
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DMSO enhances the biosynthesis of epoxyquinols in Pestalotiopsis sp. (strain IQ-011) and yields new [4 + 2] cycloaddition dimers†
Enrique
Aguilar-Ramírez
 a,
José
Rivera-Chávez
*a,
Mario Yair
Miranda-Rosas
a and
Diego
Martínez-Otero
b
a,
José
Rivera-Chávez
*a,
Mario Yair
Miranda-Rosas
a and
Diego
Martínez-Otero
b
aDepartment of Natural Products, Institute of Chemistry, Universidad Nacional Autónoma de México, Mexico City, 04510, Mexico. E-mail: jrivera@iquimica.unam.mx
bJoint Research Center for Sustainable Chemistry UAEM-UNAM, Toluca, 50200, Mexico
First published on 15th April 2025
Abstract
Pestalotiopsis sp. (strain IQ-011) produces cuautepestalorin (10), a 7,8-dihydrochromene-oxoisochromane adduct featuring a spiro-polycyclic (6/6/6/6/6/6) ring system. Additionally, it yields its proposed biosynthetic precursors: cytosporin M (1) and oxopestalochromane (11) when cultured under standard conditions (fermentation in solid media). Following an OSMAC approach guided by metabolomic studies (PCA and molecular networks), it was established that the epigenetic modulator DMSO dramatically increases the production of 1 up to 50 times according to feature-based molecular networking (FBMN) analysis, and triggers the production of other derivatives from the epoxyquinol family. Chemo-targeted isolation resulted in the discovery of four new compounds: 19-hydroxycytosporin M (2) and three [4 + 2] cycloaddition products: ent-eutyscoparol J (4), ent-pestaloquinol A (6) and ent-pestaloquinol B (8). The structures of all isolates were established based on spectroscopic, spectrometric, chiroptical, and X-ray diffraction analyses. This study demonstrates the potential of combining metabolomic tools with DMSO as an epigenetic modulator to enhance fungal metabolite diversity and highlights the importance of chiroptical methods for accurate compound identification.
Introduction
Fungi of the Pestalotiopsis genus hold great potential for producing specialized metabolites with diverse bioactivities, including cytotoxic/antitumor, antimicrobial, anti-inflammatory, antiviral, antifungal, and antimalarial properties. These compounds often display remarkable structural diversity and, in some cases, unusually complex architectures.1–5 Among these elaborated structures, epoxyquinols have attracted the attention of various research groups focusing on both organic synthesis and biosynthesis.6–18 The interest in these compounds stems from their highly functionalized framework, which displays a high degree of reactivity. This allows for numerous intermolecular combinations, leading to the production of fascinating molecules that can be further modified and refined through various chemical reactions, both enzymatic and non-enzymatic.11,19 Notably, prenylation and the Diels–Alder-type cycloaddition are of particular interest.11,20Natural prenyl-epoxyquinols are recognized for their antiangiogenic, anti-inflammatory, and cytotoxic activities, as well as for their role as inhibitors of angiotensin II.21–30 Previously, our group reported the isolation of cuautepestalorin (10) from Pestalotiopsis sp. (strain IQ-011), a unique and elaborated prenyl-epoxyquinol metabolite resulting from the heterodimerization between oxopestalochromane (11) and cytosporin M (1), presumably via a Diels–Alder cycloaddition.31 In recent years, many analogues of meroterpenoid 1 have been reported, with most of them exhibiting a 3S5R6S7R10S configuration in the tetrahydro-oxirane-chromene-diol core.24–29,32 In contrast, the secondary metabolites isolated from IQ-011 consistently exhibit a 3R5S6R7S10R configuration.31
In response to the challenge of rediscovering specialized metabolites and recognizing the untapped biosynthetic gene reservoirs in ascomycetes,33 our group recently reported the effective application of an OSMAC (One Strain Many Compounds) approach integrated with molecular networking and PCA in Talaromyces sp. IQ-313.34 This strategy has positioned DMSO as a promising tool for harnessing the biosynthetic potential of fungi.35,36 Using a similar approach in Pestalotiopsis sp. (strain IQ-011), this study has successfully isolated four new epoxyquinols, including three [4 + 2] cycloaddition dimers and enantiomers of the known compounds eutyscoparol J (5), pestaloquinol A (7), and pestaloquinol B (9).30,37
Results and discussion
By applying an OSMAC approach to Pestalotiopsis sp. (strain IQ-011), including UV radiation and several chemical agents (xylenol orange, colchicine, DMSO, rhodamine, acrolein and lead), and combinations, intended to induce abiotic stress, and conducting a metabolomic study based on PCA of LC-MS features from these extracts, it was revealed that treatment with 5% DMSO produced a significant alteration in the chemical profile compared to the other tested conditions and a control group grown under standard conditions (Fig. 1). | ||
| Fig. 1 Principal component analysis (PCA) based on LC-MS features from the extracts of Pestalotiopsis sp. (strain IQ-011) exposed to UV radiation and several stress agents. | ||
Subsequently, using the same data collected for PCA, a feature-based molecular networking (FBMN) analysis was performed. This analysis revealed the production of specific compounds that are exclusively biosynthesized under DMSO or colchicine treatment conditions (Fig. 2).38 Interestingly, this measurement revealed an increased production of cytosporin M (1) (identified within the molecular network through comparison with a standard reference) in Pestalotiopsis sp. (strain IQ-011) treated with 5% DMSO.
 | ||
| Fig. 2 (A) Molecular network created by FBMN (positive mode), showing nodes linked to compounds produced by Pestalotiopsis sp. (strain IQ-011) under standard conditions (in orange) and using several stress agents (B). The connections between nodes indicate the structural similarity of the specialized metabolites based on their fragmentation patterns, and the width of the edges is proportional to the cosine score (ranging from 0.65 to 1). Each node displays a label indicating the m/z value of the MS1 precursor ion. Mass spectrometry data were analysed to include only ions from the positive ionization mode, using a mass-to-charge ratio (m/z) range of 100–1500 and acquiring MS/MS data for the top five ions from the parent mass. The node associated with cytosporin M (1) is indicated based on the comparison with an authentic reference. | ||
Following this, the FBMN analysis focused on comparing the chemical profile of the control fungus with that treated with DMSO (Fig. 3), revealing an upregulation of cytosporin M (1) by up to 50-fold, as determined by relative quantification based on peak area features. Additionally, presumed analogues of 1 (m/z = 594.477, 622.5100, and 357.1847) were identified based on their fragmentation patterns (Fig. S4 and S49–S55†). These compounds are also less polar than 1, according to their retention times (Fig. S48†).38 Moreover, due to the suspicion that other molecular families detected exclusively under DMSO conditions might be structurally related to cytosporin M (1)—even though they do not cluster within the same molecular family (as per the analysis parameters)—the molecular formula of the MS1 ion was predicted.
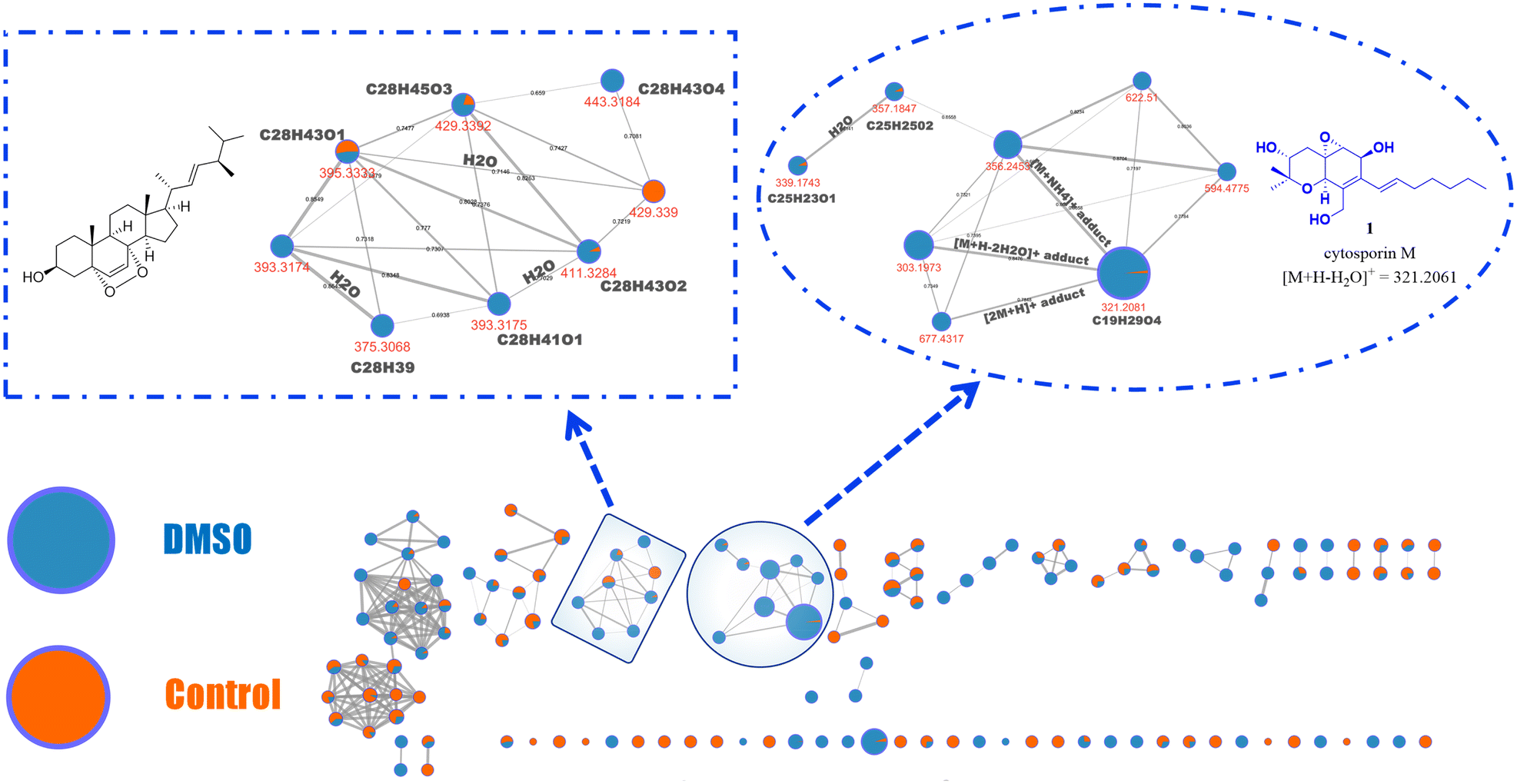 | ||
| Fig. 3 Molecular network created by FBMN, showing nodes linked to compounds produced by Pestalotiopsis sp. (strain IQ-011) under standard conditions (in orange) and those biosynthesized by the fungus grown in 5% DMSO (in blue). The connections between nodes indicate the structural similarity of the specialized metabolites based on their fragmentation patterns, and the width of the edges is proportional to the cosine score (ranging from 0.65 to 1). The size of each node is proportional to its relative abundance. Mass spectrometry data were analysed to include only ions from the positive ionization mode, using a mass-to-charge ratio (m/z) range of 100–1500 and acquiring MS/MS data for the top five ions from the parent mass. | ||
The fragmentation pattern was then compared with that of compound 1, revealing notable similarities in several ionic fragments (Fig. S4, S47, S54, and S55†). This information suggested that the detected ions upregulated under DMSO treatment are indeed cytosporin M (1) derivatives.
Additionally, through dereplication analysis, a family of ergosterol-like derivatives was identified, mainly consisting of compounds detected under DMSO conditions (Fig. 3 and Fig. S57–S65†).34
Following these preliminary analyses, the fungal culture treated with 5% DMSO was scaled up, incorporating independent replicates to ensure consistency between the controls and treatments (Fig. S66–S68†). Interestingly, the DMSO-treated cultures consistently produced a significantly higher yield of extracts compared to the untreated cultures (∼7×, Fig. S69†). Chemical investigation of the extracts obtained from the DMSO-treated fungus led to the isolation of four novel compounds (2, 4, 6 and 8), identified as prenyl-epoxyquinols (Scheme 1).
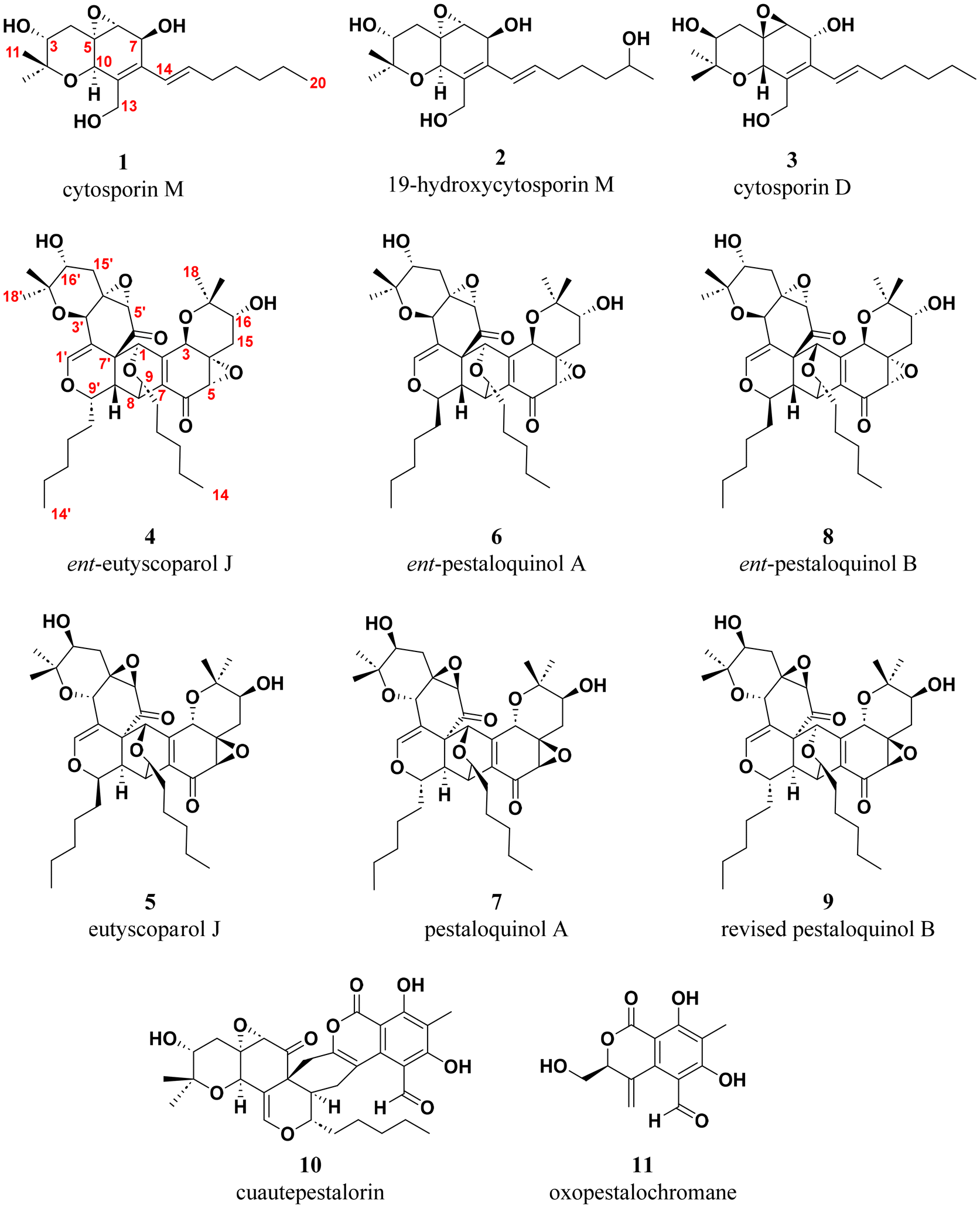 | ||
| Scheme 1 Chemical structures of the isolated molecules and previously reported epoxyquinols. Note: the numbering for dimeric compounds is adjusted to align with previously reported structures, differing from the scheme used for monomeric molecules. | ||
Compound 2 was isolated as a white, optically active solid ([α]25D = +17.5; c = 0.28 in MeOH). Its molecular formula was determined to be C19H30O6 by HRESIMS (m/z = 355.2136 for [M + H]+, calculated for C19H31O6, 355.2121), indicating five degrees of unsaturation. The UV spectrum exhibited a maximum absorption at λmax 240 nm. The NMR spectra (1H and 13C; Table 1) showed significant similarity between 2 and cytosporin M (1), with the most notable differences being the downfield shift of H-19 and C-19, as well as the doublet observed for the methyl group of 2 (C-20) instead of the triplet seen in 1 (Table 1). This analysis demonstrated that 2 is a hydroxylated derivative of 1 at C-19.
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δ H, mult. (J in Hz) | δ C | δ H, mult. (J in Hz) | δ C | |
| 1 | — | 76.4 | — | 76.4 |
| 2 | 3.69, dt (11.4, 4.5) | 73.0 | 3.69 (11.7, 4.9) | 72.9 |
| 3-OH | 1.65, d (5.5) | — | — | — |
| 4α | 2.25, dd (11.8, 11.8) | 35.7 | 2.24, dd (13.2, 11.7) | 35.7 |
| 4β | 1.77, dd (13.1, 4.9) | 1.77, dd (13.2, 4.9) | ||
| 5 | — | 59.7 | — | 59.7 |
| 6 | 3.46, dd (3.4, 1.1) | 62.0 | 3.46, dd (3.4, 1.0) | 62.0 |
| 7 | 4.72, dd (9.6, 3.3) | 65.8 | 4.71, brs. | 65.8 |
| 7-OH | 1.92, d (9.7) | — | — | — |
| 8 | — | 133.6 | — | 133.7 |
| 9 | — | 129.3 | — | 129.5 |
| 10 | 4.43, s | 68.7 | 4.42, s | 68.7 |
| 11 | 1.31, s | 27.7 | 1.31, s | 27.7 |
| 12 | 1.35, s | 16.3 | 1.35, s | 16.3 |
| 13α | 4.39, dd (12.7, 5.8) | 62.4 | 4.38, d (12.2) | 62.3 |
| 13β | ||||
| 4.14, d (12.3) | 4.13, d (12.2) | |||
| 13-OH | 2.24, brs. | — | — | — |
| 14 | 6.20, d (16.0) | 123.7 | 6.22, d (15.9) | 124.2 |
| 15 | 6.12, dt (15.9, 6.8) | 137.8 | 6.11, dt (15.9, 6.9) | 137.1 |
| 16 | 2.15, tdd (7.1, 5.5, 1.4) | 33.8 | 2.19, qd (6.9, 1.4) | 33.6 |
| 17 | 1.42, p (7.5) | 29.0 | 1.47–1.56, m | 25.3 |
| 18 | 1.30, m | 31.6 | 1.47, m | 38.8 |
| 19 | 1.30, m | 22.6 | 3.80, m | 68.1 |
| 20 | 0.88, t (7.0) | 14.2 | 1.18, d (6.1) | 23.7 |
Furthermore, analysis of the NOESY interactions revealed correlations between H-3 (δH 3.69 ppm) and H-4β (equatorial; δH 1.77 ppm), H-4β (equatorial) and H-6 (δH 3.46 ppm), as well as CH3-11 (δH 1.31 ppm) and H-3, indicating that these protons are situated on the same face of the molecule. Additionally, NOESY interactions were observed between H-4α (axial; δH 2.24 ppm) and CH3-12 (δH 1.35 ppm) and H-10 (δH 4.42 ppm), as well as between H-10 and H-7 (4.71 ppm), suggesting that this group of hydrogen atoms is located on the other face of the structure. Moreover, the experimental ECD data for 2 revealed a negative Cotton effect around 255 nm—like that observed for compound 1, which shows a negative Cotton effect around 240 nm (conversely to its enantiomer 329)—suggesting that the absolute configuration of the tetrahydro-oxirane-chromene-diol core in compound 2 matches that of its precursor, with a (3R5S6R7S10R) configuration (Fig. 4A).29,31 However, the configuration at C-19 remains undetermined.
 | ||
| Fig. 4 (A) Experimental ECD spectra for cytosporin M (1, 3R5S6R7S10R, red line) and 19-hydroxycytosporin M (2, 3R5S6R7S10R, blue line); (B) experimental ECD spectra for ent-eutyscoparol J (4, black line); calculated ECD spectra for ent-eutyscoparol J (4, blue dashed line) and its enantiomer eutyscoparol J (5, red dashed line); (C) experimental ECD spectra for ent-pestaloquinol A (6, black line), calculated ECD spectra for ent-pestaloquinol A (6, blue dashed line) and its enantiomer pestaloquinol A (7, red dashed line); (D) experimental ECD spectra for ent-pestaloquinol B (8, black line), calculated ECD spectra for a truncated model for 8 (8t, blue dashed line) and its enantiomer (9t, red dashed line). | ||
Additionally, three molecules (4, 6, and 8), which are expected to be analogues of compound 1 based on their UV profiles (λmax = 240–250 nm, Fig. S20, S29 and S38†) observed during chromatographic analysis, were isolated. After acquiring NMR data (Table 2 and Fig. S23–S26, S32–S35, S41–S44†), it was confirmed that the two-dimensional structures matched those previously reported for the dimeric epoxyquinols, eutyscoparol J (5), pestaloquinol A (7) and pestaloquinol B (9), isolated from Eutypella scoparia SCBG-8 and Pestalotiopsis sp., respectively.30,37 These compounds are proposed to originate from cytosporin D (the enantiomer of compound 1) through a series of reactions involving oxidation, a reversible 6π-electrocyclization, and a Diels–Alder [4 + 2] cycloaddition, similar to the biosynthetic pathway described for cuautepestalorin (10).30,31 According to these biosynthetic considerations, and taking into account the absolute configuration of the plausible precursor 1, it was inferred that the three diastereomers (4, 6 and 8) should be enantiomers of compounds 5, 7 and 9.30,37
| 4 | 6 | 8 | ||||
|---|---|---|---|---|---|---|
| No. | δ H, mult. (J in Hz) | δ C | δ H, mult. (J in Hz) | δ C | δ H, mult. (J in Hz) | δ C |
| 1 | 5.29, s | 71.4 | 5.26, s | 71.7 | 5.50, brs | 70.4 |
| 2 | — | 151.9 | — | 151.4 | — | 150.8 |
| 3 | 4.50, d (1.4) | 65.2 | 4.51, d (1.4) | 65.2 | 4.31, d (1.5) | 65.7 |
| 4 | — | 62.0 | — | 61.9 | — | 62.8 |
| 5 | 3.30, d (1.3) | 60.2 | 3.31, d (1.3) | 60.2 | 3.34, d (1.3) | 60.2 |
| 6 | — | 189.5 | — | 189.7 | — | 190.9 |
| 7 | — | 134.8 | — | 134.8 | — | 132.9 |
| 8 | 3.39, t (1.7) | 30.3 | 3.15, t (1.8) | 37.5 | 3.16, dd (2.8, 1.4) | 35.7 |
| 9 | 4.02, td (7.0, 1.5) | 72.1 | 4.15, td (6.8, 1.8) | 71.4 | 3.81, td (7.2, 1.3) | 76.1 |
| 10 | 0.91, m | 35.2 | 0.92, m | 35.0 | 0.91, m | 34.3 |
| 1.11, m | 1.10, m | 1.03, m | ||||
| 11 | 1.17, m | 25.1 | 1.26–1.30, m | 25.1 | 1.18–1.28, m | 24.8 |
| 1.49, m | ||||||
| 12 | 1.17–1.32, m | 31.9 | 1.15–1.25, m | 31.8 | 1.14–1.24, m | 31.4 |
| 13 | 1.24–1.33, m | 22.7 | 1.24–1.34, m | 22.7 | 1.22–1.31, m | 22.6 |
| 14 | 0.83, t (7.1) | 14.2 | 0.82, t (7.2) | 14.1 | 0.83, t (7.1) | 14.1 |
| 15α | 1.72, dd (13.2, 4.8) | 34.6 | 1.72, dd (13.4, 5.0) | 34.6 | 1.82, dd (13.5, 4.8) | 34.7 |
| 15β | 2.28, dd (13.3, 11.9) | 2.29, dd (13.8, 10.7) | 2.28, dd (13.5, 11.4) | |||
| 16 | 3.68, m | 72.5 | 3.69, dd (10.7, 4.6) | 72.5 | 3.78, m | 72.7 |
| 17 | — | 76.6 | — | 76.7 | — | 76.6 |
| 18 | 1.35, s | 16.0 | 1.35, s | 16.0 | 1.40, s | 16.0 |
| 19 | 1.24, s | 26.8 | 1.25, s | 26.8 | 1.33, s | 27.3 |
| 1′ | 6.90, d (2.0) | 147.7 | 6.70, d (1.9) | 143.9 | 6.51, d (1.8) | 145.5 |
| 2′ | — | 113.8 | — | 111.8 | — | 110.9 |
| 3′ | 4.91, brs. | 64.6 | 4.85, dd (2.0, 1.0) | 64.6 | 4.69, dd (1.9, 1.0) | 65.0 |
| 4′ | — | 67.8 | — | 68.4 | — | 68.1 |
| 5′ | 3.24, d (1.0) | 61.1 | 3.22, d (1.0) | 61.1 | 3.36, d (1.0) | 60.5 |
| 6′ | — | 200.2 | — | 200.2 | — | 201.0 |
| 7′ | — | 53.5 | — | 50.9 | — | 53.8 |
| 8′ | 2.48, dd (4.1, 1.9) | 37.1 | 2.53, d (1.9) | 36.1 | 3.13, dd (5.7, 2.7) | 40.2 |
| 9′ | 3.36, m | 76.9 | 4.06, dd (9.3, 4.8) | 78.8 | 3.35, m | 77.7 |
| 10′ | 1.17, m | 32.6 | 1.17, m | 33.1 | 1.15–1.25, m | 32.6 |
| 1.32, m | 1.42, m | 1.31–1.38, m | ||||
| 11′ | 1.18–1.22, m | 25.7 | 1.28–1.35, m | 25.8 | 1.28–1.36, m | 25.1 |
| 12′ | 1.17–1.32, m | 31.8 | 1.20–1.25, m | 31.5 | 1.14–1.24, m | 31.8 |
| 13′ | 1.24–1.33, m | 22.6 | 1.24–1.34, m | 22.6 | 1.22–1.31, m | 22.6 |
| 14′ | 0.90, t (7.1) | 14.1 | 0.85, t (6.9) | 14.2 | 0.85, t (6.9) | 14.1 |
| 15′α | 2.35, dd (14.5, 3.0) | 34.6 | 2.27, dd (14.0, 3.3) | 34.7 | 2.34, dd (14.4, 2.9) | 34.5 |
| 15′β | 1.76, dd (14.4, 6.7) | 1.80, dd (14.3, 7.2) | 1.77, dd (14.4, 6.8) | |||
| 16′ | 3.90, dd (6.7, 3.1) | 72.1 | 3.87, dd (7.2, 3.3) | 72.2 | 3.89, dd (6.7, 2.9) | 71.9 |
| 17′ | — | 75.9 | — | 75.7 | — | 76.0 |
| 18′ | 1.38, s | 18.9 | 1.35, s | 18.7 | 1.33, s | 18.5 |
| 19′ | 1.27, s | 28.0 | 1.25, s | 28.0 | 1.23, s | 27.9 |
To test this hypothesis, the ECD curve for compound 4 was recorded, revealing that it is a mirror image of the one reported for eutyscoparol J (5) (Fig. 4B).37 Compound 4 showed two negative Cotton effects at 260 nm and 350 nm, with a positive effect at around 215 nm, exactly opposite to what has been reported for 5 (Fig. 4B).37 Additionally, ECD predictions for ent-5 (4) based on the crystal structure of 5 (CCDC: 2045736) aligned with the experimental data, confirming the absolute configuration of compound 4 as 1R3R4S5S8R9S16R3′R4′S5′S7′S8′S9′S16′R, the enantiomer of eutyscoparol J (5), which was designated as ent-eutyscoparol J.
Similarly, using a crystal of pestaloquinol A (compound 7, CCDC deposition number: 2045735),37 ECD curve predictions were performed for compound 6 and its enantiomer (7). Comparison with the experimental ECD data, which displayed a positive Cotton effect near 220 nm and two negative effects at 265 nm and 350 nm, confirmed that compound 6 is the enantiomer of 7 (Fig. 4C). Based on this information, compound 6 was designated as ent-pestaloquinol A.
A crystal suitable for analysis was obtained for ent-pestaloquinol A (6) (Fig. 5), with a Flack parameter of 0.21(10). The crystalline structure exhibited significant disorder, particularly in the aliphatic chains, causing diffuse reflections in single-crystal X-ray diffraction at resolutions beyond 1 Å. This disorder resulted in high R1 values (0.12569/0.1321) and reduced accuracy in the absolute structure parameter calculation. The Pearson–Flack parameter for compound 6 (0.21(10)) was higher than that reported for compound 7 (0.07(6)),37 which exhibited lower R values (0.0515/0.0535). Notably, compound 6 crystallizes in the enantiomorphic group P3221, whereas compound 7 crystallizes in space group 3121.
 | ||
| Fig. 5 ORTEP drawing for compound 6 (CCDC: 2403947†). Ellipsoids are shown at the 50% probability level. | ||
In summary, the orthogonal experimental methods (theoretical and experimental ECD and X-ray diffraction) complement each other and strongly support the absolute configuration of 6 as 1R3R4S5S8R9S16R3′R4′S5′S7′S8′S9′R16′R.
Since there is no available crystal structure for pestaloquinol B (9), NOESY interactions were meticulously examined to confirm the relative configuration of compounds 8 and 9. Notably, a clear NOESY correlation was observed between H-8′ (δH 3.13 ppm) and H-9 (δH 3.81 ppm), which is more consistent with H-9 in α-orientation (9S, Fig. 6A) rather than the β-orientation (9R, Fig. 6B), as originally proposed for compound 9.30 Consequently, ECD predictions were conducted based on the configuration: 1R3S4R5R8R9S16S3′S4′R5′R7′R8′R9′S16′S for a truncated model (9t) of 9 (revised pestaloquinol B) and its enantiomer (8t), utilizing the conformations inferred from the crystal structures of compounds 5 and 7.
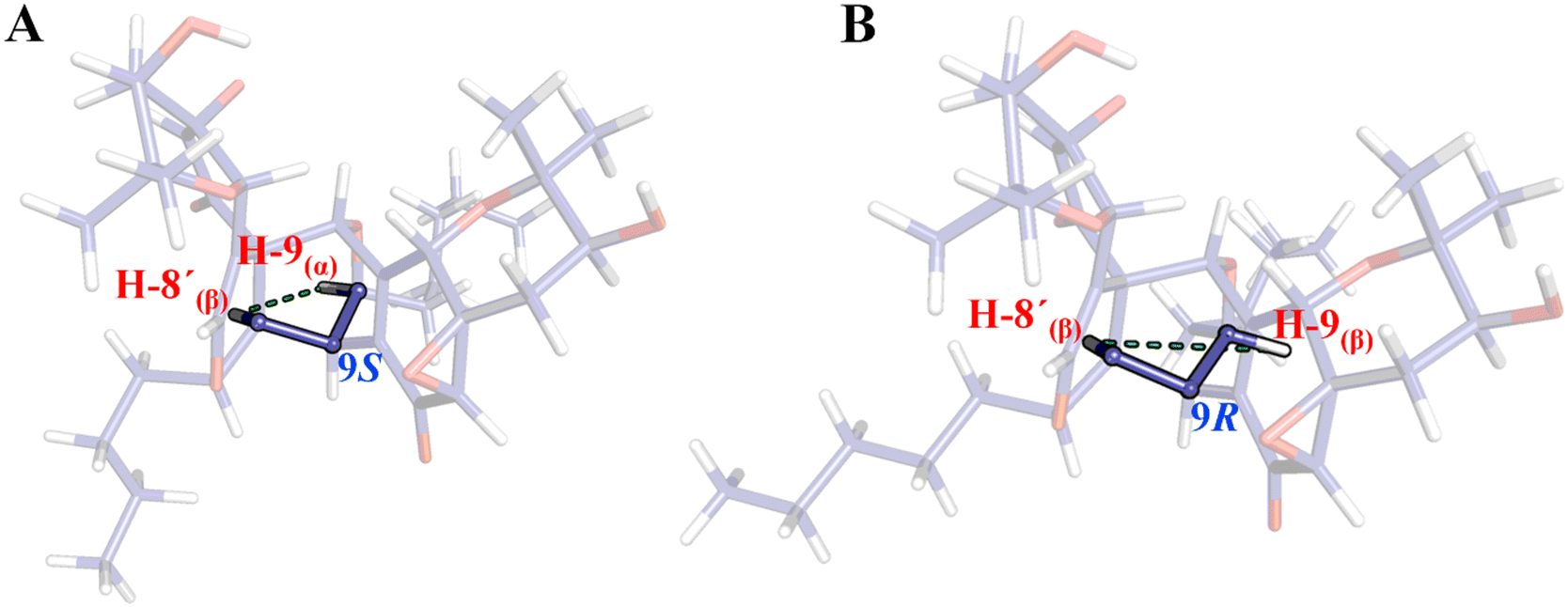 | ||
| Fig. 6 Revised configuration for pestaloquinol B (A, 1R3S4R5R8R9S16S3′S4′R5′R7′R8′R9′S16′S) based on NOESY interactions between H-8′ and H-9, compared to the originally proposed configuration for the same compound (B, 1R3S4R5R8R9R16S3′S4′R5′R7′R8′R9′S16′S). | ||
By comparing the calculated curves with the experimental ECD (Fig. 4D), which shows a positive Cotton effect at 215 nm and two negative effects at 265 and 350 nm, the absolute configuration of 8 was determined to be 1S3R4S5S8S9R16R3′R4′S5′S7′S8′S9′R16′R, the enantiomer of revised pestaloquinol B (9), and was assigned the trivial name ent-pestaloquinol B.
Based on recent findings regarding the biosynthesis of salicylaldehyde-derived epoxyquinol-like metabolites and proposed pathways for [4 + 2] cycloaddition products, the following biosynthetic route for compounds 1, 2, 4, 6, and 8 (Fig. 7) is proposed.
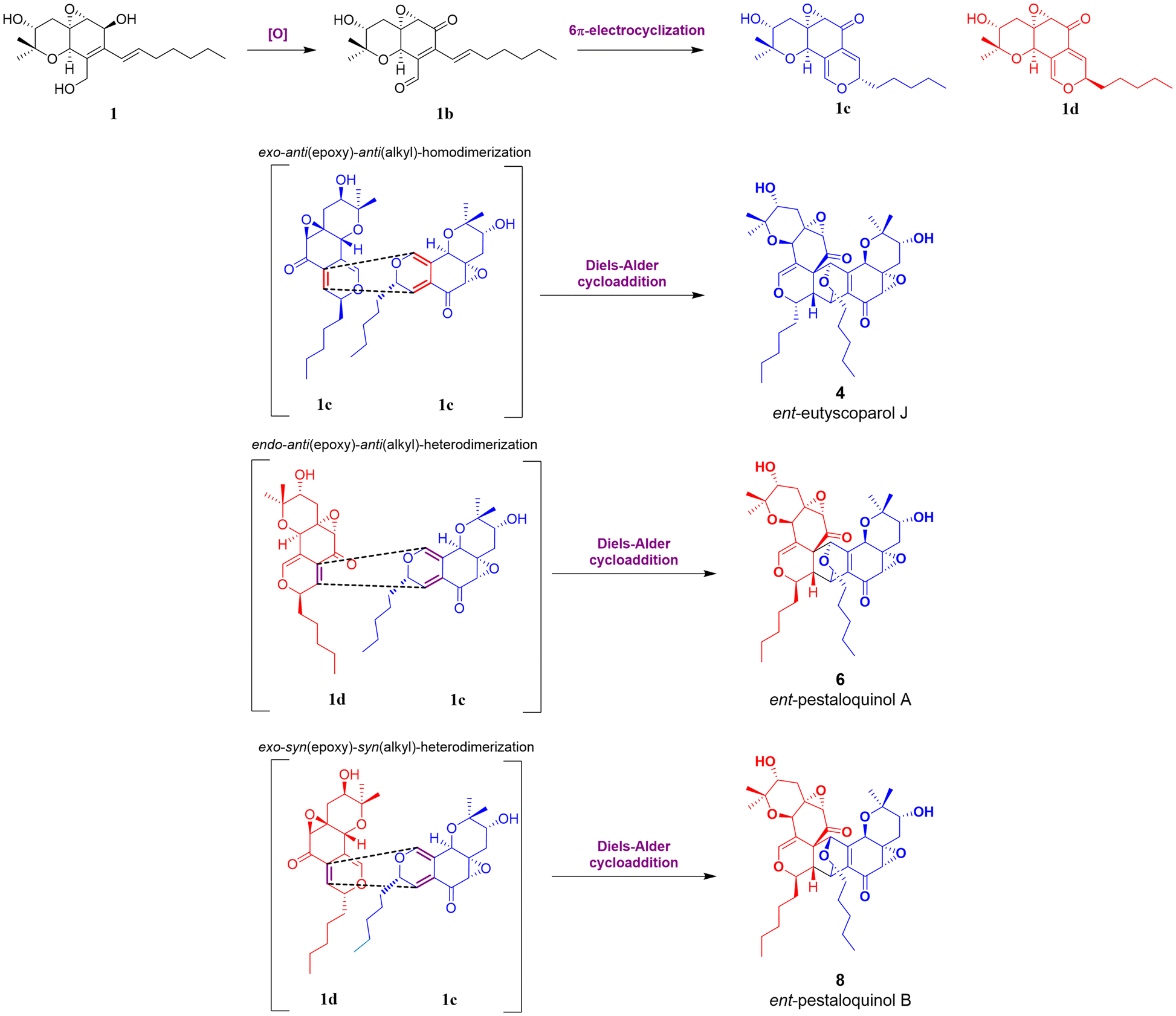 | ||
| Fig. 7 Plausible biosynthetic pathway for 4, 6 and 8, starting from epimer 1c and 1d with the aliphatic chain oriented in alpha (blue) or beta (red). | ||
The process initiates the biosynthesis of a reduced polyketide by an HRPKS enzyme, where selective oxidation of β-hydroxy groups to β-ketones and subsequent intramolecular aldol condensation and dehydration yield the aromatic intermediate salicylaldehyde.12,17,18 According to Ling Liu et al., this biosynthetic logic effectively generates specialized metabolites with both highly reduced and non-reduced fragments.12 Subsequently, the aldehyde group is reduced to yield an intermediate, which undergoes prenylation by DMATS-type enzymes.11,13,39 Then, oxidation and cyclization of the prenylated intermediate lead to the formation of a chromene intermediate, which, after epoxidation, produces cytosporin M (1).10,16 Finally, oxidation of the aliphatic chain at C-19 in 1 yields 19-hydroxycytosporin M (2).
After 1 is generated, it can be oxidized to produce 1b, which undergoes a reversible 6 π-electrocyclization to form one of the epimers 1c or 1d. These intermediates are well-suited for dimerization via the Diels–Alder cycloaddition, ultimately yielding products 4, 6, and 8.9 The Diels–Alder reaction modes of 1c and 1d can be categorized as follows according to Shoji et al.: “starting with the diene, its reactive face can be oriented either anti or syn relative to the epoxide, as well as anti or syn to the alkyl group. These orientations are labelled as anti(epoxide) or syn(epoxide) and anti(alkyl) or syn(alkyl) additions, respectively”.9 Corresponding to this classification, the configurations at C-1, C-8, C-9, and C-9′ depend on the specific epimer and whether the cycloaddition follows an endo or exo pathway. In this case, the endo pathway is notably more favourable to the formation of ent-pestaloquinol A (6), as suggested by higher yields (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 for 6, 4 and 8, respectively), in agreement with what has been reported for the synthesis of epoxyquinols A and B.7
:1:1 for 6, 4 and 8, respectively), in agreement with what has been reported for the synthesis of epoxyquinols A and B.7
Conclusions
In summary, following an OSMAC approach, guided by metabolomic studies such as PCA and molecular networking, it was determined that the epigenetic modulator DMSO significantly enhances the production of cytosporin M (1). Additionally, DMSO induces the biosynthesis of other related compounds within the epoxyquinol family. This approach enabled the chemo-targeted isolation of several natural products whose production was increased, leading to the discovery of four novel compounds: 19-hydroxycytosporin M (2), ent-eutyscoparol J (4), ent-pestaloquinol A (6), and ent-pestaloquinol B (8). The novelty of the compounds is significant, considering the 14 chiral centers in each reported dimer and the uniqueness of the tetrahydro-oxirane-chromene-diol core (exclusively observed in secondary metabolites from Pestalotiopsis sp. (strain IQ-011) with a 3R5S6R7S10R configuration). Additionally, we conducted a detailed revision of the configuration at C-9 of pestaloquinol B (9). The importance of the three dimeric compounds lies in the challenge of establishing their absolute configuration with solid evidence. Moreover, the observed configurations at C-1, C-8, C-9, and C-9′ of the three cycloaddition products, along with the yield and the configuration of their precursor cytosporin M (1), together support the biosynthetic proposals made by various groups for this kind of cycloaddition and complement studies on the dimerization modes of 2H-pyrans.Experimental
General experimental procedures
NMR experiments, encompassing both 1D and 2D, were conducted utilizing CDCl3 as solvent. The NMR instrumentation included either a Bruker Ascend III 700 MHz NMR spectrometer, equipped with a cryoprobe, operating at 700 MHz for 1H and 175 MHz for carbon 13C nuclei, or a Bruker Ascend 500 spectrometer, operating at 500 MHz for 1H and 125 MHz for 13C. Chemical shifts (δ) are reported in parts per million (ppm) relative to the solvent resonances, which serve as the internal standard (CDCl3δH/δC 7.26/77.16). High-resolution mass spectrometry data were acquired using either an AccuTOF JMS-T100LC mass spectrometer (HR-DART-MS), an Agilent 6530 Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) LC/MS system or a Q-Exactive Hybrid Quadrupole-Orbitrap LC/MS system. Electronic Circular Dichroism (ECD) spectra were obtained using a J-1500 circular dichroism spectrophotometer. Dimethyl sulfoxide (DMSO) utilized in the experiments was bought from Sigma-Aldrich. Flash chromatography was performed using a Buchi Pure C-810 system with silica gel as the stationary phase. Chromatographic analyses were conducted in HPLC reversed-phase mode, employing either a C18 column (Gemini-NX) or a Luna PFP column (Phenomenex, Torrance, CA), both with dimensions of 4.6 mm × 250 mm and a particle size of 5 μm. For compound purification, semipreparative HPLC was utilized, using the corresponding columns (10.0 mm × 250 mm).Fermentation of Pestalotiopsis sp. (strain IQ-011) (OSMAC)
In 10 mL of potato dextrose broth (PDB), various stress-inducing agents were added either individually or in combination, including DMSO (5% v/v), colchicine (100 ppm), xylenol orange (1 mM), lead salt (100 ppm), and rhodamine B (1 mM). The media were then inoculated with Pestalotiopsis sp. (strain IQ-011) and incubated for seven days at 120 rpm under regular light/dark cycles. Additionally, two control cultures were prepared without stress agents: one was exposed to UV radiation for 60 minutes, and the other remained untreated as a control. After incubation, each culture was transferred to media containing 12 g of Cheerios and allowed to grow for four weeks.Extraction and isolation
Each culture was macerated with 60 mL of a 1:1 mixture of CH2Cl2/MeOH for 24 hours. The extract was then filtered and washed with 90 mL of CH2Cl2. The organic phase was partitioned with 75 mL of H2O. The organic phase was collected and concentrated under vacuum. Subsequently, the extract was defatted with 100 mL of the 1:1 MeCN/hexane mixture, and the polar phase was retained for further analysis.
For the isolation of new metabolites, the extracts obtained from the replicates treated with DMSO (∼1.2 g) were combined. Subsequently, normal phase flash fractionation was conducted, which involved three minutes of isocratic elution with 100% hexanes, followed by 30 minutes of a gradient from 0 to 100% hexane/ethyl acetate, nine minutes of isocratic elution with 100% ethyl acetate, 10 minutes of a gradient from 0 to 10% ethyl acetate/MeOH, 10 minutes of a gradient from 10 to 25% ethyl acetate/MeOH, eight minutes of a gradient from 25 to 50% ethyl acetate/MeOH, and eight minutes of a gradient from 50 to 100% ethyl acetate/MeOH, at a flow rate of 25 mL min−1. This procedure resulted in the collection of seven fractions (F1–F7), which were then analyzed by HPLC. Cytosporin M (1, 370 mg) was obtained as a pure compound from fraction F3. From fraction F5 (109.2 mg), 30 mg underwent further fractionation by HPLC using reverse phase chromatography (PFP column) with a gradient of 15 to 40% MeCN:H2O with 0.1% formic acid over 30 minutes, from which 19-hydroxycytosporin M (2, 3.1 mg) was isolated. From fraction F2 (143.6 mg), 40.2 mg were further fractionated by HPLC, yielding ent-eutyscoparol J (4, 4.8 mg), ent-pestaloquinol A (6, 11.6 mg), and ent-pestaloquinol B (8, 4.1 mg) using a gradient of 70 to 100% MeCN:H2O 0.1% formic acid on a reverse phase C18 column.
19-Hydroxycytosporin M (2)
White solid. [α]25D = +17.5 (c 0.28, MeOH). UV λmax = 240 nm. HRESIMS m/z = 355.2136 [M + H]+ (calcd for C19H31O6, 355.2121, Δ +4.2 ppm). 1H NMR (CDCl3, 700 MHz) and 13C NMR (CDCl3, 175 MHz). See Table 1.ent-Eutyscoparol J (4)
Vitreous solid. [α]25D = −51.5 (c 0.13, MeOH). UV λmax = 240 nm. HRESIMS m/z = 669.3652 [M + H]+ (C38H53O10, 669.3639, Δ +1.9 ppm). 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz). See Table 2.ent-Pestaloquinol A (6)
Colorless crystalline solid. [α]25D = −17.2 (c 0.25, MeOH). UV λmax = 240 nm. HRESIMS m/z = 669.3656 [M + H]+ (C38H53O10, 669.3639, Δ +2.5 ppm). 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz). See Table 2.The crystallographic data and refinement details for 6 are summarized in Table S2 (ESI).† The crystallographic data have been deposited at the Cambridge Crystallographic Data Center (CCDC) with number 2403947.†
ent-Pestaloquinol B (8)
Vitreous solid. [α]25D = −6.0 (c 0.10, MeOH). UV λmax = 250 nm. HRESIMS m/z = 669.3614 [M + H]+ (C38H53O10, 669.3639, Δ −3.7 ppm). 1H NMR (CDCl3, 500 MHz) and 13C NMR (CDCl3, 125 MHz). See Table 2.Metabolomic studies
Each fungal extract was analyzed using an ultra-performance liquid chromatography (UPLC) system coupled to a mass spectrometer (Q-Exactive Plus). A gradient from 15 to 100% acetonitrile (MeCN:H2O with 0.1% formic acid) was applied over 8 minutes, with a flow rate of 0.3 mL min−1, alternating ion detection in positive-negative mode, within a mass-to-charge (m/z) range of 100–1500. The raw mass spectral files were subsequently converted to mzML format using MSConvert software, applying the peak-picking filter for both MS1 and MS2. Then, the spectra were processed using MZmine v4.0.8,40 starting with mass detection via the centroid algorithm, with noise thresholds set at 105 for MS1 and 103 for MS2. A chromatogram was constructed using the chromatogram builder interface, considering only positive polarity, with parameters set for a minimum of two consecutive scans, a minimum intensity of 105, a minimum absolute height of 2 × 105, and a tolerance of 0.005 m/z. This was followed by deconvolution using the local minimum feature resolver, isotopic grouping with the 13C isotope filter, and spectral alignment with the Join aligner algorithm (0.2 min tolerance, weight of 3 for m/z, and weight of 1 for retention time). A duplicate peak filter was then applied (tolerance of 0.005 for m/z and 0.15 min for retention time). Utilizing the feature list generated through this workflow (retention time, m/z values and peak area), principal component analysis (PCA) was performed, and two files were exported for molecular networking analysis: a feature quantification table (.csv) and an MS2 spectral summary (.mgf). Finally, FBMN was executed with a fragment ion mass tolerance of 0.01 Da, a minimum cosine score of 0.65, a maximum of 10 neighboring nodes per single node, a minimum of six matched fragment ions, and a maximum precursor shift of 500 Da. Molecular networks were visualized and analyzed using the GNPS platform or Cytoscape software v3.10.2. PCA was projected using Prism software v9.34,38
Computational analysis
The geometry of compounds eutyscoparol (5) and pestaloquinol A (7) was obtained from the corresponding crystals reported (deposition number on CCDC: 2045736 and 2045735, respectively). For revised pestaloquinol B (9), a truncated model was generated (9t), considering the conformation observed for crystals of compounds 6 and 7, and was then optimized using a semi-empirical PM3 method with Spartan 10. Subsequently, a conformational analysis was conducted with the same software, filtering out redundancies. Finally, the geometry of the three conformers derived from 9t was optimized using Gaussian 09 under a DFT force field at the B3LYP/DGDZVP level of theory, with MeCN as the default solvent model. ECD calculations for the single conformer 5, 7 and the set of conformers of 9t were performed using a TD-SCF force field at the B3LYP/6-31G(d) level of theory, employing MeCN as the default solvent model and performing calculations for 50 excited-states. The energy predicted was transformed into ECD spectra with a Lorentz/Gaussian approximation, using the following equation:where E0k and R0k represent the transition energy and the rotatory strength of the kth electronic transition, respectively, and σ is half the bandwidth at 1/e peak height.41 Finally, for the set of conformers optimized from 9t, ECD spectra were calculated by applying the Boltzmann weighting to the overall ECD curves generated from the respective predictions.41 All calculations were performed on the HP Cluster Platform 3000SL “Miztli”, a parallel supercomputer with a Linux operating system, containing 25
 312 cores and a total of 15000 GB of RAM.
312 cores and a total of 15000 GB of RAM.
Crystal structure analysis
Data were collected on a Bruker APEX II CCD diffractometer at 100 K using Cu-Kα radiation (k = 1.54178 Å) from an Incoatec ImuS source and a Helios optic monochromator. A suitable single crystal was coated with hydrocarbon oil, picked up with a nylon loop, and mounted in the cold nitrogen stream of the diffractometer. Frames were collected using ω scans and integrated with SAINT.42 Multi-scan absorption correction (SADABS) was applied.42 The structures were solved by direct methods and refined using full-matrix least-squares on F2 with SHELXL-2018 using the SHELXLE GUI.43,44 The hydrogen atoms of the C–H bonds and from O–H moieties were placed at idealized positions using AFIX148 Instruction and Uiso = 1.5 × Ueq tied to the oxygen atom. The crystal presents low intensity reflections at high resolutions (1.0 to 0.8 Å), therefore the absolute structural parameter could not be calculated accurately from the anomalous dispersion of reflections. The calculated Parsons–Flack parameter is 0.20 (10), and the Flack parameter is 0.15 (81). The structure of compound 6 presents positional disorder in two of the central rings and in the alkyl chains of 6 carbon atoms, which was modeled using the SIMU, RIGU and SAME instructions implemented in SHELXLE GUI, and the 56/46 ratio between both positions was calculated using a free variable.44Author contributions
E. A-R., J. R-C., and M. Y. M-R. executed the isolation and structural elucidation of natural compounds. E. A-R. and J. R-C. performed ECD calculations. D. M-O. recorded and analysed X-ray data. J. R-C. designed and supervised the project and edited the manuscript. All authors discussed the results and commented on the manuscript.Data availability
The data supporting this article are included in the ESI.† Copies of 1H, 13C and 2D NMR spectra and HR-MS data for 1, 2, 4, 6 and 8. UV spectra for 1, 2, 4, 6 and 8. Crystallographic data for 6 have been deposited at the CCDC under the accession number CCDC: 2403947.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research received financial support from CONACyT/CONAHCyT through the Borderline Science project CF-2019-263977 and by Dirección General de Asuntos del Personal Académico (DGAPA) of the Universidad Nacional Autónoma de México (UNAM) through the Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT) IN224424. The authors would like to thank Dr Beatriz Quiroz-García, Dr Manuel Rangel-Grimaldo, M.Sc. E. Huerta-Salazar, B.Sc. M.A. Peña-González, and Dr Francisco Javier Pérez Flores (Instituto de Química, UNAM) for collecting spectroscopic and spectrometric data. We also thank Dr A. Romo-Pérez for their help collecting spectroscopic data. J.R-C. is indebted to Dirección General de Cómputo y de Tecnologías de Información y Comunicación (DGTIC), UNAM, for providing the resources to carry out computational calculations in the Miztli System, through the project LANCAD-UNAM-DGTIC-374. This study used UNAM's NMR Lab: LURMN at IQ-UNAM, funded by CONAHCyT-Mexico (Project: 0224747) and UNAM. E.A.-R. is indebted to CONACyT/CONAHCyT for the fellowship provided to pursue graduate studies (CONACyT CVU 809192).References
- C. Wu, Y. Wang and Y. Yang, Molecules, 2022, 27, 8088 CrossRef CAS PubMed.
- S. K. Deshmukh, V. Prakash and N. Ranjan, Phytochem. Rev., 2017, 16, 883–920 CrossRef CAS.
- P. Jiang, X. Fu, H. Niu, S. Chen, F. Liu, Y. Luo, D. Zhang and H. Lei, Arch. Pharmacal Res., 2023, 46, 449–499 CrossRef CAS PubMed.
- J. Xu, X. Yang and Q. Lin, Fungal Diversity, 2014, 66, 37–68 CrossRef.
- X. L. Yang, J. Z. Zhang and D. Q. Luo, Nat. Prod. Rep., 2012, 29, 622–641 RSC.
- C. Li, R. P. Johnson and J. A. Porco, J. Am. Chem. Soc., 2003, 125, 5095–5106 CrossRef CAS PubMed.
- M. Shoji, H. Imai, M. Mukaida, K. Sakai, H. Kakeya, H. Osada and Y. Hayashi, J. Org. Chem., 2005, 70, 79–91 CrossRef CAS PubMed.
- L. Li, Y. Wang, N. Chen, X. Li, H. Li, L. Jin, Y. Ou, X. J. Kong, S. Cao, Q. Xu, X. Wu, J. Han and X. Deng, Org. Lett., 2023, 25, 4016–4021 CrossRef CAS PubMed.
- M. Shoji, H. Imai, I. Shiina, H. Kakeya, H. Osada and Y. Hayashi, J. Org. Chem., 2004, 69, 1548–1556 CrossRef CAS PubMed.
- J. M. Lv, Y. H. Gao, H. Zhao, T. Awakawa, L. Liu, G. D. Chen, X. S. Yao, D. Hu, I. Abe and H. Gao, Angew. Chem., Int. Ed., 2020, 59, 13531–13536 CrossRef CAS PubMed.
- Y. Pan, L. Liu, F. Guan, E. Li, J. Jin, J. Li, Y. Che and G. Liu, ACS Chem. Biol., 2018, 13, 703–711 CrossRef CAS PubMed.
- L. Liu, M. C. Tang and Y. Tang, J. Am. Chem. Soc., 2019, 141, 19538–19541 CrossRef CAS PubMed.
- J. Nies, H. Ran, V. Wohlgemuth, W. B. Yin and S. M. Li, Org. Lett., 2020, 22, 2256–2260 CrossRef CAS PubMed.
- L. Comméiras, J. E. Moses, R. M. Adlington, J. E. Baldwin, A. R. Cowley, C. M. Baker, B. Albrecht and G. H. Grant, Tetrahedron, 2006, 62, 9892–9901 CrossRef.
- D. Y. Li, X. L. Zhang and Z. L. Li, J. Asian Nat. Prod. Res., 2023, 25, 641–649 CrossRef CAS PubMed.
- Y. L. Yang, M. Zhou, L. Yang, M. Gressler, J. Rassbach, J. M. Wurlitzer, Y. Zeng, K. Gao and D. Hoffmeister, Angew. Chem., Int. Ed., 2023, 62, e202313817 CrossRef CAS PubMed.
- H. Wang, C. Peng, X. X. Chen, H. Y. Wang, R. Yang, H. Xiang, Q. F. Hu, L. Liu, L. W. Chung, Y. Matsuda and W. G. Wang, ACS Catal., 2024, 14, 10796–10805 Search PubMed.
- R. Yang, J. Feng, H. Xiang, B. Cheng, L. D. Shao, Y. P. Li, H. Wang, Q. F. Hu, W. L. Xiao, Y. Matsuda and W. G. Wang, J. Am. Chem. Soc., 2023, 145, 11293–11300 Search PubMed.
- T. Asai, K. Tsukada, S. Ise, N. Shirata, M. Hashimoto, I. Fujii, K. Gomi, K. Nakagawara, E. N. Kodama and Y. Oshima, Nat. Chem., 2015, 7, 737–743 Search PubMed.
- L. Liu, S. Niu, X. Lu, X. Chen, H. Zhang, L. Guo and Y. Che, Chem. Commun., 2010, 46, 460–462 Search PubMed.
- H. Kakeya, R. Onose, H. Koshino, A. Yoshida, K. Kobayashi, S. I. Kageyama and H. Osada, J. Am. Chem. Soc., 2002, 124, 3496–3497 CrossRef CAS PubMed.
- H. Kamiyama, T. Usui, H. Sakurai, M. Shoji, Y. Hayashi, H. Kakeya and H. Osada, Biosci. Biotechnol. Biochem., 2008, 72, 1894–1900 CAS.
- H. Kamiyama, T. Usui, M. Uramoto, H. Takagi, M. Shoji, Y. Hayashi, H. Kakeya and H. Osada, J. Antibiot., 2008, 61, 94–97 CAS.
- H.-X. Liao, D.-W. Sun, C.-J. Zheng and C.-Y. Wang, Nat. Prod. Res., 2017, 31, 1640–1646 CAS.
- Y. X. Zhang, H. B. Yu, W. H. Xu, B. Hu, A. Guild, J. P. Zhang, X. L. Lu, X. Y. Liu and B. H. Jiao, J. Nat. Prod., 2019, 82, 3089–3095 CAS.
- H. B. Yu, L. Du, Z. Ning, X. L. Lu, B. H. Jiao, B. Hu and X. Y. Liu, Chem. Biodivers., 2024, 21, e202401097 CrossRef CAS PubMed.
- Y. H. Zhang, H. F. Du, W. B. Gao, W. Li, F. Cao and C. Y. Wang, Mar. Drugs, 2022, 20, 486 CAS.
- X. Yu, W. E. G. Müller, D. Meier, R. Kalscheuer, Z. Guo, K. Zou, B. O. Umeokoli, Z. Liu and P. Proksch, Mar. Drugs, 2020, 18, 129 CAS.
- H. B. Yu, Z. Ning, B. Hu, Y. P. Zhu, X. L. Lu, Y. He, B. H. Jiao and X. Y. Liu, Mar. Drugs, 2023, 21, 382 CAS.
- G. Ding, F. Zhang, H. Chen, L. Guo, Z. Zou and Y. Che, J. Nat. Prod., 2011, 74, 286–291 CAS.
- J. Rivera-Chávez, J. Zacatenco-Abarca, J. Morales-Jiménez, B. Martínez-Aviña, S. Hernández-Ortega and E. Aguilar-Ramírez, Org. Lett., 2019, 21, 3558–3562 CrossRef PubMed.
- S. R. Lee, M. Dayras, J. Fricke, H. Guo, S. Balluff, F. Schalk, J. S. Yu, S. Y. Jeong, B. Morgenstern, B. Slippers, C. Beemelmanns and K. H. Kim, Commun. Chem., 2024, 7, 129 CrossRef CAS PubMed.
- L. K. Caesar, R. Montaser, N. P. Keller and N. L. Kelleher, Nat. Prod. Rep., 2021, 38, 2041–2065 RSC.
- A. T. Aron, E. C. Gentry, K. L. McPhail, L. F. Nothias, M. Nothias-Esposito, A. Bouslimani, D. Petras, J. M. Gauglitz, N. Sikora, F. Vargas, J. J. J. van der Hooft, M. Ernst, K. B. Kang, C. M. Aceves, A. M. Caraballo-Rodríguez, I. Koester, K. C. Weldon, S. Bertrand, C. Roullier, K. Sun, R. M. Tehan, C. A. P. Boya, M. H. Christian, M. Gutiérrez, A. M. Ulloa, J. A. Tejeda Mora, R. Mojica-Flores, J. Lakey-Beitia, V. Vásquez-Chaves, Y. Zhang, A. I. Calderón, N. Tayler, R. A. Keyzers, F. Tugizimana, N. Ndlovu, A. A. Aksenov, A. K. Jarmusch, R. Schmid, A. W. Truman, N. Bandeira, M. Wang and P. C. Dorrestein, Nat. Protoc., 2020, 15, 1954–1991 CrossRef CAS PubMed.
- E. Aguilar-Ramírez, J. Rivera-Chávez, V. Abogado-Aponte, B. Quiroz-García and A. Romo-Pérez, Tetrahedron, 2024, 167(7), 134283 CrossRef.
- E. Aguilar-Ramírez, V. Reyes-Pérez, C. A. Fajardo-Hernández, C. D. Quezada-Suaste, M. Carreón-Escalante, V. Merlin-Lucas, B. Quiroz-García, V. Granados-Soto and J. Rivera-Chávez, J. Med. Chem., 2023, 66, 16222–16234 CrossRef PubMed.
- W. Zhang, X. Lu, H. Wang, Y. Chen, J. Zhang, Z. Zou and H. Tan, Tetrahedron Lett., 2021, 79, 153314 CrossRef CAS.
- L. F. Nothias, D. Petras, R. Schmid, K. Dührkop, J. Rainer, A. Sarvepalli, I. Protsyuk, M. Ernst, H. Tsugawa, M. Fleischauer, F. Aicheler, A. A. Aksenov, O. Alka, P. M. Allard, A. Barsch, X. Cachet, A. M. Caraballo-Rodriguez, R. R. Da Silva, T. Dang, N. Garg, J. M. Gauglitz, A. Gurevich, G. Isaac, A. K. Jarmusch, Z. Kameník, K. B. Kang, N. Kessler, I. Koester, A. Korf, A. Le Gouellec, M. Ludwig, C. H. Martin, L. I. McCall, J. McSayles, S. W. Meyer, H. Mohimani, M. Morsy, O. Moyne, S. Neumann, H. Neuweger, N. H. Nguyen, M. Nothias-Esposito, J. Paolini, V. V. Phelan, T. Pluskal, R. A. Quinn, S. Rogers, B. Shrestha, A. Tripathi, J. J. J. van der Hooft, F. Vargas, K. C. Weldon, M. Witting, H. Yang, Z. Zhang, F. Zubeil, O. Kohlbacher, S. Böcker, T. Alexandrov, N. Bandeira, M. Wang and P. C. Dorrestein, Nat. Methods, 2020, 17, 905–908 CrossRef CAS PubMed.
- E. T. Miller, O. V. Tsodikov and S. Garneau-Tsodikova, Nat. Prod. Rep., 2024, 41, 113–147 RSC.
- R. Schmid, S. Heuckeroth, A. Korf, A. Smirnov, O. Myers, T. S. Dyrlund, R. Bushuiev, K. J. Murray, N. Hoffmann, M. Lu, A. Sarvepalli, Z. Zhang, M. Fleischauer, K. Dührkop, M. Wesner, S. J. Hoogstra, E. Rudt, O. Mokshyna, C. Brungs, K. Ponomarov, L. Mutabdžija, T. Damiani, C. J. Pudney, M. Earll, P. O. Helmer, T. R. Fallon, T. Schulze, A. Rivas-Ubach, A. Bilbao, H. Richter, L. F. Nothias, M. Wang, M. Orešič, J. K. Weng, S. Böcker, A. Jeibmann, H. Hayen, U. Karst, P. C. Dorrestein, D. Petras, X. Du and T. Pluskal, Nat. Methods., 2020, 17, 905–908 CrossRef PubMed.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- BrukerSAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 2D NMR spectra and HR-MS data for 1, 2, 4, 6 and 8. UV spectra for 1, 2, 4, 6 and 8. CCDC 2403947. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00115c |
| This journal is © The Royal Society of Chemistry 2025 |