
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licencecis-Amide promotion in α-ABpeptoid foldamers via triazolium side chains†
Jungyeon
Kim‡
 a,
Ganesh A.
Sable‡
a,
Kang Ju
Lee
a,
Hyun-Suk
Lim
*a and
Min Hyeon
Shin
*b
a,
Ganesh A.
Sable‡
a,
Kang Ju
Lee
a,
Hyun-Suk
Lim
*a and
Min Hyeon
Shin
*b
aDepartment of Chemistry and Division of Advanced Materials Science, Pohang University of Science and Technology (POSTECH), Pohang 37673, South Korea. E-mail: hslim@postech.ac.kr
bDepartment of Science Education, Daegu National University of Education, Daegu 42411, South Korea. E-mail: mhshin@dnue.ac.kr
First published on 22nd April 2025
Abstract
Precise control of amide bond rotation is crucial for the construction of well-defined three-dimensional structures in peptidomimetic foldamers. We previously introduced α-ABpeptoids as a new class of peptoid foldamers incorporating backbone chirality and demonstrated their folding propensities. However, the rotational isomerism of their backbone amide bonds remains largely unregulated. Here, we report the development of α-ABpeptoids functionalized with triazolium side chains that promote cis-amide bond formation. A series of α-ABpeptoid oligomers bearing neutral triazole or cationic triazolium side chains were synthesized and analyzed by NMR and circular dichroism spectroscopy. The triazolium-functionalized α-ABpeptoids exhibited a strong preference for cis-amide geometry, resulting in enhanced conformational homogeneity. These findings establish triazolium substitution as an effective strategy for conformational control in α-ABpeptoid foldamers, expanding their utility in the design of structured, functional peptidomimetics.
Introduction
Proteins derive their functions from their three-dimensional structures. To mimic protein functionality, researchers have sought to develop synthetic oligomers with predictable, well-defined conformations.1–3 These molecules, termed foldamers by Gellman,4 have emerged as a diverse class of synthetic architectures, enabling applications in molecular recognition, organocatalysis, and materials science.3,5–10 Notably, foldamers offer significant advantages over native peptides, as they exhibit superior proteolytic stability and allow access to a vast chemical space by incorporating both proteinogenic and non-proteinogenic side chain building blocks. Over the years, a variety of synthetic foldamers have been developed, including β- and γ-peptides,11–14 oligoureas,15,16 oligotriazoles,17 γ-AApeptides,18,19 aromatic oligoamides,3,20 and peptoids.21–26Peptoids, oligomers of N-substituted glycine, are a class of peptidomimetic foldamers with advantages due to their synthetic accessibility, side-chain diversity,27 resistance to proteolytic degradation,28,29 and enhanced cell permeability30–32 compared to natural peptides (Fig. 1a). However, peptoids generally fail to adopt well-defined folding structures due to low rotational barrier of tertiary amide bonds and the absence of intramolecular hydrogen bonding, which is typically facilitated by amide protons in natural peptides. Therefore, developing strategies to control peptoid conformation, particularly amide bond geometry, has become a central focus in peptoid research.
 | ||
| Fig. 1 (a) General structures of α- and β-peptoids, peptoids with chiral side chains, α-ABpeptoids, and β-ABpeptoids. (b and c) Peptoids with triazolium-type side chain (b) and α-ABpeptoid with triazolium-type side chain (c). | ||
A well-known strategy for controlling amide bond geometry in peptoids is the incorporation of α-chiral side chains (Fig. 1a).33–35 These α-chiral side chains impart a handedness to the peptoid backbone, leading to the formation of polyproline type-I (PPI) helical structure in peptoid oligomers. Beyond chirality, steric36–39 and stereoelectronic40–43 effects also play crucial roles in determining peptoid folding propensity. Steric hindrance from bulky side chains destabilizes the trans amide geometry due to steric repulsion between peptoid backbone and the bulky substituents in the trans configuration. Conversely, side chains with electron-deficient moieties such as pyridinium and triazolium (Fig. 1b) could promote cis-amide geometry via a stereoelectronic n → π* interaction.44 Some side chains leverage both steric and stereoelectronic effects to precisely regulate peptoid amide bond geometry.45–47 These side chains strongly bias peptoids toward cis-amide geometry at the monomer level, thereby enhancing their conformational homogeneity and structural predictability.
Inspired by the strategy of incorporating α-chiral side chains into peptoids, we developed α- and β-ABpeptoids as a novel class of peptidomimetic foldamers (Fig. 1a).48–52 These structures feature a β-peptoid backbone with a chiral methyl group positioned at either α- or β-carbon, which we hypothesized would enhance the folding propensity of the oligomers, similar to the effect of α-chiral side chains in α-peptoids. Consistent with this expectation, circular dichroism (CD) studies confirmed that α- and β-ABpeptoids adopt folded structures in solution. However, NMR studies revealed that α- and β-ABpeptoids exist as mixtures of cis and trans amide isomers, indicating that amide bond geometry remains uncontrolled in these systems.
As observed in the case of peptoids, we anticipate that precise control of amide bond rotation would be crucial for achieving stable and well-defined three-dimensional structures in α-ABpeptoids. Recently, Taillefumier and colleagues demonstrated that triazolium-containing side chains exhibit a remarkable cis-directing effect in both monomer model studies and peptoid oligomers (Fig. 1b).42,43 The triazolium side chain stabilizes cis-amide geometry through a strong n → π*Ar interaction, where the amide carbonyl of the neighboring residue donates electron density to the electron-deficient triazolium group. Inspired by Taillefumier's findings, we hypothesized that triazolium-functionalized side chains would effectively restrict amide bond rotation in α-ABpeptoids, thereby promoting the formation of a stable, homogeneous three-dimensional structure without compromising chemical diversity (Fig. 1c). Here, we report the design, solid-phase synthesis of a series of α-ABpeptoid oligomers containing triazole and triazolium side chains, along with their structural characterization using NMR and CD spectroscopy.
Results and discussion
To investigate whether the introduction of triazolium side chains promotes the folding of α-ABpeptoids by stabilizing the cis-amide conformation, we synthesized a series of α-ABpeptoid oligomers with triazolium side chains using solid phase synthesis. For comparison, α-ABpeptoids with triazole side chains were prepared. Initially, we synthesized a nosyl-protected α-ABpeptoid monomer 5 which carries a benzyl-substituted triazole, following Scheme 1. First, compound 2 was synthesized from the commercially available compound 1 as according to our previous report.48 Then, N-propargylation of compound 2 was carried out via Fukuyama–Mitsunobu reaction, yielding compound 3 in 91% yield. Next, compound 3 underwent copper-catalyzed azide–alkyne cycloaddition (CuAAC) with benzyl azide, affording compound 4 in 96% yield. Importantly, α-ABpeptoid monomers with various functional groups can be prepared by replacing benzyl azide with other azides in this step, enabling further chemical diversity. Finally, the ethyl ester of compound 4 was hydrolyzed using lithium hydroxide (LiOH) to give the α-ABpeptoid monomer 5 in 89% yield. This synthetic route is straightforward, high-yielding, and cost-effective, utilizing readily available and inexpensive starting materials. | ||
| Scheme 1 Synthesis of a α-ABpeptoid monomer with N-triazole side chain (5). | ||
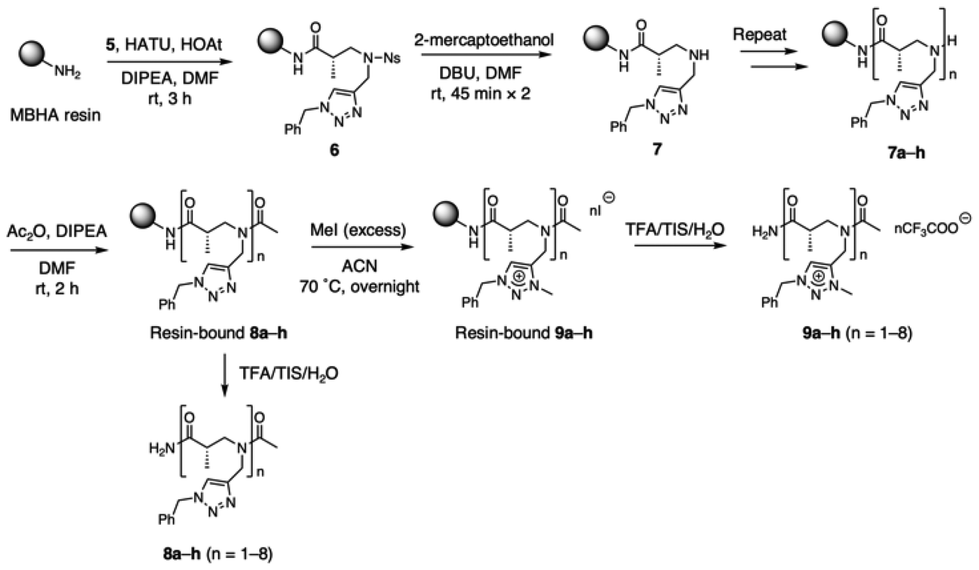
For the solid-phase synthesis of α-ABpeptoids oligomers containing triazole and triazolium side chains, monomer 5 was loaded on Rink amide MBHA resin via coupling reaction using hexafluorophosphate azabenzotriazole tetramethyl uranium (HATU)/1-hydroxy-7-azabenzotriazole (HOAt)/N,N-diisopropylethylamine (DIPEA) in DMF (Scheme 2). Next, the nosyl protecting group of resin-bound 6 was removed using 2-mercaptoethanol and 1,8-diazabicycloundec-7-ene (DBU) in DMF, yielding the resin-bound secondary amine 7. This coupling reaction-and-deprotection sequence was then repeated to obtain oligomers ranging from monomer to octamer (7a–h). The N-termini of the oligomers were acetylated using acetic anhydride (Ac2O) and DIPEA in DMF, resulting in acetylated α-ABpeptoids (8a–h). Finally, the triazole groups of 8a–h were quaternized by treatment of excess methyl iodide (MeI) at 70 °C overnight, affording α-ABpeptoid oligomers with triazolium side chains (9a–h). The α-ABpeptoid oligomers 8a–h and 9a–h were then cleaved from the resin by treating with the cleavage cocktail trifluoroacetic acid (TFA)/triisopropylsilane (TIS)/H2O (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5). The resulting α-ABpeptoid oligomers, functionalized with triazole and triazolium side chains, were then purified by reverse-phase HPLC and characterized by mass spectrometry (Table 1 and Fig. S1†). As the oligomers were treated with a TFA solution during cleavage from resin, all iodide counterions (I−) of the quaternary salts were exchanged for trifluoroacetate ions (CF3COO−). Notably, the α-ABpeptoids displayed excellent crude purity, as exemplified by the longest oligomers 8h and 9h, which showed crude purities of 86% and 93%, respectively (Fig. S2 and Table S1†). This result underscores the robustness of the synthetic method.
:2.5:2.5). The resulting α-ABpeptoid oligomers, functionalized with triazole and triazolium side chains, were then purified by reverse-phase HPLC and characterized by mass spectrometry (Table 1 and Fig. S1†). As the oligomers were treated with a TFA solution during cleavage from resin, all iodide counterions (I−) of the quaternary salts were exchanged for trifluoroacetate ions (CF3COO−). Notably, the α-ABpeptoids displayed excellent crude purity, as exemplified by the longest oligomers 8h and 9h, which showed crude purities of 86% and 93%, respectively (Fig. S2 and Table S1†). This result underscores the robustness of the synthetic method.
 | ||
| Scheme 2 Solid-phase synthesis of α-ABpeptoid oligomers containing triazole or triazolium side chains. | ||
|
|
||||
|---|---|---|---|---|
| Compound | Chain length | % puritya | Calcd massb | Obsd massc |
| a Purity was determined by analytical HPLC chromatogram of purified products. b Calculated mass for [M + H]+ (8a–h) or [M − CF3COO−]+ (9a–h) are shown. c High resolution mass spectra (HRMS) were acquired using Electrospray ionization (ESI) techniques. | ||||
| 8a | 1 | 96 | 316.1773 | 316.1773 |
| 8b | 2 | 96 | 572.3098 | 572.3096 |
| 8c | 3 | 98 | 828.4422 | 828.4419 |
| 8d | 4 | 98 | 1084.5746 | 1084.5747 |
| 8e | 5 | 99 | 1340.7070 | 1340.7067 |
| 8f | 6 | 97 | 1596.8394 | 1596.8394 |
| 8g | 7 | 96 | 1852.9718 | 1852.9706 |
| 8h | 8 | 98 | 2109.1042 | 2109.1052 |
| 9a | 1 | 96 | 330.1930 | 330.1932 |
| 9b | 2 | 99 | 714.3339 | 714.3337 |
| 9c | 3 | 97 | 1098.4749 | 1098.4745 |
| 9d | 4 | 99 | 1482.6152 | 1482.6160 |
| 9e | 5 | 99 | 1866.7567 | 1866.7566 |
| 9f | 6 | 99 | 2250.8976 | 2250.8965 |
| 9g | 7 | 97 | 2635.0386 | 2635.0339 |
| 9h | 8 | 97 | 3019.1795 | 3019.1868 |

To investigate the impact of the triazolium side chain on the amide bond geometry of α-ABpeptoids, we performed conformational analysis of α-ABpeptoid monomer models using NMR spectroscopy. 1H NMR spectra of α-ABpeptoid monomers carrying a triazole side chain (8a) and a triazolium side chain (9a) were recorded in CD3CN, CDCl3, and CD3OD (Fig. S3 and S5†). The 1H NMR spectra of both 8a and 9a displayed two sets of peaks, corresponding to cis and trans amide rotamers. The assignment of cis and trans isomers was performed using homonuclear correlation spectroscopy (COSY) and nuclear Overhauser effect spectroscopy (NOESY) (Fig. S4 and S6†). The cis/trans equilibrium constants (Kcis/trans) were determined from the integration of acetyl protons of cis and trans isomers on 1H NMR spectra (Table 2). For 8a bearing a triazole side chain, cis and trans isomers were present in nearly equal proportions in CD3CN and CD3OD with Kcis/trans values of 1.40 and 1.65, respectively. In contrast, 9a, which carries a triazolium side chain, exhibited a strong preference for the cis amide isomer with the cis-amide population exceeding 90% in all tested solvents. This demonstrates that the triazolium side chain strongly directs the amide bond of α-ABpeptoids toward cis geometry at the monomer level. Additionally, 9a showed a higher Kcis/trans value in CDCl3 compared to CD3CN and CD3OD, indicating an enhanced cis-amide preference in non-polar solvents. The significant difference in Kcis/trans values between 8a and 9a, along with the increased cis-inducing effect of the triazolium side chain in the non-polar solvent, suggests the presence of a stereoelectronic effect, such as n → π*Ar interaction between the carbonyl group of the acetamide and the electron-deficient triazolium ring.
|
|
|||
|---|---|---|---|
| Compound | CD3CN (% cis) | CDCl3 (% cis) | CD3OD (% cis) |
| a K cis/trans values were determined based on the integration of 1H NMR signals of acetyl protons. | |||
| 8a | 1.40 (58) | 3.54 (78) | 1.65 (62) |
| 9a | 10.14 (91) | 18.43 (95) | 10.07 (91) |

The C–H group of the triazolium group is capable of interacting with electronegative groups. For example, triazolium can recognize anions by acting as a hydrogen bond donor.53 Moreover, triazolium C–H in α-peptoid forms an intra-residue hydrogen bond with carbonyl group.42 To determine whether a similar triazolium C–H interaction is present in α-ABpeptoids, we compared the chemical shift of triazolium C–H in 9a in CDCl3 (a non-polar solvent) with those in CD3CN and CD3OD (polar solvents), as intramolecular hydrogen bonds are often disrupted in polar environments. The chemical shifts of triazolium C–H in 9a were 8.97 ppm in CDCl3, 8.35 ppm in CD3CN, and 8.65 ppm in CD3OD (Fig. S5†). The small difference in chemical shift between polar and non-polar solvents implies that no significant intramolecular hydrogen bond involving triazolium C–H is present in α-ABpeptoids. This observation contrasts with the substantial downfield shift of the triazolium C–H in α-peptoids in CDCl3, compared to that in CD3CN (Δδ = 1.61 ppm). We speculate that, unlike α-peptoid backbone, which adopts a conformation favorable for intra-residue hydrogen bonding, the elongated α-ABpeptoid backbone may prevent the triazolium and the carbonyl group from coming into close proximity, thereby disrupting the potential hydrogen bonding interaction.
Next, we investigated the amide rotameric preference of dimers 8b and 9b. The 1H NMR spectra of both compounds in CD3CN, CDCl3, and CD3OD exhibited four distinct isomers, corresponding to the cis–cis, cis–trans, trans–cis, and trans–trans amide geometries. For 8b, the 1H NMR spectra in CD3CN and CD3OD showed all four isomers present in nearly equal proportions, while in CDCl3, two major isomers accounted for more than 70% of the total population (Fig. 2a and Fig. S7†). In contrast, the 1H NMR spectra of 9b revealed a single predominant amide bond rotamer in all tested solvents (Fig. 2b and Fig. S8†), indicating a strong cis-directing effect of the triazolium side chain. To determine the conformation of the major rotamer of 9b, we carefully assigned peaks in its 1H NMR spectrum in CD3CN (Fig. 3). Detailed analysis of the COSY, 1H–13C heteronuclear multiple bond correlation (HMBC), and NOESY spectra enabled us to successfully distinguish the backbone protons as well as the triazolium C–H protons of the two residues (Fig. S9†). Further analysis of the NOESY spectrum allowed us to determine the amide bond geometry of 9b (Fig. 3 and Fig. S9†). Specifically, NOE correlations between the backbone α- and β-protons of the C-terminal residue (residue 2) and the backbone α-proton of the N-terminal residue (residue 1) support a cis geometry for the amide bond connecting these two residues. Additionally, NOEs involving the acetyl protons indicate that the acetamide adopts a cis conformation. Collectively, these observations demonstrate that the major rotamer of 9b adopts a cis–cis amide geometry, suggesting that the triazolium side chain strongly biases the α-ABpeptoid dimer 9b toward a locally defined conformation in solution through its cis-stabilizing effect.
 | ||
| Fig. 2 1H NMR spectra of α-ABpeptoid dimers with (a) triazole side chains (8b) and (b) triazolium side chains (9b) in CD3CN. | ||
 | ||
| Fig. 3 Structural analysis of α-ABpeptoid dimer carrying triazolium side chains (9b). (a) NOEs of 9b observed in CD3CN. NOEs indicating the cis amide configuration in the major isomer are shown in blue arrows. (b) Close-up view of the 2D NOESY spectrum of 9b in CD3CN, where cross-peaks indicating cis amide geometry are marked with blue circles. | ||
To assess whether the cis-directing effect of the triazolium side chain enhances the conformational homogeneity of α-ABpeptoid oligomers, we recorded 1H NMR spectra of 8c–h and 9c–h (Fig. S10 and S11†). Due to severe signal overlap in 1H NMR spectra, assignment of the NMR spectra was challenging. However, triazole/triazolium C–H peaks allowed us to evaluate conformational homogeneity of the α-ABpeptoids. The 1H NMR spectra of α-ABpeptoids with triazole side chains (8c–h) displayed a greater number of triazole C–H peaks than expected based on the numbers of triazole groups, indicating the presence of multiple amide bond rotamers. In contrast, for α-ABpeptoids with triazolium side chains (9c–h), a single major rotamer was observed in the 1H NMR spectra up to 5mer (9e). This result suggests that the triazolium side chains significantly enhance the amide rotameric preference of α-ABpeptoids, promoting a well-defined conformation in solution.
To evaluate the folding propensity and gain structural insights into α-ABpeptoids carrying triazole (8b–h) and triazolium side chains (9b–h), we obtained their CD spectra in trifluoroethanol (TFE). The CD spectral profiles of α-ABpeptoids varied depending on the chain length (Fig. 4a and b). The CD spectra of 8b–h exhibited a maximum at 207 nm and a minimum at 228 nm, with increasing signal intensity as the chain length increased. Similarly, α-ABpeptoids with triazolium side chains (9b–h) displayed CD spectral resembling those of 8b–h, but with an overall blue-shift. For example, the CD spectrum of 8-mer α-ABpeptoid 9h exhibited a maximum at 204 nm and a minimum at 224 nm. A notable difference between the CD spectra of 8b–h and 9b–h was that the CD signals of 9b–h reached saturation at shorter oligomer lengths. Specifically, the maximum around 204 nm and the minimum around 224 nm were saturated at 4-mer (9d) and 6-mer (9f), respectively. This finding suggests that α-ABpeptoids with triazolium side chains (9b–h) may exhibit stronger folding propensities than their triazole-containing counterparts (8b–h), likely due to the cis-directing effect of the triazolium moiety.
 | ||
| Fig. 4 (a and b) CD spectra of 8b, 8d, 8f, and 8h (a) and 9b, 9d, 9f, and 9h (b) in TFE at 20 °C. (c) Temperature dependence of the CD spectrum of 9h in TFE. (d) CD spectra of 9h after recooling. All spectra were recorded at a concentration of 60 μM. | ||
Next, we sought to evaluate the thermal stability of the conformation adopted by 9h. To this end, we recorded CD spectra of 9h in TFE at temperatures ranging from 5 to 65 °C. While the overall spectral shape remained unchanged, the intensity of the minimum at 224 nm gradually decreased in a temperature-dependent manner (Fig. 4c). This change was reversible, as the spectrum recovered both its shape and intensity upon recooling (Fig. 4d), indicating that 9h undergoes a temperature-dependent reversible folding process. Based on the CD intensity change at 224 nm, the melting temperature (Tm) of 9h was estimated to be 30 °C (Fig. S12†). We also investigated the temperature dependence of the CD spectrum of 8h (Fig. S13†). Although 8h exhibited temperature-dependent spectral changes, its transition followed a primarily linear trend rather than a sigmoidal transition, making it difficult to accurately determine its melting temperature. This difference suggests that 9h adopts a thermodynamically more stable folded structure compared to 8h, presumably due to the cis-stabilizing effect of the triazolium side chains.
Conclusions
In summary, we have described the synthesis and conformational evaluation of α-ABpeptoids functionalized with triazole and triazolium side chains. We established robust synthetic methods, including the efficient synthesis of the α-ABpeptoid monomers carrying triazole side chains, solid-phase synthesis of α-ABpeptoid oligomers, and on-resin triazole quaternization for triazolium formation. Using these methods, we successfully prepared α-ABpeptoids with triazole/triazolium side chains ranging from monomers to octamers in high yields. NMR studies demonstrated that the triazolium side chain exert a strong cis-directing effect on the amide geometry in α-ABpeptoid monomers and dimers. Additionally, NMR analysis of longer α-ABpeptoids revealed that triazolium substitution enhances rotameric homogeneity at amide bonds, promoting a more defined structural arrangement. Further insights were obtained from CD spectroscopy, which indicated chain length- and temperature-dependent spectral changes, suggesting that the formation of an ordered structure in triazolium-functionalized α-ABpeptoids. Collectively, our findings suggest that fixing the amide geometry on the cis conformation via triazolium side chain significantly improves the ability of α-ABpeptoids to adopt a well-defined conformation. Beyond its role in cis-amide stabilization, the triazolium side chain provides synthetic versatility, as the azide building blocks used in the click chemistry can be readily modified, enabling access to diverse chemical structures. Given their strong folding propensity and synthetic versatility, and the inherent advantages of peptoid-like structures, including enhanced cell permeability and proteolytic stability, α-ABpeptoids with cis-directing triazolium side chains represent a novel class of peptoid foldamers with promising applications in biomedical research and materials science. Although the precise conformation of the α-ABpeptoids studied here remains unresolved, future structural investigations, such as advanced NMR spectroscopy and X-ray crystallographic analysis, are expected to offer deeper insights into their overall architectures, particularly with respect to their secondary structures and handedness.Author contributions
J. K.: formal analysis, investigation, validation, visualization, and writing – original draft; G. A. S.: conceptualization, formal analysis, investigation, visualization, and writing – original draft; K. J. L.: conceptualization; H.-S. L. and M. H. S.: project administration, supervision, writing – review & editing, and funding acquisition. J. K. and G. A. S. contributed equally.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (Grants NRF-2022R1A2C2008460, NRF-2022M3A9G8084563, NRF-2022M3A9G8017710, NRF-RS-2023-00260005, NRF-RS-2024-00411069, RS-2023-00249191, and RS-2023-00274113).References
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef CAS PubMed.
- T. A. Martinek and F. Fulop, Chem. Soc. Rev., 2012, 41, 687–702 RSC.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252–262 CrossRef CAS PubMed.
- P. Sang and J. Cai, Chem. Soc. Rev., 2023, 52, 4843–4877 RSC.
- W. S. Horne, Expert Opin. Drug Discovery, 2011, 6, 1247–1262 CrossRef CAS PubMed.
- Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2020, 142, 17211–17223 CrossRef CAS PubMed.
- E. J. Robertson, A. Battigelli, C. Proulx, R. V. Mannige, T. K. Haxton, L. Yun, S. Whitelam and R. N. Zuckermann, Acc. Chem. Res., 2016, 49, 379–389 CrossRef CAS PubMed.
- Z. Li, B. Cai, W. Yang and C.-L. Chen, Chem. Rev., 2021, 121, 14031–14087 CrossRef CAS PubMed.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- S. Kwon, B. J. Kim, H.-K. Lim, K. Kang, S. H. Yoo, J. Gong, E. Yoon, J. Lee, I. S. Choi, H. Kim and H.-S. Lee, Nat. Commun., 2015, 6, 8747 CrossRef CAS PubMed.
- S. Shin, M. Lee, I. A. Guzei, Y. K. Kang and S. H. Choi, J. Am. Chem. Soc., 2016, 138, 13390–13395 CrossRef CAS PubMed.
- B. Legrand and L. T. Maillard, ChemPlusChem, 2021, 86, 629–645 CrossRef CAS PubMed.
- A. Violette, M. C. Averlant-Petit, V. Semetey, C. Hemmerlin, R. Casimir, R. Graff, M. Marraud, J.-P. Briand, D. Rognan and G. Guichard, J. Am. Chem. Soc., 2005, 127, 2156–2164 CrossRef CAS PubMed.
- G. W. Collie, K. Pulka-Ziach, C. M. Lombardo, J. Fremaux, F. Rosu, M. Decossas, L. Mauran, O. Lambert, V. Gabelica, C. D. Mackereth and G. Guichard, Nat. Chem., 2015, 7, 871–878 CrossRef CAS PubMed.
- N. G. Angelo and P. S. Arora, J. Am. Chem. Soc., 2005, 127, 17134–17135 CrossRef CAS PubMed.
- Y. Shi, P. Teng, P. Sang, F. She, L. Wei and J. Cai, Acc. Chem. Res., 2016, 49, 428–441 CrossRef CAS PubMed.
- P. Sang, Y. Shi, L. Wei and J. Cai, RSC Chem. Biol., 2022, 3, 805–814 RSC.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS PubMed.
- R. J. Simon, R. S. Kania, R. N. Zuckermann, V. D. Huebner, D. A. Jewell, S. Banville, S. Ng, L. Wang, S. Rosenberg and C. K. Marlowe, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9367–9371 CrossRef CAS PubMed.
- B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714–721 CrossRef CAS PubMed.
- R. Zuckermann and T. Kodadek, Curr. Opin. Mol. Ther., 2009, 11, 299–307 CAS.
- M. Oh, J. H. Lee, H. Moon, Y.-J. Hyun and H.-S. Lim, Angew. Chem., Int. Ed., 2016, 55, 602–606 CrossRef CAS PubMed.
- M. H. Shin, K. J. Lee and H.-S. Lim, Bioconjugate Chem., 2019, 30, 2931–2938 CrossRef CAS PubMed.
- K. J. Lee, G. Bang, Y. W. Kim, M. H. Shin and H.-S. Lim, Bioorg. Med. Chem., 2021, 48, 116423 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS.
- Y. Wang, H. Lin, R. Tullman, C. F. Jewell Jr., M. L. Weetall and F. L. Tse, Biopharm. Drug Dispos., 1999, 20, 69–75 CrossRef CAS PubMed.
- P. Yu, B. Liu and T. Kodadek, Nat. Biotechnol., 2005, 23, 746–751 CrossRef CAS PubMed.
- Y.-U. Kwon and T. Kodadek, J. Am. Chem. Soc., 2007, 129, 1508–1509 CrossRef CAS PubMed.
- M.-K. Shin, Y.-J. Hyun, J. H. Lee and H.-S. Lim, ACS Comb. Sci., 2018, 20, 237–242 CrossRef CAS PubMed.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS PubMed.
- P. Armand, K. Kirshenbaum, R. A. Goldsmith, S. Farr-Jones, A. E. Barron, K. T. V. Truong, K. A. Dill, D. F. Mierke, F. E. Cohen, R. N. Zuckermann and E. K. Bradley, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4309–4314 CrossRef CAS PubMed.
- C. W. Wu, T. J. Sanborn, K. Huang, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 6778–6784 CrossRef CAS PubMed.
- O. Roy, C. Caumes, Y. Esvan, C. Didierjean, S. Faure and C. Taillefumier, Org. Lett., 2013, 15, 2246–2249 CrossRef CAS PubMed.
- O. Roy, G. Dumonteil, S. Faure, L. Jouffret, A. Kriznik and C. Taillefumier, J. Am. Chem. Soc., 2017, 139, 13533–13540 CrossRef CAS PubMed.
- J. Morimoto, Y. Fukuda, D. Kuroda, T. Watanabe, F. Yoshida, M. Asada, T. Nakamura, A. Senoo, S. Nagatoishi, K. Tsumoto and S. Sando, J. Am. Chem. Soc., 2019, 141, 14612–14623 CrossRef CAS PubMed.
- J. Morimoto, J. Kim, D. Kuroda, S. Nagatoishi, K. Tsumoto and S. Sando, J. Am. Chem. Soc., 2020, 142, 2277–2284 CrossRef CAS PubMed.
- N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, J. Am. Chem. Soc., 2008, 130, 16622–16632 CrossRef CAS PubMed.
- B. C. Gorske, B. L. Bastian, G. D. Geske and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129, 8928–8929 CrossRef CAS PubMed.
- C. Caumes, O. Roy, S. Faure and C. Taillefumier, J. Am. Chem. Soc., 2012, 134, 9553–9556 CrossRef CAS PubMed.
- H. Aliouat, C. Caumes, O. Roy, M. Zouikri, C. Taillefumier and S. Faure, J. Org. Chem., 2017, 82, 2386–2398 CrossRef CAS PubMed.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed.
- J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef CAS PubMed.
- J. S. Laursen, P. Harris, P. Fristrup and C. A. Olsen, Nat. Commun., 2015, 6, 7013 CrossRef CAS PubMed.
- J. Oh, Y. Lee, G. Noh, D. Park, H. Lee, J. Lee, C. Kim and J. Seo, Small Struct., 2024, 5, 2300396 CrossRef CAS.
- K. J. Lee, W. S. Lee, H. Yun, Y.-J. Hyun, C. D. Seo, C. W. Lee and H.-S. Lim, Org. Lett., 2016, 18, 3678–3681 CrossRef CAS PubMed.
- G. A. Sable, K. J. Lee, M.-K. Shin and H.-S. Lim, Org. Lett., 2018, 20, 2526–2529 CrossRef CAS PubMed.
- G. A. Sable, K. J. Lee and H.-S. Lim, Molecules, 2019, 24, 178 CrossRef PubMed.
- K. J. Lee, G. A. Sable, M.-K. Shin and H.-S. Lim, Biopolymers, 2019, 110, e23289 CrossRef PubMed.
- S. Lee, H. Kwon, E.-K. Jee, J. Kim, K. J. Lee, J. Kim, N. Ko, E. Lee and H.-S. Lim, Org. Lett., 2024, 26, 1100–1104 CrossRef CAS PubMed.
- J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization of organic compounds, analytical data, and NMR spectra. See DOI: https://doi.org/10.1039/d5ob00355e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |