CAS PubMed; (c) Y. Lin, M. Zhou, X. Tai, H. Li, X. Han and J. Yu, Matter, 2021, 4, 2309–2339 CrossRef CAS.
- (a) Y. Yang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 25, 6039–6049 CrossRef CAS PubMed; (b) A. Nitti and D. Pasini, Adv. Mater., 2020, 32, 1908021 CrossRef CAS PubMed; (c) Y. Yang, L. Liu and Z. Wei, Acc. Mater. Res., 2024, 5, 329–346 CrossRef CAS.
- (a) L. A. P. Kane-Maguire and G. G. Wallace, Chem. Soc. Rev., 2010, 39, 2545–2576 RSC; (b) M. Verswyvel and G. Koeckelberghs, Polym. Chem., 2012, 3, 3203–3216 RSC; (c) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (d) F. García, R. Gómez and L. Sánchez, Chem. Soc. Rev., 2023, 52, 7524–7548 RSC; (e) S. Jia, T. Tao, Y. Xie, L. Yu, X. Kang, Y. Zhang, W. Tang and J. Gong, Small, 2024, 20, 2307874 CrossRef CAS PubMed.
- C. Zhang, Y. Yan, Y. S. Zhao and J. Yao, Acc. Chem. Res., 2014, 47, 3448–3458 CrossRef CAS PubMed.
- (a) M. Hu, H.-T. Feng, Y.-X. Yuan, Y.-S. Zheng and B. Z. Tang, Coord. Chem. Rev., 2020, 416, 213329 CrossRef CAS; (b) C. Liu, J.-C. Yang, J. W. Y. Lam, H.-T. Feng and B. Z. Tang, Chem. Sci., 2022, 13, 611–632 RSC.
- (a) C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671–683 RSC; (b) I. Danila, F. Pop, C. Escudero, L. N. Feldborg, J. Puigmartí-Luis, F. Riobé, N. Avarvari and D. B. Amabilino, Chem. Commun., 2012, 48, 4552–4554 RSC.
- (a) Z. Wang, C.-C. Lin, K. Murata, A. S. A. Kamal, B.-W. Lin, M.-H. Chen, S. Tang, Y.-L. Ho, C.-C. Chen, C.-W. Chen, H. Daiguji, K. Ishii and J.-J. Delaunay, Adv. Mater., 2023, 35, 2303203 CrossRef CAS PubMed; (b) H. Zhu and B. I. Yakobson, Nat. Mater., 2024, 23, 316–322 CrossRef CAS PubMed.
- (a) R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS; (b) R. Naaman, Y. Paltiel and D. H. Waldeck, Acc. Chem. Res., 2020, 53, 2659–2667 CrossRef CAS PubMed; (c) R. Naaman, Y. Paltiel and D. H. Waldeck, J. Phys. Chem. Lett., 2020, 11, 3660–3666 CrossRef CAS PubMed; (d) Z. Shang, T. Liu, Q. Yang, S. Cui, K. Xu, Y. Zhang, J. Deng, T. Zhai and X. Wang, Small, 2022, 18, 2203015 CrossRef CAS PubMed; (e) A. Chiesa, A. Privitera, E. Macaluso, M. Mannini, R. Bittl, R. Naaman, M. R. Wasielewski, R. Sessoli and S. Carretta, Adv. Mater., 2023, 35, 2300472 CrossRef CAS PubMed; (f) A. Stefani, T. Salzillo, P. R. Mussini, T. Benincori, M. Innocenti, L. Pasquali, A. C. Jones, S. Mishra and C. Fontanesi, Adv. Funct. Mater., 2024, 34, 2308948 CrossRef CAS.
- Y. Liang, K. Banjac, K. Martin, N. Zigon, S. Lee, N. Vanthuyne, F. A. Garcés-Pineda, J. R. Galán-Mascarós, X. Hu, N. Avarvari and M. Lingenfelder, Nat. Commun., 2022, 13, 3356 CrossRef CAS PubMed.
- (a) L. Wan, J. Wade, X. Shi, S. Xu, M. J. Fuchter and A. J. Campbell, ACS Appl. Mater. Interfaces, 2020, 12, 39471–39478 CrossRef CAS PubMed; (b) K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Chem. Sci., 2021, 12, 5522–5533 RSC; (c) L. Wan, Y. Liu, M. J. Fuchter and B. Yan, Nat. Photonics, 2023, 17, 193–199 CrossRef CAS; (d) F. Furlan, J. M. Moreno-Naranjo, N. Gasparini, S. Feldmann, J. Wade and M. J. Fuchter, Nat. Photonics, 2024, 18, 658–668 CrossRef CAS.
- (a) Y. Yang, R. C. da Costa, M. J. Fuchter and A. J. Campbell, Nat. Photonics, 2013, 7, 634–638 CrossRef CAS; (b) L. Zhang, I. Song, J. Ahn, M. Han, M. Linares, M. Surin, H.-J. Zhang, J. H. Oh and J. Lin, Nat. Commun., 2021, 12, 142 CrossRef CAS PubMed.
- (a) M. Schulz, F. Balzer, D. Scheunemann, O. Arteaga, A. Lützen, S. C. J. Meskers and M. Schiek, Adv. Funct. Mater., 2019, 29, 1900684 CrossRef; (b) X. Shang, L. Wan, L. Wang, F. Gao and H. Li, J. Mater. Chem. C, 2022, 10, 2400–2410 RSC.
- (a) C. Train, M. Gruselle and M. Verdaguer, Chem. Soc. Rev., 2011, 40, 3297–3312 RSC; (b) F. Pop, N. Zigon and N. Avarvari, Chem. Rev., 2019, 119, 8435–8478 CrossRef CAS PubMed; (c) G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2022, 13, 3564 CrossRef CAS PubMed.
- (a) L. Dong, Y. Zhang, X. Duan, X. Zhu, H. Sun and J. Xu, Anal. Chem., 2017, 89, 9695–9702 CrossRef CAS PubMed; (b) S. Arnaboldi, B. Gupta, T. Benincori, G. Bonetti, R. Cirilli and A. Kuhn, Anal. Chem., 2020, 92, 10042–10047 CrossRef CAS PubMed.
- (a) C. Wang, H. Fei, Y. Qiu, Y. Yang, Z. Wei, Y. Tian, Y. Chen and Y. Zhao, Appl. Phys. Lett., 1999, 74, 19–21 CrossRef CAS; (b) Y. He, S. Lin, J. Guo and Q. Li, Aggregate, 2021, 2, e141 CrossRef CAS.
- (a) J. R. Brandt, F. Salerno and M. J. Fuchter, Nat. Rev. Chem., 2017, 1, 0045 CrossRef CAS; (b) J. Crassous, M. J. Fuchter, D. E. Freedman, N. A. Kotov, J. Moon, M. C. Beard and S. Feldmann, Nat. Rev. Mater., 2023, 8, 365–371 CrossRef.
- (a) G. Pescitelli, L. Di Bari and N. Berova, Chem. Soc. Rev., 2014, 43, 5211–5233 RSC; (b) G. Pescitelli, Chirality, 2022, 34, 333–363 CrossRef CAS PubMed.
- (a) G. Albano, M. Lissia, G. Pescitelli, L. A. Aronica and L. Di Bari, Mater. Chem. Front., 2017, 1, 2047–2056 RSC; (b) G. Albano, F. Salerno, L. Portus, W. Porzio, L. A. Aronica and L. Di Bari, ChemNanoMat, 2018, 4, 1059–1070 CrossRef CAS; (c) M. Bertuolo, F. Zinna, L. A. Aronica, G. Pescitelli, L. Di Bari and G. Albano, Chem. – Asian J., 2025, 20, e202401160 CrossRef CAS PubMed.
- (a) G. Albano, M. Górecki, T. Jávorfi, R. Hussain, G. Siligardi, G. Pescitelli and L. Di Bari, Aggregate, 2022, 3, e193 CrossRef CAS; (b) G. Albano, A. Taddeucci, G. Pescitelli and L. Di Bari, Chem. – Eur. J., 2023, 29, e202301982 CrossRef CAS PubMed.
- (a) Y. Shindo and M. Nakagawa, Rev. Sci. Instrum., 1985, 56, 32–39 CrossRef CAS; (b) Y. Shindo, M. Nakagawa and Y. Ohmi, Appl. Spectrosc., 1985, 39, 860–868 CrossRef CAS.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed.
- Y. Shindo and Y. Ohmi, J. Am. Chem. Soc., 1985, 107, 91–97 CrossRef CAS.
- M. R. Craig, P. Jonkheijm, S. C. J. Meskers, A. P. H. J. Schenning and E. W. Meijer, Adv. Mater., 2003, 15, 1435–1438 CrossRef CAS.
- A. von Weber, D. C. Hooper, M. Jakob, V. K. Valev, A. Kartouzian and U. Heiz, ChemPhysChem, 2019, 20, 62–69 CrossRef CAS PubMed.
- (a) G. Albano, L. Portus, E. Martinelli, G. Pescitelli and L. Di Bari, ChemPlusChem, 2024, 89, e202300667 CrossRef CAS PubMed; (b) G. Albano, M. Bertuolo, F. Zinna, A. Taddeucci, T. Jávorfi, R. Hussain, G. M. Farinola, G. Pescitelli, A. Punzi, G. Siligardi and L. Di Bari, Nanoscale, 2025, 17, 5128–5140 RSC.
- (a) Y. Yao, T. J. Ugras, T. Meyer, M. Dykes, D. Wang, A. Arbe, S. Bals, B. Kahr and R. D. Robinson, ACS Nano, 2022, 16, 20457–20469 CrossRef CAS PubMed; (b) P. Li, B. Zhao, K. Pan and J. Deng, Nanoscale, 2023, 15, 5345–5359 RSC; (c) T. J. Ugras, Y. Yao and R. D. Robinson, Chirality, 2023, 35, 846–855 CrossRef CAS PubMed; (d) T.-L. Chen, A. Salij, K. A. Parrish, J. K. Rasch, F. Zinna, P. J. Brown, G. Pescitelli, F. Urraci, L. A. Aronica, A. Dhavamani, M. S. Arnold, M. R. Wasielewski, L. di Bari, R. Tempelaar and R. H. Goldsmith, Nat. Commun., 2024, 15, 3072 CrossRef CAS PubMed; (e) A. H. Salij, R. H. Goldsmith and R. Tempelaar, Nat. Commun., 2024, 15, 340 CrossRef CAS PubMed; (f) S. Ji, Y. Zhou, L. Xiong, X. Liu, T. Zhu, X. Zhan, Y. Yan, J. Yao, K. Wang and Y. S. Zhao, J. Am. Chem. Soc., 2025, 147, 16674–16680 CrossRef CAS PubMed; (g) L.-Z. Lin, L.-Q. Huang, S.-W. You, Y.-J. Huang, F. Zinna, A. Salij, L. Di Bari, R. H. Goldsmith, R. Tempelaar, C.-Y. Huang and T.-L. Chen, ACS Photonics, 2025, 12, 2557–2565 CrossRef CAS PubMed; (h) G. A. Turner, C. E. Dunlap, A. J. Higgins and G. J. Simpson, J. Phys. Chem. Lett., 2025, 16, 1403–1408 CrossRef CAS PubMed; (i) R. Zhang, L. Song, W. Yin, B. Zhao and J. Deng, Sci. China: Chem., 2025 DOI:10.1007/s11426-024-2546-y.
- A. Salij, R. H. Goldsmith and R. Tempelaar, J. Am. Chem. Soc., 2021, 143, 21519–21531 CrossRef CAS PubMed.
- G. Albano, G. Pescitelli and L. Di Bari, ChemNanoMat, 2022, 8, e202200219 CrossRef CAS.
- (a) R. Kuroda, T. Harada and Y. Shindo, Rev. Sci. Instrum., 2001, 72, 3802–3810 CrossRef CAS; (b) H. Arwin, S. Schoeche, J. Hilfiker, M. Hartveit, K. Järrendahl, O. R. Juárez-Rivera, A. Mendoza-Galván and R. Magnusson, Appl. Sci., 2021, 11, 6742 CrossRef CAS; (c) R. Kuroda, T. Harada and H. Takahashi, Chirality, 2024, 36, e3622 CrossRef CAS PubMed.
- (a) A. Thomas, T. Chervy, S. Azzini, M. Li, J. George, C. Genet and T. W. Ebbesen, J. Phys. Chem. C, 2018, 122, 14205–14212 CrossRef CAS; (b) O. Arteaga and B. Kahr, J. Opt. Soc. Am. B, 2019, 36, F72–F83 CrossRef CAS.
- (a) A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 1988, 97, 615–644 CrossRef CAS; (b) N. Luo, G. Zhang and Z. Liu, Org. Chem. Front., 2021, 8, 4560–4581 RSC.
- (a) Z. Hao and A. Iqbal, Chem. Soc. Rev., 1997, 26, 203–213 RSC; (b) O. Wallquist and R. Lenz, Macromol. Symp., 2002, 187, 617–630 CrossRef CAS.
- (a) M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS; (b) M. Kaur and D. H. Choi, Chem. Soc. Rev., 2015, 44, 58–77 RSC; (c) A. Abhervé, M. Mastropasqua Talamo, S. Boi, V. Poupard, F. Gendron, B. L. Guennic, N. Avarvari and F. Pop, Inorg. Chem., 2021, 60, 7351–7363 CrossRef.
- (a) G. Gogoi, L. Bhattacharya, S. Rahman, N. S. Sarma, S. Sahu, B. K. Rajbongshi and S. Sharma, Mater. Today Commun., 2020, 25, 101364 CrossRef CAS; (b) C. Liu, L. Yin, Y. Guo, B. Xie, X. Wang and Y. Li, Mater. Adv., 2024, 5, 762–776 RSC.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- W. Li, L. Wang, H. Tang and D. Cao, Dyes Pigm., 2019, 162, 934–950 CrossRef CAS.
- (a) E. H. Ghazvini Zadeh, M. V. Bondar, I. A. Mikhailov and K. D. Belfield, J. Phys. Chem. C, 2015, 119, 8864–8875 CrossRef CAS; (b) T. He, Y. Gao, S. Sreejith, X. Tian, L. Liu, Y. Wang, H. Joshi, S. Z. F. Phua, S. Yao, X. Lin, Y. Zhao, A. C. Grimsdale and H. Sun, Adv. Opt. Mater., 2016, 4, 746–755 CrossRef CAS; (c) A. Kovalenko, M. Vala, M. Ciganek, M. Weiter and J. Krajcovic, Chem. Pap., 2018, 72, 3033–3042 CrossRef CAS.
- (a) M. A. Naik, N. Venkatramaiah, C. Kanimozhi and S. Patil, J. Phys. Chem. C, 2012, 116, 26128–26137 CrossRef CAS; (b) C. Kanimozhi, N. Yaacobi-Gross, E. K. Burnett, A. L. Briseno, T. D. Anthopoulos, U. Salzner and S. Patil, Phys. Chem. Chem. Phys., 2014, 16, 17253–17265 RSC.
- Z. Wang, Z. Liu, L. Ning, M. Xiao, Y. Yi, Z. Cai, A. Sadhanala, G. Zhang, W. Chen, H. Sirringhaus and D. Zhang, Chem. Mater., 2018, 30, 3090–3100 CrossRef CAS.
- L. Sharma and H. Bronstein, RSC Adv., 2021, 11, 5276–5283 RSC.
- (a) K. Dhbaibi, L. Favereau, M. Srebro-Hooper, M. Jean, N. Vanthuyne, F. Zinna, B. Jamoussi, L. Di Bari, J. Autschbach and J. Crassous, Chem. Sci., 2018, 9, 735–742 RSC; (b) M. Mastropasqua Talamo, T. Cauchy, F. Zinna, F. Pop and N. Avarvari, Chirality, 2023, 35, 805–816 CrossRef CAS PubMed.
- T. Gao, Z. Jiang, B. Chen, Q. Sun, Y. Orooji, L. Huang and Z. Liu, Dyes Pigm., 2020, 173, 107998 CrossRef CAS.
- P. A. Hume, J. P. Monks, F. Pop, E. S. Davies, R. C. I. MacKenzie and D. B. Amabilino, Chem. – Eur. J., 2018, 24, 14461–14469 CrossRef CAS PubMed.
- J. Humphreys, C. E. Killalea, F. Pop, E. S. Davies, G. Siligardi and D. B. Amabilino, Chirality, 2023, 35, 281–297 CrossRef CAS PubMed.
- G. Albano, F. Zinna, A. Taddeucci, M. A. M. Capozzi, G. Pescitelli, A. Punzi, L. Di Bari and G. M. Farinola, Chem. – Eur. J., 2023, 29, e202300291 CrossRef CAS PubMed.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS PubMed.
- T. He, P. Leowanawat, C. Burschka, V. Stepanenko, M. Stolte and F. Würthner, Adv. Mater., 2018, 30, 1804032 CrossRef PubMed.
- (a) S. Militzer, N. Nishimura, N. R. Ávila-Rovelo, W. Matsuda, D. Schwaller, P. J. Mésini, S. Seki and A. Ruiz-Carretero, Chem. – Eur. J., 2020, 26, 9998–10004 CrossRef CAS PubMed; (b) M. Mastropasqua Talamo, F. Pop and N. Avarvari, Chem. Commun., 2021, 57, 6514–6517 RSC; (c) G. Albano, F. Zinna, F. Urraci, M. A. M. Capozzi, G. Pescitelli, A. Punzi, L. Di Bari and G. M. Farinola, Chem. – Eur. J., 2022, 28, e202201178 CrossRef CAS PubMed; (d) C. E. Killalea, M. Samperi, G. Siligardi and D. B. Amabilino, Chem. Commun., 2022, 58, 4468–4471 RSC; (e) M. Mastropasqua Talamo, T. Cauchy, F. Pop, F. Zinna, L. Di Bari and N. Avarvari, J. Mater. Chem. C, 2023, 11, 5701–5713 RSC.
- (a) M. Boukhris, M. S. J. Simmonds, S. Sayadi and M. Bouaziz, Phytother. Res., 2013, 27, 1206–1213 CrossRef CAS PubMed; (b) J. López-Fernández, M. D. Benaiges, X. Sebastian, J. M. Bueno and F. Valero, Catalysts, 2022, 12, 639 CrossRef.
- (a) A. Kovalenko, C. Yumusak, P. Heinrichova, S. Stritesky, L. Fekete, M. Vala, M. Weiter, N. S. Sariciftci and J. Krajcovic, J. Mater. Chem. C, 2017, 5, 4716–4723 RSC; (b) M. Cigánek, P. Heinrichová, A. Kovalenko, J. Kučerík, M. Vala, M. Weiter and J. Krajčovič, Dyes Pigm., 2020, 175, 108141 CrossRef; (c) D. Rais, P. Toman, J. Pfleger, U. Acharya, Y. R. Panthi, M. Menšík, A. Zhigunov, M. A. Thottappali, M. Vala, A. Marková, S. Stříteský, M. Weiter, M. Cigánek, J. Krajčovič, K. Pauk, A. Imramovský, A. Zaykov and J. Michl, ChemPlusChem, 2020, 85, 2689–2703 CrossRef CAS PubMed.
- G. Albano, L. A. Aronica, G. Pescitelli and L. Di Bari, Chirality, 2024, 36, e23608 CrossRef CAS PubMed.
- (a) A. Tamayo, T. Kent, M. Tantitiwat, M. A. Dante, J. Rogers and T.-Q. Nguyen, Energy Environ. Sci., 2009, 2, 1180–1186 RSC; (b) M. E. Farahat, H.-Y. Wei, M. A. Ibrahem, K. M. Boopathi, K.-H. Wei and C.-W. Chu, RSC Adv., 2014, 4, 9401–9411 RSC.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- (a) Q. Chu and Y. Pang, Macromolecules, 2003, 36, 4614–4618 CrossRef CAS; (b) R.-H. Chien, C.-T. Lai and J.-L. Hong, J. Phys. Chem. C, 2011, 115, 5958–5965 CrossRef CAS; (c) O. Hassan Omar, M. Falcone, A. Operamolla and G. Albano, New J. Chem., 2021, 45, 12016–12023 RSC.
- (a) G. Pescitelli, O. H. Omar, A. Operamolla, G. M. Farinola and L. Di Bari, Macromolecules, 2012, 45, 9626–9630 CrossRef CAS; (b) D. Aranda, J. Cerezo, G. Pescitelli, F. J. Avila Ferrer, J. Soto and F. Santoro, Phys. Chem. Chem. Phys., 2018, 20, 21864–21880 RSC.
- P. Pracht, S. Grimme, C. Bannwarth, F. Bohle, S. Ehlert, G. Feldmann, J. Gorges, M. Müller, T. Neudecker, C. Plett, S. Spicher, P. Steinbach, P. A. Wesołowski and F. Zeller, J. Chem. Phys., 2024, 160, 114110 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- P. Kocán, B. Pieczyrak, S. Umachi, M. Cigánek, P. Sobotík, I. Ošťádal, L. Jurczyszyn, J. Krajčovič and K. Sakamoto, Nanoscale Horiz., 2025, 10, 915–921 RSC.
- A. E. Pashenko, A. Gaidai, N. Hryhoriev, O. Volovenko, I. Levandovskiy, O. Maksymenko, D. M. Volochnyuk and S. V. Ryabukhin, Org. Process Res. Dev., 2023, 27, 477–487 CrossRef CAS.
- B. S. Dyer, J. D. Jones, G. D. Ainge, M. Denis, D. S. Larsen and G. F. Painter, J. Org. Chem., 2007, 72, 3282–3288 CrossRef CAS PubMed.
- P. Larpent, A. Jouaiti, N. Kyritsakas and M. W. Hosseini, Chem. Commun., 2013, 49, 4468–4470 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) T. Lu, J. Chem. Phys., 2024, 161, 082503 CrossRef CAS PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI Figures. 1H-NMR spectra of intermediates. 1H-NMR and 13C-NMR spectra of final products. Atomic coordinates of optimized geometries. See DOI: https://doi.org/10.1039/d5ob00782h |
| This journal is © The Royal Society of Chemistry 2025 |