
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron-deficient hemicryptophanes for the recognition of anions through anion–π interactions†
Louise
Miton
 a,
Pierre-Antoine
Dupin
a,
Elise
Antonetti
a,
Marion
Jean
b,
Paola
Nava
a,
Alexandre
Martinez
*a and
Yoann
Cotelle
*a
a,
Pierre-Antoine
Dupin
a,
Elise
Antonetti
a,
Marion
Jean
b,
Paola
Nava
a,
Alexandre
Martinez
*a and
Yoann
Cotelle
*a
aAix Marseille Univ, CNRS, Centrale Med, ISM2, Marseille, France. E-mail: alexandre.martinez@centrale-marseille.fr; yoann.cotelle@univ-amu.fr
bAix Marseille Université, CNRS, FSCM, Chiropole, F-13397 Marseille, France
First published on 14th April 2025
Abstract
The selective recognition and encapsulation of anions are crucial in many biological and environmental processes. Anion–π interactions, characterized by attractive non-covalent interactions between anions and electron-deficient aromatic surfaces, have gained significant attention for their potential in anion binding. However, due to the weak nature of these interactions, receptors that rely solely on anion–π interactions are scarce. To promote anion binding via electron-deficient surfaces, we constructed a cavity within the hemicryptophane framework using tetrafluoroxylylene arms and a benzene triimide unit, thereby displaying multiple electron-deficient surfaces. Binding studies revealed that the electron-deficient receptor exhibits significantly higher anion binding constants compared to a less electron-deficient hemicryptophane. For example, the affinity for chloride (Ka = 207) is ten times higher for the most electron-deficient hemicryptophane. More impressive is the unique affinity for triiodide (Ka = 416), representing the first example of triiodide recognition by a BTI-based receptor. The enhanced binding properties of this receptor result from the combination of different π-acidic surfaces in a single cage molecule.
Introduction
Anions are involved in many biological and environmental processes, making their selective recognition and encapsulation highly interesting.1–5 Among others, the anion–π interaction has recently emerged as a promising mode of interaction.6–10 Nonetheless, only a limited number of receptors able to bind anions solely through anion–π interactions have been reported.11–16 Anion–π interactions are defined as attractive non-covalent interactions between an electron-deficient aromatic surface with a positive quadrupolar moment (QZZ) and an anion.17 In order to increase the strength of this interaction, we aim to explore the synergy of several π-acidic aromatic surfaces within a hemicryptophane to create a well-defined electron-deficient confined space.18 Hemicryptophanes are composed of a preorganizing cyclotriveratrylene unit covalently linked to another unit having a C3 symmetry axis.19 To maximize interactions with anions, our attention turned towards the use of benzene triimides (BTIs),20–22 which possess a high quadrupolar moment (QZZ = 17.2 B, computed at the MP2/aug-cc-pVTZ level) and a suitable geometry. BTIs have recently been used in the design of molecular cages by bridging two BTI units with three meta-xylylene linkers, showing strong affinity and selectivity for azide.23 Moreover, increasing the length of the spacers using naphthalene units has an impact on the selectivity for anions with different geometries.24 We have recently described the synthesis of the BTI-based hemicryptophane 2 with xylylene linkers, showing promising lone pair–π interactions in the solid state.25 Consequently, we introduced additional electron-withdrawing groups on the linkers to increase the binding for anions. Herein, we report the synthesis and characterization of an electron-deficient hemicryptophane and its increased anion binding ability compared to the hemicryptophane lacking additional electron-deficient linkers (Fig. 1).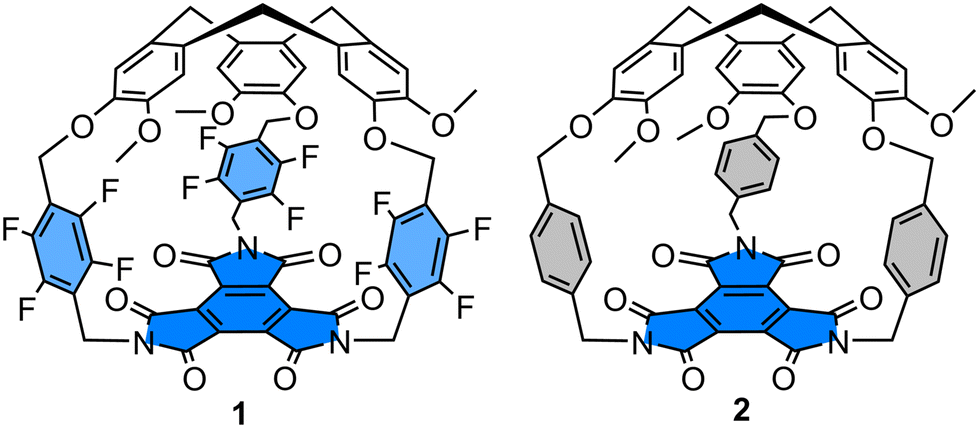 | ||
| Fig. 1 Structure of the electron-deficient hemicryptophane 1 and the parent hemicryptophane 2 lacking electron-deficient linkers. | ||
Results and discussion
Hemicryptophane 1 was synthesized in three steps from the vanillyl alcohol precursor 3. The first step involved the mono-alkylation of precursor 3 using an excess of bis(bromomethyl)-2,3,5,6-tetrafluorobenzene reagent and K2CO3 in acetone, resulting in compound 4 with a reasonable yield (49%). Then, the formation of the cyclotriveratrylene (CTV) unit was achieved through a triple Friedel–Crafts reaction, catalyzed by Sc(OTf)3 in acetonitrile, giving the desired product 5 with a moderate yield (36%). Finally, a nucleophilic substitution on precursor 5 using BTI and DIPEA in DMF gave hemicryptophane 1 (38% yield) (Scheme 1). | ||
| Scheme 1 Synthesis of hemicryptophane 1. Conditions: (a) 1,4-bis(bromomethyl)-2,3,5,6-tetrafluorobenzene, K2CO3, acetone, 70 °C, 18 h. (b) Sc(OTf)3, CH3CN, 65 °C, 48 h. (c) BTI, DIPEA, DMF, 60 °C, 72 h. | ||
The final compound was fully characterized by 1H NMR, 13C NMR and MS techniques (see ESI†). The 1H NMR spectrum of receptor 1 is consistent with C3 symmetry in solution (Fig. 2). Protons He and Ha are observed as doublets at 3.52 and 4.73 ppm, respectively. Methoxy protons appear as a singlet at 3.74 ppm, while the methylene bridges of the tetrafluoroparaxylylene spacers appear as doublets at 4.88 and 5.21 ppm for protons H3 and at 5.26 and 5.53 for protons H4. Finally, the aromatic protons of the CTV unit appear as singlets at 6.81 and 7.14 ppm for H1 and H2.
 | ||
| Fig. 2 1H NMR of hemicryptophane 1 (CDCl3, 400 MHz, 298 K). | ||
It was previously demonstrated that hemicryptophane 2 is a solvent-dependent chiral molecular switch, with an open CTV configuration in common organic solvents (including chloroform) and an inverted configuration in bulky polar aromatic solvents (such as 2-t-butylphenol).25 Thus, we evaluated the configurational stability of hemicryptophane 1. The racemic mixture of hemicryptophane 1 was resolved by chiral stationary phase HPLC on analytical and semi-preparative scales using a Chiralpak IA stationary phase and an ethanol/dichloromethane mixture (40/60) as the eluent (Fig. 3a). Contrary to the chromatogram of hemicryptophane 2, we could observe only two peaks in the chromatogram of hemicryptophane 1. Hence, no inverted configuration was observed in the chromatogram of cage 1. This behavior strongly differs from that of cage 2, which gives a mixture of in and out configurations under similar conditions, supporting the higher configurational stability of cage 1 compared to cage 2. Subsequently, ECD spectra were recorded (Fig. 3b). The ECD spectra show two mirror images typical of the open configuration, confirming their enantiomeric nature. The absolute configuration of each enantiomer can be assigned by referring to the work of André Collet and Giovanni Gottarelli published in 1985.26 Their study on cyclotriveratrylenes demonstrated that the absolute configuration of a CTV moiety could be determined by analyzing the sign changes of bands in the ECD spectrum, particularly at the 1La transition (around 240 nm), as this transition was less sensitive to the substituents on the CTV. Thus, based on the ECD spectrum of hemicryptophane 1, we propose that the first eluted enantiomer (green spectrum) is associated with the stereodescriptor M, while the second eluted enantiomer (red spectrum) is linked to the stereodescriptor P.
 | ||
| Fig. 3 (a) Analytical chiral HPLC chromatogram. (b) ECD spectra of the first eluted compound (M)-1 (in green, c = 0, 140 mmol L−1, CH3CN) and the second eluted (P)-1 (in red, c = 0.125 mmol L−1, CH3CN). | ||
Next, DFT calculations were performed to propose a structure as well as to determine the volume and ESP (electrostatic potential) maps of hemicryptophanes 1 and 2. Hemicryptophane 1 was optimized as a C3 symmetric object, in agreement with the 1H NMR spectrum (Fig. 2), and has a large cavity (V = 327 Å3), comparable to that of hemicryptophane 2 (V = 310 Å3). Thus, both cages have a very similar shape and inner volume, and the change in binding affinity should arise from their differences in electron distribution. The ESP maps depict a strong difference in electron distribution between the two receptors. Hemicryptophane 1 with tetrafluoroxylylene linkers has more electron-deficient aromatic spacers, which should increase the binding for anionic species through anion–π interactions (Fig. 4c).
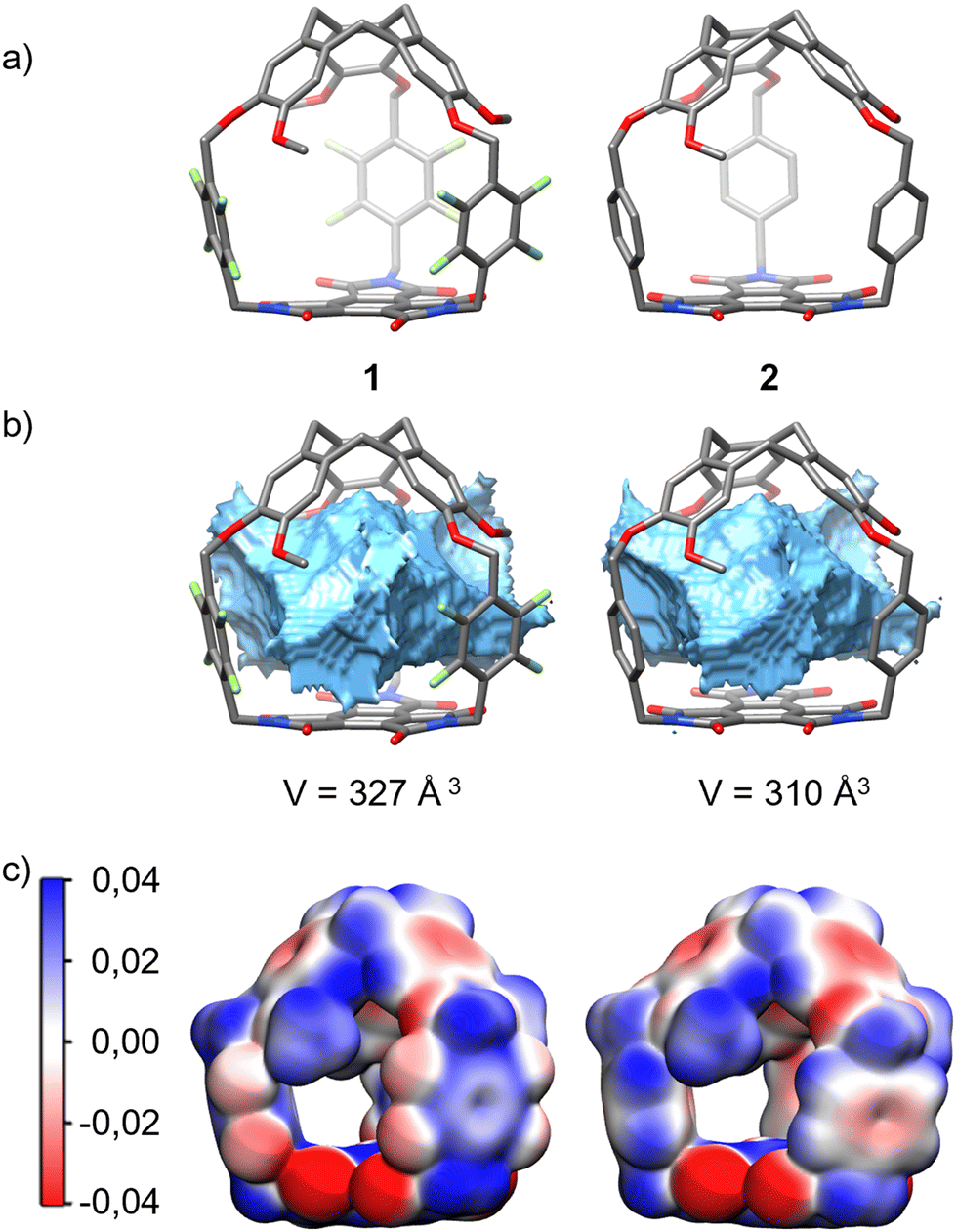 | ||
| Fig. 4 (a) DFT optimized structure of hemicryptophanes 1 and 2, together with their (b) calculated cavity volumes (MoloVol) and (c) electrostatic potential (ESP) maps. The ESP is mapped on the electron density iso-surface of 0.003 a.u. The red colour corresponds to a negative region of the electrostatic potential (−0.04 au), and the blue colour corresponds to the region where the potential is positive (+0.04 au). | ||
Finally, the recognition of anions by hemicryptophanes 1 and 2 was investigated. 1H NMR titrations were performed in CDCl3 by adding aliquots of tetrabutylammonium salts, to avoid inclusion of the counter ion, to a solution of the host.27 All the constants were obtained by plotting the chemical shifts of the methylenic protons (H4) of the linkers and fitting the data using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry with the BindFit program.28 Receptor 1 with tetrafluorolinkers has higher binding constants for anions than receptor 2 (except for nitrate), showing the efficiency of the π-acidic surfaces’ additivity within the confined space of our receptor. For halides, it can be observed that receptor 1 exhibits the strongest association constant with chloride (Ka = 207, Table 1, entry 1). The binding constant is better than those previously reported for BTI receptors and more than ten times higher compared to that for hemicryptophane 2, presenting less π-acidic surfaces.23,24 These observations are also supported by 19F NMR (Fig. S20†) as well as ESI-MS (Fig. S41†). Bromide and iodide affinities are weaker (Table 1, entries 2 and 3), which demonstrate slight selectivity. As postulated, hemicryptophane 2 shows much lower affinities for halides, 5 to 17 times lower for bromide and iodide, respectively. Next, we investigated the linear guest triiodide (Table 1, entry 4). Hemicryptophane 2 has no affinity for this guest, while hemicryptophane 1 shows the highest affinity (Ka = 416) among the tested anions. The highest polarizability of I3− could lead to stronger London and anion–π interactions, which might account for these experimental results. Moreover, this is the first example of the recognition of triiodide by a BTI-based receptor. Finally, we set out to investigate trigonal planar anions that might better fit the geometry of our molecular cages. Nitrate was evaluated since it is known to have a good affinity for electron-deficient receptors.29 However, the titrations show very low affinity for our hosts, with the best binding obtained for receptor 2 (Ka = 66, Table 1, entry 5). The complex between hemicryptophane 2 and NO3− was also observed by ESI-MS (Fig. S44†). It could be suggested that the lack of recognition ability of 1 toward the nitrate anion might be due to electronic repulsion between nitrate and the fluorine atoms on the xylylene linkers. Acetate was also tested, and again higher binding constants were observed for the most electron-deficient receptor 1 (Ka = 99) (Table 1, entry 6). Anion screening was continued with larger volume anions such as hexafluorophosphate, perrhenate, dihydrogenphosphate and picrate, but these showed low or no affinity for hemicryptophanes 1 and 2 (Ka = 22, 28, <1, <1 for 1, respectively, Table 1, entries 7–10).
:1 stoichiometry with the BindFit program.28 Receptor 1 with tetrafluorolinkers has higher binding constants for anions than receptor 2 (except for nitrate), showing the efficiency of the π-acidic surfaces’ additivity within the confined space of our receptor. For halides, it can be observed that receptor 1 exhibits the strongest association constant with chloride (Ka = 207, Table 1, entry 1). The binding constant is better than those previously reported for BTI receptors and more than ten times higher compared to that for hemicryptophane 2, presenting less π-acidic surfaces.23,24 These observations are also supported by 19F NMR (Fig. S20†) as well as ESI-MS (Fig. S41†). Bromide and iodide affinities are weaker (Table 1, entries 2 and 3), which demonstrate slight selectivity. As postulated, hemicryptophane 2 shows much lower affinities for halides, 5 to 17 times lower for bromide and iodide, respectively. Next, we investigated the linear guest triiodide (Table 1, entry 4). Hemicryptophane 2 has no affinity for this guest, while hemicryptophane 1 shows the highest affinity (Ka = 416) among the tested anions. The highest polarizability of I3− could lead to stronger London and anion–π interactions, which might account for these experimental results. Moreover, this is the first example of the recognition of triiodide by a BTI-based receptor. Finally, we set out to investigate trigonal planar anions that might better fit the geometry of our molecular cages. Nitrate was evaluated since it is known to have a good affinity for electron-deficient receptors.29 However, the titrations show very low affinity for our hosts, with the best binding obtained for receptor 2 (Ka = 66, Table 1, entry 5). The complex between hemicryptophane 2 and NO3− was also observed by ESI-MS (Fig. S44†). It could be suggested that the lack of recognition ability of 1 toward the nitrate anion might be due to electronic repulsion between nitrate and the fluorine atoms on the xylylene linkers. Acetate was also tested, and again higher binding constants were observed for the most electron-deficient receptor 1 (Ka = 99) (Table 1, entry 6). Anion screening was continued with larger volume anions such as hexafluorophosphate, perrhenate, dihydrogenphosphate and picrate, but these showed low or no affinity for hemicryptophanes 1 and 2 (Ka = 22, 28, <1, <1 for 1, respectively, Table 1, entries 7–10).
| Entry 1 | Guest | 1 | 2 |
|---|---|---|---|
| 1 | Cl− | 207 | 20 |
| 2 | Br− | 21 | 4 |
| 3 | I− | 84 | 5 |
| 4 | I3− | 416 | 2 |
| 5 | NO3− | <1 | 66 |
| 6 | AcO− | 99 | <1 |
| 7 | PF6− | 22 | 14 |
| 8 | ReO4− | 28 | 21 |
| 9 | H2PO4− | <1 | <1 |
| 10 | Picrate | <1 | <1 |
We then performed DFT geometry optimization for the complex between chloride and hemicryptophane 1. It was observed that the anion is positioned close to the BTI surface with a short distance to the carbon of the benzene moiety (d = 2.82 Å), supporting functional anion–π interactions (Fig. 5). Moreover, the electron-deficient perfluoroxylylene linker exposes its aromatic surface towards the chloride, which could increase anion recognition.
 | ||
| Fig. 5 Optimized structure of the complex Cl−@hemicryptophane 1. | ||
Conclusions
In conclusion, we have designed and synthesized an electron-deficient hemicryptophane receptor capable of effectively recognizing anions solely through enhanced anion–π interactions. By incorporating perfluoroxylylene arms and a benzene triimide (BTI) unit into the hemicryptophane structure, we created an electron-deficient confined environment that significantly improved anion affinity. Our binding studies revealed improved association constants for the hemicryptophane equipped with tetrafluoroxylylene linkers, particularly for chloride (Ka = 207), and a unique affinity for triiodide, representing the first instance of triiodide recognition by a BTI-based receptor. These results underscore the effectiveness of combining π-acidic surfaces within the receptor's cavity to strengthen binding affinities with anions. Moving forward, this strategy could be applied to develop more selective and efficient systems for complex biological and environmental applications where precise anion recognition is crucial.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. C. and A. M. thank the Agence Nationale de la Recherche for funding the APICOCAT project (grant ANR-21-CE07-0011) and the Co-LAB project (grant ANR-19-CE07-0024). We would like to thank Sabine Chevallier Michaud for low-resolution mass spectrometry and the Spectropole in Marseille for characterization. We also acknowledge the ‘Centre de Calcul Intensif d'Aix-Marseille’ for granting us access to its high-performance computing resources.References
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716 CrossRef CAS.
- H. Li, H. Valkenier, P. R. Brotherhood, S. Hussain, J. A. Cooper, O. Jurček, H. A. Sparkes, D. N. Sheppard and A. P. Davis, Nat. Chem., 2016, 8, 24 CrossRef CAS PubMed.
- P. Ballester, Chem. Soc. Rev., 2010, 39, 3810 RSC.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038 CrossRef CAS.
- K. A. Jolliffe and P. A. Gale, Org. Biomol. Chem., 2022, 20, 713 RSC.
- I. A. Rather, S. A. Wagay and R. Ali, Coord. Chem. Rev., 2020, 415, 213327 CrossRef CAS.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255 CrossRef CAS PubMed.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564 CrossRef CAS PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Rev., 2015, 115, 8867 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, Acc. Chem. Res., 2020, 53, 1364 CrossRef CAS.
- J. Luo, J. Zhu, D.-H. Tuo, Q. Yuan, L. Wang, X.-B. Wang, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, Chem. – Eur. J., 2019, 25, 13275 CrossRef CAS.
- S.-Y. Zhuang, Y. Cheng, Q. Zhang, S. Tong and M.-X. Wang, Angew. Chem., Int. Ed., 2020, 59, 23716 CrossRef.
- M.-D. Gu, Y. Lu and M.-X. Wang, J. Org. Chem., 2020, 85, 2312 CrossRef.
- H.-B. Liu, Q. Zhang and M.-X. Wang, Angew. Chem., Int. Ed., 2018, 57, 6536 CrossRef CAS PubMed.
- D.-X. Wang, Q.-Q. Wang, Y. Han, Y. Wang, Z.-T. Huang and M.-X. Wang, Chem. – Eur. J., 2010, 16, 13053 CrossRef CAS PubMed.
- S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100 CrossRef CAS PubMed.
- D. Quinonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Dey, Angew. Chem., Int. Ed., 2002, 41, 3389 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2005, 109, 9341 CrossRef CAS.
- D. Zhang, A. Martinez and J.-P. Dutasta, Chem. Rev., 2017, 117, 4900 CrossRef CAS PubMed.
- J. B. Carroll, M. Gray, K. A. McMenimen, D. G. Hamilton and V. M. Rotello, Org. Lett., 2003, 5, 3177 CrossRef CAS.
- K. A. McMenimen and D. G. Hamilton, J. Am. Chem. Soc., 2001, 123, 6453 CrossRef CAS PubMed.
- D.-H. Tuo, Q. He, Q.-Q. Wang, Y.-F. Ao and D.-X. Wang, Chin. J. Chem., 2019, 37, 684 CrossRef CAS.
- D.-H. Tuo, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, Org. Lett., 2019, 21, 7158 CrossRef CAS.
- D.-H. Tuo, Y.-F. Ao, Q.-Q. Wang and D.-X. Wang, CCS Chem., 2021, 3, 2876 Search PubMed.
- L. Miton, E. Antonetti, D. García-López, P. Nava, V. Robert, M. Albalat, N. Vanthuyne, A. Martinez and Y. Cotelle, Chem. – Eur. J., 2024, 30, e202303294 CrossRef CAS PubMed.
- J. Canceill, A. Collet, J. Gabard, G. Gottarelli and G. P. Spada, J. Am. Chem. Soc., 1985, 107, 1299 CrossRef CAS.
- M. Delecluse, C. Colomban, D. Moraleda, I. de Riggi, F. Duprat, S. Michaud-Chevallier, J.-P. Dutasta, V. Robert, B. Chatelet and A. Martinez, Chem. – Eur. J., 2019, 25, 3337 CrossRef CAS.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792 RSC.
- L. Adriaenssens, C. Estarellas, A. Vargas Jentzsch, M. Martinez Belmonte, S. Matile and P. Ballester, J. Am. Chem. Soc., 2013, 135, 8324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00245a |
| This journal is © The Royal Society of Chemistry 2025 |