
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and stability of collagen mimetic peptides featuring δ-heteroatom-substituted prolines†
Taylor A.
Gerrein‡
,
Madison M.
Wright‡
,
Natalia
Cano-Sampaio
and
Juan R.
Del Valle
 *
*
Department of Chemistry & Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: jdelval2@nd.edu
First published on 25th February 2025
Abstract
We describe the first investigation of collagen mimetic peptides harboring proline surrogates with heteroatoms at the δ-position. While dehydro-δ-azaproline and (N-methyl)-δ-azaproline destabilized the parent structure, replacement of the Xaa proline residue with δ-oxaproline resulted in a faster-folding collagen mimetic peptide with equivalent thermal stability.
The collagen family of proteins is the most abundant component of mammalian connective tissue and is composed of polyproline II (PPII) chains that assemble into a right-handed triple helix.1,2 The single chains in collagen feature a repeating Gly–Xaa–Yaa tripeptide motif, where Xaa and Yaa are most often proline (Pro; P) and its post-translationally modified variant 4(R)-hydroxyproline (Hyp; O), respectively.3 Efforts to study the impact of residue substitution on collagen stability have typically relied on collagen mimetic peptides (CMPs) composed of these tripeptide repeats. Information gleaned from decades of research using CMPs has enabled the development of biomaterials, imaging and diagnostic agents, and nanoscale assemblies with unique properties.3–7 The rapidly expanding set of unnatural amino acids that can be incorporated into proteins offers continued opportunity for the design of novel functional collagen mimetics.
As the only cyclic residue among the 20 canonical amino acids, Pro plays a unique role in stabilizing the collagen triple helix. The pyrrolidine ring enforces backbone ϕ and ψ constraints favoring PPII conformation while the tertiary amide it forms exhibits a reduced propensity to adopt the trans rotamer (ω) geometry required in the PPII fold.8,9 These characteristics suggest that cyclic analogues of Pro with enhanced trans amide propensity will enhance collagen stability. Indeed, replacement of Pro (at Xaa) with pyrrolidine-substituted analogues has been extensively studied, with γ-halo,3,10 γ-alkoxy,3 γ-(acyl)amino,11,12 γ-(acyl)aza,13 γ-thia,14 and α-azaprolines15 each maintaining or increasing the thermal stability of CMPs in the Xaa position. Interestingly, several N-alkyl glycine (peptoid) residues also stabilize CMPs despite significantly increased cis amide rotamer propensity relative to Pro.16,17 In these cases, enhanced stability was attributed to the strong PPII-promoting effect of the acyclic peptoid residues, thus reducing the entropic penalty of CMP folding and outweighing the cost of reduced trans rotamer bias.
Our long-standing interest in N-heteroatom-substituted peptides prompted us to investigate the impact of amide-hydroxamate and amide−hydrazide replacement within the collagen backbone.18 We specifically sought to study CMPs featuring cyclic δ-heteroatom-substituted Pro analogues that retain native-like ϕ and ψ constraint as well as increased trans rotamer propensity (Fig. 1).
 | ||
| Fig. 1 Structures of δ-heteroatom-substituted proline surrogates, crystal structure of a CMP triple helix (pdb 3B0S), and PPII strand alignment. Proposed Pro11 substitution sites are highlighted in red. | ||
Although δ-azaproline (aPro) was initially deemed a suitable Xaa positional probe, we previously observed that aPro-containing peptides can undergo rapid air oxidation to their dehydro-δ-azaproline (ΔaPro) analogues.19 Our finding that the unpuckered ΔaPro readily adopts PPII backbone conformation led us to instead explore its incorporation into a collagen folding model. We also sought to investigate (N-methyl)-δ-azaproline ((Me)aPro) and δ-oxaproline (oPro), two additional Pro surrogates incapable of undergoing air oxidation. The (Me)aPro monomer has not previously been described. Although oPro has been widely employed in protein chemical ligation20–22 and is found in some biologically active peptidomimetics,23–25 it has not yet been explored in a model of peptide or protein folding.
Two orthogonally-protected N-amino dipeptide building blocks were synthesized for the incorporation of ΔaPro and (Me)aPro into CMPs (Scheme 1). Sidechain redox adjustment of 1 was followed by dimethyl acetal protection to afford 2 in 55% overall yield. Fmoc deprotection and electrophilic amination of the resulting primary amine with t-butyl-diethyloxaziridine tricarboxylate (TBDOT)26,27 provided protected α-hydrazino ester 3. Nα-Acylation with Fmoc-protected Gly acid chloride and subsequent benzyl ester hydrogenolysis then gave N-aminated aminodimethoxybutyric acid (aAdb) derivative 5 in 56% yield over 2 steps. Synthesis of the (Me)aPro dipeptide building block proceeded via methylation under Mitsunobu conditions followed by tandem deprotection and reduction in the presence of TFA and triethylsilane to give 6. Benzyl ester hydrogenolysis afforded (Me)aPro dipeptide building block 7 in 89% yield.
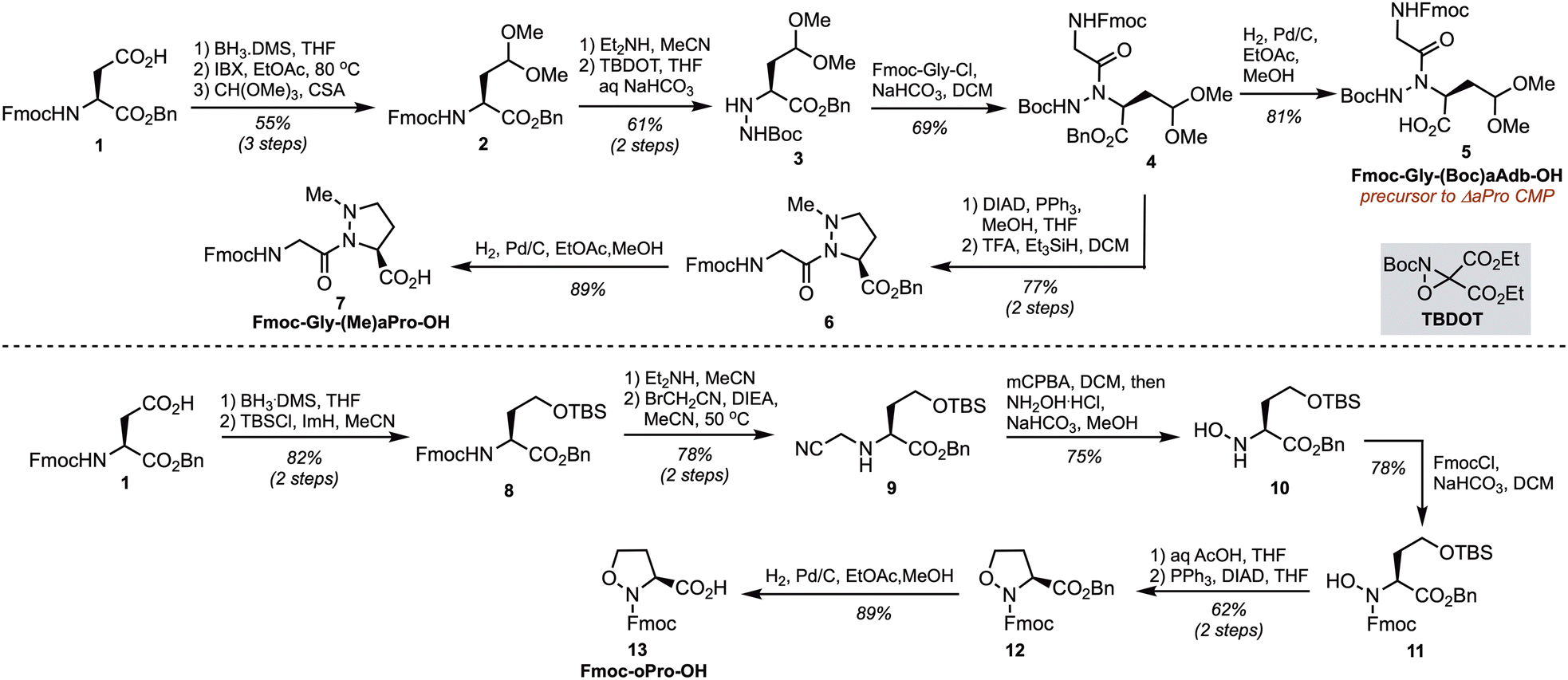 | ||
| Scheme 1 Synthesis of ΔaPro, (Me)aPro, and oPro building blocks for SPPS. | ||
The oPro residue has been synthesized previously in enantiopure form via an auxiliary-mediated dipolar cycloaddition route20,28 and, more recently, through catalytic asymmetric conjugate addition to generate a key β-amino aldehyde intermediate.29,30 As shown in Scheme 1, we developed a chiral pool approach toward Fmoc-oPro-OH from commercially available aspartate derivative 1. This route commenced with side chain reduction, silyl etherification, and N-hydroxylation using Fukuyama's procedure31 to provide intermediate 10. Fmoc-protection, mild silyl ether cleavage, and Mitsunobu cyclization provided protected oPro 12 in 48% yield over 3 steps. Finally, benzyl ester hydrogenolysis gave monomer 13 suitable for Fmoc SPPS.
To assess the impact of ΔaPro, (Me)aPro, and oPro on CMP stability, we chose a parent sequence comprised of seven Gly-Pro-Hyp (GPO) repeats. This peptide features N-terminal acetylation and a C-terminal amide, and exhibits a reported melting temperature of approximately 53 °C.16,32 CMPs with substitutions at position 11 (Xaa) were synthesized on Rink amide MBHA resin using standard Fmoc-based protocols and HCTU/NMM activation (Table 1). ΔaPro11-CMP and (Me)aPro11-CMP were prepared by incorporation of dipeptide building blocks 5 and 7, respectively. Formation of the dihydropyrazole ring of ΔaPro occurred upon acidic deprotection and cleavage from the resin. In the case of oPro11-CMP, condensation with the subsequent Gly10 residue was carried out using pre-formed Fmoc-protected Gly acid chloride to ensure complete reaction with the isoxazolidine nitrogen. All peptides were purified by RP-HPLC and their identities were confirmed by HRMS.
| Peptide | Sequence | Yield (%) |
|---|---|---|
| Pro11-CMP | Ac-(GPO)3-GPO-(GPO)3-NH2 | 5 |
| ΔaPro11-CMP | Ac-(GPO)3-G[ΔaPro]O-(GPO)3-NH2 | 12 |
| (Me)aPro11-CMP | Ac-(GPO)3-G[(Me)aPro]O-(GPO)3-NH2 | 16 |
| oPro11-CMP | Ac-(GPO)3-G[oPro]O-(GPO)3-NH2 | 17 |
All Pro11-substituted analogues were first analyzed by far-UV CD at pH 7.4 to compare their spectral signatures to that of the parent peptide. As shown in Fig. 2A, the CD spectra of oPro11-CMP and (Me)aPro11-CMP exhibited significant overlap, including less intense and red-shifted negative bands (∼203–205 nm) as well as diminished maxima (∼225 nm) relative to Pro11-CMP. The spectrum of ΔaPro11-CMP also showed the emergence of a new minimum at 239 nm. Although these differences might indicate a destabilized triple helix, modifications to prolyl amide bonds within PPII folds have been shown to give rise to unusual CD signatures.33–35 To investigate this, we synthesized single-strand PPII model octapeptides36 incorporating each of the δ-heteroatom proline surrogates. As shown in Fig. 2B, each variant displayed a significantly altered spectral signature, with ΔaPro4-PP2 again showing the emergence of a new minimum band near 235 nm. These spectra are in stark contrast to the prototypical random coil signature obtained upon substitution of Pro4 for a PPII-disrupting Gly residue.37 These results suggest unique chromophoric properties associated with replacement of the native tertiary amide with acyl hydrazone, hydrazide, or hydroxamate bonds in the CMP series.
 | ||
| Fig. 2 Far UV CD spectra of (A) CMPs analyzed at 150 μM in aq PBS (pH 7.4) and (B) PP2 octapeptides analyzed at 150 μM in 5 mM aq Na3PO4, 25 mM KF (pH 7.0). | ||
Thermal denaturation was then carried out for each CMP by monitoring mean residue ellipticity (MRE) at 225 nm as a function of temperature and fitting of the data to a two-state unfolding model.38 The parent peptide, Pro11-CMP, exhibited a clear cooperative melting transition and Tm of 53.2 °C (Fig. 3), in agreement with previously reported values.16,32 Each of the substituted variants also showed cooperative unfolding despite lacking a clear and pronounced maximum band at the tracked wavelength. The overall changes in mean residue ellipticity for ΔaPro11-CMP and oPro-CMP across the temperature range were roughly equivalent to that of Pro11-CMP, suggesting that these variants adopt a collagen fold. While substitution of Pro11 for (Me)aPro resulted in severe destabilization of the triple helix (ΔTm = −18.4 °C), ΔaPro11-CMP exhibited only a modest reduction in thermal stability (ΔTm = −2.3 °C) relative to the parent peptide. oPro11-CMP exhibited the same thermal stability as Pro11-CMP (Tm = 54.0 °C).
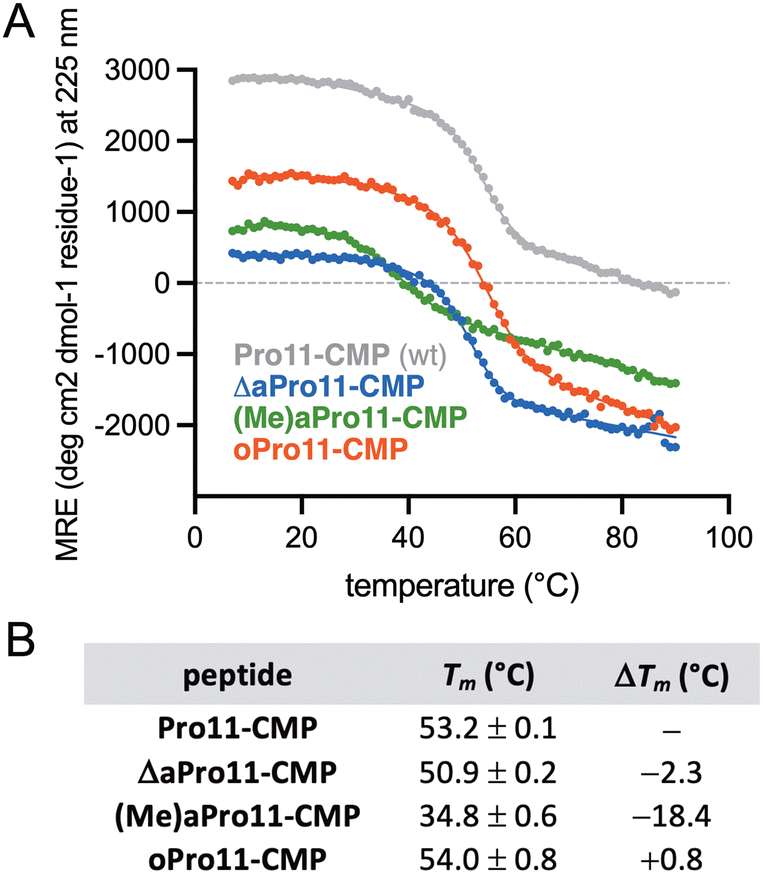 | ||
| Fig. 3 Thermal denaturation of CMPs at 150 μM in aq PBS (pH 7.4). (A) Representative plot of MRE at 225 nm as a function of temperature determined by CD. (B) Melting temperatures (Tm) and change in Tm (ΔTm) relative to Pro11-CMP derived from non-linear regression. Tm values represent the mean and standard deviation from two separate experiments. | ||
Based on our previous results with backbone N-oxidized peptides, we expected that the N-acyl bonds formed by oPro, (Me)aPro, and ΔaPro would exhibit increased trans rotamer population relative to Pro.19 To determine if CMP stability trends in this series correlate with trans rotamer propensity, we synthesized N-acetyl methyl esters of each monomer and calculated trans/cis equilibrium constants (Kt/c) on the basis of 1H NMR integrations in D2O (Fig. 4). The trans propensity of the hydroxamate bond in Ac-oPro-OMe (Kt/c = 6.7) was slightly higher than that of the Pro amide19 (Kt/c = 4.5). More dramatic increases in trans rotamer population were observed for Ac-(Me)aPro-OMe (Kt/c = 8.3) and Ac-ΔaPro-OMe19 (Kt/c = 44). Despite these increases, ΔaPro and (Me)aPro destabilized the collagen model peptide, whereas oPro was accommodated in the Xaa position without energetic penalty. These results suggest that the hydrazone and N-methyl hydrazide moieties may engage in disruptive interactions within the triple helix or preclude the adoption of optimal ϕ and ψ backbone torsions.
 | ||
| Fig. 4 Trans/cis rotamer structures and equilibrium constants (Kt/c) at 25 °C in D2O (derived from 1H NMR peak integrations). | ||
To further parse the factors that allow for accommodation of oPro, we synthesized a CMP variant that harbors (O-methyl)-N-hydroxyalanine at position 11 (see ESI† for details). The (Me)hAla residue serves as an isoelectronic analogue of oPro that lacks cyclic constraint of the ϕ and ψ backbone torsions. As shown in Fig. 5, thermal denaturation of (Me)hAla11-CMP revealed a Tm of 48.8 °C, which was ∼5 °C lower than that of oPro11-CMP and Pro11-CMP. This result demonstrates that the presence of a hydroxamate bond is not sufficient to fully maintain thermal stability and that 5-membered cyclic constraint is important for folding.
 | ||
| Fig. 5 Representative plot of MRE at 225 nm as a function of temperature for (Me)hAla11-CMP and oPro11-CMP determined by CD (150 μM in aq PBS at pH 7.4). Tm values represent the mean and standard deviation from 2 separate experiments (SD for (Me)hAla11-CMP was ±0.5 °C). | ||
Finally, we explored how δ-heteroatom substitution in our most stable variant affected the rate of triple helix assembly. The hydroxamate bond in oPro-CMP was expected to exhibit a lower isomerization barrier than the Pro tertiary amide due to the electron-withdrawing Oδ. Since the overall rate of collagen folding is limited by prolyl cis–trans isomerization, we hypothesized that oPro11-CMP would refold faster than Pro11-CMP.39,40 Hysteresis experiments, wherein the peptides were denatured and re-cooled while monitoring by CD (at 225 nm), suggested enhancement in the refolding rate upon Pro11 → oPro11 substitution (Fig. 6A). We then quantified the relative rates of refolding using a temperature jump experiment. After holding at 95 °C for 20 min the peptides were quickly reintroduced into a pre-cooled cuvette and the recovery of ellipticity monitored by CD (at 7 °C). The time required to achieve a 0.5 folded fraction (t1/2) was determined by fitting to a 3rd order kinetic model.40 As shown in Fig. 6B, oPro11-CMP exhibited slightly faster refolding than Pro-11-CMP (10.9 vs. 17.1 min), consistent with the hysteresis data.
 | ||
| Fig. 6 Refolding kinetics of Pro11-CMP and oPro11-CMP. (A) Hysteresis plots of MRE at 225 nm upon heating and re-cooling as determined by CD. (B) Plot of folded fraction as a function of time upon re-cooling. Third-order non-linear fit (black line) and time of recovery for a 0.5 folded fraction (t1/2) are indicated on each graph. Values for t1/2 represent the mean and standard deviation from 2 separate experiments. | ||
Conclusions
In summary, we explored the impact of δ-heteroatom-substituted proline surrogates on collagen triple helix folding and stability. As part of this work, we developed syntheses of ΔaPro, (Me)aPro, and oPro building blocks suitable for incorporation into peptides by SPPS. Thermal denaturation studies showed that introduction of oPro at the Xaa position of a model CMP affords an analogue that is isoenergetic with the parent peptide. In contrast, ΔaPro and (Me)aPro substitution at Xaa led to compromised thermal stability, despite the enhanced trans amide rotamer propensities of these residues relative to Pro. The amide-hydroxamate substitution associated with oPro incorporation also resulted in accelerated triple helix assembly. This study lays the groundwork for further investigation of oPro as a conformational surrogate of Pro in related models of protein folding.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the National Science Foundation (CHE2109008).References
- G. N. Ramachandran and G. Kartha, Nature, 1954, 176, 593–595 CrossRef PubMed.
- P. Fratzl, Collagen: Structure and Mechanics, Springer, New York, NY, 2008 Search PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- J. A. Fallas, L. E. R. O'Leary and J. D. Hartgerink, Chem. Soc. Rev., 2010, 39, 3510–3527 RSC.
- V. Kubyshkin, Org. Biomol. Chem., 2019, 17, 8031–8047 RSC.
- S. A. H. Hulgan and J. D. Hartgerink, Biomacromolecules, 2022, 23, 1475–1489 CrossRef CAS PubMed.
- J. Xiao, Collagen mimetic peptides and their biophysical characterization, Springer, Singapore, Singapore, 2022nd edn, 2024 Search PubMed.
- D. E. Stewart, A. Sarkar and J. E. Wampler, J. Mol. Biol., 1990, 214, 253–260 CrossRef CAS PubMed.
- M. W. MacArthur and J. M. Thornton, J. Mol. Biol., 1991, 218, 397–412 CrossRef CAS PubMed.
- Y. Nishi, S. Uchiyama, M. Doi, Y. Nishiuchi, T. Nakazawa, T. Ohkubo and Y. Kobayashi, Biochemistry, 2005, 44, 6034–6042 CrossRef CAS PubMed.
- R. S. Erdmann and H. Wennemers, J. Am. Chem. Soc., 2012, 134, 17117–17124 CrossRef CAS PubMed.
- J. Egli, C. Siebler, M. Köhler, R. Zenobi and H. Wennemers, J. Am. Chem. Soc., 2019, 141, 5607–5611 CrossRef CAS PubMed.
- M. R. Aronoff, J. Egli, M. Menichelli and H. Wennemers, Angew. Chem., Int. Ed., 2019, 58, 3143–3146 CrossRef CAS PubMed.
- T.-L. Hsu and J.-C. Horng, Protein Sci., 2023, 32, e4650 CrossRef CAS PubMed.
- Y. Zhang, R. M. Malamakal and D. M. Chenoweth, Angew. Chem., Int. Ed., 2015, 54, 10826–10832 CrossRef CAS PubMed.
- J. L. Kessler, G. Kang, Z. Qin, H. Kang, F. G. Whitby, T. E. Cheatham 3rd, C. P. Hill, Y. Li and S. M. Yu, J. Am. Chem. Soc., 2021, 143, 10910–10919 CrossRef CAS PubMed.
- R. Qiu, X. Li, K. Huang, W. Bai, D. Zhou, G. Li, Z. Qin and Y. Li, Nat. Commun., 2023, 14, 7571 CrossRef CAS PubMed.
- I. J. Angera, M. M. Wright and J. R. Del Valle, Acc. Chem. Res., 2024, 57, 1287–1297 CrossRef CAS PubMed.
- Y. M. Elbatrawi, K. P. Pedretty, N. Giddings, H. L. Woodcock and J. R. Del Valle, J. Org. Chem., 2020, 85, 4207–4219 CrossRef CAS PubMed.
- A. O. Ogunkoya, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9693–9697 CrossRef CAS PubMed.
- T. G. Wucherpfennig, F. Rohrbacher, V. R. Pattabiraman and J. W. Bode, Angew. Chem., Int. Ed., 2014, 53, 12244–12247 CrossRef CAS PubMed.
- J. Farnung, H. Song and J. W. Bode, Methods Mol. Biol., 2021, 2355, 151–162 CrossRef CAS PubMed.
- A. Vasella, R. Voeffray, J. Pless and R. Huguenin, Helv. Chim. Acta, 1983, 66, 1241–1252 CrossRef CAS.
- V. Günzler, D. Brocks, S. Henke, R. Myllylä, R. Geiger and K. I. Kivirikko, J. Biol. Chem., 1988, 263, 19498–19504 CrossRef.
- K. Abrahamsson, P. Andersson, J. Bergman, U. Bredberg, J. Brånalt, A.-C. Egnell, U. Eriksson, D. Gustafsson, K.-J. Hoffman, S. Nielsen, I. Nilsson, S. Pehrsson, M. O. Polla, T. Skjaeret, M. Strimfors, C. Wern, M. Ölwegård-Halvarsson and Y. Örtengren, MedChemComm, 2016, 7, 272–281 RSC.
- A. Armstrong, L. H. Jones, J. D. Knight and R. D. Kelsey, Org. Lett., 2005, 7, 713–716 CrossRef CAS PubMed.
- B. M. Rathman, J. L. Rowe and J. R. Del Valle, Methods Enzymol., 2021, 656, 271–294 CAS.
- A. Vasella and R. Voeffray, J. Chem. Soc., Chem. Commun., 1981, 97–98 RSC.
- T. J. Harmand, C. E. Murar, H. Takano and J. W. Bode, Org. Synth., 2019, 142–156 Search PubMed.
- C. E. Murar, T. J. Harmand and J. W. Bode, Bioorg. Med. Chem., 2017, 25, 4996–5001 CrossRef CAS PubMed.
- H. Tokuyama, T. Kuboyama, A. Amano, T. Yamashita and T. Fukuyama, Synthesis, 2000, 1299–1304 CrossRef CAS.
- T. Fiala, E. P. Barros, M.-O. Ebert, E. Ruijsenaars, S. Riniker and H. Wennemers, J. Am. Chem. Soc., 2022, 144, 18642–18649 CrossRef CAS PubMed.
- Z. Miao and J. P. Tam, J. Am. Chem. Soc., 2000, 122, 4253–4260 CrossRef CAS.
- C. Cayrou, A. Walrant, D. Ravault, K. Guitot, S. Noinville, S. Sagan, T. Brigaud, S. Gonzalez, S. Ongeri and G. Chaume, Chem. Commun., 2024, 60, 8609–8612 RSC.
- R. W. Newberry, B. VanVeller and R. T. Raines, Chem. Commun., 2015, 51, 9624–9627 RSC.
- A. M. Brown and N. J. Zondlo, Biochemistry, 2012, 51, 5041–5051 CrossRef CAS PubMed.
- B. H. Rajewski, M. M. Wright, T. A. Gerrein and J. R. Del Valle, Org. Lett., 2023, 25, 4366–4370 CrossRef CAS PubMed.
- D. Shortle, A. K. Meeker and E. Freire, Biochemistry, 1988, 27, 4761–4768 CrossRef CAS PubMed.
- J. M. Davis and H. P. Bächinger, J. Biol. Chem., 1993, 268, 25965–25972 CrossRef CAS PubMed.
- K. Mizuno, S. P. Boudko, J. Engel and H. P. Bächinger, Biophys. J., 2010, 98, 3004–3014 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds. See DOI: https://doi.org/10.1039/d5ob00176e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |