
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Red-shifted photoredox generation and trapping of alkyl radicals towards bioorthogonality†
David
Montoto
 ,
Uxía
Deus-Lorenzo
,
María
Tomás-Gamasa
*,
José L.
Mascareñas
* and
Mauro
Mato
*
,
Uxía
Deus-Lorenzo
,
María
Tomás-Gamasa
*,
José L.
Mascareñas
* and
Mauro
Mato
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS) and Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15705, Santiago de Compostela, Spain. E-mail: mauro.mato@usc.es; joseluis.mascarenas@usc.es; maria.tomas@usc.es
First published on 16th April 2025
Abstract
The photocatalytic generation and trapping of alkyl radicals is a powerful synthetic tool in organic chemistry, but it remains underexplored in biological settings. Here, we present two photoredox systems that leverage green- or red-light irradiation for the activation and subsequent Giese coupling of redox-active alkyl phthalimide esters. Besides utilizing mild low-energy light sources, these reactions operate with biocompatible BnNAH or NADH as electron donor. Notably, they display compatibility with air, water and biologically relevant conditions, including cell-culture media or even cell lysates. This work marks a significant step towards integrating synthetic alkyl-radical chemistry into biological settings.
Performing artificial synthetic reactions in living settings without disrupting the native cellular machinery—the fundamental principle of bioorthogonal chemistry1—has enabled diverse applications, from the selective manipulation of biomolecules to the controlled release of drugs.2 While early bioorthogonal transformations relied mostly on Staudinger ligations3 or on copper-catalyzed azide–alkyne click cycloadditions,4 recent developments have been increasingly focused on strain-driven cycloaddition reactions.5 There have also been notable advances in the development of cell-compatible reactions promoted by transition-metal complexes,6 although progress in this area remains limited by the rapid deactivation of the catalysts. An appealing alternative for performing non-natural synthetic transformations in biorelevant settings could be based on the use of radical chemistry. Radical-based reactions have been successfully applied for the chemoselective manipulation of peptides or proteins in aqueous media.7 However, their use in biorelevant habitats remains largely unexplored.8 A first step towards the implementation of bioorthogonal reactions based on alkyl radicals is the development of strategies to generate these species under mild and biologically compatible conditions (tolerance to air, water, biomolecules, high dilution, etc. Scheme 1A).9 In this context, photocatalysis serves as an ideal platform10 owing to its intrinsic mildness and spatiotemporal controllability.11
 | ||
| Scheme 1 Challenges in translating artificial alkyl-radical synthetic chemistry into living systems (A). Comparison between the photocatalyst-free generation of alkyl radicals by direct excitation with high-energy blue light (B) and a biocompatible low-energy light photocatalytic generation of alkyl radicals (C). EY = Eosin Y disodium salt; ZnTPP = Zn(II) tetraphenylporphyrin; BnNAH = 1-benzyl-1,4-dihydronicotinamide. | ||
Photoredox catalysis is widely employed as a mild strategy to generate alkyl radicals: versatile intermediates with a rich downstream chemistry.12 In particular, redox-active alkyl phthalimide esters (RAE) are well-known alkyl-radical sources upon single-electron transfer (SET) and irreversible mesolytic fragmentation,13 a sequence of events that can be easily triggered through photoredox manifolds,14 following pioneering work by Barton,15 Okada16 and co-workers. Most of these reactions have been explored in synthetic-chemistry contexts, under conditions that are far away from those required for biological uses. An exception to this is the recent work reported by Mendoza and co-workers on the use of stoichiometric photoexcited NADH, a biologically relevant reductant, for the efficient generation and trapping of alkyl radicals from RAEs (Scheme 1B).17 However, the reaction requires high-energy blue-light irradiation, which is not ideal in terms of selectivity and potential use in biological settings.18 With that in mind, we wondered if we could circumvent this requirement and trigger the reaction with less energetic light through the use of photoredox catalysis. Herein, we report the development of two photocatalytic systems for the red-shifted generation and subsequent Giese-type trapping of alkyl-radical fragments from RAEs 1 (Scheme 1C). Beyond the use of non-toxic red light,10,11,19 the reactions perform well under biologically relevant settings (aqueous buffers, cell lysates, etc.), using biocompatible reductants (NADH analogues) and with the spatiotemporal control inherent to photocatalytic systems.10 The combination of these features represents a significant step towards the future development of alkyl-radical bioorthogonal chemistry.
We started our exploration using tertiary RAE 1a and benzyl acrylate (2a) to probe the generation and intermolecular Giese-type trapping of alkyl radicals under air (Scheme 2). To promote the overall photoreductive process, we selected 1-benzyl-1,4-dihydronicotinamide (BnNAH, 4) as model electron and H-donor, as it is a cheap, readily available analogue of cell-endogenous NADH. As reported by the group of Mendoza, the reaction of 1a (0.025 mmol, 12 mM) with 2a (2.0 equiv.) and 4 (1.5 equiv.) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMSO/water mixture afforded Giese-coupling product 3a after 90 min of high-energy blue-light irradiation (467 nm, 61 kcal mol−1, entry 13), while no reaction was observed in the dark (entry 14).17 However, evaluation of the same reaction using lower-energy light sources (green: 525 nm, 54 kcal mol−1 or red: 660 nm, 43 kcal mol−1) without photocatalysts resulted in only traces of 3a (entries 4 and 10), which is consistent with BnNAH not absorbing light beyond 400 nm.22 Contrastingly, when 5 mol% of Eosin Y disodium salt (EY, 5) was added to the same mixture of 1a (0.050 mmol, 25 mM), 2a (2.0 equiv.) and 4 (1.5 equiv.) in 1:1 DMSO/water, we observed the formation of 3a in 75% yield (67% isolated yield) after 90 min of green-light irradiation (entry 1). A significant conversion to the product (55%) can already be observed after only 15 min of irradiation (entry 5). As expected, no product was observed in the absence of light (entry 2) or reductant (entry 3), while only traces (<3%) were detected when photocatalyst 5 was omitted (entry 4). In order to further red-shift the photoreactivity and potentially supress this minimal background reaction, we selected Zn(II) tetraphenylporphyrin (ZnTPP, 6) as red-light-harvesting photocatalyst, since porphyrins have been shown to photoactivate redox-active radical precursors in different synthetic contexts.20 We observed that using 15 mol% of ZnTPP as photocatalyst product 3a was formed in 94% yield after 240 min of red-light irradiation (entry 7), while 45% was obtained after only 60 min (entry 11). Control experiments showed no detectable amounts of product in the absence of light (entry 8), reductant (entry 9) or porphyrin photocatalyst (entry 10). Furthermore, experiments carried out using only DMSO demonstrate that the addition of water as a co-solvent is not a requirement for the reaction (entries 6 and 12).
:1 DMSO/water mixture afforded Giese-coupling product 3a after 90 min of high-energy blue-light irradiation (467 nm, 61 kcal mol−1, entry 13), while no reaction was observed in the dark (entry 14).17 However, evaluation of the same reaction using lower-energy light sources (green: 525 nm, 54 kcal mol−1 or red: 660 nm, 43 kcal mol−1) without photocatalysts resulted in only traces of 3a (entries 4 and 10), which is consistent with BnNAH not absorbing light beyond 400 nm.22 Contrastingly, when 5 mol% of Eosin Y disodium salt (EY, 5) was added to the same mixture of 1a (0.050 mmol, 25 mM), 2a (2.0 equiv.) and 4 (1.5 equiv.) in 1:1 DMSO/water, we observed the formation of 3a in 75% yield (67% isolated yield) after 90 min of green-light irradiation (entry 1). A significant conversion to the product (55%) can already be observed after only 15 min of irradiation (entry 5). As expected, no product was observed in the absence of light (entry 2) or reductant (entry 3), while only traces (<3%) were detected when photocatalyst 5 was omitted (entry 4). In order to further red-shift the photoreactivity and potentially supress this minimal background reaction, we selected Zn(II) tetraphenylporphyrin (ZnTPP, 6) as red-light-harvesting photocatalyst, since porphyrins have been shown to photoactivate redox-active radical precursors in different synthetic contexts.20 We observed that using 15 mol% of ZnTPP as photocatalyst product 3a was formed in 94% yield after 240 min of red-light irradiation (entry 7), while 45% was obtained after only 60 min (entry 11). Control experiments showed no detectable amounts of product in the absence of light (entry 8), reductant (entry 9) or porphyrin photocatalyst (entry 10). Furthermore, experiments carried out using only DMSO demonstrate that the addition of water as a co-solvent is not a requirement for the reaction (entries 6 and 12).
 | ||
| Scheme 2 Development, optimization and control experiments for the two photocatalytic systems based on green or red light. Standard conditions, agreen light: 1a (0.050 mmol, 25 mM), 2a (2.0 equiv.), 4 (1.5 equiv.) and 5 (5 mol%) in DMSO/H2O (1:1) under 525 nm light irradiation for 90 min; bred light: 1a (0.025 mmol, 12 mM), 2a (2.0 equiv.), 4 (1.5 equiv.) and 6 (15 mol%) in DMSO/H2O (1:1) under 660 nm light irradiation for 240 min. Unless stated otherwise, yields were determined either by 1H NMR or GC-MS using 1,3,5-trimethoxybenzene as internal standard. Isolated yield in parenthesis. n/d = not detected. | ||
Then, we sought to explore the range of alkyl radicals that could be generated and intermolecularly trapped under air. We compared side-by-side the performance of both photocatalytic systems for each substrate (Scheme 3). First, we evaluated the behavior of several Giese-type trapping agents using tertiary RAE 1a as model substrate. We could successfully carry out radical additions to α,β-unsaturated esters (3a, 3e), sulfones (3b), amides (3c) or nitriles (3d) using both our green- and red-light-based systems. Subsequently, we set forth to evaluate the scope of alkyl radicals that could be generated under the two sets of reaction conditions, using phenyl vinyl sulfone (which performed similarly well with both systems) as model Giese acceptor.
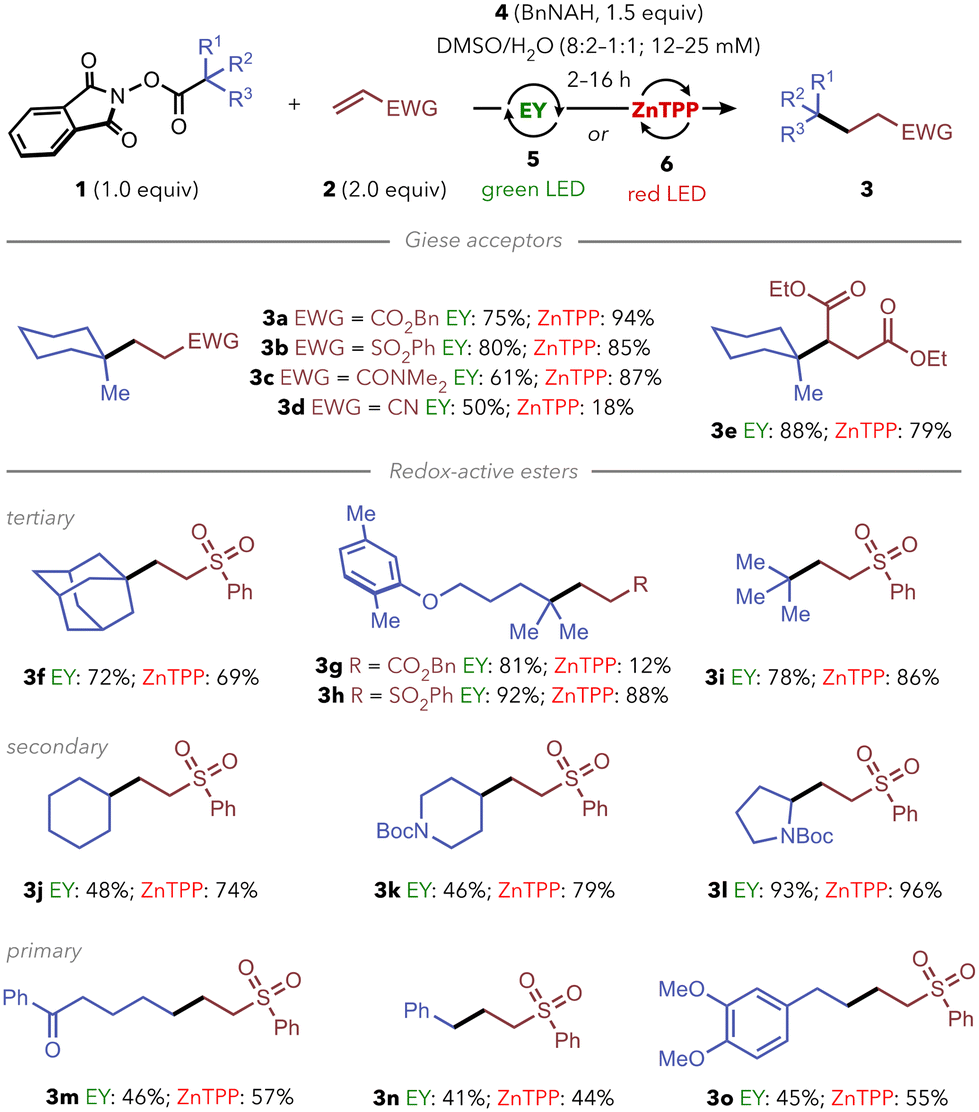 | ||
| Scheme 3 Reaction scope with the two photoredox systems. For a direct comparison, yields shown here were determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. General reaction conditions: 1 (12–25 mM, 1.0 equiv.), 2 (2.0 equiv.) and 4 (1.5 equiv.) in DMSO/H2O (1:1–8:2) with 5 (5 mol%)/green LED or 6 (15 mol%)/red LED for 2–16 h. For detailed experimental conditions, isolated yields, and results with other alkene partners, see ESI.† | ||
Beyond 1a, the reaction worked with other tertiary RAEs (3f–i), giving good yields of the corresponding radical-addition products using both photocatalytic systems. Notably, we could assemble products 3g–h starting from the RAE of Gemfibrozil (a drug used for high-cholesterol treatment), demonstrating the applicability of the method in the derivatization of relevant bioactive products. Then, we explored the reactivity with precursors of secondary and primary radicals, often considered more challenging to generate and trap due to their inherent instability. Secondary unbiased RAEs led to the expected products 3j and 3k in good yields. Furthermore, product 3l could be obtained almost quantitatively upon generation and trapping of the α-amino radical derived from N-Boc-protected proline. It is worth noting that previous methods for the green-light photocatalytic generation and trapping of alkyl radicals perform well for α-oxo or α-amino radicals, but often struggle when unbiased alkyl radicals are targeted.14a On the other hand, more stable secondary benzylic radicals resulted in low yields of Giese adducts due to competing radical dimerization.22 Finally, the feasibility of transferring simple primary alkyl radicals was also successfully demonstrated with the assembly of products 3m–o in moderate to good yields. Remarkably, the red light-based system was found to outperform the green-light photoredox manifold for the generation and trapping of less stable primary and secondary radicals.
Once we confirmed the robustness of the two protocols, we assessed their viability under biologically relevant conditions (Scheme 4). First, we extrapolated the use of cell-endogenous NADH as reductant instead of model BnNAH 4: with green light and Eosin Y, product 3a was formed in 76% yield (entry 2), while the reaction with red light and ZnTPP was unsuccessful under the evaluated conditions (entry 8). The model reactions with BnNAH were also efficient using a higher proportion of water (1:4 DMSO/water) and more dilute conditions (10 mM, entries 3 and 10). Further dilution of the substrate down to 5 mM led to a marginal decrease in yield for the red-light system (entry 11), while the reaction still performed well using Eosin Y and green LED (entry 4). The use of a 1:4 mixture of DMSO and a PBS buffered solution was well tolerated using Eosin Y at 10 mM (64%, entry 5) while a significant drop in yield was observed for ZnTPP (25%, entry 12). Importantly, we found that the reaction was still operative in a 1:4 mixture of DMSO and DMEM (Dulbecco's Modified Eagle's Medium), a widely used basal medium for supporting the growth of mammalian cells, containing a variety of biomolecules (entries 6 and 13). Furthermore, the reaction could also be carried out in a 1:4 mixture of DMSO and HeLa cell lysates with PBS buffer, obtaining comparable results to those using simpler 1:4 DMSO/PBS media for both systems (entries 7 and 14). This shows that the presence of trace amounts of the myriad of biomolecules found in living cells has no significant inhibitory effect on the studied reaction. Importantly, blank experiments without photocatalyst in these two biologically complex reaction media (DMEM or cell lysates) led to only traces of product (entries 6, 7 and 13, 14, between red brackets), highlighting the spatiotemporal control that can be achieved through photocatalysis.10
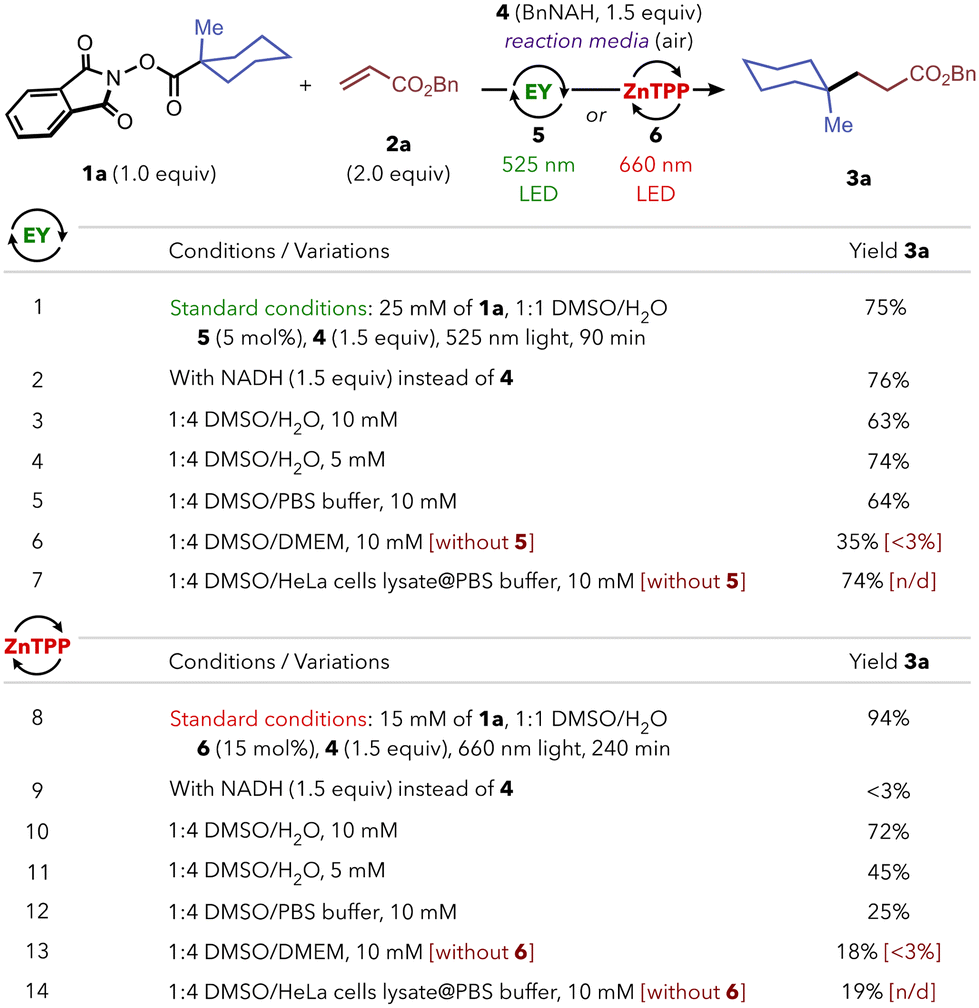 | ||
| Scheme 4 Evaluation of the performance of both photoredox systems in biologically relevant reaction media. Yields were determined either by 1H NMR or GC-MS using 1,3,5-trimethoxybenzene as internal standard. n/d: not detected. For detailed experimental conditions, see ESI.† | ||
Mechanistically, redox-active phthalimide esters 1 can be activated via single-electron reduction (half-wave potentials ranging between −1.0 and −1.6 V vs. SCE),13c,21 triggering an irreversible mesolytic cleavage to release alkyl radicals 7, CO2 and phthalimide anion (Scheme 5A). This process can be promoted by excited (oxidative quenching) or reduced (reductive quenching) blue-light-active photocatalysts, such as [Ru(bpy)3]2+.14,16 Contrastingly, Mendoza and co-workers described the direct photoexcitation of NADH or BnNAH (4) with blue light, boosting their reducing capability to trigger the activation of RAEs 1, via either inner-sphere or outer-sphere SET.17 In our case, since neither the RAE (1) nor the reductant (4) absorb green or red light,22 the addition of an appropriate light-harvesting catalyst is indispensable for the activation (see Scheme 2, entries 4 and 9). Stern–Volmer fluorescence-quenching analysis with Eosin Y (5) indicates an interaction between the excited photocatalyst and BnNAH (which would account for a bimolecular quenching constant of kq ≈ 1.6 × 1010 M−1 s−1, hinting at either static quenching or diffusion-controlled dynamic quenching), while emission of the photocatalyst remains unaffected by the presence of RAE 1a (Scheme 5B).23 Even though steady-state fluorescence quenching can only describe interactions between the singlet excited state of a photocatalyst, the observed affinity suggests a reductive-quenching photoredox manifold to be the most likely mechanistic scenario (Scheme 5A).24 This is consistent with the well-established energetically favourable SET from amine reductants (BnNAH)25 to the excited photocatalysts.24,26 Additional circumstantial evidence in favour of a reductive-quenching mechanism lies in the fact that control experiments with 15 mol% of ZnTPP (6) but without reductant (Scheme 2, entry 9) result in full recovery of the starting material (>95% of 1a), while an operative oxidative-quenching mechanism would lead to at least partial consumption of 1a. Subsequent SET from the reduced photocatalyst to 1 and irreversible fragmentation delivers radical 7. Then, 7 undergoes Giese-type addition to acceptor 2 followed by formal hydrogen-atom abstraction from BnNAH˙+ or BnNAH (4), giving reaction product 3.13,14,21 It must also be acknowledged that alternative pathways (Scheme 5, grey arrows), such as further activation of 1 by BnNAH˙+, or a free-radical propagation mechanism (hydrogen-atom abstraction by radical 8 from BnNAH, generating product 3 and nicotinyl radical BnNA˙, which would in turn activate another molecule of RAE 1 to give 7 and BnNA+) may also be effective, as previously identified for similar systems.17 However, further interrogation of these mechanistic intricacies falls beyond the intent of this work.
 | ||
| Scheme 5 (A) Mechanistic proposal for the low-energy-light photoredox activation of redox-active esters using BnNAH as reductive quencher and terminal H-donor. (B) Steady-state fluorescence quenching studies with Eosin Y (14.3 μM in DMSO). HAT = hydrogen-atom transfer; Phthal = Phthalimide. | ||
In summary, we have developed two photoredox systems for the potentially biocompatible generation and Giese-type intermolecular trapping of alkyl radicals. The systems are based on the reductive quenching of low energy light-harvesting photocatalysts (Eosin Y for green and ZnTPP for red) with BnNAH, subsequently resulting in the SET activation of a broad variety of redox-active alkyl phthalimide esters. The two systems were found to be complementary: while the green-light conditions are more robust and perform better across a wider range of Giese acceptors, the red-light system proved to be more suitable for challenging primary and secondary alkyl-radical couplings. Preliminary experiments showcase the possibility of controlling these relatively complex intermolecular radical processes under biologically relevant conditions (in aqueous buffers or cell-culture media, in the presence of cell lysates, under biocompatible low-energy light irradiation, etc.) or even using cell-endogenous NADH as the reductant. This paves the way for the further development of the nascent field of bioorthogonal radical photocatalysis.
Author contributions
M.M. conceived the project and performed conceptualization experiments. D.M. and U.D.L. designed, performed and analysed experiments. M.M., J.L.M. and M.T.G. directed the project. M.M. wrote the manuscript with contributions from all authors.Data availability
All data supporting this study, including detailed experimental procedures and characterization, are provided in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support for this work was provided by the Spanish Agencia Estatal de Investigación (AEI) (Ramón y Cajal RYC2023-043998-I and RYC2020-029150-I; Grants PID2022-137318OB-I00, IHRC22-00009 and ORFEO–CINQA network RED2022-134287-T), the Xunta de Galicia (Grant ED431C 2021/25 and Grant ED431G 2023/03: Centro de investigación do Sistema universitario de Galicia accreditation 2023–2027) and the European Union (European Regional Development Fund-ERDF 2014–2020). D.M. thanks CiQUS–USC and Xunta de Galicia for an initiation to research contract. The embedded images in Scheme 1A were created with BioRender.com. We thank Dr Arcadio Guerra for technical assistance and the Mass Spectrometry and Proteomics Unit from the RIADT–USC.References
- (a) C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 651–653 CrossRef CAS PubMed; (b) M. M. A. Mitry, F. Greco and H. M. I. Osborn, Chem. – Eur. J., 2023, 29, e202203942 CrossRef CAS PubMed.
- (a) W. Wang, X. Zhang, R. Huang, C.-M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed; (b) R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS PubMed; (c) Z. Liu, M. Sun, W. Zhang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2023, 62, e202308396 CrossRef CAS.
- (a) E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef CAS PubMed; (b) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; (b) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- (a) M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed; (b) N. K. Devaraj and R. Weissleder, Acc. Chem. Res., 2011, 44, 816–827 CrossRef CAS PubMed.
- (a) M. I. Sánchez, C. Penas, M. E. Vázquez and J. L. Mascareñas, Chem. Sci., 2014, 5, 1901–1907 RSC; (b) C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1903 CrossRef PubMed; (c) C. Vidal, M. Tomás-Gamasa, A. Gutiérrez-González and J. L. Mascareñas, J. Am. Chem. Soc., 2019, 141, 5125–5129 CrossRef CAS PubMed; (d) S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2021, 60, 22017–22025 CrossRef PubMed.
- (a) T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Ng, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M.-L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, aag1465–aag1461 CrossRef PubMed; (b) S. J. McCarver, J. X. Qiao, J. Carpenter, R. M. Borzilleri, M. A. Poss, M. D. Eastgate, M. M. Miller and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2016, 56, 728–732 CrossRef PubMed; (c) B. Josephson, C. Fehl, P. G. Isenegger, S. Nadal, T. H. Wright, A. W. J. Poh, B. J. Bower, A. M. Giltrap, L. Chen, C. Batchelor-McAuley, G. Roper, O. Arisa, J. B. I. Sap, A. Kawamura, A. J. Baldwin, S. Mohammed, R. G. Compton, V. Gouverneur and B. G. Davis, Nature, 2020, 585, 530–537 CrossRef CAS PubMed.
- X. Fernández-González, J. Miguel-Ávila, J. L. Mascareñas and M. Tomás-Gamasa, ChemCatChem, 2025, 17, e202401778 CrossRef.
- (a) A. Seoane and J. L. Mascareñas, Eur. J. Org. Chem., 2022, e202200118 CrossRef CAS PubMed; (b) S. Gutiérrez, M. Tomás-Gamasa and J. L. Mascareñas, Chem. Sci., 2022, 13, 6478–6495 RSC; (c) A. Gutiérrez-González, F. López and J. L. Mascareñas, Helv. Chim. Acta, 2023, 106, e202300001 CrossRef; (d) J. Miguel-Ávila, M. Tomás-Gamasa and J. L. Mascareñas, Trends Chem., 2023, 5, 474–485 CrossRef.
- (a) M. Mato, X. Fernández-González, C. D'Avino, M. Tomás-Gamasa and J. L. Mascareñas, Angew. Chem., Int. Ed., 2024, 63, e202413506 CrossRef CAS PubMed; (b) C. D'Avino, S. Gutiérrez, M. J. Feldhaus, M. Tomás-Gamasa and J. L. Mascareñas, J. Am. Chem. Soc., 2024, 146, 2895–2900 CrossRef PubMed.
- S. Jia and E. M. Sletten, ACS Chem. Biol., 2022, 17, 3255–3269 CrossRef CAS PubMed.
- (a) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563–2575 CrossRef CAS PubMed; (b) G. E. M. Crisenza and P. Melchiorre, Nat. Commun., 2020, 11, 803 CrossRef PubMed.
- (a) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef CAS PubMed; (b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed; (c) M. Mato, D. Spinnato, M. Leutzsch, H. W. Moon, E. J. Reijerse and J. Cornella, Nat. Chem., 2023, 15, 1138–1145 CrossRef CAS PubMed.
- (a) J. Schwarz and B. König, Green Chem., 2016, 18, 4743–4749 RSC; (b) K. N. Tripathi, M. Belal and R. P. Singh, J. Org. Chem., 2020, 85, 1193–1201 CrossRef CAS PubMed; (c) P. Niu, J. Li, Y. Zhang and C. Huo, Eur. J. Org. Chem., 2020, 5801–5814 CrossRef CAS; (d) Y. Zhang, D. Ma and Z. Zhang, Arabian J. Chem., 2022, 15, 103922 CrossRef CAS; (e) C. R. Azpilcueta-Nicolas and J.-P. Lumb, Beilstein J. Org. Chem., 2024, 20, 346–378 CrossRef CAS PubMed.
- (a) D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC; (b) M. F. Saraiva, M. R. C. Couri, M. L. Hyaric and M. V. de Almeida, Tetrahedron, 2009, 65, 3563–3572 CrossRef CAS.
- (a) K. Okada, K. Okamoto and M. Oda, J. Am. Chem. Soc., 1988, 110, 8736–8738 CrossRef CAS; (b) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS.
- R. Chowdhury, Z. Yu, M. L. Tong, S. V. Kohlhepp, X. Yin and A. Mendoza, J. Am. Chem. Soc., 2020, 142, 20143–20151 CrossRef CAS PubMed.
- For a review of the use of red-light photocatalysis in organic synthesis, see: (a) A. H. Schade and L. Mei, Org. Biomol. Chem., 2023, 21, 2472–2485 RSC; (b) For selected examples, see: B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343 CrossRef CAS PubMed; (c) B. D. Ravetz, N. E. S. Tay, C. L. Joe, M. Sezen-Edmonds, M. A. Schmidt, Y. Tan, J. M. Janey, M. D. Eastgate and T. Rovis, ACS Cent. Sci., 2020, 6, 2053–2059 CrossRef CAS PubMed; (d) F. Glaser and O. S. Wenger, JACS Au, 2022, 2, 1488–1503 CrossRef CAS PubMed; (e) M. M. Hossain, A. C. Shaikh, R. Kaur and T. L. Gianetti, J. Am. Chem. Soc., 2024, 146, 7922–7930 CrossRef CAS PubMed; (f) N. Lal, Deepshikha, P. Singh and A. C. Shaikh, Chem. Commun., 2025, 61, 3005–3008 RSC; (g) A. Stamoulis, M. Mato, P. C. Bruzzese, M. Leutzsch, A. Cadranel, M. Gil-Sepulcre, F. Neese and J. Cornella, J. Am. Chem. Soc., 2025, 147, 6037–6048 CrossRef CAS PubMed.
- (a) N. E. S. Tay, K. A. Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyi and T. Rovis, Nat. Chem., 2023, 15, 101–109 CrossRef CAS PubMed; (b) B. F. Buksh, S. D. Knutson, J. V. Oakley, N. B. Bissonnette, D. G. Oblinsky, M. P. Schowoerer, C. P. Seath, J. B. Geri, F. P. Rodriguez-Rivera, D. L. Parker, G. D. Scholes, A. Ploss and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6154–6162 CrossRef CAS PubMed.
- (a) K. Rybicka-Jasińska, T. Wdowik, K. Łuczak, A. J. Wierzba, O. Drapała and D. Gryko, ACS Org. Inorg. Au, 2022, 2, 422–426 CrossRef PubMed; (b) H. Yamamoto, K. Yamaoka, A. Shinohara, K. Shibata, K.-I. Takao and A. Ogura, Chem. Sci., 2023, 14, 11243–11250 RSC; (c) Y. Okanishi, O. Takemoto, S. Kawahara, S. Hayashi, T. Takanami and T. Yoshimitsu, Org. Lett., 2024, 26, 3929–3934 CrossRef CAS PubMed.
- (a) H. Li, C. P. Breen, H. Seo, T. F. Jamison, Y.-Q. Fang and M. M. Bio, Org. Lett., 2018, 20, 1338–1341 CrossRef CAS PubMed; (b) I. Bosque and T. Bach, ACS Catal., 2019, 9, 9103–9109 CrossRef CAS.
- For details, see ESI.†.
- Preliminary steady-state quenching experiments with ZnTPP show fluorescence to be unaffected by the presence of either 4 or 1a, suggesting the exclusive involvement of excited triplet states in the observed reactivity.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (c) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 58, 3730–3747 CrossRef PubMed.
- (a) A. Anne, J. Moiroux and J.-M. Savéant, J. Org. Chem., 2000, 65, 7213–7214 CrossRef CAS PubMed; (b) X.-Q. Zhu, H.-R. Li, A. Li, T. Ai, J.-Y. Lu, Y. Yang and J.-P. Cheng, Chem. – Eur. J., 2003, 9, 871–880 CrossRef CAS PubMed.
- (a) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC; (b) N. C. M. Magdaong, M. Taniguchi, J. R. Diers, D. M. Niedzwiedzki, C. Kirmaier, J. S. Lindsey, D. F. Bocian and D. Holten, J. Phys. Chem. A, 2020, 124, 7776–7794 CrossRef CAS PubMed; (c) Y. Kuramochi and A. Satake, Catalysis, 2023, 13, 282 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: For detailed experimental procedures, characterization data, and copies of NMR spectra of all new compounds. See DOI: https://doi.org/10.1039/d5ob00476d |
| This journal is © The Royal Society of Chemistry 2025 |