DOI:
10.1039/D5OB00202H
(Review Article)
Org. Biomol. Chem., 2025,
23, 4808-4827
Recent advances in the synthesis of 3,4-fused tricyclic indoles
Received
4th February 2025
, Accepted 14th April 2025
First published on 16th April 2025
Abstract
The 3,4-fused tricyclic indole framework is a key structural motif in numerous bioactive natural products and pharmaceuticals, thus, it has drawn much attention in synthetic organic chemistry. Synthetic organic chemists have expended substantial effort in developing efficient methods for constructing this privileged molecular framework. In this review, we highlight the advances made in this area, particularly since 2018.
 Tetsuhiro Nemoto | Tetsuhiro Nemoto was born in Chiba, Japan in 1976. He received his B.S. (1999), M.S. (2001), and Ph.D. (2005) degrees from The University of Tokyo under the direction of Prof. Masakatsu Shibasaki. In 2003, he started his professional academic career as an assistant professor in Prof. Yasumasa Hamada's group at Chiba University, and promoted to lecturer in 2009, and associate professor in 2012. In this period, he worked as a visiting researcher in Prof. Darren J. Dixon's group at The University of Oxford in 2011. Since 2015, he has been a professor of pharmaceutical chemistry at Chiba University. His research interests are catalytic organic synthesis, asymmetric catalysis, and natural product synthesis. |
 Shingo Harada | Shingo Harada was born in Osaka, Japan, in 1985. He received his Ph.D. degree under the supervision of Prof. Kiyosei Takasu. In 2013, he started his professional academic career as an assistant professor at Chiba University. He worked as a visiting scientist in Prof. Harald Gröger's group at Bielefeld university in 2017 and in Prof. Timothy R. Newhouse's group at Yale university in 2019. Since 2024, he has been serving as an associate professor in Prof. Tetsuhiro Nemoto's group at Chiba university. He specializes in carbene chemistry and theoretical chemistry. |
 Takahito Kuribara | Takahito Kuribara was born in Saitama, Japan in 1995. He received his B.S. (2020) and Ph.D. (2023) degrees from Chiba University under the supervision of Prof. Tetsuhiro Nemoto. He got started his professional academic career as an assistant professor in Prof. Tetsuhiro Nemoto's group at Chiba University in 2023. His research interests are catalytic organic synthesis, redox chemistry, photochemistry, and computational chemistry. |
 Shinji Harada | Shinji Harada graduated from the University of Tokyo in 2002 and received his Ph.D. in Pharmaceutical Sciences at the University of Tokyo in 2007, under the supervision of Prof. Masakatsu Shibasaki. He then moved to Prof. Atsushi Nishida's group at Chiba University as an assistant professor. In 2010, he pursued postdoctoral studies in the laboratory of Prof. Tobias Ritter at Harvard University. Since 2023, he has been an associate professor in Institute for Advanced Academic Research, Chiba University. His research interests include lanthanide chemistry and the development of new synthetic methods. |
1. Introduction
Tryptophan-derived indole alkaloids contain numerous bioactive compounds that are valuable in medicinal chemistry research, rendering them an important class of compounds for drug discovery. Monoterpene indole alkaloids have diverse molecular structures. These alkaloids are biosynthesized from strictosidine, which bears a tetrahydro-β-carboline framework constructed via the Pictet–Spengler reaction between tryptamine and secologanin. Consequently, many indole alkaloids have fused molecular structures at the C2 and C3 positions of the indole.1 By contrast, ergot alkaloids, such as lysergic acid, feature a functionalized six-membered ring bridging the C3 and C4 positions of the indole (Fig. 1).2 Numerous natural products and derivatives containing this framework exhibit remarkable biological activities, including ergotamine, which is used to treat migraines by constricting blood vessels, and ergometrine, a uterine contraction agent. The structure of the fused ring moiety is modified via various molecular transformations during the biosynthetic process, leading to a diverse range of natural products such as chanoclavin and cycloclavine. The tetracyclic structure of lysergic acid is biosynthesized from 4-dimethylallyltryptophan, which also serves as an intermediate in the biosynthesis of clavicipitic acid and its derivatives such as griseofamine B, which are natural products featuring a fused seven-membered ring bridging the C3 and C4 positions of the indole.
 |
| | Fig. 1 Representative 3,4-fused indole alkaloids biosynthesized from 4-dimethylallyl tryptophan. | |
On the other hand, there are 3,4-fused indole natural products that contain an oxygen functional group at the C5 or C7 positions of the indole. Because of the electron density distribution of the indole ring, the C4 position is less reactive than the C2, C3, and C5 positions, making it challenging to introduce a substituent at the C4 position via Friedel–Crafts-type reactions. The oxygen functional group on the benzene ring is believed to facilitate substitution at the C4 position, leading to the biosynthesis of various natural products with 3,4-seven- or eight-membered ring-fused indole structures. Examples include hyrtimomines A and F, which exhibit antimicrobial activity; dragmacidin E, which acts as a serine-threonine protein phosphatase inhibitor; decursivine, which possesses antimalarial activity; and serotobenine, as shown in Fig. 2. Additionally, 3,4-fused indole natural products, such as ht-13-A and ht-13-B, which feature a seven-membered ring bridged via an ether oxygen, are known.
 |
| | Fig. 2 Representative 3,4-fused indole alkaloids bearing an oxygen substituent on the benzenoid ring of the indole. | |
Recently, new 3,4-fused indole natural products have been isolated and their structures elucidated (Fig. 3). In 2020, Klein-Júnior et al. isolated nemorosines A and B and nemorosinoside A and its derivatives, from the leaves of Psychotria nemorosa; the compounds comprise a hydroxyl group at the C6 position of the indole core.3 Nemorosinoside A and its derivatives have a distinctive structure with a secologanin unit connected to a seven-membered ring. Notably, nemorosine A reportedly inhibits MAO-A activity with an IC50 value of 0.9 μM. In 2021, Wu et al. reported the isolation and identification of clonorosin A, a 3,4-six-membered ring-fused indole with a 2,5-diketomorpholine motif, isolated from the soil-derived fungus Clonostachys rosea YRS-06.4 This alkaloid exhibited antimicrobial activity against Fusarium oxysporum with a minimum inhibitory concentration value of 50 μg mL−1. More recently, Arnold et al. isolated 7-hydroxytryptophan-derived azepinoindole alkaloids, purpurascenines A–C, from the fruiting bodies of Cortinarius purpurascens Fr. (Cortinariaceae).5 Purpurascenine A binds to the 5-HT2A serotonin receptor and potentially exerts antagonistic effects.
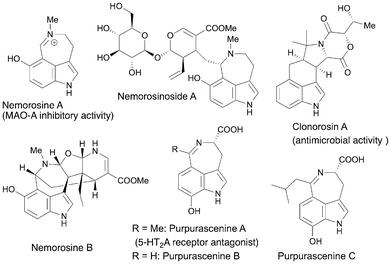 |
| | Fig. 3 Recently isolated and structurally elucidated 3,4-fused indole alkaloids. | |
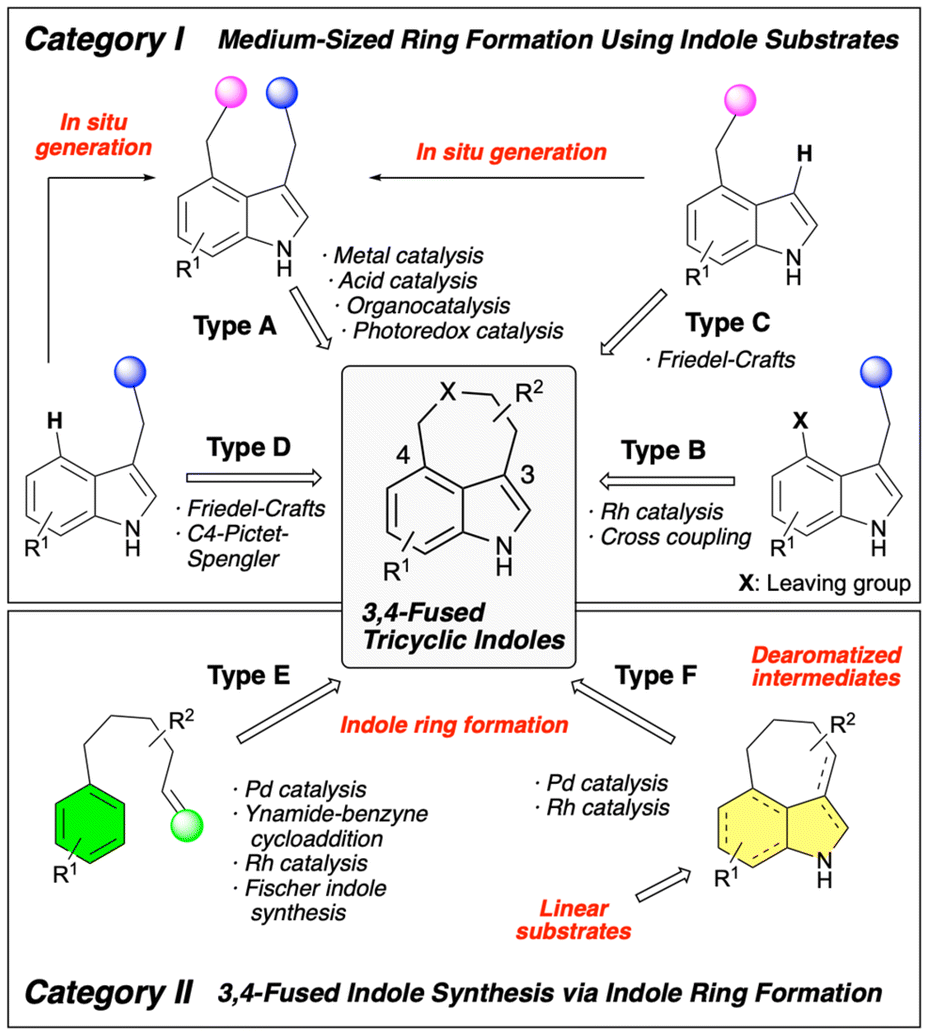
Building on this structural and biological background, considerable effort has been directed toward the development of synthetic methods for 3,4-fused tricyclic indole derivatives. The synthetic approaches to this molecular framework have been reviewed several times. Wipf,6 Shibata,7 and Jia8 have independently published comprehensive reviews on the synthesis of ergot alkaloids. Additionally, Guiry reviewed synthetic methods that utilize one-pot or multi-step synthesis,9 and Fan, Xu, and Yang reviewed synthetic approaches that use domino reactions with Pd catalysis.10 In 2018, we published a review of this topic, focusing on the synthetic methods for constructing 3,4-fused tricyclic indole skeletons via indole ring formation.11 In that review, we classified the types of reactions used for the synthesis of this framework into two categories. Category I involves methods that use functionalized indole derivatives as starting materials to construct fused medium-sized rings, whereas category II involves the construction of a 3,4-fused tricyclic indole skeleton via indole ring formation. In this review, we summarize the recent advances in this field of research since 2018 based on this classification. Each category is further subdivided as shown in Scheme 1. The use of 3,4-difunctionalized indoles as substrates to construct medium-sized rings (Type A) is the most typical strategy in category I. Another conventional approach is the construction of medium-sized rings using substrates with a leaving group at the C4 position of the indole (Type B). Reaction systems using 4-substituted indoles (Type C) or 3-substituted indoles (Type D) as starting materials have also been reported, wherein medium-sized rings were constructed via Friedel–Crafts-type reactions. However, some reaction systems that appear to follow Type C or Type D mechanisms generate a Type A substrate in situ by introducing a substituent at the C4 or C3 position, resulting in the formation of a target skeleton. We categorize these reactions as “In situ generation of substrates for Type A reaction” in this review. The synthetic processes in category II are subdivided into two. Type E reactions involve the synthesis of target tricyclic skeletons from linear substrates or intermediates via direct indole ring formation. By contrast, Type F reactions first generate fused-ring molecules with a dearomatized indole framework, followed by a rearomatization step to construct the target 3,4-fused tricyclic indole skeleton. Although many of the synthetic methods discussed in this review have demonstrated broad substrate generality, we have minimized the discussion on the substrate scope to focus on strategies for constructing the target 3,4-fused tricyclic indole framework and explore their mechanistic insights. Please refer to the original research articles for more detailed information on substrate generality.
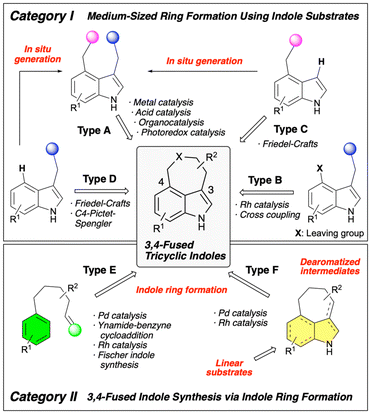 |
| | Scheme 1 Classification of synthetic methods for 3,4-fused tricyclic indoles discussed in this review. | |
2. Type A synthetic methods
2.1. Fused medium-sized ring formation via transition metal catalysis
N-(Pivaloyloxy)arylamide derivatives such as 1 react with 1,5-enynes or 1,6-enynes in the presence of a Rh(III) catalyst, yielding fused polycyclic compounds.12 Building on these previous findings, in 2020, Reddy et al. reported the construction of polycyclic 3,4-fused indole skeletons via a Rh(III)-catalyzed domino reaction using 4-alkynyl 3-alkenyl indoles, such as 2, as their 1,6-enyne counterparts (Scheme 2).13 When 1 and 2 were treated with 5 mol% of [RhCp*Cl2]2 and CsOAc in acetone at room temperature, the 3,4-fused polycyclic indole derivative 3 was obtained in 78% yield. The reaction occurs via the following mechanism: the reaction between 1 and [RhCp*Cl2]2 activates the ortho C–H bond, forming the rhodacycle intermediate 4. Subsequent insertion of the alkyne in 2 into the Rh–C bond of 4 generates intermediate 5. The reductive elimination of 5 followed by protonation, produces cyclized product 6. Finally, the intramolecular aza-Michael reaction proceeds in the presence of CsOAc as the base, yielding 3. Regarding substrate generality, the catalytic domino reaction system was applicable to various substituted N-(pivaloyloxy)benzamides and to N-(pivaloyloxy)thiophene-2-carboxamide and indole-2-carboxamide derivatives. Additionally, methyl ketones, phenyl ketones, and nitrile groups are suitable for alkene terminal substitutions.
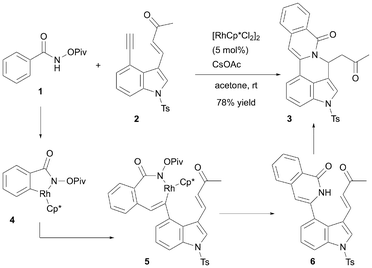 |
| | Scheme 2 Reddy's synthetic method based on the Rh-catalyzed domino reaction. | |
In 2023, Olson et al. reported synthetic strategies for lysergic acid diethylamide derivatives based on transition metal catalysis for the construction of the fused six-membered rings. Although several strategies have been examined using Cu and Ir catalysis, these methods are impractical because of product stability. The researchers discovered that a method utilizing Rh-catalyzed C–H insertion (Scheme 3) was the most efficient for constructing the target framework.14 After the Suzuki–Miyaura cross-coupling between compound 7 and 4-Bpin indole 8, an α-keto ester unit was introduced at the C3 position of the resulting indole derivative 9, yielding 10. After condensation with hydrazine, oxidation using MnO2 in the presence of Rh2(OAc)4 generates the Rh carbene in situ. This facilitates the desired C–H insertion reaction to produce 11 with the target molecular framework as a single diastereomer in 60% yield over both steps.
 |
| | Scheme 3 Olson's synthetic method based on the Rh-catalyzed C–H insertion. | |
Another example of the synthesis of a lysergic acid framework using a Type A reaction with a transition metal-catalyst was reported by Garner et al.15 The researchers uesd an intramolecular azomethine ylide cycloaddition reaction to construct a fused six-membered ring. 3,4-Disubstituted indole derivative 12 and chiral glycylsultam 13 were treated with 20 mol% of AgOAc in THF at room temperature to produce 15 in 63% yield. This reaction was expected to proceed via concerted transition state 14 (Scheme 4). The resulting product, 15, was successfully transformed into a lysergic acid framework via ring expansion process of the functionalized pyrroline moiety.
 |
| | Scheme 4 Garner's synthetic method based on the Ag-catalyzed intramolecular azomethine ylide cycloaddition. | |
In addition to these examples, Pan, Liu, and co-workers reported the asymmetric total synthesis of griseofamine B,16 which utilized a Pd-catalyzed cyclization reaction as an application of the Type A synthetic method previously developed by Park et al.17 and Jia et al.18
2.2. Fused medium-sized ring formation via acid-promoted reactions
In 2019, Jia et al. accomplished the total synthesis of speradine C using a bioinspired method for constructing the 3,4-fused indole framework based on an Brønsted acid-catalyzed intramolecular [3 + 2]-annulation (Scheme 5).19 The reaction of aldehyde 16 with glycine derivative 17 was performed in the presence of LiHMDS at −78 °C, yielding 18 as a mixture of diastereomers. Subsequent treatment of 18 with 0.2 equivalent of Tf2NH in CH2Cl2 produced 22, 23, and 24 in 53% yield, with a diastereomeric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.3:0.5. The mechanism for the formation of major product 22 is believed to occur as follows: the dehydration of 18 generates intermediate 19, which is activated by an acidic proton, promoting intramolecular [3 + 2]-annulation. This annulation proceeds through transition state 20 to minimize the steric repulsion between the indole ring and the dimethyl groups of the prenyl unit, leading to the predominant formation of intermediate 21. Owing to the influence of the tosyl group on the nitrogen and hydrogen atoms on the fused ring, the concave face of intermediate 21 is less sterically hindered than the convex face. Consequently, the protonation of the enol occurs preferentially from the concave face, forming 22 as the major diastereomer. Jia et al. successfully accomplished the total synthesis of speradine C via a five-step transformation from 22via25.
:0.3:0.5. The mechanism for the formation of major product 22 is believed to occur as follows: the dehydration of 18 generates intermediate 19, which is activated by an acidic proton, promoting intramolecular [3 + 2]-annulation. This annulation proceeds through transition state 20 to minimize the steric repulsion between the indole ring and the dimethyl groups of the prenyl unit, leading to the predominant formation of intermediate 21. Owing to the influence of the tosyl group on the nitrogen and hydrogen atoms on the fused ring, the concave face of intermediate 21 is less sterically hindered than the convex face. Consequently, the protonation of the enol occurs preferentially from the concave face, forming 22 as the major diastereomer. Jia et al. successfully accomplished the total synthesis of speradine C via a five-step transformation from 22via25.
 |
| | Scheme 5 Jia's synthetic method demonstrated in the total synthesis of speradine C. | |
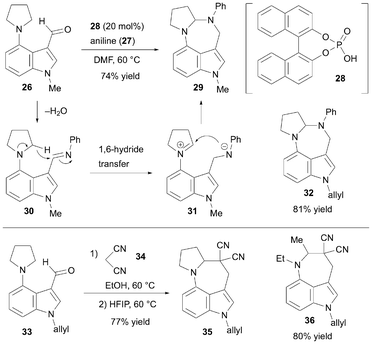
In 2023, Qiu et al. developed an acid-catalyzed [6 + 1]-annulation reaction to synthesize 3,4-fused azepinoindoles via a 1,6-hydride transfer/cyclization cascade (Scheme 6).20 Sequential intramolecular hydride transfer/cyclization is an effective strategy for rapid construction of fused cyclic molecules. The researchers hypothesized that a 3,4-azepine ring-fused indole skeleton could be constructed using the 4-aminoindole-3-carbaldehyde derivatives as six-atom synthons in a 1,6-hydride transfer/cyclization reaction. The designed reaction cascade was first examined using the indole derivative 26 with aniline 27 as a one-atom synthon. Detailed optimization revealed that the postulated transformation proceeded in the presence of 20 mol% of 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate 28 in DMF at 60 °C, producing the diazepinoindole derivative 29 in 74% yield. The reaction begins with the formation of imine 30 between compounds 26 and 27. The vicinal hydrogen on the nitrogen atom of the pyrrolidine ring in 30 undergoes 1,6-hydride transfer to the internal imine, yielding the iminium intermediate 31. Finally, cyclization occurs, producing 29. The highest yield was obtained when the substituent on the indole nitrogen was an allyl group, as observed in 32. In addition, malononitrile 34 can be used as a one-atom synthon. Specifically, the Knoevenagel condensation in ethanol between 33 and 34 produced the corresponding indole derivative with an electron-deficient alkene moiety. The 1,6-hydride transfer/cyclization cascade of the obtained product proceeded after replacing the solvent with hexafluoroisopropanol (HFIP), producing the azepinoindole derivative 35 in 77% yield. Moreover, as shown for 36, indole substrates containing a diethylamine unit were applicable when malononitrile was used as the one-carbon synthon.
 |
| | Scheme 6 Qiu and Xiao's synthetic method based on the phosphoric acid-catalyzed 1,6-hydride transfer/cyclization cascade. | |
In addition to this example, the construction of the fused-ring system via an acid-catalyzed intramolecular addition of a nitrogen nucleophile to imine has also been utilized in the 3,4-fused indole synthesis method developed by Link et al.21
In 2024, Trushkov et al. developed a synthetic method for tropane-fused indole derivatives based on Yb(OTf)3-catalyzed intramolecular [3 + 2]-cross-cycloaddition between an imine and a donor–acceptor cyclopropane to afford bridged bicyclic compounds (Scheme 7).22 To accomplish this transformation, the researchers designed 37 as a substrate. By heating 37 with aniline 27 in the presence of 10 mol% of Yb(OTf)3, 38, which has a bridged bicyclic structure, was obtained in 85% yield. The reaction mechanism begins with the formation of imine 39via a reaction between 37 and 27. Subsequently, the donor–acceptor cyclopropane unit is activated by Yb(OTf)3, which facilitates an intramolecular nucleophilic attack by the imine nitrogen (40), leading to the formation of intermediate 41. Finally, the intramolecular cyclization of 41 occurs, resulting in the formation of 38. This reaction can accommodate various aromatic amine derivatives as substrates; however, the reactivity is significantly influenced by the steric factors of aromatic amines.
 |
| | Scheme 7 Trushkov's synthetic method based on the Yb(OTf)3-catalyzed intramolecular [3 + 2]-cross-cycloaddition. | |
Recently, Harada, Nemoto, and co-workers developed a novel approach for the synthesis of oxazabicyclo ring-fused indoles via a regioselective intramolecular nitrone-alkene cycloaddition (Scheme 8).23 The researchers conducted a computational analysis of previously reported nitrone-alkene cycloadditions using density functional theory (DFT) calculations.24 A minimal energy difference was observed between the transition states corresponding to the two possible regioisomers (bridged vs. fused adducts, 1.0 kcal mol−1), indicating the inherent difficulty in controlling the regioselectivity. Computational predictions identified the addition of an acid as a potentially effective strategy to induce an energy difference of more than 20 kcal mol−1 between transition states leading to bridged vs. fused products, strongly favoring the former. Therefore, the reaction conditions were optimized using nitrone 43 prepared via the condensation of the indole derivative 42 and hydroxylamine 44. The regioselective cycloaddition proceeded in the presence of In(OTf)3, enabling the synthesis of the complex polycyclic compound 45 in a single step without generating fused compound 46. A gram-scale reaction was performed involving a series of transformations, including N–O bond cleavage, to yield amino alcohol 47. This work represents an elegant example of how modern computational methods can be applied to solve challenging selectivity problems in complex heterocyclic synthesis.
 |
| | Scheme 8 Harada and Nemoto's synthetic method based on the regioselective intramolecular nitrone-alkene cycloaddition inspired by computational chemistry. | |
2.3. Formation of fused medium-sized rings via NHC-catalysis
In 2024, Suresh et al. developed an N-heterocyclic carbene (NHC)-catalyzed umpolung-driven intramolecular cyclization to construct 3,4-cycloheptanone annulated indoles.25 As shown in Scheme 9, compound 48, which bears an aldehyde and a 1,6-conjugate acceptor, was transformed into the α-aryl-3,4-fused tetracyclic indole 50 in 96% yield in the presence of 20 mol% of thiazolium-based NHC precatalyst 49 and 60 mol% of K2CO3 in acetonitrile at 80 °C. The reaction occurs based on the intramolecular vinylogous Stetter reaction: treatment of 50 with a base generates a free NHC catalyst, which reacts with the aldehyde moiety in 48; subsequent proton transfer affords Breslow intermediate 51. Thereafter, the intramolecular 1,6-conjugate addition of 51 produces 50via intermediate 52, while generating a free NHC catalyst. When methanol was used as the solvent, 48 was transformed into the oxidized product 53 in 93% yield because of the dissolved oxygen in methanol. The NHC-catalyzed method was applied to a substrate bearing a 1,4-conjugate acceptor to afford the α-alkyl-3,4-fused tetracyclic indole 54 in 95% yield. In addition, intramolecular benzoin condensation of a dialdehyde-containing substrate yielded the hydroxy cycloheptanone-annulated indole 55 in 90% yield.
 |
| | Scheme 9 Suresh's synthetic method based on the NHC-catalyzed intramolecular cyclization. | |
2.4. Fused medium-sized ring formation via photoredox catalysis
In 2023, Piersanti et al. reported an Ir photoredox catalyst-enabled decarboxylative radical cyclization of γ,γ-dimethylallyltriptophane derivatives 56 and 62 for access to 3,4-six-, seven-, and eight-membered ring-fused tricyclic indoles (Scheme 10).26 The researchers examined two decarboxylation strategies: reductive single-electron transfer (SET) of redox-active esters and oxidative SET of carboxylic acids. In the former, the reaction of the N-hydroxyphthalimide-derived redox-active ester 56 with 2 mol% of an Ir photoredox catalyst produced 3,4-eight-membered ring-fused tricyclic indole 58 in 33% yield at room temperature under light irradiation with 34 W blue LEDs. The reaction starts with the single-electron oxidation of the indole ring to generate the indole radical cation 59. Subsequent sequential electron and proton transfers afford intermediate 60 with an allyl radical at the C4 side chain. Thereafter, the single electron reduction of the redox-active ester produces the α-amino radical 61, which traps the resonance-stabilized allyl radical, yielding product 58. When 62 was treated with 4 mol% of an Ir photoredox catalyst and K2HPO4 under the same light irradiation conditions, 3,4-six- and seven-membered ring-fused tricyclic indole derivatives 63 and 64 were obtained in 59% yield, with a ratio of 1:0.7. The reaction most probably begins with the single electron oxidation of carboxylic acid, generating α-amino radical 65. Subsequently, 6-exo-trig and 7-endo-trig radical cyclizations yield secondary radicals 66 and 67, respectively. Finally, hydrogen atom transfer from DMF to intermediates 66 and 67 affords products 63 and 64, respectively, as confirmed by deuterium experiments using DMF-d7 as a hydrogen atom donor.
 |
| | Scheme 10 Piersanti's synthetic method based on the photoredox-catalyzed decarboxylative strategy. | |
2.5. Synthetic methods based on C–H activation at the C4 position of the indole derivatives
In 2020, Harada, Nemoto, and co-workers reported a Rh-catalyzed, site-selective C–H functionalization of indoles at the C4 position utilizing α,β-unsaturated enones (Scheme 11).27 Their mechanistic analysis, based on experimental and computational investigations, indicated that the enone-directed C–H metalation process is reversible (71 ⇄ 72) and that C–H activation at the C2 position of the indoles can also occur. Nevertheless, the subsequent generation of the Rh–carbene species 73 was irreversible, controlling the overall site selectivity. Subsequent migratory carbene insertion and demetallation via intermediate 74 afforded 70, which could serve as a versatile scaffold for the synthesis of 3,4-fused tricyclic indoles. Additional mechanistic studies indicated that Cu(OAc)2 plays two important roles in this transformation. First, it provides the source of the acetate anion necessary for C–H bond cleavage. Second, it forms a complex with the C–H insertion product, regenerating the active Rh catalyst. Furthermore, chiral squaramide 75 promoted the intramolecular C–C bond formation of 70via transition state 76, furnishing 3,4-seven-membered ring-fused indole 77 with 97% ee.
 |
| | Scheme 11 Harada and Nemoto's synthetic method based on the site-selective C–H activation followed by catalytic enantioselective C–C bond formation. Ar: p-trifluoromethylphenyl group. | |
In 2021, Ge, Chen, and co-workers successfully accomplished the total synthesis of (±)-festuclavine and (±)-pyroclavine via a biomimetic synthetic strategy (Scheme 12).28 One of the key steps was the 4-nitrobenzenesulfonyl (Ns) group-directed C–H activation of the L-tryptophan derivative 78. This Pd-catalyzed transformation was performed on a gram scale, affording 80 in 67% yield. The researchers converted 80 to 81 in several steps of the Giese coupling reaction. After preparing the N-hydroxyphthalimide ester 82, decarboxylative cyclization was examined under nickel catalysis in N,N-dimethylacetamide (DMA). The desired product 83 was obtained in good yield with good diastereoselectivity (72% yield, 5.5:1 dr); however, racemization was observed, which is a common occurrence in radical coupling reactions. Finally, (±)-festuclavine and (±)-pyroclavine were synthesized from (±)-83via functional group interconversions and piperidine formation. In a related approach, Prabhu reported Rh-catalyzed C–H bond activation using a simple substrate set comprising indole-3-carbaldehyde derivatives such as 85 and allyl alcohol derivatives such as 84.29 Product 86, bearing two carbonyl groups, is a versatile precursor for the synthesis of 3,4-fused indoles. Specifically, 86 was treated with a base and a fused six-membered ring was constructed in 87, which is a core structure found in the frameworks of ergot and hapalindole alkaloids.
 |
| | Scheme 12 Ge, Chen, and Prabhu's synthetic method based on the site-selective C–H activation followed by the ring closure. DIC: N,N′-diisopropylcarbodiimide. NHPI: N-hydroxyphthalimide. | |
2.6. Other synthetic methods in this category
In 2021, Thiery et al. reported the construction of a 3,4-fused tricyclic indole skeleton via an intramolecular iodolactonization reaction (Scheme 13a).30 The reaction of carboxylic acid derivative 88 with iodine 89 in the presence of a stoichiometric amount of AgNO3 afforded the iodinated lactone-fused indole product 90 in 75% yield. The iodine moiety of 90 was functionalized via nucleophilic substitution with sodium azide 91 or thiophenol 93, producing the corresponding products 92 and 94 in 76% and 56% yields, respectively. Notably, the azide-substituted derivative 92 enabled effective side-chain diversification through click chemistry. The potential utility of this approach in medicinal chemistry was demonstrated by evaluating the antibacterial activity of the synthesized derivatives.
 |
| | Scheme 13 Synthesis of 3,4-seven-membered ring-fused tricyclic indoles via iodolactonization and oxidative cyclization. | |
In 2023, Chein et al. accomplished the biomimetic total synthesis of clavicipitic acid using a DDQ-mediated cross-dehydrogenative coupling reaction to construct the 3,4-fused indole skeleton. (Scheme 13b).31 After the preparation of L-tryptophan derivative 95 from 4-bromoindole, prenyl chloride, and L-serine, compound 95 was reacted with DDQ in nitromethane at 0 °C and then warmed to room temperature. Azepinoindole derivatives 96 and 97 were obtained in 77% yield at a 6.5:1 ratio. These products were separable using column chromatography, and the trans-isomer 96 could be converted into clavicipitic acid via sequential deprotection of the two Boc groups. From a mechanistic point of view, DDQ first interacts with 95, inducing hydride transfer through charge transfer complex formation, thereby generating ion pair intermediate 98. The subsequent diastereoselective construction of the seven-membered ring proceeded via transition state 99, in which the carbamate nitrogen was more likely to attack the benzylic position from the upper side of the resonance plane because of the reduced steric interactions between the Boc and prenyl groups. Compound 96 was also utilized in the synthesis of griseofamine B by Pan et al.,32 wherein the researchers used a Pd-catalyzed aminocyclization reaction developed by Jia et al.33
3.
In situ generation of substrates for Type A reaction
Substrates for Type A reactions can be synthesized by introducing a substituent at the C3 position of appropriately modified C4-substituted indole derivatives. Therefore, reaction systems have been developed in which substrates for Type A reactions are generated in situ from those used for Type C reactions, enabling a sequential cyclization process that leads to the formation of 3,4-fused indoles. Although these reactions may appear to be Type C reactions, they can be classified as Type A reactions based on the reaction mode used to construct the fused ring structure.
3.1. Introduction of C3 substituent via a 1,4-addition of indole derivatives to α,β-unsaturated carbonyl compounds
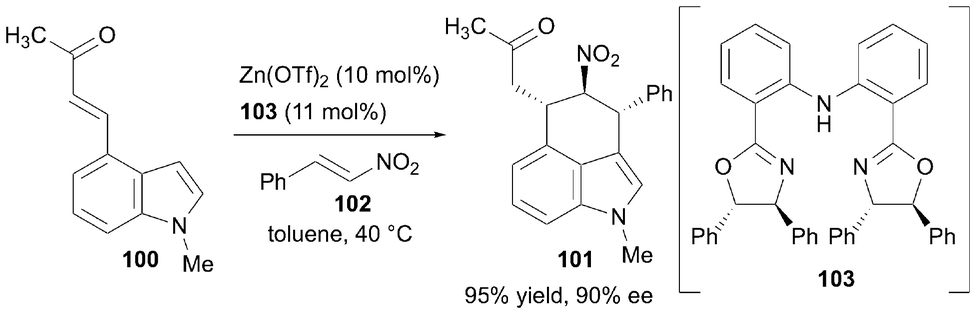
In 2019, Guiry et al. reported the synthesis of six-membered ring-bridged 3,4-fused indoles through tandem asymmetric Friedel–Crafts alkylation/Michael addition (Scheme 14). Although the synthetic methods for constructing the target framework using this strategy had already been reported,34 the researchers further explored the reaction by using indole derivatives with an α,β-unsaturated ketone at the C4 position and trans-β-nitrostyrene derivatives.35 The treatment of 100 and 102 with 10 mol% of Zn(OTf)2 in toluene led to a highly diastereoselective reaction (dr = 49:1), where a Friedel–Crafts-type 1,4-addition to nitrostyrene was followed by an intramolecular Michael addition, yielding 101. When 103 was used as the chiral ligand, the tricyclic product 101 was obtained in 95% yield with 90% ee.
 |
| | Scheme 14 Guiry's synthetic method based on the tandem asymmetric Friedel–Crafts alkylation/Michael addition. | |
On the other hand, Xie and Guo et al. developed an asymmetric synthetic method for seven-membered ring-bridged 3,4-fused tricyclic indoles via regio- and enantio-selective Friedel–Crafts alkylation/N-hemiacetalization and dehydration cascades (Scheme 15).36 The researchers used 4-aminoindole derivative 104 as a bis-nucleophile and aimed to synthesize 3,4-disubstituted indole derivatives in situ via conjugate addition to compound 105. The target conjugate addition proceeded in the presence of 5 mol% of Ni(OTf)2 and 5 mol% of chiral tridentate ligand 107, yielding compound 109. Subsequent intramolecular N-hemiacetalization, followed by the dehydration of 110 afforded the 3,4-seven-membered ring-fused tricyclic indole 106 in 90% yield and 91% ee. Compound 108 was obtained as a by-product in only 5% yield, indicating that high regioselectivity between the C3 and C5 positions was accomplished in the initial Friedel–Crafts-type reaction step.
 |
| | Scheme 15 Xie and Guo's synthetic method based on the regio- and enantioselective Friedel–Crafts alkylation/N-hemiacetalization and dehydration cascade. | |
3.2. Introduction of C3 substituent via a metal-catalyzed process
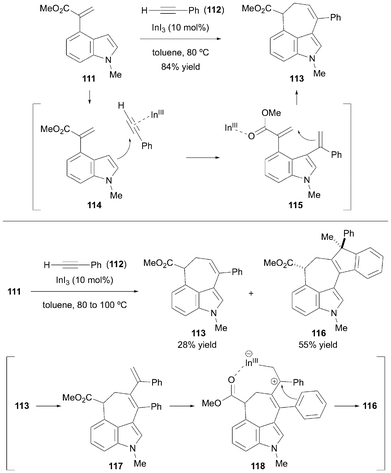
In 2018, Harada and Nishida et al. reported the synthesis of seven-membered carbocycles via the [5 + 2]-cycloaddition of indoles and alkynes (Scheme 16).37 This method utilizes In(III) as a bifunctional catalyst, activating both the alkyne and α,β-unsaturated ester moieties of indoles to form two new carbon–carbon bonds in a single step. The authors demonstrated that their methodology could be extended to the synthesis of 3,4-fused tricyclic indoles. The desired cyclohepta[c,d]indole product 113 was obtained in 84% yield using 4-substituted indole substrate 111 and phenylacetylene 112 under optimized reaction conditions. In this transformation, the indium salt activates the triple bond owing to its π-Lewis acidity (114). Hence, Friedel–Crafts alkenylation proceeded and subsequent rearomatization/protonation afforded the alkenylated indole 115. In(III) activated the ester moiety as a σ-Lewis acid, and a second C–C bond was formed to construct a seven-membered ring. Pentacyclic compound 116 was synthesized at higher reaction temperatures, probably via the following mechanism: compound 116 was generated from 113via compound 117 followed by intramolecular cyclization (118).
 |
| | Scheme 16 Harada and Nishida's cyclohepta[c,d]indole synthesis by bifunctional In(III) catalyst. | |
Yu et al. recently developed an efficient synthetic route for azepino[5,4,3-cd]indoles via Rh-catalyzed [4 + 3]-annulation of N-sulfonyl-1,2,3-triazoles with 4-vinyl indoles (Scheme 17).38 Using triazoles as aza-[3C] synthons and their ability to generate Rh carbenes has been reported previously. Based on this precedent, Yu et al. hypothesized that incorporating a Michael acceptor into the indole skeleton would facilitate intramolecular amino cyclization. Using Rh2(OAc)4 as the catalyst in the presence of DBU as a base, the reaction between triazole derivative 119 and indole derivative 120 proceeds to afford the desired azepino[5,4,3-cd]indole 121 in 95% yield. Additionally, several transformations were performed to modify the azepinoindole framework. This method involves a reaction cascade. First, indole 120 attacked the α-imino Rh(II) carbene 122, leading to intermediate 123. Thereafter, catalyst dissociation afforded enamine 124. Subsequently, an intramolecular aza-Michael addition led to the formation of the product. Additionally, Yu et al. demonstrated a one-pot synthesis of 121 from phenylacetylene and tosyl azide via Cu–Rh sequential catalysis.
 |
| | Scheme 17 Yu's azepinoindole synthesis by the Rh-catalyzed [4 + 3]-annulation. | |
3.3. Construction of medium-sized rings using donor–acceptor cyclopropanes/aziridines
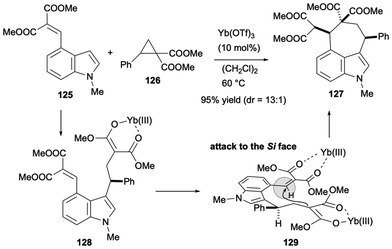
Donor–acceptor cyclopropanes have been utilized as versatile 1,3-zwitterion precursors in organic synthesis.39 In 2020, Zhang, Xu and their co-workers demonstrated that [4 + 3]-cyclization between indole derivatives bearing a Michael acceptor at the C4 position and a donor–acceptor cyclopropane is effective for synthesizing 3,4-seven-membered ring-fused indole derivatives.40 As shown in Scheme 18, 125 reacted with donor–acceptor cyclopropane 126 in dichloroethane at 60 °C in the presence of 10 mol% of Yb(OTf)3, yielding 127 in 95% yield with 13:1 diastereoselectivity. After alkylation at the C3 position of the indole to generate 128, Michael addition subsequently proceeds via transition state 129 to avoid steric hindrance between the diester units, resulting in high diastereoselectivity. Various substitution patterns on the aromatic ring of the donor–acceptor cyclopropane are applicable.
 |
| | Scheme 18 Zhang, Xu's method based on the [4 + 3]-cyclization between C4-substituted indole derivatives and a donor–acceptor cyclopropane. | |
The synthesis of 3,4-fused tricyclic indoles using donor–acceptor cyclopropanes was also reported by Kerr et al. in 2022,41 who utilized 4-ethynyl indole 130 as a coupling partner with donor–acceptor cyclopropane 126 to achieve the target synthesis. C3 alkylation of 130 with 126, followed by an intramolecular Conia-ene reaction, proceeded in the presence of 2 equiv. of ZnBr2 and 1 equiv. of 2,6-lutidine in benzene under reflux, producing 131 in 86% yield (Scheme 19a). Punniyamurthy and Trivedi also reported the synthesis of 3,4-fused azepinoindoles via a C3-alkylation–aza-Michael reaction cascade using 2-aryl 1-sulfoxyaziridines as 1,3-zwitterion precursors.42 As shown in Scheme 19b, the reaction between 125 and 2-phenyl 1-tosylaziridine 132 in the presence of 5 mol% Cu(OTf)2, followed by the addition of K2CO3 as a base to promote an intramolecular aza-Michael reaction, yielded the 3,4-seven-membered ring-fused tricyclic indole 133 in 75% yield with high diastereoselectivity.
 |
| | Scheme 19 Synthesis of 3,4-seven-membered ring-fused tricyclic indoles using 1,3-zwitterion precursors. | |
4. Type B synthetic methods
4.1. Synthetic methods using intramolecular nucleophilic addition of arylrhodium(I) species
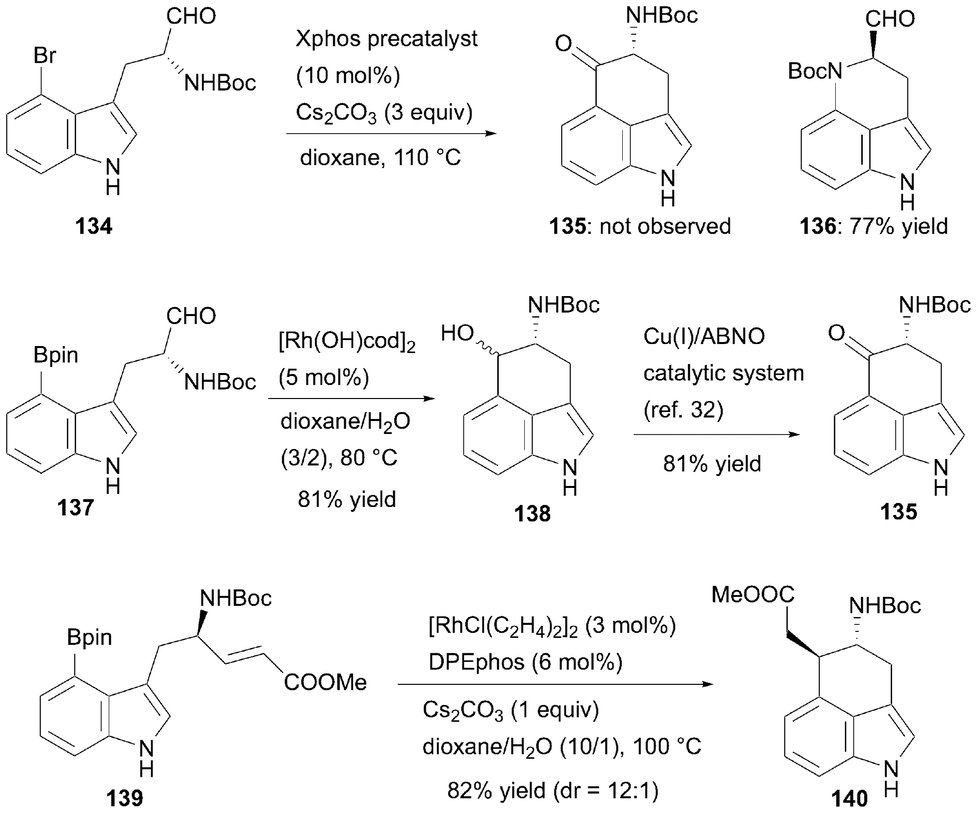
Optically active 4-amino-5-oxo-1,3,4,5-tetrhydrobenz[cd]indole, also known as 4-amino Uhle's ketone, is a valuable intermediate in the synthesis of 3,4-six-membered ring-fused ergot alkaloid derivatives.43 However, owing to facile racemization through enolization under both acidic and basic conditions,44 an efficient method for synthesizing this compound in its optically active form is highly desirable. To overcome this challenge, Piersanti et al. developed an efficient conversion method in 2021 using optically active 4-substituted tryptophan derivatives as starting materials (Scheme 20).45 The researchers first examined transition-metal-catalyzed carbonyl-arylation/acylation using the bromoindole derivative 134. However, extensive examination revealed that the desired six-membered ring-fused product 135 was not obtained, and an unexpected six-membered NH carbamate-arylated cyclization product 136 formed in 77% yield when 10 mol% (2-dicyclohexylphosphino-2′,4′,6′-triisopropyl-1,1′-biphenyl)[2-(2-aminoethyl)phenyl]palladium(II) chloride (Xphos precatalyst) was used. Piersanti et al. overcame this challenge by generating a more nucleophilic aryl rhodium(I) species. When pinacolboronic acid derivative 137 was treated with 5 mol% of [Rh(OH)cod]2 in a dioxane/H2O mixed media at 80 °C, the cyclization reaction afforded the corresponding alcohol adduct 138 in 81% yield as a diastereomixture (cis:trans = 1:2.6). Oxidation of this alcohol proceeded smoothly using the Cu(I)/ABNO catalytic system developed by Stahl et al.,46 yielding the 4-amino Uhle's ketone derivative 135 in 81% yield without any loss of optical purity. The developed approach provides a solution for synthesizing the desired 4-amino Uhle's ketone derivatives in an optically active form, which can be successfully applied to the asymmetric total synthesis of all rugulovasine stereoisomers47 and (−)-6,7-secoagroclavine.48 Furthermore, Piersanti and Bartoccini et al. recently developed a diastereoselective synthetic method for 3,4-six-membered ring-fused indole derivatives, such as 140, by harnessing the conjugate addition of an arylrhodium(I) species generated from pinacolboronic acid derivative 139 to an internal α,β-unsaturated ester.49
 |
| | Scheme 20 Piersanti and Bartoccini's synthetic method based on the intramolecular nucleophilic addition of arylrhodium(I) species. | |
4.2. Construction of the medium-sized ring based on the Pd- or Ni-catalyzed cross coupling strategy
The construction of medium-sized rings using an intramolecular Heck reaction with substrates bearing a leaving group at the C4 position of the indole is a representative method for building ergoline frameworks. A recent example is the work of Wipf et al., who utilized the Heck reaction as a key step in the short synthesis of ergot alkaloids (Scheme 21).50 Using compound 141, which can be synthesized in two steps from commercially available materials, as a substrate, Wipf et al. performed the Heck reaction in the presence of a 10 mol% Pd catalyst. The 3,4-seven-membered ring-fused indole derivative 142 was obtained in 80% yield. However, after isomerization of the double bond, the resulting compound 143 was treated under Heck reaction conditions to produce a mixture of compounds 144 and 145 in 50% yield (144:145 = 2:1). These compounds were possibly formed via a Heck coupling–alkene isomerization sequence. In addition to this work, Smith et al. reported the synthesis of lysergic acid, utilizing a Heck reaction as the key step for constructing the fused six-membered ring.51 More recently, Opatz et al. successfully constructed a lysergic acid framework using a Ni-catalyzed cross-coupling process.52
 |
| | Scheme 21 Wipf's synthetic method based on the intramolecular Heck coupling. | |
5. Type C synthetic methods
5.1. Fused medium-sized ring formation via enantioselective [4 + 3]-cycloaddition with azomethine ylides
Azomethine ylides are valuable building blocks in the synthesis of optically active azaheterocycles. However, achieving catalytic asymmetric [4 + 3]-cycloadditions presents challenges owing to unfavorable entropic factors, transannular interactions, and the presence of multiple stereocenters. In 2021, Deng et al. reported an Ir-catalyzed asymmetric [4 + 3]-cycloaddition for the synthesis of azepino[3,4,5-cd]indole derivatives (Scheme 22).53 The researchers used racemic 4-indolyl allylic alcohol (±)-146 and an azomethine ylide precursor 147 to produce 3,4-fused indole 149 in 83% yield, exhibiting excellent enantioselectivity and diastereoselectivity (>99% ee and >20:1 dr) in the presence of [Ir(cod)Cl]2, Carreira's P/olefin ligand (S)-148, and Zn(OTf)2. The proposed reaction mechanism for the [4 + 3]-cycloaddition is described below. The Zn(II)-stabilized azomethine ylide species 150 enantioselectively attacks the cationic π-allyl iridium(III) intermediate 151, which is generated in the presence of Zn(OTf)2 as an acid promoter. Subsequently, the addition of the C3 position of indole 152 to the electrophilic imine, activated by coordination with Zn(II), forms product 149. The diastereoselectivity may be attributed to steric repulsion between the aryl imine and diester moiety. Deng et al. focused on the required amount of indole (2 equiv.) and explored the kinetic resolution of compound 146, yielding (R)-146 with an enantiomeric excess greater than 99%.
 |
| | Scheme 22 Deng's synthetic method based on the Ir-catalyzed [4 + 3] cycloaddition reaction. | |
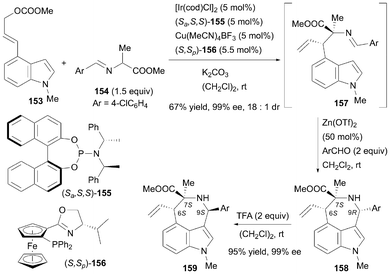
In addition, Dang, Dong, Wang, and their coworkers reported a stereodivergent synthetic method that uses a cooperative Ir- and Cu-catalyzed formal asymmetric [4 + 3]-cycloaddition (Scheme 23).54 By combining Ir(I)/(Sa,S,S)-155 and Cu(I)/(S,Sp)-156 catalysts, the reaction of (E)-4-indolyl allyl carbonate 153 with an alanine-derived aldimine ester 154 formed the first coupling intermediate 157. The subsequent intramolecular Friedel–Crafts reaction, catalyzed by Zn(OTf)2, furnished the final product 158 in 67% overall yield with 99% ee and 18:1 dr. In this reaction, the π-allyl iridium(III) complex generated from 153 determined the C6 stereochemistry: using Ir(I)/(Sa,S,S)-155 led to the formation of the 6S stereocenter, while Ir(I)/(Ra,R,R)-155 produced the 6R stereocenter. Similarly, the Cu(I)-stabilized azomethine ylide formed from 154 controlled the C7 stereochemistry: Cu(I)/(S,Sp)-156 generated a 7S stereocenter and Cu(I)/(R,Rp)-156 yielded a 7R stereocenter. Moreover, C9 epimerization of 158 was accomplished via a trifluoroacetic acid (TFA)-promoted ring-opening-cyclization reaction to yield product 159 in 95% yield and 99% ee. Therefore, the synergistic effect of the Ir/Cu catalysts, along with TFA-promoted epimerization, facilitated the stereodivergent synthesis at the C6, C7, and C9 positions of the azepino[3,4,5-cd]indole derivatives.
 |
| | Scheme 23 Dang, Dong, and Wang's synthetic method based on cooperative Cu(I)/Ir(I)-catalyzed [4 + 3] cycloaddition reaction. | |
5.2. Other catalytic synthetic methods in this category
In 2019, Gu et al. reported the synthesis of 3,4-seven-membered ring-fused indole derivatives using 4-aminoindole 160 as the 1,4-bisnucleophile (Scheme 24).55 Their approach involved initial formation of a nucleophilic enamine between 160 and diethyl acetylenedicarboxylate 161, generating the 1,6-bisnucleophile intermediate 164. Subsequent condensation with aldehydes such as 162 facilitates the formation of a seven-membered ring structure. Detailed optimization of the reaction conditions revealed that using 5 mol% of Cu(OTf)2 as the catalyst, with ethanol as the solvent, afforded product 163 in 87% yield under air. When the reaction was performed under an argon atmosphere, a mixture of enamine-type compounds 165 and 163 was obtained (163:165 = 1:4). Therefore, compound 163 was formed via air oxidation of 165. Additionally, because the conversion of 164 to 163 was not observed in the absence of Cu(OTf)2, the Cu catalyst probably played a key role in the condensation step between 164 and 162. In addition to aromatic aldehydes, aliphatic aldehydes could be used as the aldehyde components, demonstrating the broad substrate generality of the reaction.
 |
| | Scheme 24 Gu's synthetic method based on the Cu-catalyzed three-component condensation. | |
In 2019, Stokes et al. developed an efficient acid-catalyzed method for the synthesis of tetrahydrobenzo[cd]indoles via the intramolecular hydroindolation of cis-configured methindolylstyrenes (Scheme 25a).56 The researchers used cis-β-(α′,α′-dimethyl)-4′-methindolylstyrene 166 as a substrate and benzenesulfonic acid as a catalyst, in toluene, at 130 °C. This setup facilitated selective cyclization at the C3 position of the indole, yielding the desired tetrahydrobenzo[cd]indole 167, although some substrates underwent a competitive minor cyclization pathway at the C5 position, producing regioisomer 168. The observed regioselectivity was attributed to the dispersive interactions between the indole and styrene moieties, which preorganized the substrate structure to that of 169 for efficient six-membered ring formation. Notably, the cis-alkene configuration and geminal dimethyl substitution were critical for controlling the regioselectivity and reaction efficiency, as trans-configured substrates favored undesired oligomerization.
 |
| | Scheme 25 Synthesis of 3,4-seven-membered ring-fused tricyclic indoles using Brønsted acid-catalyzed intramolecular Friedel-Craft reaction. | |
Recently, Das et al. reported a similar reaction system using 1,1-diarylalkanols as starting materials.57 As shown in Scheme 25b, 170 reacted with 10 mol% of p-TsOH·H2O in HFIP at room temperature, forming the corresponding cationic intermediate, which was then trapped by an indole nucleophile to afford 171 in 82% yield. Additionally, alkene 172 was generated via a formal E1 elimination of the tertiary alcohol. Construction of the 3,4-fused tricyclic indole framework via cation formation from 172 under reflux conditions highlighs the advantage of the cyclization reaction of 170, which proceeds at room temperature.
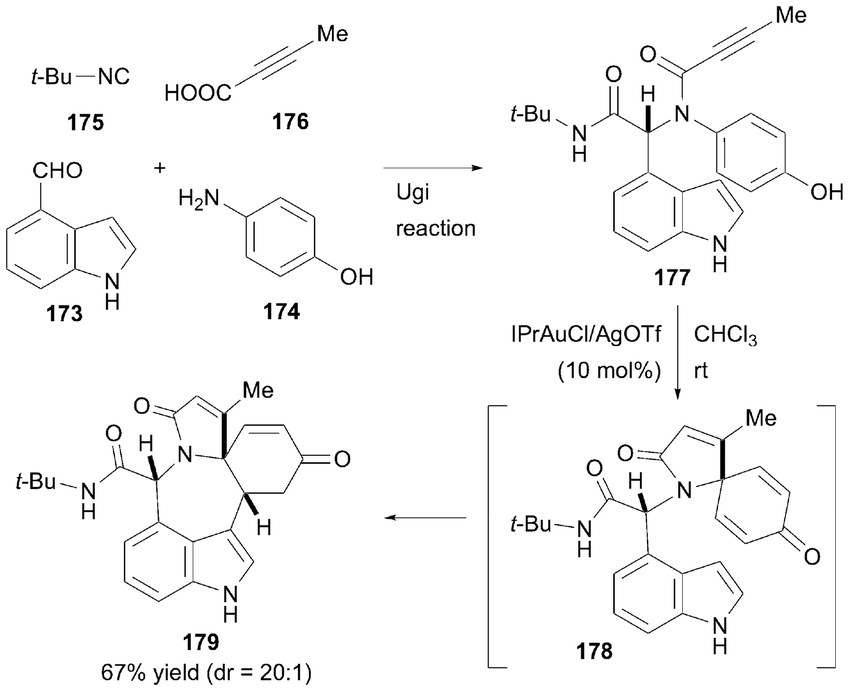
In 2020, Li, Van der Eycken, and co-workers developed an Au-catalyzed method for synthesizing polycyclic azepino[5,4,3-cd]indoles via a post-Ugi dearomatization cascade (Scheme 26).58 This process begins with the formation of Ugi adducts using indole-4-carboxaldehyde 173, 4-aminophenol 174, isocyanide 175, and alkyne carboxylic acid 176, producing versatile substrate 177. The key reaction was initiated by activating the triple bond with a cationic Au catalyst, generated in situ from IPrAuCl and AgOTf, which underwent dearomative ipso-cyclization in a 5-endo-dig manner to yield spiro compound 178. Subsequently, Michael addition at the C3 position of the indole selectively formed the azepino[5,4,3-cd]indole core 179. The substrate design was crucial because terminal alkynes proved to be unsuitable for the reaction. Furthermore, substituents on the indole nitrogen influenced the pathway, with electron-withdrawing groups on the nitrogen reducing the nucleophilicity at C3, thereby hindering Michael addition.
 |
| | Scheme 26 Li and Van der Eycken's synthetic method based on the Au-catalyzed post-Ugi cyclization. | |
6. Type D synthetic methods
Type D reactions are less common than the other types of reactions. In our previous review, we categorized the reactions listed in Section 2–6 as Type D reactions. However, in this review, we classified them as Type A reactions based on the reaction mode of the medium-sized ring formation step.
A recently reported synthetic method in this category involves the construction of azepinoindole frameworks via a C4-Pictet–Spengler reaction. This methodology was designed to mimic the proposed biosynthetic pathway of hyrtiazepine-type alkaloids, such as hyrtimomine A, which feature a hydroxy group at the C5 position of the indole ring. Accordingly, it represents a goal-oriented strategy for the synthesis of related natural products and their derivatives. In 2017, Abe and Yamada reported the total synthesis of hyrtioreticulins C and D via a C4-Pictet–Spengler reaction (Scheme 27a).59 They aimed to construct a 3,4-fused tricyclic indole framework via a C4-Pictet–Spengler reaction between a 5-hydroxytryptophan derivative 180 and acetaldehyde 181. However, controlling the chemoselectivity was crucial, as competitive reaction at the C2-position could lead to the formation of undesired β-carboline-type products. Optimization of the reaction conditions revealed that conducting the reaction under microwave irradiation effectively suppressed the formation of β-carboline-type byproducts. Furthermore, when the reaction was carried out in a solvent mixture of N,N-diisopropylethylamine (DIPEA) and methanol (1:1), the 3,4-fused tricyclic indole derivative trans-182 was obtained in 59% yield with a 15:1 trans-selectivity. The observed trans-selectivity was attributed to the steric bias between two plausible post-cyclization intermediates 183 and 184, with intermediate 184 being favored due to reduced steric hindrance in the transition state. Hydrolysis of trans-182 afforded (–)-hyrtioreticulin C in 72% yield.
 |
| | Scheme 27 Synthesis of azepinoindole frameworks via a C4-Pictet–Spengler reaction. | |
Abe, Yamada et al. also applied this strategy to the synthesis of the core structure of hyrtiazepine alkaloids (Scheme 27b).60 In the C4-Pictet–Spengler reaction between compound 180 and an aldehyde derivative 185, the use of trifluoroethanol (TFE) as the solvent and 1,4-diazabicyclo[2.2.2]octane (DABCO) as the base enabled the trans-selective formation of 186 in 85% yield.
A similar transformation was demonstrated by Yu and Liu et al. in 2021 (Scheme 27c).61 Following C2-selective arylation of a tryptamine derivative using palladium catalysis to afford compound 187, the subsequent C4-Pictet–Spengler reaction with isatin 188 in the presence of TFA in HFIP furnished the 3,4-fused tricyclic indole derivative 189 in 77% yield.
In addition to the C4-Pictet–Spengler reactions, the key step in the synthesis of (±)-decursivine, as reported by Manetsch et al. in 2021, can be categorized under this reaction type.62 To accomplish an efficient synthesis of (±)-decursivine, the researchers investigated the conversion from 190 to (±)-decursivine (Scheme 28). After screening the oxidative transformation conditions, they discovered that the desired reaction proceeded in 47% yield using [bis(trifluoroacetoxy)iodo]benzene (PIFA) in HFIP. Furthermore, the addition of 5 mol% BINOL phosphoric acid 28 improved the yield, successfully synthesizing (±)-decursivine in 74% yield. The reaction initially proceeds with the oxidation of the 5-hydroxyindole unit by PIFA to afford the dearomatized intermediate 191. Subsequent eight-membered ring formation is facilitated by the involvement of BINOL phosphoric acid, which is converted into intermediate 192. Finally, an intramolecular C–O bond formation results in the formation of the furan ring, effectively affording (±)-decursivine.
 |
| | Scheme 28 Manetsch's synthetic method demonstrated in the total synthesis of (±)-decursivine. | |
7. Type E synthetic methods
7.1. Synthetic method based on Pd Catalysis
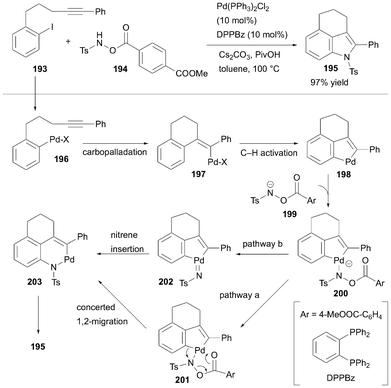
In 2020, Luan et al. developed a one-step synthetic method to produce diverse 3,4-fused tricyclic indole derivatives using secondary hydroxylamines as bifunctional nitrogen sources in a Pd-catalyzed reaction.63 Their strategy for constructing the indole ring relied on the hypothesis that fine-tuned hydroxylamine derivatives could enable two sequential C–N bond formations. The first step involves electrophilic amination of a palladacycle intermediate, and the second step proceeds via reductive elimination. This design leverages the bifunctional nature of hydroxylamine derivatives in a sequential Pd-catalyzed process, facilitating the streamlined one-step synthesis of diverse 3,4-fused tricyclic indole derivatives. Luan et al. successfully obtained 195 by heating an iodobenzene derivative with an alkyne side chain 193 and a hydroxylamine derivative, using 10 mol% of a Pd catalyst, Cs2CO3 as a base, and PivOH as an additive, in toluene. After optimizing the structure, hydroxylamine 194, comprising a Ts group on nitrogen and benzoate derivative on oxygen, performed best, affording 195 in 97% yield (Scheme 29). Using this reaction system, 3, 4-fused tricyclic indole derivatives with six-, seven-, and eight-membered rings were synthesized. Additionally, 3,5-fused and 3,6-fused tricyclic indole derivatives were synthesized using the same catalytic system. This reaction system demonstrated high substrate generality and enabled the construction of the desired framework in a single step from simple 2-substituted iodobenzene and hydroxylamine derivatives via Pd-catalyzed C–H activation. Given its efficiency and simplicity, it can be regarded as a state-of-the-art methodology among the reported synthetic approaches. The reaction mechanism begins with the reaction of the Pd(0) catalyst with 193, generating intermediate 196. This was followed by carbopalladation to yield intermediate 197. Subsequent C–H activation then forms palladacycle intermediate 198. Following the formation of intermediate 200 from the reaction between 199, derived from 194 with a base, and palladacycle intermediate 198, two possible reaction pathways were proposed. In pathway a, the electron flow, as shown in intermediate 201 triggers concerted 1,2-migration, yielding intermediate 203. In pathway b, insertion of the Pd-nitrene intermediate 202 also led to the formation of intermediate 203. Finally, the reductive elimination afforded 195. This method is limited to the synthesis of indole derivatives with a Ts protecting group on nitrogen. In 2022, Bai, Luan, and co-workers expanded their approach to include the synthesis of 3,4-fused tricyclic indoles featuring a removable alkyl group on the nitrogen.64 The researchers used hydroxylamines with bulky alkyl substituents as the nitrogen source. This process relies on the selective cleavage of large t-butyl or benzyl groups during palladacycle formation via an SN1 mechanism, which allows the incorporation of smaller alkyl groups into the product.
 |
| | Scheme 29 Luan's synthetic method based on the Pd-catalyzed sequential carbopalladation, C–H activation, and two C–N bonds formation using a bifunctional secondary hydroxylamine derivative as the nitrogen source. | |
After the Luan's report, Zhang et al., and Jiang and Yu et al. independently reported the synthesis of 3,4-fused tricyclic indoles in 2020, following the same concept, using N,N-di-t-butyldiaziridinone 205 as the nitrogen source (Scheme 30). Zhang et al. successfully synthesized the desired 3,4-fused tricyclic indole 206 in 96% yield by reacting iodobenzene derivative 204 with 205 under conditions in which Pd(OAc)2 was used as the catalyst and P(o-tol)3 served as the ligand.65 By contrast, Jiang and Yu et al. used 10 mol% Pd(OAc)2 and 1 equivalent of PPh3 as the ligand, and successfully produced 207 in 83% yield from 205 and 193.66 Mechanistically similar to that of the system developed by Luan et al., the reaction proceeded with the formation of palladacycle intermediate 198. Subsequently, the oxidative addition of 205 to palladacycle intermediate 198 occurred, resulting in the formation of Pd(IV) intermediate 208. The transformation of intermediate 208 to 212 involves two pathways. One route proceeds via the formation of the Pd-nitrene intermediate 210, which is generated by the release of t-butyl isocyanate 209. The other pathway suggests that intermediate 208 undergoes reductive elimination to form eight-membered palladacycle intermediate 211, which then undergoes subsequent β-N elimination to yield the key palladacycle intermediate 212.
 |
| | Scheme 30 Zhang's, and Jiang and Yu's synthetic methods based on the Pd-catalyzed sequential carbopalladation, C–H activation, and two C–N bonds formation using N,N-di-t-butyldiaziridinone as the nitrogen source. | |
Furthermore, Zhang, Liang, Li, and Quan, and their co-workers reported a synthetic method for 3,4-fused tricyclic indoles via an ortho-C–H activation using the palladium/norbornene chemistry.67,68 The researchers discovered that after ortho-C–H activation of iodoaniline derivative 213, facilitated by a Pd catalyst and norbornene 216, incorporation of 214 as an ortho-C–H alkylating agent triggered cyclization involving N–S bond cleavage, yielding the corresponding tricyclic indole derivative 215 in 63% yield (Scheme 31). The reaction proceeds according to the following mechanism. First, the three-component reaction of the Pd catalyst, 213, and norbornene 216 forms intermediate 217. This intermediate then undergoes ortho-C–H activation, transforming into 218. The subsequent reaction of 218 with 214 facilitates ortho-alkylation, resulting in intermediate 219. The following β-carbon elimination leads to the extrusion of norbornene, forming intermediate 220. This intermediate undergoes an intramolecular migratory insertion reaction with an alkyne to afford intermediate 221. Subsequent C–N bond coupling, accompanied by N–S bond cleavage to liberate 222, afforded the corresponding 3,4-six-membered ring-fused indole derivative 215. This reaction mechanism is supported by control experiments and DFT calculations. This reaction system could also be applied to the synthesis of 3,4-seven-membered ring-fused indoles using one-carbon unit-extended alkylating reagents.
 |
| | Scheme 31 Zhang, Liang, Li, and Quan‘s synthetic method via an ortho-C–H activation using the palladium/norbornene chemistry. | |
7.2. Synthetic method based on the intramolecular ynamide-benzyne cycloaddition
In 2023, ynamide-benzyne [3 + 2]-cycloaddition reactions were developed by Takasu, Takikawa, and co-workers to synthesize silicon-containing 3,4-fused tricyclic indoles (Scheme 32).69 The proposed reaction mechanisms and unique intermediates are of particular interest. The authors proposed that the intramolecular [3 + 2]-cycloaddition of 223 proceeds through transition state 225via benzyne intermediate 224, generating indolium ylide 226. The subsequent hydrolysis of 226 affords 227 in 79% yield. Deuterium labeling experiments indicate that residual water contaminated with 18-crown-6 and/or Cs2CO3 serves as a potential proton source at the C2 position of the indole variants. Notably, no literature precedent exists for using ynamides as three-atom components in [3 + 2]-cycloaddition reactions. Computational studies have suggested that simple combinations of ynamides and benzynes preferentially undergo intermolecular [2 + 2]-cycloaddition reactions over [3 + 2]-cycloaddition reactions. The transformation had a broad substrate scope and afforded various fused products in good yields. Furthermore, the synthesized product could be converted into potentially useful 3,4-fused indole derivatives via C–H functionalization and into indoles with functionalities at the C4-positions through ring-opening.
 |
| | Scheme 32 Takasu and Takikawa's synthetic method based on the intramolecular [3 + 2]-cycloaddition reactions between ynamides and benzynes. | |
7.3. Application of the previously developed Type E methods
Reddy et al. applied 3,4-fused tricyclic indole synthesis, based on acetyl-amide-directed Rh-catalyzed C–H activation developed by Xu and Liu et al.,70 as the key step in the synthesis of 3,4-oxepino-fused indole derivatives (Scheme 33).71 As part of their efforts to develop synthetic methods using Morita-Baylis–Hillman (MBH) products, the researchers investigated the Rh-catalyzed cyclization reaction of linear substrate 230 derived from MBH adduct 228 and 3-acetoamido phenol 229. The subsequent intramolecular annulation reaction proceeded in the presence of 2 mol% of Rh catalyst in t-amyl alcohol at 120 °C, producing the corresponding 3,4-oxepino-fused indole 231 in 90% yield. In addition to various aryl groups, alkyl groups are applicable as substituents at the alkyne terminal, demonstrating that this reaction system has wide substrate generality.
 |
| | Scheme 33 Application of the Xu and Liu's synthetic method to the synthesis of 3,4-oxepino-fused indole by Reddy and co-workers. | |
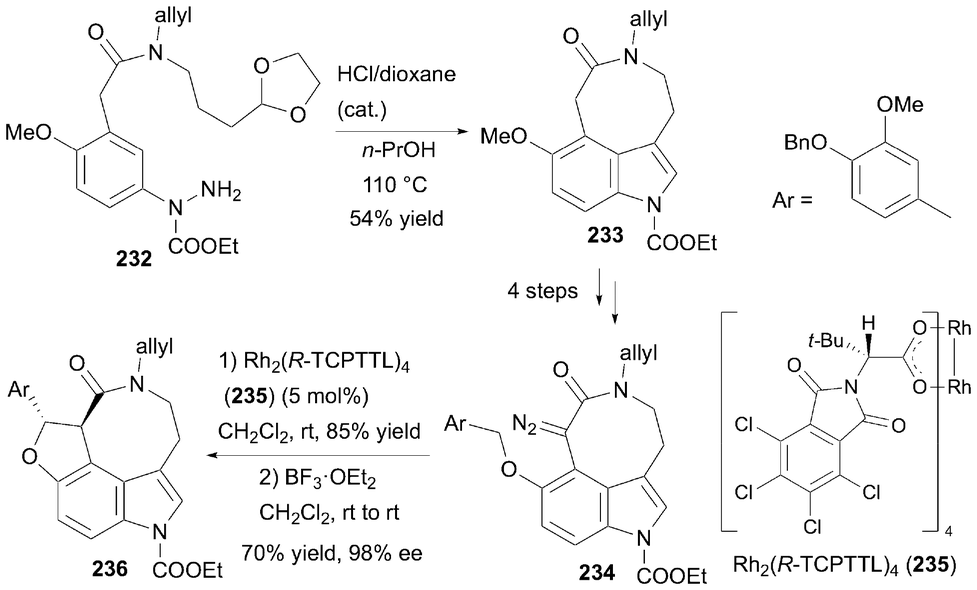
In 2022, Cho et al. succeeded in accomplishing the total synthesis of (+)-decursivine and (+)-serotobenine based on a previously developed method using intramolecular Fisher indole synthesis (Scheme 34).72 Starting from compound 232, intramolecular Fisher indolization was performed in n-PrOH at 110 °C in the presence of a catalytic amount of HCl, resulting in the formation of 3,4-eight-membered ring-fused tricyclic indole derivative 233 in 54% yield. After a 4-step transformation to obtain diazoamide 234, an intramolecular C–H insertion reaction was performed using chiral Rh catalyst 235, successfully constructing the dihydrofuran ring in 85% yield. At this stage, the cis-isomer was favored (cis:trans = 27:1), but treatment with BF3 enabled isomerization to the trans-isomer in 70% yield. Intermediate 236 with 98% ee was subsequently transformed in three steps to yield (+)-serotobenine. Compound 233 was also used in the enantioselective total synthesis of (+)-decursivine using a similar strategy.73
 |
| | Scheme 34 Cho's synthetic method demonstrated in the enentioselective toral synthesis of (+)-serotobenine and (+)-decursivine. | |
8. Type F synthetic methods
8.1. Synthetic method based on the tandem palladium catalysis strategy
Assisted tandem catalysis is defined as a reaction system in which the catalytic cycle switches to another catalytic cycle upon the addition of a trigger reagent. This approach enables sequential transformations within the same reaction vessel, allowing efficient synthesis by shifting the reaction pathways as needed. In 2020, He, Fan and their co-workers succeeded in the rapid construction of 3,4-fused tricyclic indoles based on this strategy (Scheme 35).74 The reaction began with a three-component coupling reaction involving 2-alkynylcyclohexadienimine derivative 237, alkynyl iodide 238, and 2-iodoaniline 239 in the presence of a Pd catalyst and K2CO3 at 50 °C, which initially produced compound 240. After removing acetonitrile, the solvent was replaced with DMF, and sodium formate 241 was added as the trigger reagent. Upon heating to 90 °C, the catalytic cycle switched to the next stage. The reaction proceeds via the oxidative addition of iodoarene to the Pd catalyst, followed by a reaction with sodium formate, which generates palladium hydride species 243via intermediate 242. Next, insertion of the Pd–H bond into the internal alkyne unit yields palladacycle intermediate 244, which undergoes reductive elimination to afford tricyclic intermediate 245. Finally, rearomatization of intermediate 245 furnished 3,4-fused tricyclic indole derivative 246 in 65% yield. In a control experiment, when the reaction was conducted without the addition of sodium formate, 240 was obtained in 74% yield, demonstrating that sodium formate acts as a trigger reagent for this process.
 |
| | Scheme 35 He and Fan's synthetic method based on the tandem Pd catalysis. | |
8.2. Application of the previously developed Type F methods
Zong, Wang and co-workers applied the Rh-catalyzed 3,4-fused tricyclic indole synthesis developed by Miura and Murakami et al.75 as a key step in the synthesis of (+)-isolysergol (Scheme 36a).76 Compound 248, featuring a tetracyclic ergot alkaloid framework, was obtained in 64% yield by heating chiral intermediate 247 in the presence of 1 mol% of Rh2(OCOt-Bu)4, followed by the addition of MnO2. Compound 248 was then converted into (+)-isolysergol via a three-step transformation.
 |
| | Scheme 36 Application of the previously developed Type F methods to synthetic studies of natural products. | |
On the other hand, Harada, Nemoto and their collaborators conducted a synthetic study on dragmacidin E, utilizing their previously developed method.77 Nemoto et al. introduced a Pd-catalyzed cascade process for the synthesis of 3,4-fused tricyclic indoles (Scheme 36b).78 This methodology was successfully applied to linear substrate 249 for the construction of the 3,4-fused tricyclic indole framework. Treatment of 249 with 30 mol% of Pd catalyst in DMSO at 80 °C yielded tricyclic 3-alkylideneindoline derivative 250 in 60% yield. This intermediate was successfully transformed into synthetic intermediate 251, which comprised contiguous stereocenters on the fused seven-membered ring, through a seven-step process that included isomerization of the double bond to form the indole skeleton. The utility of synthesizing 3,4-fused indoles via the acid-promoted isomerization of 3-alkylideneindoline derivatives was also demonstrated by Rawal in 2022.79 In addition, Nakajima, Nemoto and co-workers conducted a detailed mechanistic study of their Pt-catalyzed reaction system for the synthesis of 3,4-fused tricyclic indoles, which were initially developed by the Nemoto group.80 By combining DFT calculations with experimental validation, the researchers obtained an in-depth understanding of the reaction pathways.81
9. Conclusions
This review highlights the advances made in the synthesis of 3,4-fused tricyclic indoles, particularly the advances published since 2018. We had previously11 classified the method of constructing a tricyclic framework based on the sequential building of the indole ring as Type G. To the best of our knowledge, no reports of Type G reactions have been published since 2018. This suggests a shift toward more direct approaches for constructing the target framework, moving away from stepwise synthetic methods.
Many of the reported reactions were designed to synthesize natural product frameworks, and future studies should explore the synthesis of natural products using these methods. Additionally, given the wide range of bioactive natural products containing the 3,4-fused tricyclic indole framework, these reactions have immense potential for application in medicinal chemistry. Representative examples of synthetic pharmaceuticals based on a 3,4-fused tricyclic indole framework include rucaparib and pergolide (Fig. 4). Rucaparib, a poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor,82 has been approved by the FDA as Rubraca for the treatment of ovarian cancer in 2016. Pergolide, a dopamine receptor agonist, is used to treat Parkinson's disease, and mimics the action of dopamine, helping to compensate for the dopamine deficiency characteristic of Parkinson's disease.83 The development of molecules with significant biological activity from such relatively simple molecular structures underscores the potential of 3,4-fused tricyclic indole frameworks for drug discovery. To fully harness the drug discovery potential of this core structure, interdisciplinary research must be promoted alongside synthetic chemistry, including the investigation of the biological activities of synthetic derivatives. In addition, the development of more refined and environmentally friendly synthetic methods that can be scaled up for large-scale industrial production is crucial.
 |
| | Fig. 4 Examples of synthetic pharmaceuticals with 3,4-fused tricyclic indole frameworks. | |
Additionally, reaction development based on reactivity predictions using DFT calculations has been reported.23 While further improvements in predictive accuracy remain a challenge, the approach of virtually designing reactions and validating them through experimental verification is expected to gain increasing attention as a research strategy.
The 3,4-fused tricyclic indole framework is a key structural motif in numerous bioactive natural products and pharmaceuticals and has garnered significant interest from synthetic organic chemists. The development of practical synthetic methods targeting this structure has the potential to contribute significantly to advances in both organic synthesis and medicinal chemistry. We anticipate further progress will be made in this area.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI [Grant Numbers: 23K24004 (T.N.)], JSPS Bilateral Program Number JPJSBP120249908 (T.N.), the FUGAKU Trust for Medicinal Research (T.N.).
References
-
(a) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC.
-
N. R. Tasker and P. Wipf, The Alkaloids: Chemistry and Biology, 2021, vol. 85, p. 1 Search PubMed.
- L. C. Klein-Júnior, S. Cretton, Y. V. Heyden, A. L. Gasper, S. Nejad-Edrahimi, P. Christen and A. T. Henriques, J. Nat. Prod., 2020, 83, 852 CrossRef PubMed.
- C.-X. Jiang, B. Yu, Y.-M. Miao, H. Ren, Q. Xu, C. Zhao, L.-L. Tian, Z.-Q. Yu, P.-P. Zhou, X. Wang, J. Fang, J. Zhang, J. Z. Zhang and Q.-X. Wu, J. Nat. Prod., 2021, 84, 2468 CrossRef CAS PubMed.
- Y. T. H. Lam, J. Hoppe, Q. N. Dang, A. Porzel, A. Soboleva, W. Brandt, R. Rennert, H. Hussain, M. D. Davari, L. Wessjohann and N. Arnold, J. Nat. Prod., 2023, 86, 1373 CrossRef CAS PubMed.
- S. R. McCabe and P. Wipf, Org. Biomol. Chem., 2016, 14, 5894 RSC.
- M. Ito, Y. Tahara and T. Shibata, Chem. – Eur. J., 2016, 16, 5468 CrossRef PubMed.
-
(a) H. Liu and Y. Jia, Nat. Prod. Rep., 2017, 34, 411 RSC;
(b) K. Yuan and Y. Jia, Chin. J. Org. Chem., 2018, 38, 2386 CrossRef CAS.
- R. Connon and P. J. Guiry, Tetrahedron Lett., 2020, 61, 151696 CrossRef CAS.
- L. Fan, X. Zhu, X. Liu, F. He, G. Yang, C. Xu and X. Yang, Molecules, 2023, 28, 1647 CrossRef CAS PubMed.
- T. Nemoto, S. Harada and M. Nakajima, Asian J. Org. Chem., 2018, 7, 1730 CrossRef CAS.
-
(a) Y. Fukui, P. Liu, Q. Liu, Z.-T. He, N.-Y. Wu, P. Tian and G.-Q. Lin, J. Am. Chem. Soc., 2014, 136, 15607 CrossRef CAS PubMed;
(b) S. Y. Hong, J. Jeong and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 2408 CrossRef CAS PubMed;
(c) C. R. Reddy, K. Mallesh, S. Bodasu and R. R. Donthiri, J. Org. Chem., 2020, 85, 7905 CrossRef PubMed.
- C. R. Reddy, P. Sathish, K. Mallesh and Y. L. Prapurna, ChemistrySelect, 2020, 5, 12736 CrossRef.
- J. R. Tuck, L. E. Dunlap and D. E. Olson, J. Org. Chem., 2023, 88, 13712 CrossRef CAS PubMed.
- U. Rathnayake and P. Garner, Org. Lett., 2021, 23, 6756 CrossRef CAS PubMed.
- T. Sheng, C. Ma, G. Zhang, Z. Pan and Z. Liu, J. Nat. Prod., 2022, 85, 1128 CrossRef CAS PubMed.
- J.-M. Ku, B.-S. Jeong, S.-S. Jew and H.-G. Park, J. Org. Chem., 2007, 72, 8115 CrossRef CAS PubMed.
- Z. Xu, W. Hu, Q. Liu, L. Zhang and Y. Jia, J. Org. Chem., 2010, 75, 7626 CrossRef CAS PubMed.
- H. Liu, L. Chen, K. Yuan and Y. Jia, Angew. Chem., Int. Ed., 2019, 58, 6362 CrossRef CAS PubMed.
-
(a) X.-D. An, Z. Wang, Y.-B. Shen, B. Qiu and J. Xiao, J. Org. Chem., 2023, 88, 8791 CrossRef CAS PubMed;
(b) Y.-B. Shen, Q.-H. Zhuang, X.-L. Wang, X.-D. An, B. Oiu, T. Shi and J. Xiao, Green Chem., 2024, 26, 11899 RSC.
- F. Potlitz, G. J. Palm, A. Bodtke, M. Lammers, D. Schade and A. Link, Molecules, 2024, 29, 3064 CrossRef CAS PubMed.
- S. M. Antropov, S. A. Tokmacheva, I. I. Levina, O. A. Ivanova and I. V. Trushkov, Adv. Synth. Catal., 2024, 366, 2784 CrossRef CAS.
- T. Phumjan, T. Yazawa, S. Harada and T. Nemoto, Org. Lett., 2025, 27, 1549 CrossRef CAS PubMed.
- W. Oppolzer, J. I. Grayson, H. Wegmann and M. Urrea, Tetrahedron, 1983, 39, 3695 CrossRef CAS.
- P. C. Behera, B. Anilkumar, J. B. Nanubolu and S. Suresh, Org. Lett., 2024, 26, 8654 CrossRef CAS PubMed.
- A. Regni, F. Bartoccini and G. Piersanti, Beilstein J. Org. Chem., 2023, 19, 918 CrossRef PubMed.
- S. Harada, M. Yanagawa and T. Nemoto, ACS Catal., 2020, 10, 11971 CrossRef CAS.
- Y. Ge, H. Wang, H.-N. Wang, S.-S. Yu, R. Yang, X. Chen, Q. Zhao and G. Chen, Org. Lett., 2021, 23, 370 CrossRef CAS PubMed.
- M. S. Sherikar, R. Devarajappa and K. R. Prabhu, J. Org. Chem., 2020, 85, 5516 CrossRef CAS PubMed.
- B. Champciaux, C. Raynaud, A. Viljoen, L. Chene, J. Thibonnet, S. P. Vincent, L. Kremer and E. Thiery, Bioorg. Med. Chem., 2021, 43, 116248 CrossRef CAS PubMed.
- K.-W. Chiu, Y.-H. Tseng, Y.-X. Li and R.-J. Chein, Org. Lett., 2023, 25, 3456 CrossRef CAS PubMed.
- T. Sheng, C. Ma, G. Zhang, X. Pan and Z. Liu, J. Nat. Prod., 2022, 85, 1128 CrossRef CAS PubMed.
- Z. Xu, W. Hu, Q. Liu, L. Zhang and Y. Jia, J. Org. Chem., 2010, 75, 7626 CrossRef CAS PubMed.
-
(a) S. Romanini, E. Galletti, L. Caruana, A. Mazzanti, F. Himo, S. Santoro, M. Fochi and L. Bernardi, Chem. – Eur. J., 2015, 21, 17578 CrossRef CAS PubMed;
(b) C. Despotopoulou, S. C. McKeon, R. Connon, V. Coeffard, H. Muller-Bunz and P. J. Guiry, Eur. J. Org. Chem., 2017, 6734 CrossRef CAS;
(c) J.-Q. Chen, Y. Mi, Z.-F. Shi and X.-P. Cao, Org. Biomol. Chem., 2018, 16, 3801 RSC.
- R. Connon and P. J. Guiry, Eur. J. Org. Chem., 2019, 5950 CrossRef CAS.
- B.-Y. Xue, C.-Y. Hou, X.-B. Wang, M.-S. Xie and H.-M. Guo, Org. Chem. Front., 2023, 10, 1910 RSC.
- T. Takeda, S. Harada, A. Okabe and A. Nishida, J. Org. Chem., 2018, 83, 11541 CrossRef CAS PubMed.
- Z.-H. Wang, Z.-F. Xu, J. Feng and M. Yu, Eur. J. Org. Chem., 2024, e202400532 CrossRef CAS.
- For reviews on the donor-acceptor cyclopropanes, see:
(a) K. Ghosh and S. Das, Org. Biomol. Chem., 2021, 19, 965 RSC;
(b) F. Doraghi, S. Karimian, O. H. Qareaghaj, M. J. Karimi, B. Larijani and M. Mahdavi, J. Organomet. Chem., 2004, 1005, 122963 CrossRef.
- B. Q. Li, Z.-W. Qiu, A.-J. Ma, J.-B. Peng, N. Feng, J.-Y. Du, H.-P. Pan, X.-Z. Zhang and X.-T. Xu, Org. Lett., 2020, 22, 1903 CrossRef CAS PubMed.
- S. R. Cochrane and M. A. Kerr, Org. Lett., 2022, 24, 5509 CrossRef CAS PubMed.
- S. Kar, P. K. Maharana, T. Punniyamurthy and V. Trivedi, Org. Lett., 2023, 25, 8850 CrossRef CAS PubMed.
- F. Bartoccini and G. Piersanti, Synthesis, 2021, 1396 CAS.
- Y.-A. Zhang, Q. Liu, C. Wang and Y. Jia, Org. Lett., 2013, 15, 3662 CrossRef CAS PubMed.
- F. Bartoccini, A. Regni, M. Retini and G. Piersanti, Org. Biomol. Chem., 2021, 19, 2932 RSC.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed.
- F. Bartoccini, A. Regni, M. Retini and G. Piersanti, Eur. J. Org. Chem., 2022, e202200315 CrossRef CAS.
- F. Diotallevi, F. Bartoccini and G. Piersanti, Eur. J. Org. Chem., 2024, e202400035 CrossRef CAS.
- G. Leoni, F. Bartoccini and G. Piersanti, Adv. Synth. Catal., 2024, 366, 1397 CrossRef CAS.
- N. R. Tasker and P. Wipf, Org. Lett., 2022, 24, 7255 CrossRef PubMed.
- B. J. Knight, R. C. Harbit and J. M. Smith, J. Org. Chem., 2023, 88, 2158 CrossRef CAS PubMed.
- J. Brauer, R. Wiechert, A. Hahn and T. Opatz, Org. Lett., 2024, 26, 4314 CrossRef CAS PubMed.
- W.-L. Yang, T. Ni and W.-P. Deng, Org. Lett., 2021, 23, 588 CrossRef CAS PubMed.
- L. Xiao, B. Li, F. Xiao, C. Fu, L. Wei, Y. Dang, X.-Q. Dong and C.-J. Wang, Chem. Sci., 2022, 13, 4801 RSC.
- S. Chen, P. Ravichandiran, A. El-Harairy, Y. Queneau, M. Li and Y. Gu, Org. Biomol. Chem., 2019, 17, 5982 RSC.
- X. Cai, A. Tohti, C. Ramirez, H. Harb, J. C. Fettinger, H. P. Hratchian and B. J. Stokes, Org. Lett., 2019, 21, 1574 CrossRef CAS PubMed.
- A. Gogoi, R. Chouhan and S. K. Das, Org. Lett., 2025, 27, 2461 CrossRef CAS PubMed.
- Y. He, L. Song, C. Liu, D. Wu, Z. Li, L. Van Meervelt and E. V. Van der Eycken, J. Org. Chem., 2020, 85, 15092 CrossRef CAS PubMed.
- T. Abe and K. Yamada, J. Nat. Prod., 2017, 80, 241 CrossRef CAS PubMed.
- T. Abe, T. Haruyama and K. Yamada, Synthesis, 2017, 4141 CrossRef CAS.
- S. Wang, B. Yu and H.-M. Liu, Org. Lett., 2021, 23, 42 CrossRef CAS PubMed.
- P. T. Parvatkar, E. S. Smotkin and R. Manetsch, Sci. Rep., 2021, 11, 19915 CrossRef CAS PubMed.
- L. Fan, J. Hao, J. Yu, X. Ma, J. Liu and X. Luan, J. Am. Chem. Soc., 2020, 142, 6698 CrossRef CAS PubMed.
- J. Wu, L. Li, M. Liu, L. Bai and X. Luan, Angew. Chem., Int. Ed., 2022, 61, e202113820 CrossRef CAS PubMed.
- C. Cheng, X. Zuo, D. Tu, B. Wan and Y. Zhang, Org. Lett., 2020, 22, 4985 CrossRef CAS PubMed.
- L. Zhang, J. Chen, T. Zhong, X. Zheng, J. Zhou, X. Jiang and C. Yu, J. Org. Chem., 2020, 85, 10823 CrossRef CAS PubMed.
- B.-S. Zhang, F. Wang, X.-Y. Gou, Y.-H. Yang, W.-Y. Jia, Y.-M. Liang, X.-C. Wang, Y. Li and Z.-J. Quan, Org. Lett., 2021, 23, 7518 CrossRef CAS PubMed.
- For reviews on the palladium/norbornene chemistry, see:
(a) J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863 CrossRef CAS PubMed;
(b) N. Della Ca’, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389 CrossRef PubMed;
(c) J. Wang and G. Dong, Chem. Rev., 2019, 119, 7478 CrossRef CAS PubMed.
- T. Tawatari, R. Kato, R. Kudo, K. Takasu and H. Takikawa, Angew. Chem., Int. Ed., 2023, 62, e202300907 CrossRef CAS PubMed.
- X. Zhang, Y. Li, H. Shi, L. Zhang, S. Zhang, X. Xu and Q. Liu, Chem. Commun., 2014, 50, 7306 RSC.
- C. R. Reddy, P. Sathish and R. R. Valleti, ChemistrySelect, 2019, 4, 8229 CrossRef CAS.
- I.-K. Park, J. Park and C.-G. Cho, Angew. Chem., Int. Ed., 2012, 51, 2496 CrossRef CAS PubMed.
- D.-H. Kim, H.-M. Kim, J.-S. Lim, H.-W. Lee and C.-G. Cho, Org. Lett., 2022, 24, 2873 CrossRef CAS PubMed.
- W. Wang, M. Yang, D. Han, Q. He and R. Fan, Adv. Synth. Catal., 2020, 362, 1281 CrossRef CAS.
- T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272 CrossRef CAS PubMed.
- J.-T. Lu, Y. Zong, X. Yue and J. Wang, J. Org. Chem., 2023, 88, 8761 CrossRef CAS PubMed.
- D. Choi, N. Takahashi, H. Maruoka, S. Harada, A. Nastke, H. Gröger and T. Nemoto, J. Org. Chem., 2023, 88, 7674 CrossRef CAS PubMed.
- S. Nakano, N. Inoue, Y. Hamada and T. Nemoto, Org. Lett., 2015, 17, 2622 CrossRef CAS PubMed.
- F. Taenzler, J. Xu, S. Athe and V. H. Rawal, Org. Lett., 2022, 24, 8109 CrossRef CAS PubMed.
- Y. Suzuki, Y. Tanaka, S. Nakano, K. Dodo, N. Yoda, K. Shinohara, K. Kita, A. Kaneda, M. Sodeoka, Y. Hamada and T. Nemoto, Chem. – Eur. J., 2016, 22, 4418 CrossRef CAS PubMed.
- T. Kuribara, M. Nakajima and T. Nemoto, J. Org. Chem., 2021, 86, 9670 CrossRef CAS PubMed.
-
(a) L. Porcelli, A. Quatrale, P. Mantuano, M. G. Leo, N. Silvestris, J. F. Rolland, E. Carioggia, M. Lioce, A. Paradiso and A. Azzariti, Mol. Oncol., 2013, 7, 308 CrossRef CAS PubMed;
(b) R. Plummer, P. Lorigan, N. Steven, L. Scott, M. R. Middleton, R. H. Wilson, E. Mulligan, N. Curtin, D. Wang, R. Dewji, A. Abbattista, J. Gallo and H. Calvert, Cancer Chemother. Pharmacol., 2013, 71, 1191 CrossRef CAS PubMed.
- For a review on pergolide, see: A. Markham and P. Benfield, CNS Drugs, 1997, 7, 328 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shingo
Harada
*a,
Shingo
Harada