
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperacid-catalysed α-deuteration of ketones with D2O†
Haiying
Yuan
a,
Kaibo
Xu
a,
Jinling
Li
a,
Teck-Peng
Loh
 *ab,
Zhenguo
Zhang
*ab and
Zhenhua
Jia
*a
*ab,
Zhenguo
Zhang
*ab and
Zhenhua
Jia
*a
aHenan Linker Technology Key Laboratory, College of Advanced Interdisciplinary Science and Technology (CAIST), Henan University of Technology, Zhengzhou 450001, China. E-mail: teckpeng@ntu.edu.sg
bDivision of Chemistry and Biological Chemistry, School of Chemistry, Chemical Engineering, and Biotechnology, Nanyang Technological University, Singapore 637371, Singapore
First published on 19th May 2025
Abstract
In this study, we present a superacid catalyzed protocol for the α-deuteration of ketones with D2O using [Ph3C]+[B(C6F5)4]− as a pre-catalyst to generate in situ the superacidic species [D]+[B(C6F5)4]−. The features of this catalytic process include simple manipulation, high deuteration efficiency (up to 99%), excellent functional group compatibility and a broad substrate scope, including 30 substrates comprising common building blocks and bioactive molecules like pentoxifylline. Moreover, the avoidance of toxic reagents enables sustainable access to deuterated products, demonstrating the method's practical potential for use in mechanistic studies and pharmaceutical applications.
Deuterium-labeled compounds play a pivotal role in pharmaceutical research, mechanistic studies, and synthetic chemistry due to their unique kinetic isotope effects and broad applications.1 In recent years, deuterated drugs have attracted significant attention in pharmaceutical science,2 with several deuterated drugs being approved and introduced to the market,3 including deutetrabenazine, the first deuterated drug approved by the FDA in 2017, and donafenib, first approved in 2021. In addition, numerous deuterated drug candidates are currently in clinical development, such as d1-JNJ38877605, an ATP-competitive inhibitor of c-Met.4
In modern drug discovery, bioisosteric replacement remains a fundamental strategy for lead compound optimization and bioactivity enhancement. Keto groups are ubiquitous functional motifs in natural products and drugs. Therefore, the hydrogen–deuterium exchange (H/D exchange) at the α-position of carbonyl groups is of particular significance in drug design. For instance, CTP-499, a phase II clinical drug, which is a deuterated compound derived from the metabolite of pentoxifylline (HDX), demonstrated improved safety, enhanced absorption, and reduced interindividual variability compared to HDX.5 Recent advancements in selective H/D exchange have focused on organobase-catalyzed H/D exchange6 and strong acid mediated deuteration.7 Additionally, transition metal-catalyzed H/D exchange has also been recognized as an alternative approach (Fig. 1A).8 However, these processes often suffer from several limitations, such as the need for harsh conditions, scalability challenges, poor functional group tolerance, and potential toxicity concerns associated with metal residues.
 | ||
| Fig. 1 (A) Deuteration application in medicinal chemistry. (B) Wasa's work: frustrated Lewis pair catalyzed α-deuteration of carbonyls with D2O. (C) This work: metal-free catalytic α-deuteration of ketones with D2O. | ||
More recently, Wasa and co-workers developed an efficient α-deuteration of carbonyl compounds with D2O under metal-free conditions, leveraging the catalytic ability of frustrated Lewis pairs (Fig. 1B).7a Building upon our previous research on triaryl carbenium ion-pair mediated organic transformations,9 we herein describe a metal-free catalytic protocol for H/D exchange at the α-position of keto moieties, enabling the synthesis of a series of deuterated products with potential bioactivity. In addition, this method exhibits high deuteration efficiency, broad functional group compatibility and substrate versatility, accommodating both ketones and structurally complex molecules (Fig. 1C).
Based on our work on catalytic H/D exchange of arenes with D2O,10 we started our investigation using pentoxifylline (1) as the model substrate, employing trityl tetrakis(pentafluorophenyl)borate ([Ph3C]+[B(C6F5)4]−) as the catalyst with D2O as the deuteration reagent to optimize the reaction conditions. Finally, we achieved the desired deuterated product 2 (pentoxifylline-d5) in 93% yield with up to 96% deuterium incorporation at the α-positions. The optimal reaction conditions included the use of 5 mol% [Ph3C]+[B(C6F5)4]−, 55 equiv. D2O (0.2 mL), at 100 °C in 10 hours under an argon atmosphere (Table 1, entry 1). Control experiments provided crucial insights, confirming the essential role of the ion-pair in facilitating this metal-free deuteration process efficiently (Table 1, entry 2). When [Ph3C]+[BF4]− replaced [Ph3C]+[B(C6F5)4]−, the expected deuteration was not observed due to weaker anion basicity (Table 1, entry 3), underscoring the significant influence of the anion. Furthermore, the use of K+[B(C6F5)4]− resulted in substantially reduced deuteration efficiency, affording the desired product in 86% yield (Table 1, entry 4). When dichloromethane (DCM) was tested as an alternative solvent to dichloroethane (DCE), deuterium incorporation at the α-position of the keto group was observed at 91% and 96%, respectively (Table 1, entry 5). Consequently, DCE was chosen as the optimal solvent for the subsequent reaction optimization, owing to its high boiling point that helped drive efficient and robust hydrogen–deuterium exchange. Also, deuterated methanol (CD3OD) was used as the deuterium source to conduct the hydrogen–deuterium exchange reaction under the standard conditions. Comparative analysis revealed only a marginal decrease in keto deuteration efficiency at the α-position (85% and 89%), while the isolated yield was maintained at consistently high levels of 95% (Table 1, entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Variation from ‘standard conditions’ | x (%) | y (%) | Yieldc (%) |
| a Reaction conditions: pentoxifylline (0.1 mmol, 27.8 mg), [Ph3C]+[B(C6F5)4]− (5 mol%, 0.005 mmol), D2O (55 equiv., 0.2 mL), DCE (1 mL), 100 °C, argon, in 10 hours. b Yield of deuteration was based on hydrogen, indicated in red. c All yields are NMR yields using CH3NO2 as an internal standard. d Isolated yields. n.d.: no deuteration. | ||||
| 1 | None | 95 | 96 | 93 (89d) |
| 2 | No [Ph3C]+[B(C6F5)4]− | n.d. | n.d. | 99 |
| 3 | [Ph3C]+[BF4]− instead of [Ph3C]+[B(C6F5)4]− | n.d. | n.d. | 90 |
| 4 | K+[B(C6F5)4]− instead of [Ph3C]+[B(C6F5)4]− | 40 | 34 | 86 |
| 5 | DCM instead of DCE | 91 | 96 | 86 |
| 6 | CD3OD instead of D2O | 89 | 85 | 95 |
| 7 | 90 °C instead of 100 °C | 90 | 93 | 94 |
| 8 | 8 h instead of 10 h | 89 | 93 | 86 |
| 9 | O2 instead of argon | 50 | 50 | 82 |

A slight decrease in reaction temperature reduced the deuteration efficiency, affording 2 in 90% and 93% deuterated yields (Table 1, entry 7). When the reaction time was reduced to 8 hours, the deuterated product 2 was obtained in 89% yield (Table 1, entry 8). Notably, strict atmospheric control proved essential, as oxygen-rich conditions significantly compromised reaction efficiency, leading to 50% yield of the deuterated product with lower deuterium incorporation, likely due to competing oxidative side reactions (Table 1, entry 9).11
With the optimal reaction conditions in hand, we next explored the substrate scope to demonstrate the versatility and functional group tolerance of this deuteration method across diverse molecular architectures (Scheme 1). First, natural products and drugs with keto moieties were successfully deuterated to yield the corresponding deuterated product at the α-position of keto groups (2–11). For instance, pentoxifylline (2), tonalide (3), nootkatone (4), geranylacetone (5), and dihydro-β-ionone (6) were converted into deuterated products with high levels of deuterium incorporation at expected positions, underscoring the feasibility of direct deuteration of bioactive molecules without the need for complex protection/deprotection strategies. Moreover, our strategy demonstrated exceptional performance in the regioselective deuteration of steroidal and polycyclic frameworks (7–11), achieving over 95% deuterium incorporation at diverse positions. Notably, deuterium labeling of these structurally complex scaffolds presents significant advantages for pharmaceutical development and drug discovery, highlighting the potential application of these labeled molecules while maintaining the integrity of functional groups, including hydroxyls, lactones, and alkenes.
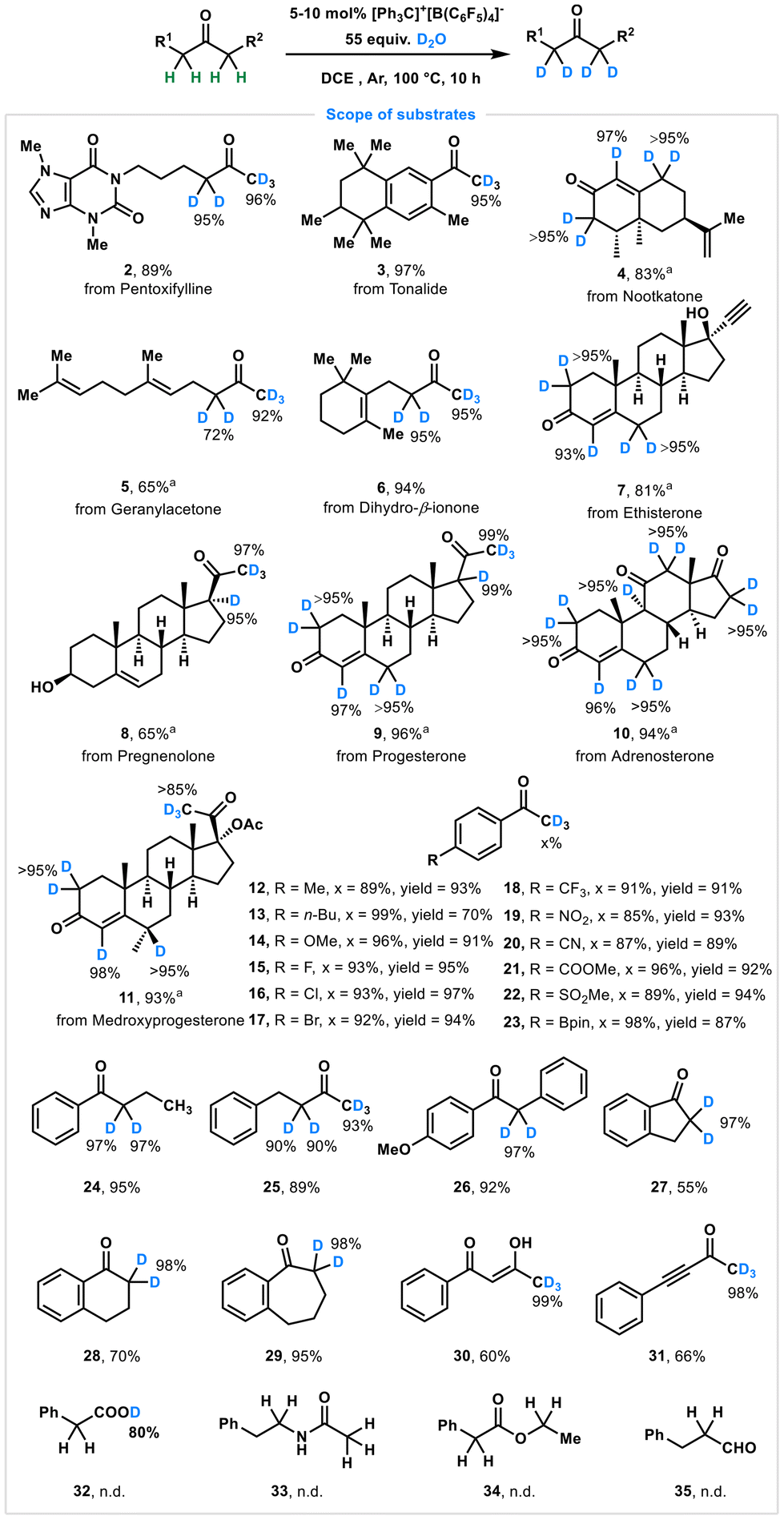 | ||
Scheme 1 Scope of substrates. Reaction conditions: substrates (0.1 mmol), [Ph3C]+[B(C6F5)4]− (5 mol%), D2O (55 equiv., 0.2 mL), DCE (1 mL), 100 °C, argon, in 10 hours. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) DCE:DMSO (1:1) as co-solvent. All yields are isolated yields. n.d.: no deuteration. DCE:DMSO (1:1) as co-solvent. All yields are isolated yields. n.d.: no deuteration. | ||
Subsequently, we evaluated the catalytic performance of this protocol using acetophenone derivatives with a wide range of electronic properties, and noted that this method exhibited excellent functional group tolerance. Electron-donating groups (–Me, –n-Bu, and –OMe), halogen groups (–F, –Cl, and –Br) and electron-withdrawing (–CF3, –NO2, –CN, –COOMe, –SO2Me, and –Bpin) substituents (12–23) were well tolerated, with 76%–99% deuterium incorporation in 70%–97% yields, highlighting the general compatibility of this method. Other ketones, including 1-phenylbutan-1-one (24), 4-phenylbutan-2-one (25), 1-(4-methoxyphenyl)-2-phenylethan-1-one (26), indanone (27), 1-tetralone (28) and 1-benzosuberone (29), underwent selective deuteriation efficiently to produce the expected products in 55%–95% yields with up to 98% deuterium incorporation. Additionally, the enolate of ketone was also compatible with our metal-free deuteration to give 30 in 60% yield with 99% deuterium labeling. Alkynones, which were recently employed in bioconjugation for protein modification by our group and others,12 underwent deuterium labeling at the methyl group to generate 31 in 98% deuteration yield. Additionally, some typical carbonyl compounds, such as carboxylic acids, esters, and aldehydes, were evaluated for H/D exchange under standard conditions. Unfortunately, none of these underwent hydrogen–deuterium exchange (32–35).
To demonstrate the practical utility and scalability of our metal-free deuteration protocol, we scaled up the synthesis of deuterated pentoxifylline-d5 at the gram level, using 10 mL of D2O as the deuterium source. The product was obtained in high yield without the need for column chromatographic purification, highlighting the potential application of our method (Scheme 2A).
 | ||
| Scheme 2 Practical application and mechanistic studies. | ||
Interestingly, the deuterium incorporation was reversible.
As shown in Scheme 2B, D/H exchange with deionized water was achieved when deuterated pentoxifylline-d5 was used as the substrate and treated under slightly modified conditions for 24 hours. Moreover, when 1.0 equiv. Brønsted acids, such as AcOH, HCl, H2SO4, TFA and TfOH, were employed instead of the catalytic amount of 5 mol% [Ph3C]+[B(C6F5)4]− ion pair catalyst, only minimal amounts of the deuterated products were observed, showcasing the distinct activity of the ion pair in facilitating the desired deuteration process (Scheme 2C). Comparative experiments revealed that the superacid-catalyzed method reported here demonstrated clear advantages over stoichiometric base-catalyzed protocols (e.g., using 1.0 equivalent of K2CO3 as the catalyst). (For further details, please refer to the ESI.†)
As shown in Scheme 3, the proposed mechanism began with the hydrolysis of the triaryl carbenium ion-pair in an aqueous medium, generating the hydrated proton complex (D2O)nD+[B(C6F5)4]− along with triphenylmethanol as a byproduct. In the presence of ketone substrates, the oxygen atom of the keto group underwent rapid protonation, leading to the formation of the corresponding enol tautomer. This reactive intermediate subsequently tautomerized back to the keto structure, simultaneously regenerating the superacid species. This catalytic cycle enabled single H/D exchange at the α-position, and through the cumulative effect of deuterium isotope exchange, ultimately achieved complete deuteration of the ketone compound at the α-position.
 | ||
| Scheme 3 Proposed mechanism. | ||
In summary, we have developed a highly efficient and selective metal-free catalytic H/D exchange strategy for precise deuterium incorporation across a broad range of keto-containing pharmaceuticals, ketones, sterically hindered frameworks, and bioactive steroids. This approach exhibits excellent functional group tolerance and scalability, making it a valuable tool for the synthesis of deuterium-labeled compounds with significant applications in pharmaceutical research, drug metabolism studies, and mechanistic investigations of organic reactions.
Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support provided by the Distinguished University Professor Grant (Nanyang Technological University) and the Agency for Science, Technology, and Research (A*STAR) under its MTC Individual Research Grant (M21K2c0114) and RIE2025 MTC Programmatic Fund (M22K9b0049) to T.-P.L.References
- (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed; (b) J. Atzrodt and V. Derdau, J. Labelled Compd. Radiopharm., 2010, 53, 674–685 CrossRef CAS.
- (a) R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Nat. Rev. Drug Discovery, 2023, 22, 562–584 CrossRef CAS PubMed; (b) S. L. Harbeson and R. D. Tung, Med. Chem., 2014, 2, 8–22 Search PubMed; (c) T. G. Gant, J. Med. Chem., 2014, 57, 3595–3611 CrossRef CAS PubMed; (d) T. Pirali, M. Serafini, S. Cargnin and A. A. Genazzani, J. Med. Chem., 2019, 62, 5276–5297 CrossRef CAS PubMed; (e) A. Mullard, Nat. Rev. Drug Discovery, 2016, 15, 219–221 CrossRef CAS PubMed.
- (a) A. Mullard, Nat. Rev. Drug Discovery, 2017, 16, 305 Search PubMed; (b) S. Qin, F. Bi, S. Gu, Y. Bai, Z. Chen, Z. Wang, J. Ying, Y. Lu, Z. Meng, H. Pan, P. Yang, H. Zhang, X. Chen, A. Xu, C. Cui, B. Zhu, J. Wu, X. Xin, J. Wang, J. Shan, J. Chen, Z. Zheng, L. Xu, X. Wen, Z. You, Z. Ren, X. Liu, M. Qiu, L. Wu and F. Chen, J. Clin. Oncol., 2021, 39, 3002–3011 CrossRef CAS PubMed; (c) A. Mullard, Nat. Rev. Drug Discovery, 2022, 21, 623–625 CrossRef CAS PubMed; (d) S. H. DeWitt and B. E. Maryanoff, Biochemistry, 2018, 57, 472–473 CrossRef CAS PubMed.
- (a) X. Li, E. K. Dowling, G. Yan, Z. Dereli, B. Bozorgui, P. Imanirad, J. H. Elnaggar, A. Luna, D. G. Menter, P. G. Pilié, T. A. Yap, S. Kopetz, C. Sander and A. Korkut, Cancer Discovery, 2022, 12, 1542–1559 CrossRef CAS PubMed; (b) K. Sanderson, Nature, 2009, 458, 269 CAS.
- (a) V. Braman, P. Graham, C. Cheng, D. Turnquist, M. Harnett, L. Sabounjian and J. Shipley, Clin. Pharmacol. Drug Dev., 2013, 2, 53–66 CrossRef CAS PubMed; (b) M. V. Perez-Gomez, M. D. Sanchez-Niño, A. B. Sanz, C. Martín-Cleary, M. Ruiz-Ortega, J. Egido, J. F. Navarro-González, A. Ortiz and B. Fernandez-Fernandez, J. Clin. Med., 2015, 4, 1325–1347 CrossRef CAS PubMed.
- (a) C. Rape and W. H. Sachs, Tetrahedron, 1968, 24, 6287–6290 CrossRef; (b) C. Sabot, K. A. Kumar, C. Antheaume and C. Mioskowski, J. Org. Chem., 2007, 72, 5001–5004 CrossRef CAS PubMed; (c) M. Rempt and G. Pohnert, Angew. Chem., Int. Ed., 2010, 49, 4755–4758 CrossRef CAS PubMed; (d) S. Peng, N. M. Okeley, A.-L. Tsai, G. Wu, R. J. Kulmacz and W. A. van der Donk, J. Am. Chem. Soc., 2002, 124, 10785–10796 CrossRef CAS PubMed; (e) K. I. Galkin, E. G. Gordeev and V. P. Ananikov, Adv. Synth. Catal., 2021, 363, 1368–1378 CrossRef CAS; (f) C. Berthelette and J. Scheigetz, J. Labelled Compd. Radiopharm., 2004, 47, 891–894 CrossRef CAS; (g) M. Zhan, T. Zhang, H. Huang, Y. Xie and Y. Chen, J. Labelled Compd. Radiopharm., 2014, 57, 533–539 CrossRef CAS PubMed; (h) T. Tanaka, Y. Koga, Y. Honda, A. Tsuruta, N. Matsunaga, S. Koyanagi, S. Ohdo, R. Yazaki and T. Ohshima, Nat. Synth., 2022, 1, 824–830 CrossRef CAS.
- (a) Y. Chang, T. Myers and M. Wasa, Adv. Synth. Catal., 2020, 362, 360–364 CrossRef CAS PubMed; (b) M. Shang, X. Wang, S. M. Koo, J. Youn, J. Z. Chan, W. Yao, B. T. Hastings and M. Wasa, J. Am. Chem. Soc., 2017, 139, 95–98 CrossRef CAS PubMed.
- (a) J. Krüger, B. Manmontri and G. Fels, Eur. J. Org. Chem., 2005, 1402–1408 CrossRef; (b) B. M. Wheatley and B. A. Keay, J. Org. Chem., 2007, 72, 7253–7259 CrossRef CAS PubMed.
- (a) Z. Zhang, X. Liu, L. Ji, T. Zhang, Z. Jia and T.-P. Loh, ACS Catal., 2022, 12, 2052–2057 CrossRef CAS; (b) Z. Zhang, L. Ji, X. Liu, Z. Li, Y. Lv, Z. Jia and T.-P. Loh, Org. Chem. Front., 2022, 9, 5154–5159 RSC.
- Z. Zhang, Y. Lv, W. Q. R. Ong, X. Zhao, Z. Jia and T.-P. Loh, Angew. Chem., Int. Ed., 2024, 63, e202408509 CrossRef CAS PubMed.
- (a) Z. Zhang, J. Gu, Y. Lv, L. Ji, X. Liu, B. Wu, F. Liu, Z. Jia and T.-P. Loh, Cell Rep. Phys. Sci., 2023, 4, 101246 CrossRef CAS; (b) Z. Zhang, J. Gu, L. Ji, X. Liu, T. Zhang, Y. Lv, F. Liu, Z. Jia and T.-P. Loh, ACS Catal., 2022, 12, 14123–14129 CrossRef CAS; (c) Z. Zhang, Y. Lv, L. Ji, P. Chen, S. Han, Y. Zhu, L. Li, Z. Jia and T.-P. Loh, Angew. Chem., Int. Ed., 2024, 63, e202406588 CrossRef CAS PubMed.
- (a) S. Teng, E. W. H. Ng, Z. Zhang, C. N. Soon, H. Xu, R. Li, H. Hirao and T.-P. Loh, Sci. Adv., 2023, 9, eadg4924 CrossRef CAS PubMed; (b) Z. Zhang, L. Li, H. Xu, C.-L. K. Lee, Z. Jia and T.-P. Loh, J. Am. Chem. Soc., 2024, 146, 1776–1782 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00683j |
| This journal is © The Royal Society of Chemistry 2025 |