
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic methodologies to access skipped dienes: a focus on the catalytic systems
Polyssena
Renzi
 *,
Alberto
Lanfranco
,
Lucia
Pazderová
,
Marco
Rusconi
,
Jacopo
Scarfiello
and
Annamaria
Deagostino
*,
Alberto
Lanfranco
,
Lucia
Pazderová
,
Marco
Rusconi
,
Jacopo
Scarfiello
and
Annamaria
Deagostino
Department of Chemistry, University of Turin, Via Giuria 7, 10125 Turin, Italy. E-mail: polyssena.renzi@unito.it
First published on 30th May 2025
Abstract
1,4-Dienes, also known as skipped dienes, are widely diffused in natural products and serve as valuable synthetic intermediates. However, their synthesis continues to pose a substantial challenge. In recent years, there has been a notable development in the field, with the emergence of highly stereoselective methodologies for the construction of the 1,4-diene moiety. This review discusses the latest advances in the synthesis of skipped dienes, with a particular emphasis on the catalytic system and reaction mechanism. Metal-mediated (Ru, Co, Rh, Ir, Ni, Pd, Cu, Au, Ca, Ti, Cr, Yb), metal-free and organocatalyzed transformations as well as synergistic/dual and metallaphotoredox-catalysed reactions, published in the last five years, are reported.
 Polyssena Renzi | Polyssena Renzi completed her PhD in 2014 at the University of Rome La Sapienza (Italy) on the development of novel strategies for asymmetric synthesis and their optimization via Design of Experiment. In 2015, she joined the group of Prof. R. M. Gschwind (Regensburg University, Germany) as a postdoctoral fellow, where she dealt with mechanistic investigations of organocatalyzed reactions by means of NMR techniques. Since October 2023, she has been assistant professor at the University of Turin where she works in the field of photocatalysis and in the synthesis of molecules for BNCT applications. |
 Alberto Lanfranco | Dr Alberto Lanfranco graduated in chemistry in 2019 at the University of Torino, presenting a thesis on the synthesis of a biotin-functionalised carborane for BNCT/MRI applications. In 2024, he obtained a PhD degree in Chemical and Materials Sciences, working in the group of Annamaria Deagostino with a doctoral fellowship (AIRC IG). His project dealt with the synthesis of sulfamido-functionalised carboranes for inhibition of carbonic anhydrase IX and BNCT applications. He is currently a postdoc researcher in the same group, working on the synthesis of carborane-based theranostic agents for the treatment of Alzheimer's disease. |
 Lucia Pazderová | Dr Lucia Pazderová is a Postdoctoral Researcher in the Department of Chemistry at the University of Turin (Italy). She received her PhD in Inorganic Chemistry from Charles University (Czechia) in 2021, following an MSc in Bioinorganic Chemistry from Palacký University Olomouc (Czechia) in 2015. The current research interest of Dr Pazderová is focused on the functionalisation of metallacarborane derivatives for synthesising antitumoral agents for boron neutron capture therapy. |
 Marco Rusconi | Marco Rusconi graduated in Chemistry from the University of Turin in 2023, obtaining his Master's degree with a thesis concerning a catalyst-free hydrothiolation reaction of alkenes mediated by visible light. In the same year, he started his Doctorate of National Interest in Annamaria Deagostino's group. His PhD project is dedicated to the development of synthetic methodologies for the photoinduced functionalization of carborane clusters. |
 Jacopo Scarfiello | Jacopo Scarfiello received his Master's degree in industrial chemistry from the University of Torino in 2019. His thesis consisted of synthesizing chalcones through micellar catalysis employing green surfactants in the aqueous phase. In 2022, he obtained a doctoral fellowship (PNRR) from the University of Perugia while working in Annamaria Deagostino's group under the guidance of Dr Polyssena Renzi. His PhD research involves UV-visible light-promoted chlorination reactions. |
 Annamaria Deagostino | Annamaria Deagostino is professor of organic chemistry at the University of Turin, where she graduated in chemistry, and after a fellowship at University of Padova, she undertook PhD studies at the University of Turin from 1994 to 1997. In 1998, she obtained a postdoctoral fellowship at the University of Caen, under the supervision of Prof. Marie-Claire Lasne, and then she returned to the University of Turin. The main interests of her research group are in the field of synthetic organic chemistry, mainly focused on organopalladium chemistry, visible light photoinduced processes and the synthesis of BNCT (boron neutron capture therapy) theranostic agents. |
Introduction
Among the structural motifs present in biologically active compounds, including polyketides, alkaloids and polyunsaturated fatty acids, the skipped diene moiety occupies a privileged position. These compounds are also highly versatile building blocks for organic synthesis,1,2 and are widely employed as functional intermediates in the construction of more complex structures.3 In contrast to the stable 1,3-diene system, 1,4-diene compounds, which contain two double bonds separated by a sp3-hybridised carbon, exhibit greater conformational flexibility and are characterised by a non-planar structure. The structural flexibility advocated for (i) stereoselective synthesis, (ii) mild conditions to avoid isomerization and (iii) stereo-divergency to selectively access all the possible isomers from the same starting materials. The methodologies for obtaining 1,4-dienes have been relatively underdeveloped; however, in recent years a flowering has been observed with a rapid advancement in synthesis in this field. The majority of the synthetic procedures developed are characterised by a high degree of stereoselectivity, and alkynes, alkenes or dienes can be exploited as the starting materials. Fig. 1 provides a summary of the most frequent substitution patterns that can be encountered in 1,4-dienes, both in acyclic and cyclic compounds. Most simple structures are characterised by a single substituent, with the introduction of up to four different groups being possible in the 1,4-diene skeleton, thereby increasing the complexity. | ||
| Fig. 1 Structure of dienes and summary of the most common substitution patterns in 1,4-dienes. | ||
This review examines the synthesis of skipped dienes over the period from 2020 to 2024. The analysis of catalysis by different metals, organic-based compounds and dual catalysis is presented as a central tool for the generation of these moieties, both in the presence and absence of light. The review is organised in the following four main sections:
(a) Metal-catalysed synthesis of skipped dienes,
(b) Metallaphotoredox-catalysed synthesis of skipped dienes,
(c) Metal-free transformations,
(d) Synergistic/dual catalysis.
The section on metal-catalysed transformations is further divided according to the nature of the metal centre (group 8: Ru; group 9: Co, Rh, Ir; group 10: Ni, Pd; group 11: Cu, Au; miscellaneous: Ca, Ti, Cr, Yb). Fig. 2 provides a graphical overview of the number of papers published in the last five years according to the type of catalysis exploited. It is evident that 1,4-diene synthesis is dominated by metal-catalysed transformations, particularly from metals belonging to groups 10 and 11, followed by group 9. Among the metals, catalysis by ruthenium is the least developed, with only two papers. Conversely, dual catalysis and metallaphotoredox catalysis are emergent areas.
 | ||
| Fig. 2 Graphical overview of the last five years’ publications of 1,4-diene synthesis. | ||
The objective of this review is to provide a comprehensive overview of the most recent strategies for synthesising 1,4-dienes, with a particular emphasis on the catalytic system and the underlying reaction mechanism. Some examples of 1,5- and 1,6-diene synthesis were also reported. This field is expanding rapidly, with an ever-increasing number of publications. In fact, more than 40 papers have been published in the last two years, which highlights the need for an updated review.
Metal-catalysed synthesis of skipped dienes
Group 8: ruthenium (Ru)
Concerning the metal-catalysed synthesis of skipped dienes, catalysts based on ruthenium (Ru) as the active metal centre are utilised to a lesser extent. A review of the literature revealed that only two papers, exploiting a Ru-based catalyst, were published between 2020 and 2024 in which the 1,4-diene was directly employed as the starting material for a subsequent transformation, thus highlighting the wide applicability of these compounds in synthetic methodologies. The group of Trost reported a Ru-catalysed alkene–alkyne coupling reaction to synthesise unsymmetrical 3-boryl-1,4-dienes 3 (Scheme 1).4 These compounds were not isolated but reacted in situ with carbonyl compounds 4 to access the 1,3-dienyl-6-oxy structural motif 5a–d, which were further exploited as a platform for the synthesis of complex polyketides. The methodology allowed the formation of contiguous stereocenters and two new carbon–carbon bonds, and it was characterised by a high stereoselectivity.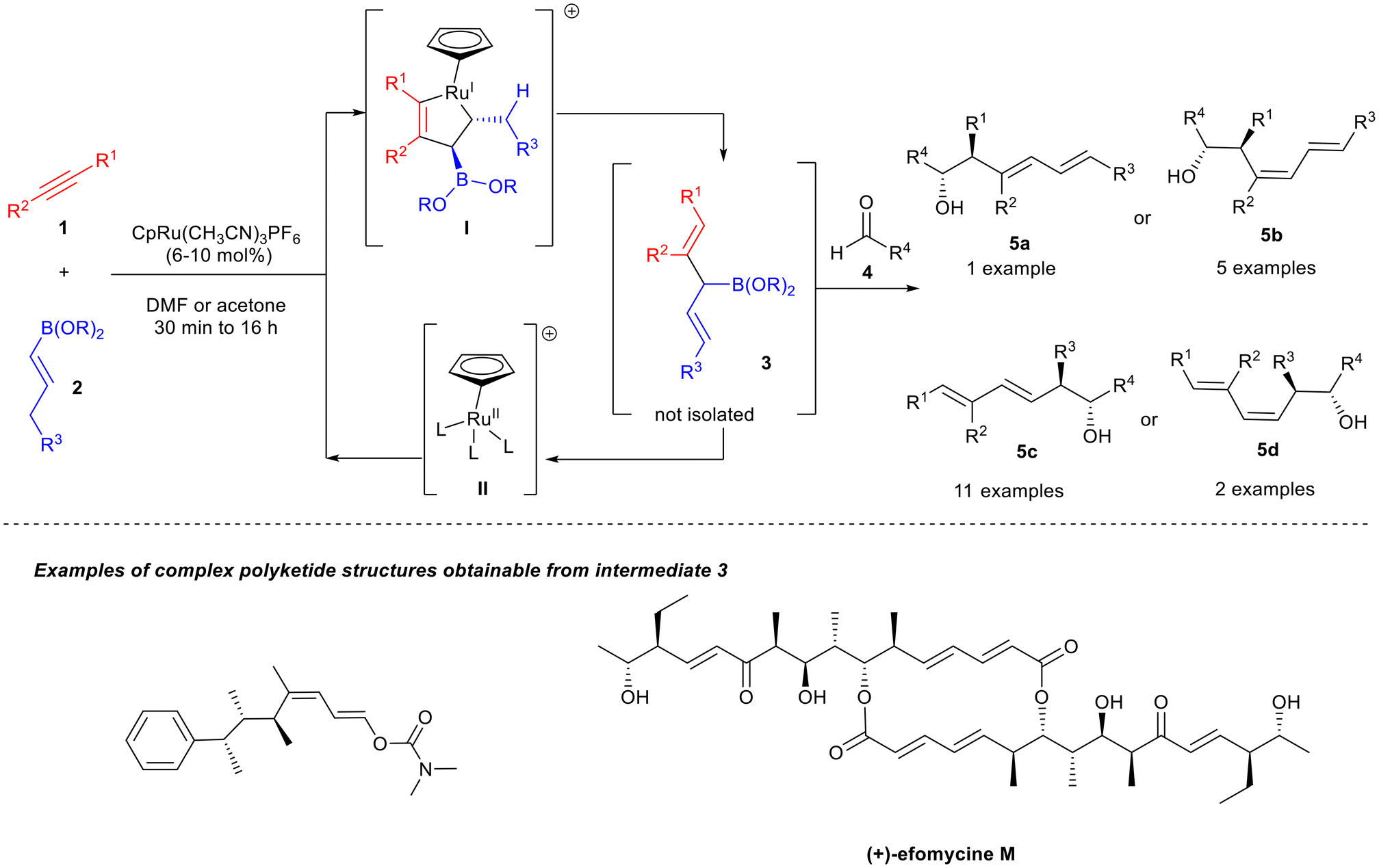 | ||
| Scheme 1 Ru-catalysed coupling reaction to access intermediates 3-boryl-1,4-dienes 3. | ||
Later, in a paper published in 2022, the group of Hirano described the synthesis of borylated 1,4-dienes 8.5 The aforementioned compounds were obtained through a Ru(0)-promoted cross-dimerisation of borylated 1,3-diene 6 with substituted alkenes 7 (Scheme 2). The reaction proceeded at 30 °C in benzene, in the presence of 10 mol% of [Ru(naphthalene)(1,5-cod)], resulting in the formation of a mixture of 1,4- and 1,5-diene products, contingent upon the substrate structure. The utility of these borylated dienes was demonstrated by their application in a variety of Suzuki–Miyaura coupling reactions, as well as in the formal synthesis of rac-bongkrekic acid.
 | ||
| Scheme 2 Ru(0)-catalysed synthesis of diborylated 1,4-dienes 8. | ||
Group 9: cobalt (Co), rhodium (Rh) and iridium (Ir)
Catalysis by the metals belonging to group 9 relies on cobalt, rhodium and iridium-based catalysts. Cobalt catalysis plays an increasingly central role in cross-electrophile coupling reactions, offering a valuable alternative to other commonly employed transition metals.6 In this field, the group of Shu recently developed an asymmetric cobalt-catalysed cross-electrophile vinylation reaction of allylic alcohols 9 with vinyl triflates 10, (Scheme 3). The use of cobalt allowed high enantiospecificity with easily accessible enantioenriched secondary alcohols, avoiding the need for chiral ligands, to access asymmetric skipped dienes 11 with an inversion of configuration.7 The reaction required BiCl3 as a Lewis acid to activate the allylic oxalate formed in situ with dimethyl oxalate (DMO), and manganese (Mn) as a metal reductant to generate the active cobalt catalytic species. This protocol was applied to several allylic carbonates bearing different electronic properties on the aromatic ring, and both cyclic and acyclic vinyl triflates in moderate to very good yields and excellent enantiospecificities. Furthermore, the authors reported the functionalisation of vinyl triflates of testosterone and (+)-nootkatone, representing an efficient method for the incorporation of enantioenriched allyl alcohols into complex biologically active molecules. | ||
| Scheme 3 Cobalt-catalysed enantiospecific cross-electrophile coupling reaction of enantioenriched allyl alcohols 9 and vinyl triflates 10. | ||
In 2022, Lu and co-workers reported a cobalt-hydride catalysed hydroallylation reaction of terminal alkynes 12 with allylic electrophiles 13 to access Markovnikov-type skipped dienes 14 with good regioselectivity.8 The described protocol accomplished great efficiency, as demonstrated by the high turn-over number (TON) up to 1160 and the short reaction time of 20 min of the model reaction (Scheme 4). Furthermore, this methodology was successfully scaled up to a gram-scale without any noticeable decrease in yield. Terminal alkynes 12 containing heterocycles were well tolerated, as well as conjugated enyne and silyl alkyne. The alkene scope comprised mostly aliphatic and aromatic bromides; nonetheless different allylic electrophiles such as allyl iodide and allyl phosphate could also be functionalised to the corresponding dienic product, with slightly lower yield and regioselectivity.
 | ||
| Scheme 4 Hydroallylation reaction of terminal alkynes 12 with allylic electrophiles 13. | ||
Based on experimental studies, the authors hypothesised that a cobalt hydride species I could be obtained from the reaction of the hydrosilane and Co(II) in the presence of t-BuOLi (Scheme 4). The coordination of the terminal alkyne 12 to I generates the alkynyl cobalt hydride complex II. Subsequently, α-selective insertion of the alkyne into the Co–H bond furnishes the alkenyl cobalt intermediate IIIa that quickly isomerises to the cobalt carbene zwitterion IIIb. Finally, an SN2- and SN2′-type reaction with the allylic electrophile 13 results in the formation of the skipped diene product 14.
Recently, the group of Hirano expanded the reactivity of the apparently simple CoBr2/phosphine/Zn/ZnI2 catalytic system on internal alkynes 1 and conjugated dienes 15 to catalyse complex divergent cycloaddition reactions (Scheme 5).9
 | ||
| Scheme 5 Divergent cobalt-catalysed cycloaddition reaction of alkynes with conjugated dienes. β: bite angle. | ||
The authors observed that the bite angle of the phosphine ligand was able to significantly contribute to the chemodivergence of the reaction, providing a practical rationale for obtaining the desired cycloaddition product. Indeed, phosphines with wide bite angles such as PPh3 (although not strictly called bite angle, the P–M–P angle with PPh3 is wider than in most diphosphines) selectively produced 3-alkenylcyclobut-1-enes 16, and meanwhile ligands with narrower bite angles, i.e. ethylenebis(diphenylphosphine) (dppe L3), provided cyclohexa-1,4-dienes 18. Conversely, diphosphines like 1,3-bis(diphenylphosphino)propane (dppp L4) with intermediate bite angles afforded bicyclo[3.1.0]-hexenes 17 as the major product (Scheme 5). Nonetheless, it was observed that the electronic properties of the conjugated dienes could also impact the outcome of the cycloaddition reaction. Indeed, 1-aryldienes preferentially yielded the bicyclic product 17; the electron-deficient dienes generally gave the cyclobutadienic product 16, and meanwhile the electron-rich dienes afforded the cyclohexa-1,4-dienes 18 as the major products. According to the proposed reaction mechanism (see Scheme 5), the active catalytic species is thought to be a cationic Co(I) complex that can coordinate with the substrates to generate I. Next, an oxidative coupling allows the formation of the cationic cobaltacycle II. From the common intermediate II, according to the P–M–P angle, the reaction can evolve towards the different products 16–18 with different rates. A fast reductive elimination is observed with a wider P–M–P angle, leading to the [2 + 2] cycloadduct product 16. In contrast, a narrow bite angle implies a slow reductive elimination from II, which instead firstly isomerises to the seven-membered cobaltacycle III and then produces the [2 + 4] cycloadduct product 18. In the intermediate cases, intramolecular insertion of the alkenyl group occurs, thus yielding the bicyclic alkene 17.
Recently, Uyeda disclosed an asymmetric cobalt-catalysed procedure to access highly functionalised skipped dienes in an enantioselective manner through the aid of a chiral ligand L5 in the presence of a Zn/ZnI2 system.10 This versatile protocol afforded acyclic organozinc compounds 21, which could be further functionalised with an electrophile, starting from vinylidenes 19 and 2,5-dihydrofuranes 20 (Scheme 6). Deep mechanistic investigations, supported by DFT calculations, suggested a Zn-mediated generation of the cobalt vinylidene species I that could undergo a [2 + 2] cycloaddition pathway, followed by a ring-opening step involving a challenging ZnI2-assisted outer-sphere β-O elimination to afford 21. The scope of the methodology was mostly explored on the substrates 19. In fact, 1,1-dichloroalkenes bearing electron-donating and electron-withdrawing aryl groups furnished skipped dienes 22 in moderate to good yields with excellent enantioselectivities. Furthermore, heterocycles including quinoline and benzofuran, as well as different functional groups potentially critical for transition metal-catalysed procedures, such as boronate esters and bromides, were well tolerated. The ring-opening reaction produced organozinc compounds 21 that, after aqueous work-up, were quenched to alcohols containing the skipped diene motif 22. Nonetheless, 21 could instead be trapped with different electrophiles, accessing a broader range of functionalised 1,4-dienes. Indeed, deuterated product 22-D was obtained with 82% deuteration by quenching the crude reaction with CD3OD. Otherwise, functionalisation with NIS (N-iodosuccinimide) furnished the iodinated C2 skipped diene 23 with 53% yield and excellent enantioselectivity. Finally, Negishi reaction conditions could be employed to obtain the cross-coupling products with methyl iodide and 2-bromopyridine (products 24 and 25, 46% and 55% yield, respectively).
 | ||
| Scheme 6 Asymmetric zinc-assisted ring-opening reaction of unstrained heterocycles using cobalt vinylidenes I. | ||
In 2023, the group of RajanBabu reported a chemodivergent asymmetric cycloaddition reaction between alkynes 1 and 1,3 dienes 15 to furnish skipped dienes employing a chiral ligand/cobalt catalytic system in the presence of zinc as the metal reductant and sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF4) as the activator.11 Notably, starting from the same set of substrates, this protocol accomplished an enantioselective cobalt-catalysed [4 + 2] or [2 + 2] cycloaddition reaction to produce 1,4-cyclohexadienes 18 or cyclobutenes 16, respectively (Scheme 7). Control experiments demonstrated that the active catalytic species is a cationic Co(I) complex, although no mechanistic studies were reported to rationalise the observed ligand-dependent chemodivergence. Nonetheless, the authors noted that chiral and achiral biphosphines with relatively narrow bite angles (<93°), such as (R,R)-BenzP* L7, were the most effective for the synthesis of 1,4-cyclohexadienes 18. Meanwhile, the cobalt complexes with (S,S)-DIOP L6 or (R)-t-Bu-PHOX L6′ ligand could be employed to selectively furnish cyclobutenes 16 containing the skipped diene unit.
 | ||
| Scheme 7 Cobalt-catalysed chemodivergent and enantioselective cycloaddition reactions between conjugated alkenes 15 and alkynes 1. | ||
Rhodium is a highly versatile transition metal catalyst widely used in organic synthesis due to its exceptional ability to assist various bond-forming reactions with high efficiency and selectivity. Its catalytic properties enable key transformations such as hydrogenation, hydroformylation, C–H activation, and cycloaddition reactions.12 In this regard, exploiting rhodium's versatility in promoting cycloadditions, Zheng and colleagues developed a method to obtain carbonyl-substituted cyclohexa-1,4-dienes 18 with up to 96% yield and >99% enantiomeric excess.13 This transformation was based on an asymmetric intermolecular [4 + 2] cycloaddition of 1,3-dienes 15 with acetylenedicarboxylates 1a, catalysed by rhodium(I)-chiral phosphoramide complex. In the optimised reaction conditions, Rh(NBD)2BF4 was employed as the catalyst in the presence of ligand L8 and the additive AgSbF6 in toluene at room temperature (Scheme 8).
 | ||
| Scheme 8 Catalytic asymmetric [4 + 2] cycloaddition of 1,3-dienes 15 with acetylenecarboxylates 1a. | ||
The best results were achieved with R = Me, R1 = n-pentyl, R2 = H. Generally, the yields decreased with more hindered substituents. The reactivity was, also, compromised in the presence of low boiling 1,3-dienes. Good tolerance was observed toward different masked alcohols, protected amines, esters and phenyl groups. Moreover, in cases of low reactivity, the reaction temperature was increased up to 110 °C, adding 1,2-dichloroethane as a co-solvent. A plausible mechanism is shown in Scheme 8. The pre-formed rhodium(I) complex coordinates with the acetylenedicarboxylates 1a and the 1,3-diene 15 forming the rhodacyclopentene species I after an oxidative cyclisation. The subsequent suprafacial 1,3-allylic migration generates the heptadiene intermediate III, through the metal-mediated η3-complex II. Finally, a reductive elimination allows the cycloaddition product 18 to be obtained, along with the regeneration of the rhodium(I) complex. 1,5-Dienes could be obtained through allyl–allyl cross coupling between allylic electrophiles and allylmetal reagents.14 Moreover, gem-difluorinated cyclopropane emerged as fluoroallyl surrogates to access fluoroallylic skeletons in presence of nucleophiles through transition-metal catalysed C–C bond activation.15 Interestingly, rhodium-based catalysts promote fluoroallylation of arenes via the aryl C–H fluoroallylation of olefins starting from gem-difluorinated cyclopropanes.16 Exploiting this potentiality, by fine-tuning the rhodium precursor and the ligand, the group of Xia reported a regio-switchable rhodium-catalysed methodology to obtain fluorinated 1,n-dienes (n = 3, 4, 5) from gem-difluorinated cyclopropanes 26 and allyl-Bpin 27.17 As shown in Scheme 9, fluorinated 1,4-dienes 28 were selectively obtained employing [Rh(C2H4)2Cl]2 in the presence of phosphine (4-ClC6H4)3P L9 using water as an additive, in dioxane at 110 °C for 24 hours. In order to obtain fluorinated 1,5-dienes 28a both the rhodium catalyst and the ligand were changed to [Rh(CO)2Cl]2 and (4-CF3C6H4)3P L10, respectively. Moreover, AgBF4 replaced water as the additive with dimethoxyethane (DME) serving as the solvent at 120 °C for 12 hours (Scheme 9b).
 | ||
| Scheme 9 Catalytic regioselective synthesis of fluorinated 1,4-dienes 28 and fluorinated 1,5-dienes 28a. | ||
Several gem-difluorinated cyclopropanes 26 were tested bearing both electron-donating, electron and di-substituted aryl moieties. The scope was also extended to different allyl-Bpin 27a bearing phenyl and benzyl groups in the R1 position.
The proposed reaction mechanism, illustrated in Scheme 10, involves the oxidative addition of the gem-difluorinated cyclopropanes 26 to the rhodium(I) complex as the first step, to give the four-membered rhodacycle I. Then, the β-F elimination gives the key allyl-Rh(III) complex II. At this stage, there are two possible pathways for the formation of intermediate II. In path a, allyl-Bpin 27 inserts into the rhodium complex II to afford intermediate IIIa, followed by β-H elimination to obtain the dienyl-Bpin-bound rhodium complex IVa. The dissociation of the rhodium complex IVa gives dienyl-Bpin Va and F–Rh(III)–H. The reductive elimination of F–Rh(III)–H would regenerate the rhodium catalyst releasing one molecule of HF. Fluorinated 1,4-diene 28 can be formed by either protodeboronation of dienyl-Bpin Va or through a sequence of intramolecular transmetallation and reductive elimination via intermediate VIa. Concerning the path b, the allyl-Rh(III) complex II undergoes transmetallation with allyl-Bpin 27a to obtain the di-allyl rhodium complex IIIb. The following allyl–allyl reductive elimination allows the fluorinated 1,5-diene 28a to be formed.
 | ||
| Scheme 10 Mechanistic proposal for the synthesis of fluorinated 1,4-dienes 28 and fluorinated 1,5-dienes 28a. | ||
To obtain 1,4-dienes from 1,3-enynes, a [3 + 3] oxidative annulation strategy leveraging 1,4-rhodium(III) migration is essential.18 Since rhodium catalysis promotes three-component carboamination reactions,19 an interesting multicomponent approach, where N-pyrimidylindoles 29, 1,3-enynes 30 and dioxazolones 31 were reacted to afford 1,4-dienes 32, was developed by Li and colleagues (Scheme 11).20 The process was characterised by high E/Z selectivity and regioselectivity and the carboamination was promoted by a Rh(III) complex. The E isomer product was, selectively, formed in the presence of [Cp*Rh(MeCN)3][SbF6]2 and 4 Å molecular sieves in 2,2,2-trifluoroethanol (TFE) at 0 °C for 12 hours without exclusion of air or moisture. On the other hand, the Z isomer was formed employing [Cp*RhCl2]2 with AgSbF6 and 4 Å molecular sieves in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DCE:MeOH at 30 °C for 12 hours under N2. For both the regioisomers, N-pyrimidinylindoles 29 bearing electron-donating, electron-withdrawing, n-hexyl, benzyl, phenyl and halogen groups in different positions of the aromatic ring were tested with successful results. 1,3-Enynes 30 bearing i-propyl and bulkier groups such as ethylbenzyl, i-pentyl, n-pentyl were well tolerated in both reactions. Acetyl-, n-hexyl-, benzyl- and phenyl-substituted dioxazolones 31 all afforded high yields and selectivity. A plausible catalytic cycle is outlined in Scheme 11. First, C–H activation of indole 29 allows the formation of the five-membered rhodacycle I with the catalyst. Subsequently, the coordination of the 1,3-enyne 30 and regioselective migratory insertion of Rh–C(aryl) delivers the rhodium alkenyl intermediate II, which evolves to the π-allyl rhodium(III) species III. Allyl-to-allyl rearrangement generates intermediates IVa and IVb, where the stereochemistry of the allyl ligand is largely dictated by the steric hindrance between the rhodium complex, the indole ring and the R group in the 1,3-enyne. Then, the dioxazolone 31 ligation followed by decarboxylation forms the reactive Rh(V) allyl nitrene species Va and Vb. Finally, the C–N reductive elimination and protodemetalation releases the product in a specific configuration, closing the catalytic cycle.
:1 mixture of DCE:MeOH at 30 °C for 12 hours under N2. For both the regioisomers, N-pyrimidinylindoles 29 bearing electron-donating, electron-withdrawing, n-hexyl, benzyl, phenyl and halogen groups in different positions of the aromatic ring were tested with successful results. 1,3-Enynes 30 bearing i-propyl and bulkier groups such as ethylbenzyl, i-pentyl, n-pentyl were well tolerated in both reactions. Acetyl-, n-hexyl-, benzyl- and phenyl-substituted dioxazolones 31 all afforded high yields and selectivity. A plausible catalytic cycle is outlined in Scheme 11. First, C–H activation of indole 29 allows the formation of the five-membered rhodacycle I with the catalyst. Subsequently, the coordination of the 1,3-enyne 30 and regioselective migratory insertion of Rh–C(aryl) delivers the rhodium alkenyl intermediate II, which evolves to the π-allyl rhodium(III) species III. Allyl-to-allyl rearrangement generates intermediates IVa and IVb, where the stereochemistry of the allyl ligand is largely dictated by the steric hindrance between the rhodium complex, the indole ring and the R group in the 1,3-enyne. Then, the dioxazolone 31 ligation followed by decarboxylation forms the reactive Rh(V) allyl nitrene species Va and Vb. Finally, the C–N reductive elimination and protodemetalation releases the product in a specific configuration, closing the catalytic cycle.
 | ||
| Scheme 11 Regioselective catalytic three-component carboamination and mechanistic proposal. | ||
Pioneeringly, merging metal–carbene and strain-release chemistry, the group of Hari developed a novel regio- and stereoselective rhodium-catalysed strain-enabled protocol (Scheme 12),21 where skipped dienes 35 were obtained from bicyclo[1.1.0]butanes (BCBs) 33. BCBs are nucleophiles, able to react with electrophilic metal–carbene species generated by diazocompounds 33 in the presence of Rh2(OAc)4. The reaction was conducted without additives in dichloromethane at room temperature for 12 hours.
 | ||
| Scheme 12 Regio- and stereo-selective catalytic synthesis of 1,4-dienes 35 through strain release and concerted mechanism. | ||
To investigate the reaction potential, a series of diazocompounds 34 were tested. The electron-withdrawing and electron-donating groups on the aryl moiety, as well as the biphenyl, naphthalene, and thiophene groups as R2, were well tolerated. The use of different diazo esters, including those with alkyl, allyl, and trichloroethyl groups, resulted in favourable outcomes. Only ethyl diazoacetate, t-butyl diazoacetate, and diazomalonate exhibited poor yields and stereoselectivities (21–31% yield, E/Z = 2:1). It is worthy of mention that diazo compounds derived from biological molecules, including menthol, borneol, and cholesterol, participated in this reaction, with yields ranging from 85 to 95% and E/Z ratios exceeding 20:1. As far as the BCBs 37 are concerned, esters bearing i-propyl, t-butyl, and vinyl cyclohexyl groups yielded the desired skipped diene. Furthermore, BCB amides substituted on the nitrogen with phenyl, electron-poor aryls, and benzyl groups also reacted well. Based on DFT and experimental studies, a concerted mechanism was proposed by the authors, where the Rh(II) catalyst forms the metallocarbene species I by reacting with the diazo compound, upon loss of N2. Since it was not possible to optimise computationally intermediates II and III, it was supposed that the concerted mechanism should proceed through a less energetic transition structure able to facilitate the formation of the trans product 35 (Scheme 12).
In an attempt to extend the reactivity to non-acceptor metal carbenes, the group of Echavarren deeply investigated the reactivity of 1,3-dienes in [4 + 3] cycloadditions, catalysed either by Rh or Au.22 As far as the rhodium catalysis is concerned, 7-vinyl-1,3,5-cycloheptatrienes were investigated as model substrates in the [4 + 3] cycloaddition with 1,3-dienes via a retro-Buchner reaction in the presence of Rh2TFA4 [rhodium(II) trifluoroacetate dimer] at 40 °C, using 1,2-dichloroethane (DCE) as the solvent. A library of cycloheptadienes was successfully synthesised in modest to excellent yield with this methodology, showing a high functional group tolerance. A detailed mechanism was proposed by both DFT calculations and kinetic experiments, starting from the rhodium(II)-catalysed retro-Buchner reaction followed by a Cope rearrangement of the cyclopropane scaffold.
Among group 9 metals, also iridium-based catalysts have been exploited for the stereo- and enantio-selective synthesis of skipped dienes.23 In 2022, the group of Li reported an unprecedented enantioselective Ir-catalysed E-selective addition of α,β-unsaturated amides 36 to terminal alkynes 12 to form 1,4-dienes 37 with a quaternary carbon center at the C-3 position (Scheme 13).24 The best catalytic system was composed of Ir(COD)2Otf and the ligand (R)-Segphos L11, in dichloroethane (DCE) at 60 °C for 24 hours.
 | ||
| Scheme 13 Regio- and stereo-selective Ir-catalysed addition of a tertiary allylic C–H bond of α,β-unsaturated amides 36 to alkynes 12. | ||
Secondary and tertiary amides were tolerated, although the latter required an increased amount of catalyst. Substrates containing ether, aryl halide, phenol, and thiophene as the R3/R4 were well tolerated. Moreover, the formation of skipped dienes was observed when the amides possessed aryl, alkyl, cyclohexyl, cyclopentyl, and alkene groups in the R1 position. In the case of substrates containing two alkenes, the reaction exhibited chemoselectivity. Additionally, silyl-substituted terminal alkynes underwent regioselective addition, and a higher catalyst loading was required for alkyl- and aryl-substituted terminal alkynes. Computational studies and control experiments helped to clarify the reaction mechanism, which is shown in Scheme 13. First, the alkyne–alkyne coupling allows the formation of the iridium dihydride intermediate I able to coordinate the amide 36. A migratory insertion of the alkyne 12 into the iridium hydride generates the corresponding vinyl iridium complex II. Then, the alkene forms an iridium–carbon bond to generate intermediate III. Finally, β-hydride elimination occurs to form the 1,4-diene iridium complex IV, releasing the desired skipped diene 37 and regenerating the iridium dihydride complex.
A methodology to synthesise 1,4-dienes bearing a nitrile group without employing volatile, carcinogenic, flammable and prone to polymerisation acrylonitriles was developed by Roy and Mukherjee (Scheme 14).25 Their strategy involved an enantioselective formal α-allylation of acrylonitriles using 4-cyano-3-oxotetrahydrothiophenes (c-THTs) 38 as an easy-to-handle surrogate of acrylonitriles. The first step consisted of a selective alkylation of racemic allyl alcohols 9 on c-THTs 38, catalysed by [Ir(COD)Cl]2 in the presence of the ligand (S)-L12 and the additive camphor sulfonic acid (CSA) in dichloromethane at room temperature, for up to 64 hours. The final skipped diene 40 was obtained in the second step where the intermediate 39 underwent a retro-Dieckmann/retro-Michael reaction after treatment with an aqueous solution of LiOH in THF at 0 °C.
 | ||
| Scheme 14 Two-step enantioselective iridium-catalysed synthesis of cyano skipped dienes 40. | ||
Several branched allylic alcohols 9 were considered to investigate the scope of the reaction. Both electron-donating and electron-withdrawing groups on the aryl ring were well tolerated. Unfortunately, alkyl, alkenyl, and highly electron-rich aryl-substitution remained unreactive. More recently, the same research group reported an iridium-catalysed allylic alkylation on phosphonates followed by a Horner–Wadsworth–Emmons olefination, to form skipped dienes bearing electron-withdrawing groups (Scheme 15).26 Diethyl ethylphosphonates 41 were firstly reacted with allylic tert-butyl carbonates 42 employing [Ir(COD)Cl]2 as the catalyst, (Sa,S,S)-L13 as the ligand and t-BuOK as the base in dichloromethane at 50 °C for up to 72 hours. Then, product 43 was reacted with paraformaldehyde in the presence of Cs2CO3 in THF at 25 °C for 2.5 hours to form the skipped diene 44. In the reaction scope, cinnamyl carbonates 42 bearing electron-withdrawing and electron-donating groups on the aryl ring generally presented high yields and enantioselectivities. Also, naphthyls and heterocycles were well tolerated. On the other hand, aliphatic substituents were not suitable because of a lack of regioselectivity. Furthermore, phosphonates 41 bearing electron-deficient moieties such as ketones, nitrile and several esters were suitable as the starting materials.
 | ||
| Scheme 15 Two-step enantioselective iridium-catalysed synthesis of 1,4-dienes 44. | ||
Another strategy to synthesise skipped dienes through iridium-catalysed olefinic C–H allylation and alkenylation in water was developed by Zhang and colleagues (Scheme 16).27 In this protocol, acrylamides 45 and 1,3-butadienes 15 were reacted in water in the presence of [IrOMe(COD)]2 at 70 °C for 24 hours. Several 1-aryl-1,3-butadienes 15 were exploited, demonstrating that halogens and methoxy groups on the aryl and anthranyl ring were well tolerated. Several aromatic N-Ts acrylamides, bearing electron-donating and -withdrawing groups and different N-substituted acrylamides, such as methanesulphonyl, were efficiently converted to the final product. The catalytic cycle proposal is illustrated in Scheme 16. First, the methoxoiridium catalyst reacts with the N-Ts acrylamide 45 to form amidoiridium species I through a ligand exchange. The following oxidative addition of the olefinic C–H bond gives the hydroiridium species II which reacts with 1,3-diene 15 to generate the π-allyliridium species III by a branch selective alkene insertion. Irreversible reductive elimination forms the amidoiridium species IV followed by a ligand exchange that allows the formation of the 1,4-diene 46 and the regeneration of the catalytic active amidoiridium species I.
 | ||
| Scheme 16 Regioselective iridium-catalysed synthesis of 1,4-dienes 46. | ||
Group 10: nickel (Ni) and palladium (Pd)
Catalysis by group 10 metals relies on nickel- and palladium-based catalysts. The nickel-catalysed approach has emerged as a significant advancement in synthetic chemistry, distinguished by its cost-effectiveness, operational simplicity, and compatibility with a variety of functional groups. As a readily available and affordable transition metal, nickel facilitates efficient catalysis also in reactions involving skipped dienes.28,29In 2023, Gao and co-workers introduced a novel method for synthesising skipped aminodienes 49 using a nickel-catalysed ring-opening and cross-coupling reaction with vinylaziridines 47 and multifunctional organoboronic acids 48 (Scheme 17).30 Optimised reaction conditions include NiBr2·bipy as the catalyst, and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) as the base in THF as the solvent, at 50 °C for 16 hours. These conditions provided the highest yield (91%) and excellent regioselectivity of the skipped aminodiene product 49. The reaction exhibited first-order kinetics with respect to the nickel catalyst, indicating its crucial role in facilitating the process. In contrast, it displays zero-order kinetics regarding the vinylaziridine substrate 47, meaning that the substrate's concentration does not significantly affect the reaction rate once it has begun.
 | ||
| Scheme 17 Nickel-catalysed ring-opening/cross-coupling reaction. | ||
The proposed mechanism (Scheme 18) begins with a sequential transmetalation and reductive elimination involving styrylboronic acid, which converts Ni(II) species into the active Ni(0) species, leading to the formation of intermediate I through coordination with substrate 47. When DBU is employed, it facilitates the formation of the η3-allyl nickel intermediate III from the intermediate II by promoting the ring-opening of vinylaziridines 47 through hydrogen bonding with styrylboronic acid 48. Concurrently, the complex IV is formed from 48 and DBU via N–B bond coordination. The presence of complex IV enhances the transmetalation between styrylboronic acid and III, resulting in intermediates Va and Vb. These intermediates, undergoing reductive elimination, yield linear intermediate VIa and branched intermediate VIb. This process releases one molecule of 48 and Ni(0) for further reactions, producing intermediates VIIa and VIIb, which subsequently undergo protonation to form products 49a and 49b.30 DBU enhances the reaction by stabilising the lower-energy intermediate II through hydrogen bonding, facilitating the formation of III. DFT calculations showed that the transition state with trans-styrene and bulky allyl is more favourable than the cis-configuration. By lowering transition state energies during transmetalation, DBU promoted the production of linear-selective products and contributed to regioselectivity, making it essential for optimising the reaction.30
 | ||
| Scheme 18 Mechanism proposal for the nickel-catalysed ring-opening/cross-coupling reaction. | ||
Liu and co-workers reported a nickel-catalyzed method for the regioselective allylic alkenylation of allylic alcohols 9 with alkenyl boronates 50, enabling efficient synthesis of 1,4-dienes 51 in excellent yields (up to 97%) under mild conditions (50 °C in acetonitrile) (Scheme 19).28 The reaction employs Ni(cod)2 (5 mol%) and a monodentate phosphine ligand (10 mol%), operating without the need for a base or alcohol activator. This streamlined system offers high efficiency, broad substrate compatibility, and excellent regioselectivity. Ligand selection played a critical role in optimizing reaction outcomes. Monodentate ligands proved significantly more effective than bidentate ones, which gave lower yields and poor selectivity. Among those tested, PBu3 delivered the highest regioselectivity (linear:branched ratio of 26:1), while PPh2Cy showed a favorable balance of steric and electronic properties. Two optimized conditions were established: t-Bu3P L14 for aryl alkenyl boronates and PPh2Cy L15 for alkyl variants and selected alcohols. In both cases, regioselectivity remained excellent, with linear products strongly favored (ratios >50:1), and the stereochemistry of the diene products was exclusively E-configured. Compared with other transition metal systems—such as palladium, iridium, or copper—the nickel-catalyzed approach excels in atom economy, mildness, and selectivity. It avoids the need for pre-activated electrophiles or strong additives and works efficiently with both aryl- and alkyl-substituted allylic alcohols and a wide range of alkenyl boronates, making it a highly practical and selective method for constructing linear 1,4-dienes.
 | ||
| Scheme 19 Nickel-catalysed coupling of allylic alcohol derivatives with alkenyl boronates. | ||
In 2023, Xi and co-workers presented an innovative method for the direct hydroallylation of terminal alkynes 12 with allylic alcohols 9, utilising a nickel catalyst and promoted by carbon dioxide (Scheme 20).31 This method is notable for its straightforward and efficient approach to the synthesis of valuable compounds under mild conditions, achieving excellent Markovnikov selectivity for both alkyl- and aryl-substituted terminal alkynes. The reaction occurred through three catalytic cycles (see Scheme 20). The proposed mechanism of catalytic cycle A includes the activation of allylic alcohol 9 and the generation of Ni(I) intermediate in three steps. The activation of the allylic alcohol 9 by CO2 allows the formation of the allyl hydrocarbonate I, which is a more reactive species compared with the original alcohol. The second step is the oxidative addition of Ni(0) with the allyl hydrocarbonate. This process results in the formation of an η1-allylnickel(II) intermediate II bearing a Ni–C bond. Next, a ligand exchange with lithium acetate (AcOLi) replaces a ligand on the nickel centre, generating intermediate III and LiHCO3. This latter is a key species that is involved in the subsequent catalytic cycle. Then the vinylzinc species IV, generated in the catalytic cycle C, reacts with the Ni(II) intermediate III leading to the formation of intermediate V, which contains a Ni–C bond and is essentially the final product in a semi-reduced form. To obtain the final 1,4-diene product 52, intermediate V undergoes reductive elimination, which entails the cleavage of the Ni–C bond and the release of the desired diene. In catalytic cycle B, formic acid was formed through the activation of LiHCO3. The LiHCO3 generated in catalytic cycle A interacts with the Ni catalyst, resulting in the release of formic acid (HCOOH) and the regeneration of the active Ni catalyst (Ni(0)L2). In catalytic cycle C, the Ni(0) catalyst undergoes oxidative addition with the newly generated HCOOH, forming a hydride complex VIII. Then, the terminal alkyne 12 inserts into the Ni–H bond of intermediate VIII, resulting in the formation of a new Ni–C bond and creating the alkenyl–nickel intermediate VI. This step is the key for the regioselectivity of the reaction, favouring the Markovnikov addition. The last step included transmetalation of the alkenyl–nickel intermediate VI with ZnBr2, producing the vinylzinc species IV and regenerating the Ni(0) catalyst (NiL2), which is ready to begin the cycle again. The presented method was scalable, allowing for gram-scale reactions that yielded significant amounts of 1,4-dienes 52, highlighting its practical utility in synthetic organic chemistry.31
 | ||
| Scheme 20 Nickel-catalysed hydroallylation of alkynes 12 with allylic alcohols 9. | ||
A nickel-catalysed allylmethylation of alkynes 1 using allylic alcohols 9 and trimethylaluminum was described in 2020 by Li and co-workers. This reaction was highly stereoselective and produced tetrasubstituted alkenes 53 with good yields (Scheme 21).29 The combination of Ni(cod)2 and PPh3 as the catalyst, along with 1.5 equivalents of allylic alcohol 9 and AlMe3, in toluene at 60 °C, yielded the highest amount of the desired product while reducing unwanted side reactions. The proposed mechanism (Scheme 21) for the nickel-catalysed allylative dicarbofunctionalisation of alkynes involves the rapid reaction of trimethylaluminum (AlMe3) with the allylic alcohol 9, leading to the formation of alkoxyaluminum species such as allyloxydimethylaluminum I (ADMAL) and/or bis(allyloxy)methylaluminum II (BAMAL). The alkoxyaluminum species I coordinates to the nickel catalyst, and then an allylnickelation takes place, i.e. the allyl group from the alkoxyaluminum species is transferred to the nickel catalyst, forming an allylnickel species III. This species then undergoes a carbonickelation reaction with the alkyne 1, forming the vinylnickel species IV. In the following step, the methyl group is transferred to the vinylnickel species from the trimethylaluminum or a newly generated alkoxyaluminum species I/II (ADMAL or BAMAL), affording a skipped diene complex V. The last step is a reductive elimination in which the skipped diene product 53 is released, regenerating the active nickel catalyst for further reaction cycles. The key advantage of the described method is the use of easily accessible and affordable reagents, making it a practical and cost-effective approach for the synthesis of the skipped dienes and trienes.
 | ||
| Scheme 21 Nickel-catalysed dicarbofunctionalisation of alkynes 1. | ||
An innovative technique for the unsymmetrical bis-allylation of alkynes, crucial for constructing complex molecular architectures in organic synthesis, was recently introduced by Ji and co-workers (Scheme 22).32 The authors utilised a nickel catalyst, specifically a robust Ni(0)/NHC (N-heterocyclic carbene) system, to facilitate this reaction under mild conditions. This method allowed for the effective use of both electrophilic trifluoromethyl alkenes 54 and nucleophilic allylboronates 27, resulting in the formation of valuable skipped triene 55 products with high regio- and stereoselectivity. The optimised reaction conditions consisted of alkyne 1, α-trifluoromethyl alkene 54, and allylboronates 27 reacted with 5 mol% Ni(cod)2, 5 mol% NHC ligand L17, and 1.5 equivalents of t-BuOK in n-hexane at 50 °C for 24 hours. The authors addressed challenges in unsymmetrical bis-allylation, which has been underexplored compared with symmetrical methods. They emphasised the importance of the selectivity due to potential complications from multiple reactive species. The mechanism (Scheme 22) begins with the oxidative cyclometallation of an alkyne 1 and α-trifluoromethyl styrene 54, creating a crucial nickel metallacycle intermediate I. This intermediate enables the selective syn-addition of the alkyne to the electrophile, ensuring desired regioselectivity. Following this, a β-fluorine elimination step occurs, releasing a fluorine atom from the trifluoromethyl group and generating a new σ-complex II which undergoes transmetalation with allylboronate 27, introducing the allyl group into the intermediate III and affording the skipped triene 55 by reductive elimination. The authors noted that the regioselectivity remained consistent despite potential competitive pathways due to the steric hindrance of the tertiary C–Ni bond, which makes certain interactions less favourable. Furthermore, the method demonstrated a wide functional group tolerance, making it versatile for various synthetic applications.32
 | ||
| Scheme 22 Nickel-catalysed difluorinative bis-allylation of alkynes 1 and proposed reaction mechanism. | ||
Chen and co-workers developed an efficient nickel-catalyzed strategy for the synthesis of gem-difluorinated 1,4-dienes 56 through a three-component coupling of trifluoromethyl alkenes 54, internal alkynes 1, and organoboronic acids 48 under mild conditions (Scheme 23).33 This protocol exhibited excellent chemo-, regio-, and stereoselectivity, providing access to structurally diverse fluorinated dienes that are otherwise difficult to obtain using traditional methods. The optimized reaction conditions involved a 1.8:1:1.5 ratio of alkyne, α-trifluoromethyl alkene, and organoboronic acid, respectively, in a 9:1 toluene:anisole solvent mixture. The catalytic system comprised Ni(cod)2 (10 mol%) and PCy3L18 (20 mol%), with K3PO4 (0.6 equivalents) as the base. The reaction was performed at 35 °C for 12 hours. These mild conditions proved broadly effective, addressing limitations of earlier methods that required more complex reagents or showed narrow scope. High yields and excellent cis/trans selectivity were achieved, even on a gram scale. The success of the reaction hinged on the Ni(cod)2/PCy3 catalytic system; replacing either component or altering the base or solvent significantly reduced reactivity and selectivity. Mechanistic studies revealed a key nickelacyclopropane intermediate, supporting a pathway involving oxidative cyclization, alkyne insertion, β-fluorine elimination, transmetalation, and reductive elimination. This sequence highlights the central role of ligand and metal in enabling selective C–F bond activation and C–C bond formation.
 | ||
| Scheme 23 Nickel-catalysed defluorinative three-component coupling reaction. | ||
The same research group presented a nickel-catalysed reductive three-component coupling of aldehydes 4, 1,3-butadiene 15, and alkenyl triflates or bromides 57 to access skipped dienes 58 (Scheme 24).34 The method accommodates a broad range of functional groups and heterocycles, and is scalable, underscoring its synthetic and industrial utility. The optimal system uses NiBr2(DME), 2,2′:6′,2′′-terpyridine ligand L19, zinc powder, and n-Bu4NI in dimethylacetamide (DMA) at room temperature, using compound 15 as a 2 M solution in THF. Ligand screening revealed the unique efficiency of L19, while bidentate or electron-rich terpyridines were ineffective. The iodide additive likely aids electron transfer from the zinc to the nickel. Mechanistically, the Ni(II) precatalyst is reduced to Ni(0), which undergoes oxidative addition to the alkenyl electrophile (Ni(II)) and is further reduced to Ni(I). Insertion of butadiene 15 forms an allyl-Ni(I) species that adds to the aldehyde, completing the cycle through reductive workup.
 | ||
| Scheme 24 Nickel-catalysed reductive carboalkenylation of 1,3-butadiene 15. | ||
The resulting skipped dienes can be further transformed into polyenes, epoxides, ketones, azides, and triazoles. Limitations include poor regioselectivity with isoprene and lower yields with electron-deficient aldehydes. This study demonstrates the importance of fine-tuned catalyst systems for achieving efficient multicomponent couplings.34
A similar protocol employing aldehydes 4, 1,3-butadiene 15, and alkenylzirconium reagents 59 was recently developed for the efficient synthesis of skipped dienes 60 (Scheme 25).35 This method provides notable advantages, including high regio- and stereoselectivity under ligand- and additive-free conditions. Alkenylzirconium reagents uniquely enabled the desired three-component coupling, in contrast to ineffective alkenylaluminum and alkenylboron reagents. Optimal conditions were established at 0 °C for 48 hours to maximize yields and chemoselectivities. The reaction likely proceeds via an electrophilic allylnickel(II) intermediate formed by oxidative cyclometalation, subsequently coupling with the alkenylzirconium reagent to form the skipped diene while avoiding premature aldehyde reactions. The substrate scope was broad, including various electron-rich, electron-poor, ortho-substituted arylaldehydes, and heterocyclic aldehydes. The skipped dienes 60 obtained featured diverse functional groups suitable for further synthetic transformations into derivatives such as 1,3,6-trienes and conjugated dienes. The reaction's practicality and scalability were demonstrated via one-pot procedures and scale-up experiments, underscoring its potential for industrial applications and further exploration of multi-component nickel-catalyzed reactions.
 | ||
| Scheme 25 Nickel-catalysed carboalkenylation of 1,3-dienes 15. | ||
Xie and co-workers reported a highly efficient nickel-catalyzed cross-electrophile allylation of vinyl bromides 65, providing a powerful strategy for the construction of 1,4-dienes 63 under mild conditions (Scheme 26).36 This transformation employs readily available allylic acetates 62 and diverse (E)-alkenyl bromides, with Zn as a terminal reductant and MgCl2 as an additive. Optimal performance was achieved using NiCl2(PPh3)2 and 4,4′-di-tert-butyl-2,2′-bipyridine L20 in DMA, affording products in up to 85% isolated yield. The methodology demonstrated broad functional group tolerance, accommodating electron-rich, electron-deficient, and sterically hindered substrates, including bioactive and structurally complex compounds. The approach was successfully applied to the site-selective modification of β-elemene, a natural anti-tumour agent, enabling the introduction of various vinyl substituents at the allylic position. Several of these modified analogs showed significantly improved anti-proliferative activity, underscoring the synthetic and medicinal value of the transformation.
 | ||
| Scheme 26 Nickel-catalysed cross-electrophile allylation of vinyl bromides 61; optimised reaction conditions. | ||
Mechanistic investigations support a Ni(0)/Ni(I)/Ni(III) catalytic cycle. Radical scavengers such as TEMPO and BHT did not suppress the reaction, while oxygen inhibited product formation, suggesting that radical pathways are unlikely and that air-sensitive nickel intermediates are involved. The proposed mechanism begins with oxidative addition of the allylic acetate to Ni(0), generating a (π-allyl)Ni(II) intermediate, which is reduced by Zn to Ni(I). Subsequent oxidative addition of the vinyl halide 61 forms a Ni(III) species that undergoes reductive elimination to afford the product. The choice of ligand was crucial, with L20 outperforming others, likely due to its ability to stabilize key nickel species. The selectivity of Ni for this transformation was highlighted by the failure of Co, Cu, or Fe, reinforcing its unique suitability in this cross-electrophile coupling strategy. Preliminary attempts at asymmetric induction using a chiral pybox ligand resulted in only 24% ee, suggesting opportunities for further development in enantioselective variants.36
The majority of the palladium-catalysed strategies exploited to prepare skipped dienes, here reported, concern the allylation or vinylation of suitable substrates, among them alkynes, alkenes and arylhydrazones.37–42
As far as strategies involving alkenes are concerned, a Stille cross-coupling was used by Tsui and co-workers to obtain fluorinated 1,4-dienes 66 starting from gem-difluorotetrasubstituted vinyl esters 64 and allylstannanes 65via a C–F bond activation in a stereoselective manner. Pd2(dba)3 and dppe L5 were demonstrated to be the best catalytic system (Scheme 27).43 The reactions were carried out in toluene at 80 °C. Several allylating reagents were tested, among them boron and silyl derivatives, although stannanes 65 gave best results, allowing 13 functionalised 1,4-dienes 66 in good to excellent yields to be recovered. The reaction was applied to benzyl, aryl-substituted vinyl esters containing both electron-withdrawing and -donating groups.
 | ||
| Scheme 27 C–F activation of difluoroalkenes 64 to prepare fluorinated 1,4-dienes 65. | ||
Un-activated cycloalkenes 68, and α-nitro ketene dithioacetals 67 were the starting materials for the synthesis of polyfunctionalised-1,4-dienes 69 in a palladium-catalysed cross-dehydrogenative-coupling (Scheme 28).44 The pre-functionalisation of the substrates was not necessary. Pd(OAc)2 in the presence of benzoquinone (BQ) was the catalyst which showed the best efficiency, although only three skipped dienes 69a–c were obtained in moderate yields.
 | ||
| Scheme 28 Pd-catalysed cross-dehydrogenative-coupling to obtain 1,4 dienes 69. | ||
Very recently, the α- and β-C–H allylation of E- and Z-styrenes 70, in order to obtain multifunctionalised 1,4-dienes and 1,4,7-trienes with excellent diastereoselectivities, was reported by the research group of Zhang (Scheme 29).45 The process was enabled by the chelation-assistance of pyridine-2-carboxamide (DG), using allyl carbonates 71 as the reagents and Pd(OAc)2/AcOH as the catalytic system in ethanol. A wide scope was described, both in α- and β-allylation, in moderate to excellent yields, and both E- and Z-arylalkenes were used to obtain 21 skipped dienes 72 (Scheme 29). meta- and para-substituents such as F, OMe, CF3, and Me successfully reacted in addition to long alkyl chain derivatives. In one case, the reaction was demonstrated to be scalable. Moreover, the reaction showed good to excellent diastereoselectivities in the α-allylations. The proposed mechanism, described in Scheme 29, involves the coordination between the substrate 70 and palladium to give a π-olefin–palladium complex I, which affords a six-membered palladacycle II by a reversible α-C–H activation. Ligand exchange by allyl carbonate 71 coordination and alkene insertion take place to produce an eight-membered palladacycle IV, followed by ligand exchange with the formation of intermediate V. The cycle is ended by a β-oxygen elimination to produce aryl 1,4-diene 72.
 | ||
| Scheme 29 Chelation-assisted α and β C–H allylation of aryl alkenes 70. | ||
The direct allylation of alkynes46,47 is an efficient and straightforward method for the preparation of skipped dienes, so many examples of this strategy have been reported in last years and are here described. Chen and co-workers reported the synthesis of spirocyclo-containing skipped dienes 75 with an all-carbon tetrasubstituted alkene by reacting aryl phenol-tethered alkynes 73 with allyl iodides 74a, in the presence of a Pd catalyst by a cascade allylative di-carbofunctionalisation (Scheme 30).48 A dearomative C-allylation instead of O-allylation of aryl phenols was the key of the process. PdCl2(PhCN)2 was revealed to be the best catalyst in the presence of t-BuOLi at 70 °C in 1,2-dichloroethane (DCE). Both the base and Pd catalyst were essential to the success of the reaction. A wide scope was presented, both in alkynes 73 and aryl iodides 74a, and 54 skipped dienes 75 were successfully recovered in moderate to high yields. Arylacetylenes with electron-donating and electron-withdrawing groups were tolerated, and examples with alkyl substituents were reported. As far as the allylating agent is concerned, iodides were the most efficient, both linear and branched. The reaction is scalable to 2 mmol. The authors proposed a mechanism in which, following a classical Tsuji–Trost reaction, a π-allyl palladium intermediate I is formed and coordinates to the alkene moiety of 74a to afford intermediate II. Subsequently, the activated triple bond is attacked by the phenol with the assistance of a base to furnish intermediate III. Finally, product 75 and Pd(0) are released by reductive elimination. Recently, starting from results reported by the same research group on the palladium-catalysed regio-selective hydroallylations of alkynes with allylborons,49 an exhaustive computational study was published. A unified mechanism called “Lewis-acid–base-interaction promoted deprotonation/3,3-rearrangement” was proposed.50
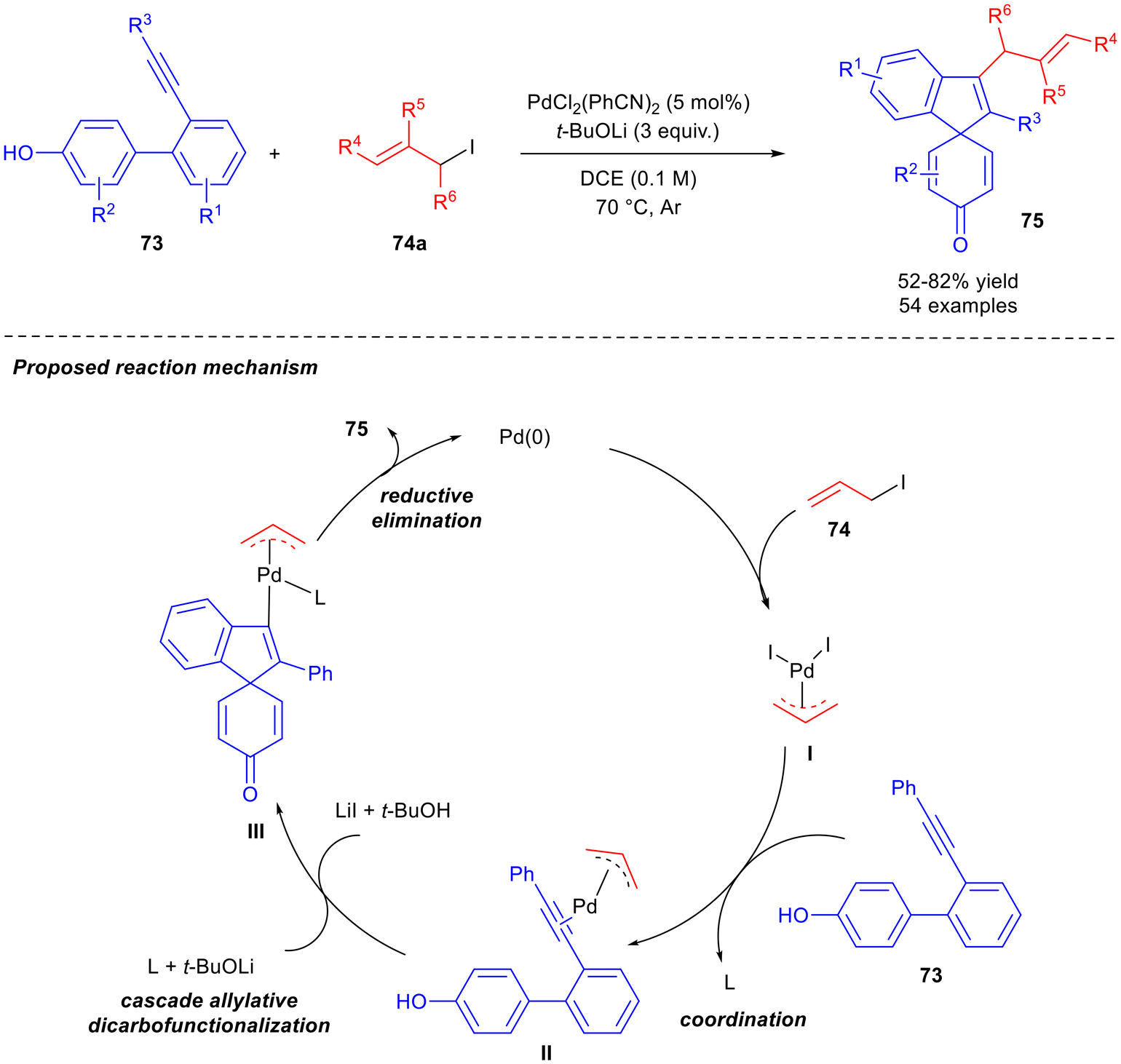 | ||
| Scheme 30 Cascade allylative dicarbofunctionalisation to obtain skipped dienes 75 and mechanism proposal. | ||
Terminal alkynes 12 and allyl halides 74/62 were exploited by Qin and collaborators to afford skipped trienes 76 in a stereoselective manner by a palladium-catalysed cascade synthesis (Scheme 31).51 Thanks to the possibility of a C–H activation, the use of toxic and unstable organometallic reagents was avoided, although high temperatures were required (120 °C). The best catalyst was shown to be {Pd[P(Ph-p-Cl)3]2Cl2}, that was used in the presence of K2CO3 as the base, in CH3CN. Several allylating agents such as allyl bromide 74b, chloride 74c and acetate 62 were successfully tested, affording the suitable skipped trienes 76.
 | ||
| Scheme 31 Cascade process to afford skipped dienes 76 from terminal alkynes 12. | ||
Both electron-rich and electron-deficient aryl terminal alkynes 12, in the meta- and para-positions, were appropriate substrates for generating the multicomponent coupling products in moderate to excellent yields, whereas the substituents at the ortho-position were less efficient. Also, heteroaryl derivatives were demonstrated to be good starting materials. Preliminary mechanistic studies indicated that both a Sonogashira reaction as the intermediate step, and a radical pathway should be involved, and the allylic dimeric {[Pd(allyl)Cl]2} could be the active catalyst.
Since allylpalladium species can be produced either by 1,2- or 1,3-dienes, results on the use of these reagents have been recently reported.52–55 An interesting multicomponent approach, where allenamides 77, alkynes 12 and aryl (alkyl)silylboronic pinacol esters 78, in the absence of the phosphine ligand, afforded skipped 1,4-dienes 79 decorated with one boryl and two silyl functionalities was described (Scheme 32).56 A very broad scope was presented both in allenyl amides 77 and alkynes 12. Notably, allyl acetates were tolerated under the reaction conditions. Many successful examples of natural complex molecules were reported, such as estrone, vitamin E or gibberellic acid. The protocol was examined for allenamides 77 bearing different chelating groups on the N atom. Substrates with different sulphonyl-based directing groups, both aryl and alkyl sulphonamides, participated in this reaction with slight variations in the standard conditions.
 | ||
| Scheme 32 Pd-catalysed reaction of allenamides 77, alkynes 12 and silylboron pinacol esters 78. | ||
As shown in Scheme 32, a mechanism, based on DFT and experimental studies, was proposed by the authors in which the allenylamide Pd complex I is obtained by the oxidative addition of PhMe2SiBpin 78, followed by the coordination of Pd to the allenamide (II). The insertion of the allenamide 77 allows the Pd-allyl complex III to be obtained. Alkyne coordination to III leads to Pd-allyl–alkyne complex IV, with the chelation of the SO2 group with Pd in η1 coordination mode. Finally, product 79 is obtained by the insertion of the PhMe2Si into the alkyne producing the complex V, followed by the reductive elimination which regenerates the Pd(0) species.
In 2023, the research group of He reported the stereodivergent asymmetric formal hydroalkenylation of 1,3-dienes 15 to produce all the four stereoisomers of 1,4-diene 80 bearing a stereocenter, with a total control of the Z/E geometry of the olefins (Scheme 33).57 A series of Josiphos-type chiral ligands were evaluated, and the most efficient is represented in Scheme 33. The geometry of internal olefin in the diene substrates did not affect the reaction, presumably due to the facile isomerisation of (Z)-15 into (E)-1, so Z/E mixtures of 15 were directly used as the substrates. A broad scope was presented, several functional groups with different steric hindrance and electronic characters being suitable for this strategy to afford di-, tri- and tetrasubstituted dienes 80. Interestingly, similar reaction conditions, and the same chiral ligand L21, were successfully applied to alkynes 1 too. In this case, following the author's hypothesis, the alkyne 1 undergoes hydrocarbonation via the formation of conjugated diene intermediate V, instead of the more conventional hydrofunctionalisation which involves the allene species III. Alkyne 1 is first converted into the allene III, which quickly produces the stable η3-Pd species IVvia irreversible hydrometallation.
 | ||
| Scheme 33 Asymmetric hydroalkenylation of 1,3-dienes 15 and alkynes 1. | ||
N-Tosylhydrazone's role in Pd(0) cross-couplings is well established, especially thanks to the pioneering work of Barluenga, Valdés and Wang.58–60 Examples of dienes obtained via Pd(0) catalysed reactions of N-tosylhydrazones have been recently developed. In 2023, a palladium-catalysed oxidative allylation of N-tosylhydrazones 82 to produce skipped 1,4-dienes 83 was reported (Scheme 34).61 The protocol demonstrated a high site selectivity, allowing the 1,4-dienes, containing a trisubstituted alkene, in a regio- and stereoselective manner to be obtained. The scope was studied both on allylaryls 81 and tosylhydrazones 82, both mono- and bicyclic, affording 1,4-dienes 83 in moderate to good yields. Whereas in the case of the alkenes, electron-rich substituents showed better efficiency, electron-withdrawing substituents on the N-tosylhydrazones aromatic ring gave the higher yields. Preliminary mechanistic studies hypothesised the π-allylpalladium carbenoid species I as the active intermediate which, upon carbene migratory insertion, delivers the alkyl palladium intermediate, producing the 1,4-diene 83 after β-H elimination.
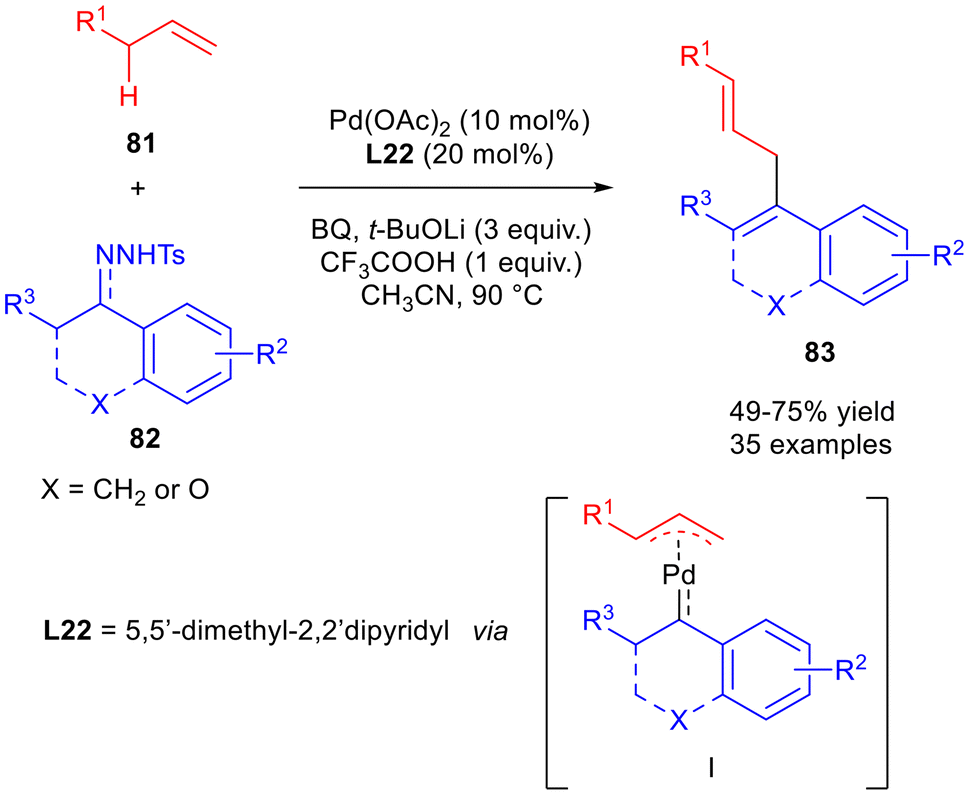 | ||
| Scheme 34 Oxidative allylation to afford skipped dienes 83. | ||
In 2022, a divergent protocol for Z-selective synthesis of 3-aryl-1,4-dienes 86 and gem-dialkylvinylcyclopropanes 87 from 2,2-dialkyl-3-(E)-alkenyl N-tosylhydrazones 85 under Pd-catalysis in an enantioselective manner was reported (Scheme 35).62 The dialkylbiaryl phosphine ligand SPhos L23 was the optimal ligand. In this case, α,α-disubstituted tosylhydrazones 85 played the role of cyclopropylcarbinyl (CPC) equivalents to produce skipped dienes. Moreover, the palladium catalysis assured a greater structural diversity of products 86 due to the wide availability of the aryl halides 84. This resulted in a controlled divergent reactivity which allowed skipped dienes 86 and cyclopropane derivatives 87 to be obtained, inaccessible by traditional vinylation methods, exploiting the palladium complex I described in Scheme 35. A tentative mechanism was illustrated, where oxidative addition of Pd(0) to an aryl bromide 84 would afford an aryl Pd(II) species, which would then react with intermediate I formed in situ producing Pd-carbene II. A subsequent 1,2-aryl migration affords Pd complex III that is subjected to a syn-carbopalladation resulting in the alkyl palladium intermediate IV. The latter is supposed to undergo a sequence of β-alkyl and β-hydride eliminations to generate 86. Alternatively, direct β-hydride elimination from IV could lead to vinylcyclopropane products 87.
 | ||
| Scheme 35 Z-Selective synthesis of 3-aryl-1,4-dienes 86 and gem-dialkylvinylcyclopropanes 87 from 2,2-dialkyl-3-(E)-alkenyl N-tosylhydrazones 85. | ||
The last example of this paragraph concerns a very peculiar strategy, reported by Marek's research group, which allows skipped dienes 90, including two congested quaternary carbons, to be obtained, exploiting the double carbometalation of cyclopropenes (Scheme 36).63 Since the synthesis of the reagents alkenyl-[1,1]-bicyclopropyl methanol derivative 88 is quite challenging, the authors firstly optimised a protocol for the synthesis of the single diastereomers, based on the Cu catalysis starting from cyclopropenyl ester. Following a Heck strategy, the regioisomer 88a, containing a hydroxymethylene function and an allylic group on each cyclopropene ring, was reacted with aryl iodide 89 to provide the corresponding skipped dienes 90 with excellent diastereoselectivity for the creation of the two distant quaternary carbon stereocenters. In order to add a stereocenter to the 1,4 dienes, the regioisomer (E)-propenyl[1,1]-bicyclopropyl methanol 88b was produced by Ru-catalysed isomerisation of 88a. In the same reaction conditions, dienes which contain three distant stereocenters 90b, including two quaternary carbons, were recovered in moderate yields (47–50%) as a single (E,E)-isomer (>99:1) and excellent diastereomeric ratio (dr 92:08:0:0).
 | ||
| Scheme 36 Synthesis of skipped dienes with two congested quaternary carbons. | ||
Group 11: copper (Cu) and gold (Au)
The synthesis of skipped dienes through metals belonging to group 11 is based on the usage of catalysts containing copper or gold as the active metal centre. The copper-catalysed synthesis of skipped dienes is usually carried out on alkynes as the substrates, exploiting the ability of copper to coordinate the triple bond.In 2020, Qing et al. published a copper-catalysed hydrodifluoroallylation of terminal alkynes in a regio- and stereo-selective manner (Scheme 37).64 The importance of the fluoroalkyl moiety has grown, especially in pharmaceuticals and agrochemicals.65,66 The authors performed the reaction on ethynylbenzenes 12 and 3-bromo-3,3-difluoropropene (BDFP) 91, using CuOTf·0.5C6H6 as the catalyst, SIPr L24 as the ligand, 1,1,3,3-tetramethyldisiloxane (TMDSO) 92 as the hydride source, CsF as the base and THF as the solvent at room temperature. Difluoroallylated (E)-alkenes 93 were obtained with total anti-Markovnikov regioselectivity. The substrate scope was then investigated by varying the substituents on the (hetero)aromatic ring. A wide variety of functional groups was tolerated, both electron-donating and electron-withdrawing, despite these latter leading to lower yields and traces of the Z-isomers. A reaction mechanism was proposed, starting from the generation of (SIPr)CuF species, which reacts with TMDSO, i.e. the hydride donor, yielding (SIPr)CuH. The syn-addition of this latter to the alkyne 12 affords the alkenyl intermediate I in a stereoselective fashion. Finally, I undergoes a regioselective difluoroallylation with 91 to give the desired skipped diene 93, thus regenerating the (SIPr)CuF species. A competing pathway (in grey) has also been proposed, leading to lower yield especially in the case of the electron-withdrawing group and internal alkynes.
 | ||
| Scheme 37 Copper-catalysed hydrodifluoroallylation of alkynes 12 and plausible mechanism. | ||
In 2022, the group of Fañanás-Mastral employed allylic gem-dichlorides 94 as the partner in the copper-catalysed allylboration reaction of alkynes 12, in an enantio- and diastereo-selective manner thanks to a chiral ligand L25, i.e. Hoveyda's sulphonate-bearing N-heterocyclic carbenes, using B2pin2 as the borylating agent, t-BuOLi as the base, in toluene as the solvent (Scheme 38).67 Skipped (E,Z)-dienes 95 were obtained in modest to high yields and with excellent regio-, enantio- and diastereo-selectivity thanks to this methodology.
 | ||
| Scheme 38 Enantioselective copper-catalysed allylboration of alkynes 12. | ||
The substrate scope was investigated on both the alkyne 12 and the allyl dichloride 94, leading to a wide tolerance of many functional groups. Only in some cases, i.e. in the presence of electron-withdrawing groups on the alkyne 12, was the reaction slower or was a slight decrease in the selectivity observed. The authors did not report the mechanism of the reaction; however a detailed rationalisation of the origin of the selectivity was achieved thanks to DFT calculations, and in particular all the possible transition states were deeply studied and compared. Very recently, the same research group applied this methodology to the enantioselective allylboration of acetylene 1b.68 The optimised conditions concerned the use of CuCl as the catalyst and the bulky N-heterocyclic carbene L26, substituted with a phenyl and a tert-butyl groups, as the ligand, t-BuONa as the base and B2pin2 as the borylating agent (Scheme 39). These conditions afforded either the skipped dienes 97, if allylic phosphates 96 were used as the partner, or chlorinated skipped dienes 98, when allylic gem-dichlorides 94 were employed. Chemo-, regio- and diastereo-selectivities were excellent in both cases, in the presence of methyl, (hetero)aryl and cyclohexyl substituents.
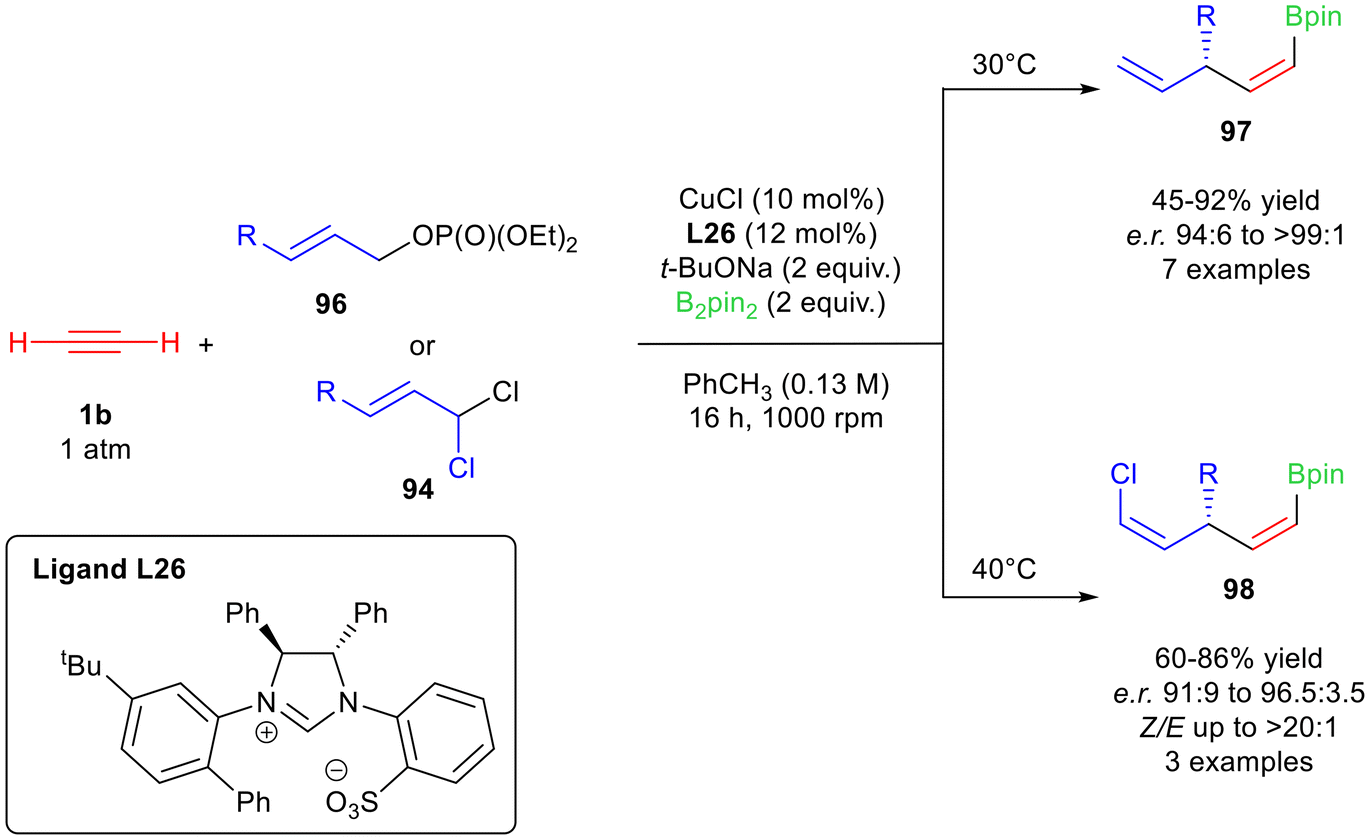 | ||
| Scheme 39 Copper-catalysed enantioselective allylboration of acetylene 1b. | ||
The abovementioned selectivities were corroborated by DFT calculations of the transition state energies. Moreover, the utility of this methodology was highlighted with several late-stage functionalisations and through the stereodivergent enantioselective total synthesis of (+)-Nyasol and (+)-Hinokiresinol, as well as the enantioselective formal synthesis of (+)-Phorbasin C and other relevant chiral compounds. A Cu(I)/NHC catalytic system was also employed by the group of Teichert in the H2-mediated C–C coupling of internal alkynes 1 and allyl chlorides 74c to access skipped dienes 99 (Scheme 40).69 The first set of reactions was carried out on aryl-substituted internal alkynes 1 and (E)-1-chlorohex-2-ene, i.e. R3 = n-Pr and R4 = H, using [SIMesCuCl] as the catalytic system, and H2 as the hydride source, in 1,4-dioxane which allowed t-BuONa to be soluble, thus favouring the heterolytic H–H bond cleavage (Scheme 40). A variety of functional groups, both electron-withdrawing and electron-donating, were well tolerated, giving the products 99 in modest to good yields and excellent regioselectivity concerning the hydrocupration reaction. Moreover, the presence of reactive groups, such as tosylate, acetate and chloride, did not affect the reaction outcome.
 | ||
| Scheme 40 H2-mediated copper-catalysed C–C coupling reactions to access skipped dienes 99 and 101. | ||
Propargylic silyl ethers 100 showed a similar reactivity in the abovementioned conditions (Scheme 40). The H2 pressure could be lowered to 10 bar with these substrates and, delightfully, 1,4-dienes 101 bearing an allylic alcohol portion could be obtained after the deprotection of the silyl ether. The substrate scope was even wider in this case, tolerating bulky substituents and heterocycles. Noteworthy, benzyl ethers were not cleaved under H2 conditions, whereas halogen-substituted aryls did not undergo protodehalogenation. A plausible mechanism was elucidated upon kinetic isotopic effect (KIE) studies and control experiments (Scheme 41). CuCl is first activated by t-BuONa to generate the active species t-BuOCu, which favours the heterolytic H2 cleavage, thus forming the Cu–H hydride species. The syn-hydrocupration reaction with alkyne 1 affords vinyl copper adduct I, which in turn reacts with allyl chloride 74c. While hypothesising this mechanism, the authors considered the methyl group as the sterically most demanding substituent, which blocks one hemisphere from the attack of I, thus favouring the formation of adduct II. (Z,Z)-1,4-Diene 99 originates from a chlorine-directed syn-carbocupration to access III, which is in equilibrium with the IV species. This latter undergoes syn-β-Cl-elimination yielding the desired diene 99, thus restoring CuCl.
 | ||
| Scheme 41 Plausible reaction mechanism for the Cu(I)-catalysed C–C coupling. | ||
In 2022, Fernández and co-workers reported a Cu(I)-catalysed allylic coupling of 1,1-diborylalkenes 102 and allyl bromides 74b to afford (Z)-skipped dienes 103 using PPh3, t-BuOLi as the base and THF as the solvent at 60 °C (Scheme 42).70 This methodology allowed the selective activation of the more hindered Bpin group on 102, leading to a (Z)-α-borylalkenyl copper(I) complex I, which behaves as α-borylalkyl copper(I) systems in nucleophilic substitutions, thus generating the desired (Z)-diene 103.
 | ||
| Scheme 42 Copper-catalysed reaction of 1,1-diborylalkenes 102 and allyl bromides 74b. | ||
The optimised conditions were set up to minimise the protodeboronation side reaction; however an increase in the steric hindrance on 102 increased the formation of the protodeboronation by-product. On the other hand, aromatic rings substituted with both electron-donating and electron-withdrawing groups on 102 afforded the (Z)-dienes 103 in modest to good isolated yields, pointing out that lower yields could be obtained because of the instability of the C(sp2)–Bpin moiety during the purification. A deep insight into the mechanism was finally reported by means of DFT calculations and free-energy profiles, whereas some control experiments were performed to justify the hypothesis of a SN2′ pathway. More recently, Yun et al. developed a diastereoselective borylative allylation of α,β-unsaturated sulfones 104, tuning the reactivity depending on the strength of the base.71 In fact, the use of MeOLi at 40 °C afforded syn-3,4-boroallylated sulfones 105 starting from alkenyl sulfones 104 and allylic phosphate 96, in the presence of CuCl as the catalyst (Scheme 43). On the other hand, the skipped dienes 106 can be selectively obtained by using t-BuOK, i.e. a stronger base, at 60 °C. The authors proposed that 106 can also be formed from 105 upon a deborylation–desulfonylation process, in the presence of t-BuOK, as also shown in the catalytic cycle (Scheme 43A).
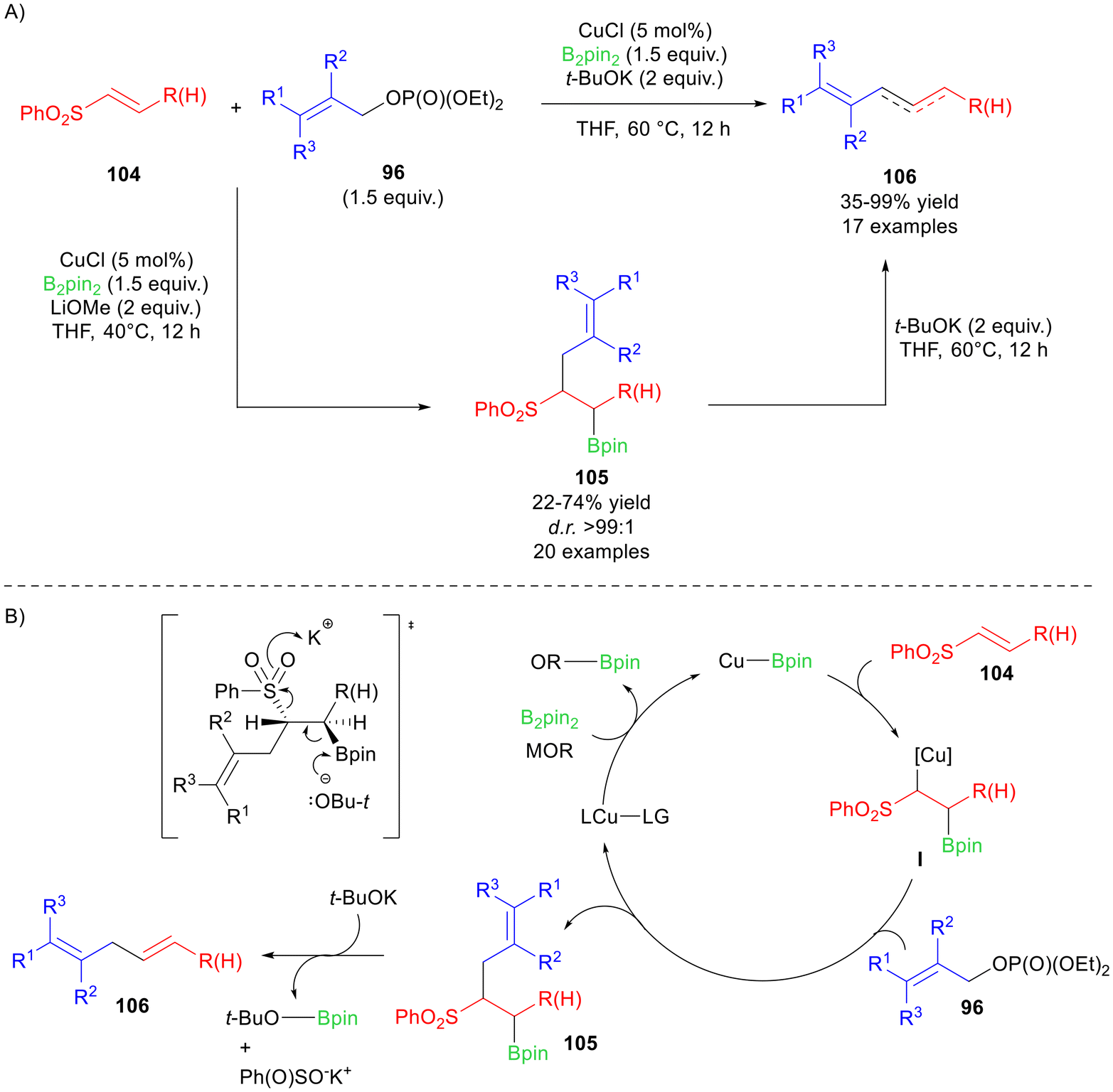 | ||
| Scheme 43 (A) Copper-catalysed reaction of alkenyl sulfones 105 with allyl phosphates 96 to access skipped dienes 106. (B) Plausible catalytic cycle for the chemical transformation. | ||
The substrate scope was investigated by coupling either terminal vinyl sulfones 104 with differently substituted allyl phosphates 96, i.e. R = H and R1, R2, R3 = H, alkyl, aryl, benzyl, naphthyl, or internal vinyl sulfones 104 with simple allyl phosphates 96, i.e. R = alkyl, aryl, benzyl and R1, R2, R3 = H or phenyl. In all cases, modest to good yields of 105 were observed, with a slight decrease in reactivity when bulkier substituents were employed. Skipped dienes 106 were smoothly obtained with un-substituted vinyl sulfones 104 and linear allyl phosphates 96, whereas branched allyl phosphates 96 ended up in the formation of 1,3-dienes due to isomerisation under basic conditions. Despite this fact, aryl-substituted vinyl sulfones 104 gave the skipped dienes 106 in moderate to good yield, if coupled with un-branched allyl phosphates. The deborylation/desulfonylation process on 105 allowed to extend the scope of the dienes 106. The catalytic cycle is shown in Scheme 43B, starting from the formation of the Cu–Bpin species, which reacts with the double bond of 104 affording the active catalytic species I. The subsequent reaction with 96 gives compound 105 that converts into the skipped diene 106 upon treatment with t-BuOK, via anti-elimination of both the boryl and the phenylsulfonyl groups.
Drawing inspiration from the research of the group of Hari on the coupling of BCBs 37 with α-diazoesters 34 in the presence of a ruthenium catalyst (see Scheme 12),20 Zhang and colleagues developed a novel methodology to obtain skipped dienes from BCBs (Scheme 44).72 The unstable diazo compound, which was exploited as the carbene precursor, was substituted with the safer and easier to handle triftosylhydrazone 107. In this case, the cross-coupling reaction was mediated by the copper-based catalyst, TpBr3Cu(NCMe), in DCE at 40 °C in the presence of an excess of NaH for 24 h. As regards the triftosylhydrazone scope, both aryl- and vinyl-substituted trifluoromethyl triftosylhydrazones were well tolerated, as well as heteroaryl rings (benzofuran, thiophene and benzothiophene). In the context of the BCB core, the utilisation of ketones, biphenyl, ester, and amide substituents was found to be effective, exhibiting no discernible influence on the E/Z selectivity, which remained at a consistent 20/1 ratio across all instances. Control experiments and DFT calculations were utilised to elucidate the reaction mechanism, excluding a concerted pathway differently from Hari because of the impossibility of locating possible transition states. The catalytic cycle starts with the generation of the copper carbene II from the catalyst and the triftosylhydrazone 107. Subsequently, the BCB 33 can attack carbene II to generate the ylide intermediate III. The latter undergoing ring opening allowed the formation of the skipped diene 108 and the regeneration of the copper catalyst. The transition state of III, formed from the E isomer, was found to be lower in energy than in the case of the Z-isomer, thus providing a rational explanation for the observed stereoselectivity.
 | ||
| Scheme 44 Copper-catalysed cross-coupling reaction of BCBs 33 with triftosylhydrazone 107. | ||
Gold catalysis is a valuable strategy for the synthesis of both cyclic and acyclic skipped dienes, commonly via the formation of gold carbene intermediates. The synthesis of 7-membered carbocycles is usually accomplished by ring expansion,73 ring-closing metathesis,74,75 cross-coupling76 or cycloaddition strategies.77,78 In particular, 1,4-cycloheptadienes can be obtained via a cyclopropanation/Cope rearrangement sequence, starting from a diazo compound.79 However, this methodology is limited to the acceptor metal carbenes. The group of Echavarren extended the reactivity to non-acceptor metal carbenes investigating both Rh or Au catalysis (see the paragraph on Rh catalysis).22 The gold(I)-catalysed [4 + 3] cycloadditions were investigated on 5-alkoxy-1,6-enynes 109 as the substrates in a cycloisomerisation/migration/cycloaddition cascade sequence (Scheme 45).22 The gold(I) catalyst played a crucial role in governing the selectivity towards either the cycloheptadiene 110 or the cyclopropane 111. Catalysts [Au1] and [Au2] were chosen upon reaction optimisation, thus leading to modest to complete selectivities and modest to good yields.
 | ||
| Scheme 45 Gold-catalysed [2 + 1] and [4 + 3] cycloadditions of alkoxyenynes 109. | ||
Furthermore, this transformation was subjected to a deep mechanistic investigation, starting from an enyne cycloisomerisation cascade, i.e. a 5-exo-dig cyclisation followed by a 1,5-migration of the OR group. Then, the reaction with 1,3-diene 15 can generate either 110 or 111via a [2 + 1] or a [4 + 3] cycloaddition, respectively, according to the energy barriers of the transition states. In 2021, López et al. reported the gold-catalysed reaction of vinyl-diazo compounds 112 with vinylsilanes 113, exploiting the β-silicon effect, thus stabilising an adjacent carbocation, to access skipped dienes 114 (Scheme 46).80 Upon optimisation, JohnPhos AuCl was chosen as the best catalytic system, using sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate as a halide scavenger.
 | ||
| Scheme 46 Gold-catalysed reaction of vinyldiazo compounds 112 with vinylsilanes 113. | ||
As far as the vinylsilane 113 is concerned, only aromatic substituents bearing both electron-withdrawing and electron-donating groups were investigated, giving the desired product 114 in good to excellent yields. The electron-withdrawing groups on the diazo compound 112 were mainly alkyl/benzyl esters but also ketones were tested, affording dienes in good yields. The authors proposed the mechanism illustrated in Scheme 46, starting from the decomposition of the diazo compound 112 which affords the gold carbene intermediate I upon loss of N2. Then, the vinylsilane 113 attacks I giving a carbocation II, stabilised by both π-conjugation from the adjacent phenyl group and hyperconjugation from the TMS group at the β-position. An intramolecular 1,4-migration of this latter yields a diene intermediate III, which in the presence of water traces affords the final skipped diene product 114.
More recently, Shin and co-workers published a gold-catalysed sulfonium-Claisen rearrangement on cinnamyl thioethers 115 and tert-butyl propiolates 116 to access skipped dienes 118 in an enantioselective fashion (Scheme 47).81 The authors pointed out the novelty of this methodology since usually cinnamyl substituents do not effectively undergo this kind of rearrangement. In fact, the sulfonium intermediate I can break down into allyl cation II in the presence of cinnamyl groups, i.e. R = Ar2, leading to the formation of 119 and 120 as by-products (Scheme 47).
 | ||
| Scheme 47 Gold(I)-catalysed sulfonium-Claisen rearrangement of cinnamyl thioethers and tert-butyl propiolates. | ||
Upon optimisation, (R,Sp)-Josiphos L27 was found to be the best ligand which gave the highest yield, thus limiting the formation of by-products. As far as the scope is concerned, only Ar2 was studied, and in particular mono-, di- and tri-substituted aromatic rings were investigated. Both electron-withdrawing and electron-donating groups on the para- and ortho-positions were well tolerated, giving the product in high yields and enantioselectivities, as well as halogens and alkoxy groups. Notably, the former can be then exploited to further functionalise the scaffold via cross-coupling reactions. On the other hand, the authors focused their attention on o-acetoxy derivatives, proposing a facile route to 2-chromanones A and 4H-chromenes B, with complete retention of the optical purity (Scheme 47). Finally, a late-stage functionalisation was done to further confirm the wide applicability of this methodology.
Miscellaneous: calcium (Ca), chromium (Cr), titanium (Ti) and ytterbium (Yb)
This section deals with the synthesis of skipped dienes by methodologies based on the use of metals, for which only one example each is known in the literature in the period analysed, in particular, calcium, chromium, titanium and ytterbium. In 2023, Li et al. presented a calcium-promoted synthesis of 1,4-dienes through the activation of the C–OH bond of Morita–Baylis–Hillman alcohol (Scheme 48).82 The catalytic system, composed of Ca(NTf2)2, KPF6 and NEt3, was able to facilitate the C–OH bond cleavage of activated allylic alcohols 9 promoting the dehydrative allylation of stabilised P-ylides 121 in dioxane at 100 °C for 12 hours. Skipped dienes 122 were obtained through a one-pot Wittig reaction. | ||
| Scheme 48 One-pot calcium-catalysed dehydrative allylation and Wittig reaction. | ||
Several aryl substituted compounds 9 with both electro-withdrawing and -donating groups on the aromatic ring were tested, and demonstrated to be suitable to obtain the final product. Also, cycloalkyl, alkyl, phenyl, fused ring, heteroaromatic and pyridine groups were tolerated. As far as P-ylides 121 are concerned, ethyl and benzyl esters were suitable substituents. Aryl aldehydes bearing electron-withdrawing groups performed with a low stereoselectivity for trisubstituted 1,4-dienes. The proposed mechanism, illustrated in Scheme 48, involves the initial interaction between calcium and the hydroxyl group of 9, enabling Ca–OH activation with the formation of complex I. Subsequently, the SN2 substitution of the P-ylides 121 occurs generating intermediate II. It follows a dehydration process delivering the desired P-ylide III with the regeneration of the catalyst. Finally, the allylic P-ylide reacts with the aldehyde 4 to generate the final 1,4-diene 122.
In 2019, the group of Robiette reported the rearrangement of substituted 1,1-dicarbonylester vinylcyclopropane 123 into skipped dienes 124 in the presence of sub-stoichiometric amounts of TiCl4 in DCM as the solvent (Scheme 49).83 An in-depth experimental and computational study revealed that the reaction mechanism leading to the skipped dienes 124 involves the cleavage of the three-membered ring followed by a 1,2-migration.83,84 Two possible 1,3-zwitterion intermediates I/I′ can be formed either via ring-opening on the benzylic or on the styryl side. In the first case, the 1,2-migration is in charge of the styryl group (intermediate I), and in the other case the phenyl group is migrating (intermediate I′). The deuteration experiment confirmed the phenyl group migration while the computational study proved that the 1,2-migration is a reversible process allowing, for prolonged reaction times, the cyclisation of the zwitterionic intermediate to a stable cyclopentene, thus revealing the skipped diene to be the kinetic product in this transformation. As far as the scope of the reaction is concerned, different R groups were tolerated in the vinylcyclopropane 123, in particular alkyl and aromatic groups. Only electron-rich aromatic groups were not tolerated, leading to the isolation of the sole cyclopentene because of the stabilisation of the zwitterion by conjugation. Suitable migrating groups R1 were aromatic ring-bearing electronic-rich substituents and fluorine atoms in the para-position, or a 2-furyl group. Electron-poor and alkyl groups did not deliver the desired skipped diene. A one-pot procedure was also reported for this transformation employing 1,3-dienes 15 and sulfonium salts 125 as the starting materials.
 | ||
| Scheme 49 TiCl4-mediated rearrangement of 1,1-dicarbonylester vinylcyclopropane 123 into skipped dienes 124. | ||
Chromium-based catalysts found applications in several reactions that involve unsaturated hydrocarbons.85–88 However, a functional methodology related to the chromium-catalysed hydroboration reaction of unsaturated hydrocarbons was still missing. In 2021, Zhao and Ge reported a chromium-catalysed dimerisation/hydroboration of allenes to obtain borylated skipped (E/Z)-dienes 128 with high chemo-, regio- and stereoselectivities (Scheme 50).89 The reactions were performed on a series of functionalised allenes 126, treated with HBpin 127 in the presence of CrCl2, mesPDI L28, i.e. a pyridine-2,6-diimine ligand, and NaBHEt3 as the activator, at room temperature and using THF as the solvent. The robustness of this methodology was emphasised by the high functional group tolerance. Indeed, moieties such as (silyl)ethers, halogens, acetals, tosylates and terminal alkynes were well tolerated, giving the desired products in modest to good yields, whereas the presence of carbonyl groups did not allow the transformation. The authors also reported examples of the cross-dimerisation/hydroboration reactions, by using two differently substituted allenes 126, and of different late-stage functionalisation of borylated skipped dienes 128. A plausible catalytic cycle was proposed based on EPR analyses, control experiments and kinetic studies (Scheme 50). CrCl2 undergoes activation in the presence of NaBHEt3, mesPDI L28 and THF as the solvent, affording the active species (L)(THF)2Cr–H I. An electronically unsaturated Cr(I) hydride II species is then formed upon the loss of a THF molecule. Allene 126 is coordinated to II giving III, which converts into the allylchromium species IV by migratory insertion. A second molecule of allene 126 coordinates to IV, forming a new C–C bond in another allylchromium intermediate VI. This latter reacts with HBpin 127 in THF to yield the desired skipped diene 128, regenerating the active Cr(I) hydride I.
 | ||
| Scheme 50 Chromium-catalysed dimerisation/hydroboration of allenes 126 to access skipped dienes 128. | ||
In 2021, Gogoi et al. developed the Yb(III)-catalysed syn-thioallylation of ynamides 129 to give tetrasubstituted thio-amino-skipped dienes 131 (Scheme 51).90 The reaction was carried out starting from N-oxazolidinone-protected ynamides 129 and allyl substituted sulfides 130 in the presence of a catalytic amount of Yb(OTf)3 in 1,2-dichloroethane as the solvent at 80 °C for 8 hours. The presence of the oxazolidinone carbonyl moiety was essential for the reactivity due to the stabilisation of the vinyl-ytterbium intermediate II thanks to the possible coordination with the putative sulfonium species (Scheme 51, bottom). The scope of the reaction for the ynamide 129 was limited to compounds bearing the oxazolidinone moiety and an aryl substituent in the alkyne terminus. In fact, no reaction was observed with the alkyl terminus, while complex mixtures were formed with N-sulfonyl protected ynamides. The range of allylsulfides 130 was quite broad, tolerating both electron-rich and halogen substituents when the R group is an aryl ring. This latter can also be disubstituted with electron-rich groups or two chlorine atoms. The corresponding skipped dienes 131 were successfully prepared with alkyl–allyl sulfides. The reaction mechanism was elucidated by DFT calculations, which identified Yb(OTf)2+ as the active species capable of coordinating to the starting materials to give I. The subsequent syn-insertion of the alkyne 129 into the S–Yb bond yields the stabilised intermediate II. The latter, which undergoes a suprafacial [3,3]-sigmatropic shift, promotes the migration of the allyl group from the sulfonium to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, yielding intermediate III. DFT calculations were corroborated by an experimental study using crossover and competition experiments which demonstrated the intramolecular allyl migration.
C bond, yielding intermediate III. DFT calculations were corroborated by an experimental study using crossover and competition experiments which demonstrated the intramolecular allyl migration.
 | ||
| Scheme 51 Yb(III)-catalysed syn-thioallylation of ynamides 129. | ||
Metallaphotoredox-catalysed synthesis of skipped dienes
Recently, visible-light mediated metallaphotoredox catalysis has been recognised as a powerful tool to access complex moieties under mild conditions, exploiting the combination of the exceptional efficiency of the C–C bond formation of transition metal catalysis with the innovative activation modes of light-promoted processes.91 In this context, Chu and co-workers reported a photoinduced stereodivergent reductive coupling reaction between vinyl triflates 10 and allylic carbonates 42 (Scheme 52) to selectively afford skipped dienes 132 both in the E- and Z-configuration from the same set of substrates.92 Through an elegant fine-tuned matching of the triplet energies of the employed photocatalysts, this cross-electrophile coupling furnished E-configured skipped dienes with a Ru-based photocatalyst, while the opposite stereoisomer was obtained with a Ir-based photocatalyst, both with excellent stereoselectivity. Indeed, the contra-thermodynamic E → Z alkene isomerisation through a photoinduced energy transfer process was feasible with [Ir(ppy)2(dtbbpy)]+, which possess a sufficiently high triplet state energy, as opposed to [Ru(bpy)3]2+. The vast application of this dual photoredox/nickel catalysis was demonstrated by its broad scope, comprising both electron-withdrawing and -donating substituents on the aromatic platform of the allylic partner, as well as both cyclic and acyclic vinyl triflates. Furthermore, specifically for the case of Z-configured skipped dienes 132, the authors noted an appreciable decrease in Z/E stereoselectivity in the functionalisation of allylic carbonates 42 with free ortho positions. | ||
| Scheme 52 Stereodivergent cross-electrophile coupling reaction of vinyl triflates 10 and allylic carbonates 42; [Ir]: Ir(ppy)2(dtbbpy)(PF6)2; [Ru]: [Ru(bpy)3](PF6)2; [Ni]: Ni(OAc)4·4H2O; bpy L29: 2,2′-bipyridine; HE: Hantzsch ester. | ||
In 2023, Chu's group developed another metallaphotoredox procedure for the stereodivergent synthesis of E- and Z-configured skipped dienes 134 (Scheme 53). This three-component reaction involved a 1,2-carboallylation of terminal alkynes 12 with allylic carbonates 42 and alkyl tetrafluoroborates 133, employing a nickel catalyst and commercially available 4CzIPN as the photocatalyst to selectively furnish either (Z,Z)-1,4-dienes or, with the aid of pyrene, (E,Z)-1,4-dienes.93 The protocol displayed great generality, as demonstrated by the functionalisation of several allylic carbonates 42 bearing useful synthetic handles such as nitrile, ester, carboxylic acid, bromide and others in good to excellent yields, with no significant effect on reaction efficiency from both the electron-withdrawing and -donating groups. A wide range of terminal alkynes with different electronic properties and steric hindrance was well tolerated by the reported methodology, although the less reactive internal alkynes were unsuitable due to competitive self-coupling of the allylic carbonates or cross-coupling of the latter with alkyl tetrafluoroborates 133.
 | ||
| Scheme 53 Three-component stereodivergent 1,2-carboallylation of alkynes 12 with allyl carbonates 42 and alkyl tetrafluoroborates 133. [Ni]: NiCl2·Phen. | ||
A deep mechanistic investigation was performed by the authors to study the reaction mechanism of the metallaphotoredox-catalysed carboallylation reported in Scheme 53. As demonstrated by Stern–Volmer fluorescence quenching experiments, photoexcited 4CzIPN* is involved in a single-electron oxidation of the alkyl tetrafluoroborate 133 (Scheme 54). This event generates the corresponding alkyl radical I, identified by trapping the t-Bu˙ radical from t-BuBF3K with the radical scavenger TEMPO, which then could add to the terminal alkyne 12 to produce the alkenyl radical intermediate II. The nickel catalytic cycle starts with Ni(0), which could undergo an oxidative addition with the allylic carbonate 42 to afford the π-allyl Ni(II) species III. The previously generated alkenyl radical II could be captured by III to form trans-(alkenyl)(allyl)Ni(III) IV. A subsequent reductive elimination generates the (E,Z)-1,4-diene 134 as the final reaction product in the presence of pyrene and a Ni(I) complex that, through a final single electron transfer, closes both catalytic cycles, regenerating 4CzIPN to its electronic ground state and Ni(0). In the absence of pyrene that acts as a triplet energy modulator, electronically excited 4CzIPN* is able to quench itself through a photoinduced energy transfer process with (E,Z)-1,4-134, producing the thermodynamically disfavoured alkene (Z,Z)-1,4-134.
 | ||
| Scheme 54 Mechanism of the carboallylation reaction reported in Scheme 53. PC: photocatalyst (4CzIPN). | ||
In the same year, He and Xia described a divergent synthesis of skipped dienes 136 and trisubstituted alkenes 138 from the same set of substrates.94 Their highly regio- and stereoselective protocol employed dual cobalt/photoredox catalysis, with a remarkably low catalyst loading (0.1 mol%) of the photocatalyst 4CzIPN (Scheme 55). Stereodefined skipped dienes 136 were obtained as the ene-type coupling product of alkynes 1 with functionalised alkenes 135 using a hemilabile P,N-ligand such as Ph-Phox L30. Meanwhile, trisubstituted alkenes were produced by simply changing the ligand to a strong bidentate one such as Xantphos L31. The authors mainly focused on the cross-coupling reaction of Tulipalin A 136a as the alkene partner, a useful synthon considered to be a cyclic analogue of methyl methacrylate (MMA) due to its exo-methylene group at the α-position of the lactone moiety. Concerning the chemoselective generation of skipped dienes, Tulipalin A was employed to install the 1,4-diene motif on a broad scope of terminal and internal alkynes, although no reaction was observed in the case of propargylic moieties. Furthermore, one example of an alkyne derived from a natural steroid was successfully converted under the Ph-Phox-controlled conditions, demonstrating the applicability to late-stage functionalisation. Under the same reaction conditions, the authors also explored the generality of the alkene partners, reporting four examples with moderate yields (44–46%). Interestingly, only activated alkenes such as acrylate and acrylonitrile were suitable coupling partners.
 | ||
| Scheme 55 Ligand-controlled dual cobalt/4CzIPN cross-coupling reaction of alkynes 1 with alkenes 135. | ||
The authors proposed a reaction mechanism (Scheme 56) in which photoexcited 4CzIPN* could initiate a single-electron oxidation of Hantzsch ester (HE) generating HE˙− and 4CzIPN˙−. Subsequently, the latter could reduce the Co(II) complex to Co(I), regenerating the photocatalyst.
 | ||
| Scheme 56 Mechanism of the cross-coupling reaction reported in Scheme 55. PC: photocatalyst (4CzIPN); HE: Hantzsch ester. | ||
Next, the substrates are coordinated to the Co(I) species, followed by an oxidative addition that generates the spirocyclic cobaltacyclopentene I, the common intermediate of this chemodivergent protocol. The presence of the hemilabile ligand Ph-Phox L30 favours an exocyclic β-Hb elimination, more competitive than the β-Ha elimination not being in a syn coplanar arrangement. Consequentially, the alkenyl Co(III)–H species II is generated and, through a reductive elimination the skipped diene 136 is produced, while regenerating the Co(I) complex. Otherwise, the strong bidentate ligand Xantphos L31 could dictate the progress of the reaction toward the protolysis of I by HE˙−, affording the Co(III)-enolate III, which through subsequent single-electron transfer by 4CzIPN˙− and protolysis could afford the trisubstituted alkene as the alternative reaction product.
Recently, Li, Gu and Xia developed an elegant stereodivergent protocol for the synthesis of (Z,Z)- and (E,Z)-configured skipped dienes obtained through the synergistic catalysis of cobalt and 4CzIPN (Scheme 57). This reaction between alkynes 1 and allenes 126 efficiently used DIPEA and water as the hydrogen source instead of the Hantzsch ester, commonly employed for such purpose.95 Under otherwise identical conditions, by changing the solvent from MeCN to 2-MeTHF, the reaction furnished (E,Z)- and (Z,Z)-1,4-dienes 137, respectively. Thorough mechanistic investigation supported by DFT calculations allowed the authors to hypothesise that the origin of the solvent-controlled stereoselectivity lay in the modulation of the triplet energy state of electronically excited 4CzIPN, involved in the photoinduced energy transfer process that triggered the E → Z alkene isomerisation. Indeed, the triplet energy levels of 4CzIPN* and the (E,Z)-1,4-diene 137 were better matched in MeCN as opposed to in THF, allowing the isomerisation to the (Z,Z)-configured product 137 to occur only in the former. The described reaction tolerated several active groups on the alkyne partner, such as hydroxyl and ester, and was successfully applied to both internal 1 and terminal alkynes 12. However, terminal aryl alkynes were not suitable substrates for this reductive coupling strategy. Finally, the authors observed a noticeable effect of the substituents of the allenic platform on the reaction efficiency, specifically with ester groups. Indeed, allenoates bearing small ester groups (methyl, ethyl) afforded the products in low yields in the MeCN conditions, differently from bulkier ester groups (isopropyl, benzyl). Furthermore, functionalised allenes containing Ts and Ac groups in place of CO2R could not be employed for this reaction, suggesting that the ester moiety might coordinate with cobalt.
 | ||
| Scheme 57 Solvent-controlled stereoselective synthesis of skipped dienes 137 from alkynes and allenes with synergistic cobalt/4CzIPN catalysis. | ||
Metal-free transformations
Among the published methodologies, only one metal-free transformation was reported for the synthesis of skipped dienes. In 2020, Harris et al. described a metal-free dialkenylation of salicylaldehydes 138 with alkenyl boronic acids 48, which was mediated by the Brønsted acid HBF4·OEt2 (Scheme 58).96 The reaction was conducted in the presence of 2.8 equivalents of boronic acids 48, 40 mol% of the acid, in dioxane at 50 °C. The skipped dienes products 139 were found to be sensitive compounds. As highlighted by the authors, this may impact the yield in instances of complete conversion of the starting materials. The scope was extended to include un-substituted and di-substituted salicylaldehydes 138 bearing electron-donating and halogen groups at the 3 and 5 positions, which yielded the corresponding dienes in yields ranging from 61 to 83%. Moreover, tolylvinyl boronic acid, as well as phenylvinyl and 1H-indene-2-boronic acids, were tested within the scope of this study, forming dienes 139 in comparable yields. It is worth noting that the formation of 2H-chromenes was observed when 6-halosalicylaldehydes were employed as the substrate. As illustrated in Scheme 58, the authors proposed the following mechanism: the acid is employed to activate the aldehyde, enabling its coordination with the boronic acid to form I. The subsequent Petasis-like transfer of the vinyl group from the borate to the carbonyl moiety yields intermediate II. The diene is formed in the last step by an 1,4-addition of II. | ||
| Scheme 58 Metal-free synthesis of skipped dienes 139 from salicylaldehydes 138 with alkenyl boronic acids 48. | ||
Synergistic/dual catalysis
Synergistic catalysis represents a robust strategy for the formation of new bonds, wherein two catalysts and two catalytic cycles operate in concert.97 Additionally, the synthesis of skipped dienes has been enhanced by this approach. In 2020, Zhuo and colleagues developed a novel approach to accessing boron and fluorine-containing molecules (Scheme 59).98 A dual catalytic system composed of CuCl/PCy3 and Pd(dppf)Cl2 was employed to obtain skipped gem-difluorodienes 141 through a regioselective boryldifluoroallylation of alkynes 1/12 with 3,3-difluorosubstituted allylic esters 140 and B2pin2 in the presence of t-BuONa, in THF at 60 °C. Regarding the scope of the transformation, both internal and terminal alkynes were suitable substrates, with lower yields observed for cyclic derivatives. The reaction exhibited high regioselectivity towards the alkyne moiety when the starting material presented both a terminal alkene and alkyne functionality, and functional groups such as amide, ether, and alkyl halide were well tolerated. The addition of electron-donating and electron-withdrawing groups, as well as halogens and heterocycles, to the allylic electrophiles proceeded smoothly, affording the diene 141 in yields ranging from good to high. The process was also proved to be scalable, and the diene 141 was subjected to a variety of transformations, demonstrating the utility of the boryldifluoroallylation reaction. A plausible mechanism was also proposed (Scheme 59, bottom). The catalytic cycle starts with the stereo- and regio-selective borylcupration of the alkyne with the complex formed between the copper-based catalyst and B2pin2, resulting in the generation of the borylalkenylcopper intermediate I. In the second catalytic cycle, palladium is exploited to activate the 3,3-difluorosubstituted allylic esters 140via an oxidative addition into the Pd(0) complex. The interaction between the two intermediates I and II permits the formation of the product 141 and the regeneration of both catalysts. The regioselectivity of this step is controlled by the palladium catalyst. | ||
| Scheme 59 Synthesis of skipped gem-difluorodienes 117 though dual copper and palladium catalysis. | ||
The synthesis of linear 1,4-dienes can be achieved through the dehydrative allylation of alkenyl sp2 C–H bonds, as outlined by the research group of Xie in 2021 (Scheme 60).99 A dual cooperative catalytic system, composed of commercially available Ca(NTf2)PF6 and Pd(PPh3)4, was employed in conjunction with feedstock starting materials, including acrylates 7 and allylic alcohols 9. The reaction exhibited sensitivity to temperature, with reduced yields observed at temperatures below 100 °C. The use of Ca(NTf2)PF6 was crucial in facilitating the cleavage of the C–OH bond. A total of 50 products was obtained, exhibiting yields ranging from low to high, using primary, secondary, and tertiary allylic alcohols. Furthermore, Morita–Baylis–Hillman alcohols were subjected to the dehydrative allylation reaction, wherein the position and electronic properties of the substituents on the aromatic ring did not affect the yields. In contrast, the stereoselectivity was influenced by the electronic properties, and a lower E/Z ratio was observed in the presence of electron-withdrawing groups. The reaction was found to be suitable for use with a variety of acrylates and alkenes, including styrenes and derivatives of biologically active compounds (estradiol, cholesterol, galactopyranose). The formation of a complex in 1:1 stoichiometry between Ca(NTf2)PF6 and the alcohol was detected by applying the method of continuous variation and DOSY experiments. The results of deuteration and kinetic isotopic effect experiments demonstrated that the alkenyl sp2 C–H bond cleavage was not the rate-determining step. Consequently, a proposed mechanism was formulated based on these findings (Scheme 60 bottom). The C–OH bond is activated by the interaction with Ca(NTf2)PF6 (I). Once the allylic alcohol has been activated, the oxidative addition of palladium enables the formation of intermediate II. The coordination of the oxygen atom to calcium and its interaction with palladium initiates the OH elimination, which results in the formation of intermediate III. This latter intermediate then coordinates the alkene, leading to the generation of intermediate IV. The 1,4-diene product 142 is obtained from IV after a Heck-like process.
 | ||
| Scheme 60 Allylation of alkenyl sp2 C–H bonds by a dual catalytic system. | ||
Bifunctional skipped dienes 144 bearing an allylic alcohol and an alkenylboronate moiety can be obtained from a three-component coupling between a substituted vinyl epoxide 143, a B2pin2 molecule, and alkynes 1/12 (Scheme 61).100 As described by the group of Fañanás-Mastral, a dual synergistic catalytic system composed of a copper and a palladium-based catalyst should be employed to observe the diene formation. A catalytic amount of t-BuONa was added to the reaction mixture in THF to increase the efficiency of the reaction. Furthermore, the authors observed that the addition of the vinyl epoxide 143 at a slow rate was essential to achieve higher yields. The use of different alkynes, including internal aryl alkyl alkynes and 1,2-diarylalkynes, as well as trimethylsilylacetylene, proved to be effective substrates, while the use of 1-hexyne or phenylacetylene did not result in the desired product formation. 1,2-Disubstituted vinyl epoxides gave yields ranging from moderate to good, albeit at temperatures exceeding 30 °C. Furthermore, the protocol was extended to cyclic vinyl carbonates. The reaction was found to be regioselective, with no addition of B2pin2 to the vinyl epoxide observed. Indeed, the LCu–Bpin complex has been shown to add in a regio- and stereoselective manner to the alkyne 1/12, thereby generating the β-borylalkenylcopper(I) intermediate I. Concurrently, the oxidative addition of epoxide 143 to the Pd(0) complex facilitates the formation of η3-allylpalladium complex II. Subsequent transmetallation of organometallic intermediates I and II allows the generation of III, which, following a reductive elimination pathway, permits the regeneration of the Pd(0) catalyst and the formation of the copper alkoxide IV. The latter is reactive enough to undergo σ-bond metathesis with B2pin2, regenerating the active LCu–Bpin complex and releasing intermediate V, the protonation of which permits the formation of diene 144.
 | ||
| Scheme 61 Cu/Pd-catalysed allylboration of alkynes with B2pin2 and vinyl epoxides 143. | ||
In 2024, the group of Han and Xie developed an enantioselective synthesis of skipped dienes 147 by the synergistic activity of a chiral pyrrolidine 146 and a Mn(I)-catalyst (Scheme 62).101 In the optimised reaction conditions, the 2,4-dienals 145 were coupled with the boronic acids 48 in the presence of 20 mol% of the aminocatalyst 146 and 5 mol% of Mn2(CO)8Br2 in TFA at 50 °C. The structure of the aminocatalyst was modified with the objective of enhancing the enantioselectivity. This was achieved by incorporating a fluorine atom in a trans arrangement with respect to the bulky silyl ether group, which improved the reactivity of the chiral iminium ion intermediate. The synthesis of skipped 1,4-dienals 147 was achieved with exclusive regioselectivity and good stereoselectivity using a range of 2,4-dienals 145, including heteroarenes and both electron-withdrawing and electron-donating groups on the different positions of the phenyl ring, with yields ranging from 49 to 78%. Furthermore, polyfluoroarenes were included in the scope, given their biological importance. Furthermore, alkenyl boronic acids with diverse substitutions, including aromatic rings, heteroarenes, and alkyl groups, were also tested. Some of the final products 147 were, also, subjected to downstream transformations, and the synthesis of (−)-blepharocalyxin D was also undertaken. According to the proposed reaction mechanism (see Scheme 62 bottom), the aminocatalyst 146 reacts with the dienal 145 to form an iminium ion I. The presence of TFA facilitates this process by lowering the LUMO energy. Concurrently, the boronic acid 48 undergoes a metallation process with Mn2(CO)8Br2, resulting in the formation of intermediate II. This is achieved through a selective migratory insertion of the C–C bond of 48 into the C–Mg bond of I, with the insertion side being influenced by the steric bulk of the catalyst. The final three steps entail the demetallation and isomerisation of intermediate III, resulting in the formation of enamine IV. The subsequent hydrolysis of V leads to the release of the diene product 147 and the regeneration of the catalysts.
 | ||
| Scheme 62 Synthesis of skipped dienes 147 from 2,4-dienals 145 and boronic acid 48 by synergistic aminocatalysis. | ||
Summary and outlook
This review presents a comprehensive overview of the latest developments in synthetic methodologies for accessing skipped dienes in the past five years. Since metal-mediated protocols play a major role in the synthesis of skipped dienes, the contribution of the different metal catalysts, classified by chemical group, is here covered. The combination of unsaturated starting materials with a metal-based catalyst, as well as metal-free transformations and synergistic catalysis, with and without light mediation, has been employed to produce 1,4-dienes with excellent regio- and stereo-selectivities. Despite copper- and palladium-promoted processes still being exploited in most of the papers, the role of group 9 elements, cobalt, rhodium and iridium, is clearly emerging, together with nickel. In many reported examples, a careful tuning of the catalytic system, by choosing the proper ligand, solvent, temperature and the presence of an additive, proved to be a crucial factor in enabling a regio-switchable approach and a rapid access to a specific stereoisomer of the diene product. Moreover, novel approaches focused on mild reaction conditions, the use of visible light photoredox protocols, and also metal free processes for the selective preparation of 1,4-dienes will, surely, increase in the next years.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge support from the Project CH4.0 under the MUR Program “Dipartimenti di Eccellenza 2023–2027” (CUP: D13C22003520001). Altair Chemical is acknowledged for co-funding a PhD scholarship of JS.References
- G. Petruncio, Z. Shellnutt, S. Elahi-Mohassel, S. Alishetty and M. Paige, Nat. Prod. Rep., 2021, 38, 2187–2213 RSC.
- T. Sato, T. Suto, Y. Nagashima, S. Mukai and N. Chida, Asian J. Org. Chem., 2021, 10, 2486–2502 CrossRef CAS.
- T. R. Pradhan and J. Kyoon Park, Chem. – Eur. J., 2022, 28, e202202120 CrossRef CAS PubMed.
- B. M. Trost, J. J. Cregg, C. Hohn, W. J. Bai, G. T. Zhang and J. S. Tracy, Nat. Chem., 2020, 12, 629–637 CrossRef CAS PubMed.
- S. Okazaki, K. Shimada, N. Komine and M. Hirano, Organometallics, 2022, 41, 390–411 CrossRef CAS.
- C. Dorval and C. Gosmini, Low-valent Cobalt Complexes in C–X Coupling and Related Reactions, in Cobalt Catalysis in Organic Synthesis, M. H. G. Hilt, Wiley, 2020, pp. 163–205 Search PubMed.
- W. Y. Ma, G. Y. Han, S. Kang, X. Pang, X. Y. Liu and X. Z. Shu, J. Am. Chem. Soc., 2021, 143, 15930–15935 CrossRef CAS PubMed.
- J. Chen, J. Ying and Z. Lu, Nat. Commun., 2022, 13, 4518 CrossRef CAS PubMed.
- Y. Tomita, N. Haraguchi, S. Kiyota, N. Komine and M. Hirano, Org. Lett., 2022, 24, 7774–7778 CrossRef CAS PubMed.
- V. V. Kanale and C. Uyeda, Angew. Chem., Int. Ed., 2023, 62, e202309681 CrossRef CAS PubMed.
- D. Singh and T. V. RajanBabu, Angew. Chem., Int. Ed., 2023, 62, e202216000 CrossRef CAS PubMed.
- K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169–196 CrossRef CAS PubMed.
- R. L.-Y. Bao, J. Yin, L. Shi and L. Zheng, Org. Biomol. Chem., 2020, 18, 2956–2961 RSC.
- H. Nakamura, M. Bao and Y. Yamamoto, Angew. Chem., Int. Ed., 2001, 40, 3208–3210 CrossRef CAS PubMed.
- J. Xu, E. A. Ahmed, B. Xiao, Q. Q. Lu, Y. L. Wang, C. G. Yu and Y. Fu, Angew. Chem., Int. Ed., 2015, 54, 8231–8235 Search PubMed.
- Z. T. Jiang, J. Huang, Y. Zeng, F. Hu and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 10626–10631 CrossRef CAS PubMed.
- Y. Zeng, H. Yang, J. Du, Q. Huang, G. Huang and Y. Xia, Chem. Sci., 2022, 13, 12419–12425 RSC.
- D. J. Burns, D. Best, M. D. Wieczysty and H. W. Lam, Angew. Chem., Int. Ed., 2015, 54, 9958–9962 CrossRef CAS PubMed.
- F. Burg and T. Rovis, J. Am. Chem. Soc., 2021, 143, 17964–17969 CrossRef CAS PubMed.
- L. Kong, X. Han, P. Hu, F. Wang and X. Li, Chem. Commun., 2023, 59, 6690–6693 RSC.
- G. A. Kadam, T. Singha, S. Rawat and D. P. Hari, ACS Catal., 2024, 14, 12225–12233 CrossRef CAS.
- H. Armengol-Relats, M. Mato and A. M. Echavarren, Angew. Chem., Int. Ed., 2021, 60, 1916–1922 CrossRef CAS PubMed.
- L. Xu, K. Meng, J. Zhang, Y. Sun, X. Lu, T. Li, Y. Jiang and G. Zhong, Chem. Commun., 2019, 55, 9757–9760 RSC.
- Z. X. Wang, P. C. Gao, E. Z. Lin and B. J. Li, Angew. Chem., Int. Ed., 2022, 61, e202200075 CrossRef CAS PubMed.
- P. Roy and S. Mukherjee, Org. Lett., 2023, 25, 4520–4524 CrossRef CAS PubMed.
- A. Seal and S. Mukherjee, Org. Lett., 2023, 25, 2253–2257 CrossRef CAS PubMed.
- Y. Huang, L. Xu, F. Yu, W. Shen, X. Lu, L. Ding, L. Zhong, G. Zhong and J. Zhang, J. Org. Chem., 2020, 85, 7225–7237 CrossRef CAS PubMed.
- X. Y. Wu, M. Yang and Y. H. Liu, Org. Lett., 2023, 25, 8782–8786 CrossRef CAS PubMed.
- J. Li, W. Li, S. Yu and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 14404–14408 CrossRef PubMed.
- L. Zhang, Q. Du, Y. Luo, Y. Wang, F. Gao, Y. Ye and W. Zhang, Chin. J. Chem., 2023, 41, 3003–3011 CrossRef CAS.
- Z. Y. Zhang, D. Y. Li and C. J. Xi, Org. Lett., 2023, 25, 698–702 CrossRef CAS PubMed.
- Y. Li, W. S. Zhang, S. N. Yang, X. Y. Wang, Y. Liu, D. W. Ji and Q. A. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300036 CrossRef CAS PubMed.
- X. Zhang, X. M. Huang, Y. Z. Chen, B. Chen and Y. H. Ma, Org. Lett., 2023, 25, 1748–1753 CrossRef CAS PubMed.
- Y. Xu, T. Su, J. R. Zhang, H. J. Ding, Y. Gao, M. Xu, P. Cao, P. Hu, B. Q. Wang and B. Chen, Org. Lett., 2023, 25, 3136–3140 CrossRef CAS PubMed.
- C. G. Wang, Y. Zhang, S. Wang, B. Chen, Y. Li, H. L. Ni, Y. Gao, P. Hu, B. Q. Wang and P. Cao, Org. Lett., 2021, 23, 535–541 CrossRef CAS PubMed.
- Y. Ye, X. Qi, B. Xu, Y. Lin, H. Xiang, L. Zou, X. Y. Ye and T. Xie, Chem. Sci., 2022, 13, 6959–6966 RSC.
- M. S. McCammant, L. Liao and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 4167–4170 CrossRef CAS PubMed.
- G. W. Kabalka and M. Al-Masum, Org. Lett., 2006, 8, 11–13 CrossRef CAS PubMed.
- F. K. Sheffy, J. P. Godschalx and J. K. Stille, J. Am. Chem. Soc., 1984, 106, 4833–4840 CrossRef CAS.
- H. Matsushita and E. Negishi, J. Am. Chem. Soc., 1981, 103, 2882–2884 CrossRef CAS.
- S. Parisotto, L. Palagi, C. Prandi and A. Deagostino, Chem. – Eur. J., 2018, 24, 5484–5488 CrossRef CAS PubMed.
- S. Parisotto and A. Deagostino, Org. Lett., 2018, 20, 6891–6895 CrossRef CAS PubMed.
- M. Li, Y. H. Wang and G. C. Tsui, Org. Lett., 2021, 23, 8072–8076 CrossRef CAS PubMed.
- Y. Li, H. Liu, Z. Huang, H. Wang and Z. Yu, Tetrahedron Lett., 2021, 82, 153396 Search PubMed.
- Y. L. Liao, X. D. Zhang, X. L. Li, X. Y. Liu, J. K. Chen, C. Shen, R. He, G. F. Zhong and J. Zhang, Org. Chem. Front., 2024, 11, 3341–3347 RSC.
- G. Cera and G. Maestri, ChemCatChem, 2022, 14, e202200295 CrossRef CAS.
- Y. Yamamoto, J. Org. Chem., 2007, 72, 7817–7831 Search PubMed.
- A. Liu, D. Chi and S. Chen, Org. Lett., 2021, 23, 8333–8337 Search PubMed.
- D.-W. Ji, Y.-C. Hu, H. Zheng, C.-Y. Zhao, Q.-A. Chen and V. M. Dong, Chem. Sci., 2019, 10, 6311–6315 RSC.
- L. J. Liu, Y. X. Liu, Q. Wu, X. F. Zhao, Y. L. Li, G. Chen and S. W. Bi, J. Org. Chem., 2023, 88, 4536–4545 CrossRef CAS PubMed.
- P. Z. Bai, Y. B. Jiang, T. B. Xiao and G. P. Qin, Eur. J. Org. Chem., 2022, e202200143 Search PubMed.
- L. Li, S. Wang, A. Jakhar and Z. H. Shao, Green Synth. Catal., 2023, 4, 124–134 CrossRef CAS.
- P. S. Wang and L. Z. Gong, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS PubMed.
- S. Parisotto and A. Deagostino, Synthesis, 2019, 1892–1912 Search PubMed.
- J. Tsuji, J. Synth. Org. Chem., Jpn., 1999, 57, 1036–1050 CrossRef CAS.
- T. R. Pradhan, M. Paudel, T. Feoktistova, P. H. Y. Cheong and J. K. Park, Angew. Chem., Int. Ed., 2022, 61, e202116154 CrossRef CAS PubMed.
- M.-Q. Tang, Z.-J. Yang and Z.-T. He, Nat. Commun., 2023, 14, 6303 CrossRef CAS PubMed.
- J. Barluenga and C. Valdés, Chem. Commun., 2010, 46, 1724–1726 RSC.
- Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560–572 RSC.
- X. Zhao, J. Jing, K. Lu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 1724–1726 RSC.
- Y. Jin, C. Li, W. Wu and H. Jiang, Adv. Synth. Catal., 2023, 365, 2338–2343 CrossRef CAS.
- N. F. C. Ritchie, A. J. Zahara and S. M. Wilkerson-Hill, J. Am. Chem. Soc., 2022, 144, 2101–2106 CrossRef CAS PubMed.
- Y. Siddaraju, J. Sabbatani, A. Cohen and I. Marek, Angew. Chem., Int. Ed., 2022, 61, e202203652 CrossRef CAS PubMed.
- N. Y. Wu, Y. G. Huang, X. H. Xu and F. L. Qing, Adv. Synth. Catal., 2020, 362, 2852–2856 CrossRef CAS.
- P. T. Lowe and D. O'Hagan, J. Fluor. Chem., 2020, 230, 109420 CrossRef CAS.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. Del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- A. Chaves-Pouso, A. M. Álvarez-Constantino and M. Fañanás-Mastral, Angew. Chem., Int. Ed., 2022, 61, e202117696 CrossRef CAS PubMed.
- A. M. Álvarez-Constantino, A. Chaves-Pouso and M. Fañanás-Mastral, Angew. Chem., Int. Ed., 2024, 63, e202407813 CrossRef PubMed.
- L. T. Brechmann, B. Kaewmee and J. F. Teichert, ACS Catal., 2023, 13, 12634–12642 CrossRef CAS.
- M. Pujol, R. J. Maza, O. Salvado, J. J. Carbó and E. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202208495 CrossRef CAS PubMed.
- M. Lim, D. Park and J. Yun, Org. Lett., 2023, 25, 8917–8921 CrossRef CAS PubMed.
- X. Zhang, T. Tian, P. Liao, Z. Liu, K. Murali and X. Bi, Org. Lett., 2025, 27, 2300–2304 CrossRef CAS PubMed.
- E. J. Kantorowski and M. J. Kurth, Tetrahedron, 2000, 56, 4317–4353 Search PubMed.
- T. Ohyoshi, S. Funakubo, Y. Miyazawa, K. Niida, I. Hayakawa and H. Kigoshi, Angew. Chem., Int. Ed., 2012, 51, 4972–4975 CrossRef CAS PubMed.
- J. A. Enquist Jr. and B. M. Stoltz, Nature, 2008, 453, 1228–1231 CrossRef PubMed.
- P. Gao and S. P. Cook, Org. Lett., 2012, 14, 3340–3343 CrossRef CAS PubMed.
- K. E. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed.
- M. Harmata, Chem. Commun., 2010, 46, 8886–8903 RSC.
- H. M. L. Davies, D. G. Stafford, B. D. Doan and J. H. Houser, J. Am. Chem. Soc., 1998, 120, 3326–3331 CrossRef CAS.
- O. Bernardo, K. Yamamoto, I. Fernández and L. A. López, Org. Lett., 2021, 23, 4452–4456 CrossRef CAS PubMed.
- J. Jang, Y. Bae and S. Shin, Org. Lett., 2023, 25, 3881–3885 CrossRef CAS PubMed.
- X. Li, D. Zhang, Y. Wang, S. Xiao, Y. Wu, P. Xie and T.-P. Loh, New J. Chem., 2023, 47, 18779–18784 RSC.
- M. Richald, A. Delbrassinne and R. Robiette, Eur. J. Org. Chem., 2019, 3779–3782 CrossRef CAS.
- A. Delbrassinne, M. Richald, J. Janssens and R. Robiette, Eur. J. Org. Chem., 2021, 2862–2868 CrossRef CAS.
- H. Mitsunuma, S. Tanabe, H. Fuse, K. Ohkubo and M. Kanai, Chem. Sci., 2019, 10, 3459–3465 RSC.
- J. Li and P. Knochel, Synthesis, 2019, 2100–2106 CAS.
- J. L. Schwarz, F. Schäfers, A. Tlahuext-Aca, L. Lückemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS PubMed.
- D. S. McGuinness, Chem. Rev., 2011, 111, 2321–2341 CrossRef CAS PubMed.
- Y. Zhao and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 2149–2154 CrossRef CAS PubMed.
- M. P. Gogoi, R. Vanjari, B. Prabagar, S. Yang, S. Dutta, R. K. Mallick, V. Gandon and A. K. Sahoo, Chem. Commun., 2021, 57, 7521–7524 RSC.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- F. Song, F. Wang, L. Guo, X. Feng, Y. Zhang and L. Chu, Angew. Chem., Int. Ed., 2020, 59, 177–181 CrossRef CAS PubMed.
- J. Qin, Z. Zhang, Y. Lu, S. Zhu and L. Chu, Chem. Sci., 2023, 14, 12143–12151 RSC.
- S. Q. Zhang, Y. L. Li, K. Cui, C. Chen, Z. Y. Gu, H. He and J. B. Xia, Org. Chem. Front., 2023, 10, 6070–6080 RSC.
- X. Y. Ren, J. J. Liu, S. Q. Zhang, Y. L. Li, K. Cui, J. Li, Z. Y. Gu and J. B. Xia, Chin. J. Catal., 2024, 61, 291–300 CrossRef CAS.
- D. H. Harris, R. O. Barichello and Y. Bolshan, Eur. J. Org. Chem., 2020, 6000–6003 CrossRef CAS.
- A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC.
- K. F. Zhuo, W. Y. Xu, T. J. Gong and Y. Fu, Chem. Commun., 2020, 56, 2340–2343 RSC.
- X. Y. Cai, H. C. Xing, J. Qiu, B. W. Li and P. Z. Xie, Org. Lett., 2021, 23, 4368–4373 CrossRef CAS PubMed.
- N. Vázquez-Galiñanes, I. Velo-Heleno and M. Fañanás-Mastral, Org. Lett., 2022, 24, 8244–8248 CrossRef PubMed.
- C. G. Zhao, J. Cai, C. Du, Q. Gao, J. Han and J. Xie, Angew. Chem., Int. Ed., 2024, 63, e202400177 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |