
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudies in enantioselective microbial deuteration†
Carl
Recsei
 *a,
Marina
Cagnes
a,
Robert A.
Russell
a,
Reilly E.
Sonstrom
b and
Tamim
Darwish
a
*a,
Marina
Cagnes
a,
Robert A.
Russell
a,
Reilly E.
Sonstrom
b and
Tamim
Darwish
a
aAustralian Nuclear Science and Technology Organisation, National Deuteration Facility, New Illawarra Rd, Lucas Heights, New South Wales, 2234, Australia. E-mail: recseic@ansto.gov.au
bBrightSpec, Inc., 770 Harris St, Suite 104b, Charlottesville, Virginia 22903, United States
First published on 11th March 2025
Abstract
This study reports methods for reductive microbial deuteration with a high degree of isotopic labelling, focusing on economical approaches using various yeast strains and inexpensive carbon sources. A strategy of α-hydrogen exchange followed by deuterative microbial reduction gave >95% backbone perdeuteration for crucial chiral building blocks for medicinal and analytical applications, without loss of the enantioselectivity demonstrated in the corresponding protiative processes. Under an air atmosphere, Saccharomyces cerevisiae strain MBG5177 outperformed baker's yeast in the synthesis of (2-2H1)solketal, while Pichia pastoris strain X-33 provided >95% deuteration in the synthesis of the two studied building blocks using methanol-d4 as an affordable carbon source. These findings emphasise the potential of microbial strains not traditionally employed by synthetic organic chemists for isotope labelling. Finally, molecular rotational resonance spectroscopy (MRR) was employed as an analytical tool. MRR was shown to provide accurate measurement of site-specific deuteration levels and enantiopurity, validating its utility for simplifying process evaluation in deuterium-labelling chemistry. This work underscores the value of diverse microbial resources and advanced spectroscopic methods in advancing isotope labelling and biocatalysis, with implications for both research and industrial applications.
1. Introduction
Selective incorporation of deuterium can significantly alter the metabolic, chemical, and physical properties of molecules, for instance by improving drug stability. New techniques for the selective deuteration of organic molecules are essential to meet the demand for deuterated compounds in various fields such as pharmaceuticals, neutron science and the provision of deuterated standards for analytical chemistry.1,2 Pharmaceuticals and the study of biological or biomimetic systems by neutron techniques generally require a particular enantiomer of a relevant chiral compound. Likewise, chiral deuterated standards are desirable for the quantitative analysis of biomolecules, particularly lipidomics. Deuterated standards simplify the quantitation of components of biological systems as addition of a known quantity of a deuterated standard to a biologically-derived mixture corrects for variability in the efficacy of analyte extraction from that mixture. Given the ubiquity of chiral effects in determining the affinity of biomolecules for one another, the production of the natural stereoisomer of deuterated mass spectrometry standards is generally desirable to ensure the accuracy of such analyses.Enantioselective deuteration is a challenge, being subject to the same limitations as other deuteration processes, namely the paucity of relevant reagents available in deuterated form. To make selective deuteration more feasible for industrial applications, there is a need for cost-effective and environmentally sustainable biocatalytic methods to access a wider range of substrates.
Microbial biosynthesis provides a solution to the paucity of deuterative chiral catalyst/reagent systems: a microbe generates the necessary redox cofactors and catalysts from a deuterated growth medium. Since microbial transformations are generally enzyme-catalysed, they offer a way to perform deuterative reactions enantioselectively. Microbial transformations are also advantageous in being considered green and permit characterisation of the constituents of formulations as being naturally produced, in conformance with consumer preference.3
For the synthetic organic chemist, the most familiar microbial transformations are reductions employing baker's yeast (Saccharomyces cerevisiae); often the reduction of carbonyls to give optically-active, secondary alcohols.4 Feeding the yeast with a carbon source such as glucose allows it to regenerate redox cofactors to keep the enzyme turning over. Unlike non-biological systems, only one enzyme enantiomer is generally available, leading to one enantiomer of the reduced product. In the case of baker's yeast, the usual selectivity is known as Prelog selectivity: if the more sterically demanding substituent on the carbonyl to be reduced also has the higher CIP (Cahn–Ingold–Prelog) priority, re-delivery of hydrogen gives the configuration of the reduced product as (S).5 This reactivity of baker's yeast is highly valuable, yet strongly substrate dependant. Despite this, synthetic organic chemistry laboratories use dried baker's yeast nearly exclusively among all extant microbes because of its functional resemblance to traditional chemical reagents. However, there is growing interest in evaluating unconventional yeasts, with recent studies demonstrating promising capabilities.6,7
In a prominent example of the promise of yeast in industrial processes, Nie and coworkers reused the whole cells of S. cerevisiae strain JUC15 in the synthesis of the building block (R)-phenyl-1,2-ethanediol by microbial reduction 40 times without loss of activity or lowering of >99.9% enantioselectivity.8 This is one example of a large number of reports detailing the versatility of microorganisms in reducing structurally-diverse substrates.4,9 The applicability of baker's yeast to enantioselective reduction is complemented by established techniques for manipulation of the system. For instance, simple additives can completely reverse the usual Prelog stereoselectivity,10 judicious choice of protecting/directing groups can enhance yields and improve or reverse stereochemical outcomes and employing non-Saccharomyces microbes offers access to complementary substrate scope, yield and enantioselectivity.11,12
A promising alternative to microbial biosynthesis is chemoenzymatic deuterative reduction. Conceptually, this group of methodologies seeks to employ only the desired cellular components for the reaction, dispensing with the use of the whole organism and adding reagents as required. Vincent and coworkers reported the reductive deuteration of carbonyl-containing substrates with very high (>99%) enantiomeric excess and isotopic purity by using H2 to drive a biocatalytic reduction – re-reducing NAD+ in the presence of D2O.13 This seminal work built upon an established technique of regeneration of deuterated nicotinamide cofactors.14 Another key study used an organic solvent-tolerant alcohol dehydrogenase enzyme from a strain of Rhodococcus ruber expressed in E. coli and the lyophilised E. coli cells used to reduce a range of carbonyl-containing substrates in low to very high enantiopurity, employing deuterated isopropanol as the deuterium source.15 Other researchers have explored enzyme-mediated deuterative synthesis by employing reducing cofactors, labelled with deuterium chemoenzymatically.16–18 Chemoenzymatic methods do suffer from drawbacks, however, including the complexity of recycling the reaction components and the inability of the system to regenerate enzymes and cofactors. It may also be more practical to conduct one-pot, multi-step transformations with suitably engineered microorganisms than with chemoenzymatic methods because of the robustness of the former.
Progress in reductive asymmetric deuterative enzymatic synthesis therefore proceeds on two fronts: the chemoenzymatic route developing alongside the use of living cultures of microorganisms. In this report we describe progress on the second front, with an emphasis on the development of cost-effective methods for high deuterium incorporation. To the best of our knowledge, high (>95%-d) levels of site-specific deuterium incorporation by a cultured microorganism have not previously been reported.
Few in vivo examples of deuterium incorporation by microbial reduction exist in the literature. The studies that do exist were aimed at elucidation of biosynthetic pathways and involve partial incorporation of deuterium as a probe rather than aiming for high levels of isotopic purity.3,19–24 The motivation for the current study is to allow for the wealth of manipulations available to optimise microbial reductions and the extensive literature about the biosynthetic capabilities of yeast to be applied to the synthesis of deuterated compounds by establishing the means to achieve relatively affordable, preparative syntheses with high, defined levels of deuterium incorporation. Further motivation for this study is the difficulty encountered in measuring the enantioselectivity of biosynthetic processes in the context of variably isotope-labelled materials. The literature tends to provide bespoke processes for enantiopurity determination of individual building blocks which are not necessarily applicable to their deuterated variants (such as 1H NMR analysis), necessitating extensive synthetic and analytical work to determine this key parameter. We anticipated that the emerging spectroscopic technique of molecular rotational resonance (MRR) might provide a significant improvement to current methods for enantioselectivity determination.
2. Choice of synthetic targets
To study the enantioselective incorporation of deuterium by microbial reduction we targeted two structures which are used in medicinal chemistry and in the synthesis of lipids for mass spectrometric standards. First, we intended to carry out reduction of the monobenzoate ester of 1,3-dihydroxyacetone (1, shown as (1,1,3,3-2H4)1 in Scheme 1), from which both enantiomers of the versatile building block solketal (2,2-dimethyl-1,3-dioxolane-4-methanol, 2, shown as (R)-(1,1,2,3,3-2H5)2 in Scheme 1) may be prepared.25 The known baker's yeast-mediated reduction of 1 may be considered a ‘best-in-class’ microbial reduction for its >97% enantiomeric excess and high (80%) yield, as well as its applicability to the synthesis of optically-active glycerol derivatives.4,26 We chose to target solketal because, aside from its role in the synthesis of medicinally-relevant compounds, it is also a building block that is applicable to the synthesis of all triglycerides and all glycerophospholipids and therefore to the production of a wide range of deuterated mass spectrometry standards for lipidomics. | ||
| Scheme 1 A combination of H/D exchange next to a carbonyl (teal deuterons) and deuterative microbial reduction (purple deuteron) can be employed for the synthesis of (1,1,2,3,3-2H5)2. | ||
An advantage of reducing a ketone is that the α-hydrogens may be pre-exchanged for deuterium. In a protiative reduction this would give rise to (1,1,3,3-2H4)2 and to (1,1,2,3,3-2H5)2 in a deuterative reduction. Equally, a deuterative reduction of the unexchanged starting material would give (2-2H1)2. These three materials have not previously been reported as single enantiomers.
The reduction of 1-benzyloxyacetone (3, shown as (1,1,3,3,3-2H5)3 in Scheme 2) was intended to produce the second target: 1,2-propanediol 1-O-benzyl ether (4, shown as (S)-(1,1,2,3,3,3-2H6)4 in Scheme 2). Chiral 1,2-propanediol derivatives are important building blocks for medicinal chemistry applications. With a lower differentiation between the steric demand of the two substituents on the ketone, this material is more challenging to produce in high enantiomeric excess by selective delivery of a hydrogen atom to one prochiral face. As for 2, access to deuterated variants of 4 is facilitated by the ease of exchange of α-hydrogen atoms prior to reduction. We are unaware of any previous reports in which a deuterated 1,2-propanediol 1-O-benzyl ether has been prepared in enantioenriched form.
 | ||
| Scheme 2 A combination of H/D exchange next to the carbonyl of 3 (teal deuterons) and subsequent deuterative microbial reduction (purple deuteron) can be employed for the synthesis of (1,1,2,3,3,3-2H6)4. | ||
Our aim was to use this two-part deuterative strategy, α-exchange and deuterative microbial reduction, to deuterate at >95%-d for the labelled positions, ensuring suitability for medicinal chemistry applications.
3. Preliminary studies
It is known that some de novo biosynthesis may occur with reasonably high (>80%) deuterium incorporation using deuterium oxide as the growth medium but feeding the microorganism with protiated carbon sources only.27 This is the ideal recipe for deuteration by biosynthetic means since deuterium oxide is several orders of magnitude cheaper per mole of deuterium atoms than, for example, deuterated glucose. Successful use of this strategy is dependent on efficient exchange between biosynthetic intermediates derived from the protiated carbon source and the deuterated medium.In previous reports, we have described the biosynthesis of terpenoids using only deuterium oxide as the deuterium source, showing that >85% of near-uniform overall deuteration in squalene and sterols is possible without feeding the organism a deuterated carbon source.27,28 This is partly because the biosynthesis of terpenoids occurs via acidic intermediates able to exchange hydrogen atoms with the water of the intracellular medium. The incorporation of deuterium into terpenoids also involves the reduction of intermediates by enzyme-mediated delivery of hydrogen atoms originating from the reducing cofactor NADPH. Given that we had previously produced biosynthetic products with near-uniform deuteration, we knew that metabolism of the protiated carbon source to produce NADPH was occurring alongside exchange processes that led to NADPD being produced and employed by the organism.
This is consistent with the known action of flavin-dependent dehydrogenases in exchanging NADPD deuterons from a deuterated carbon source with a protiated medium. This effect has been described in terms of ‘missing’ deuterium when tracing the fate of deuterated glucose in cells.18 Reports affected by the ‘missing deuterium’ effect have been characterised as follows: ‘… studies examining NADPH production with deuterated carbon sources have failed to account for roughly half of NADPH's redox active hydrogen’. Therefore, performing the microbial reduction of 1 in deuterium oxide as a study to gain insight for the development of more highly deuterative methods, we expected to find a maximum of roughly 50% deuterium incorporation without a deuterated carbon source – lower than what is achievable in de novo terpenoid biosynthesis.18Table 1 shows that initial studies to probe the limits of deuterium incorporation in the reduction of 1 using >99.8%-d deuterium oxide as the sole deuterium source and with dried baker's yeast purchased from a supermarket are indeed consistent with a roughly 50%-d limit in microbial reduction carried out in deuterium oxide.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Change(s) from standard conditions | Yield | %-d | ee (%) |
| 1 | H | 20 h reaction with 8 g of yeast in 60 mL of H2O buffer.26 | 80% | — | >97% |
| 2 | H | 4 h reaction with 8 g of yeast in 60 mL of H2O buffer, no (L)-cysteine additive.26 | 56% | — | 85% |
| 3 | H | None | 91% | 10% | N.D. |
| 4 | H | 16 h fermentation prior to addition of 1. | 90% | 40% | N.D. |
| 5 | H | No glucose, buffered at pH 5, 5 h fermentation prior to addition of 1. | 86% | 34% | N.D. |
| 6 | H | 0.2 g of baker's yeast, 16 h fermentation prior to addition of 1, then 16 h reaction. | 90% | 11% | N.D. |
| 7 | H | Buffered at pH 5, 5 h fermentation prior to addition of 1 (2 repeats). | 86% | 46% | N.D. |
| 85% | 45% | N.D. | |||
| 8 | H | Buffered at pH 8, 5 h fermentation prior to addition of 1. | 85% | 46% | N.D. |
| 9 | H | Unbuffered, 0.15 M NaCl, 5 h fermentation prior to addition of 1. | 89% | 46% | N.D. |
| 10 | D | Use of MBG5177 in modified HS medium (5 g L−1 yeast extract, 10 g L−1 peptone, 20 g L−1 glycerol-d8). 1 week culture followed by 1 week reaction. | 92% | 90% | N.D. |
| 11 | D | Use of MBG5177, growth in modified HS medium (5 g L−1 yeast extract, 10 g L−1 peptone, 20 g L−1 glycerol-d8) then transfer of cells to deficient HS medium (20 g L−1 glycerol-d8). 1 week culture followed by 1 week reaction. | 94% | 94% | 93% |

We were able to achieve higher yields than the existing reports (see entries 1 and 2 for literature results, Table 1) by reducing the cell density slightly, increasing the reaction volume and using centrifugation to break emulsions and thereby aid dual-phase extraction (entries 3–10). Our initial test reaction showed low (10%) deuterium incorporation. Presumably, the reduction of 1 was occurring rapidly, giving little time for exchange of the reducing hydrogen atoms in NADPH upon formation of the reductant. Increasing the fermentation time prior to addition of the substrate from one hour to 16 hours allowed the deuteration level to rise to 40% (entry 4). We concluded that pre-stirring in the deuterated medium to allow time for H/D exchange was a factor in increasing the deuteration level (cf. entries 3 & 5) but further experiments revealed that there was no increase in deuteration level with longer than 5 hours fermentation prior to substrate addition. Removing the glucose food source entirely but maintaining the culture for 5 hours prior to adding 1 gave a result (entry 5, 34%-d) that indicated that the large quantity of dried cells used in this baker's yeast reduction contain sufficient stored chemical energy to carry out the reduction without feeding. The unfed reaction was buffered at pH 5 (favourable to S. cerevisiae). This reaction showed high cell mortality from deprivation of a carbon source, and the resulting cell lysis caused the reaction to become almost intractably thick and the chromatographic separation of the product from lysed cell components rather difficult, ruling out the use of unfed cultures in general. The buffer seemed unimportant to the results of reduction – buffering at pH 5 (entry 7, 2 repeats) or pH 8 (entry 8) did not affect the result substantially (pH 5 and pH 8 were measured at the end of the reactions, respectively) while substituting the buffer for isomolar 0.15 M NaCl and allowing the pH to be determined by the organism gave a final pH of 4.5. For the remainder of our studies, we abandoned buffering and allowed the organism to determine the pH level.
In a bid to reduce the dependency of the result on H/D exchange by allowing the yeast to multiply in the deuterated medium we decreased the quantity of initially supplied dried yeast by a factor of 24 and extended the fermentation time prior to addition of 1. In fact, this reduced the deuterium incorporation to 11% (entry 6). In this case lesser preformed quantities of NADPH/D were presumably exhausted quickly, and the reduction began to consume NADPH/D rapidly as it was formed without time for sufficient exchange to take place. The standard conditions for the studies in Table 1 use 60 g L−1 of dried yeast cells. This very high cell density is not generally achievable (particularly in D2O) even with long fermentation times, except by addition of the pre-formed organism to the medium or isolation of cells from a culture and resuspension in a lower volume of medium, a fact confirmed by the experiment in entry 6. The lower cell density did aid purification, though: the culture could be extracted efficiently with ethyl acetate without filtration or the formation of enduring emulsions.
Typically, about 1–3% of the product was the achiral 2-O-benzoylglycerol isomer. The formation of this product was not pH dependent but did increase to about 6% of the product in the unfed culture (entry 5). The best results in terms of deuterium incorporation where deuterium oxide is the only source of deuterium were those with formation of the largest quantity of pre-exchanged reductant prior to introduction of 1. It is not clear why greater than 5 hours fermentation time prior to introducing 1 does not appear to result in further H/D exchange.
For the glycerol backbone (5 hydrogen atoms), 45% deuteration at the site of reductive deuteration (entries 7–9) and 95% deuteration by pre-exchange of the α-hydrogen atoms of 1 would give 85% overall backbone deuteration. This level of deuteration is likely sufficient for applications such as neutron studies or certain mass spectrometric techniques such as NanoSIMS but is insufficient for the goals of the current study.
Since the roughly 50%-d limit of deuteration we had observed with deuterium oxide as the sole deuterium source seemed likely to apply regardless of the mass of protiated yeast cells added or culturing (i.e. pre-exchange fermentation) time, high deuteration necessarily involves a deuterated carbon source, since the slow addition of the substrate to be reduced – allowing time for cofactor regeneration and exchange – would not overcome the roughly 50%-d limit, given that the biological mechanism imposing the limit would likely be unchanged.
Unlike the deuteration level, very high cell densities were found to be unnecessary for high yields and may even reduce yields by rendering the culture difficult-to-extract. This observation suggested that rather than using dried cells, we might achieve reasonable yields from the cell densities available from inoculating a medium with a yeast strain stored in agar and culturing it with a deuterated carbon source. This strategy would also overcome the exchange limit of ca. 50%-d. Given the high cost of deuterated glucose we turned to S. cerevisiae strain MBG5177 (entries 10 and 11), which was developed to be able to grow robustly on glycerol, a compound which is much less expensive than sugars such as glucose in deuterated form.
4. Synthesis of (2-2H1)solketal
Using a modified Hestrin & Schramm (HS) medium in which glycerol-d8 was used as the carbon source (peptone and yeast extract function as the source of nitrogen, amino acids and co-factors), in 99.8%-d deuterium oxide, the deuteration level in the microbial reduction of 1 increased to 90% (Table 1, entry 10). It was reasoned that the yeast extract and peptone were likely to have been the source of most of the incorporated protium. Given the lower cell density resulting from the culturing of the strain in liquid medium rather than dispensing dried baker's yeast, the pre-reaction culturing time as well as reaction time were increased to a week to ensure complete conversion. For practical reasons the nitrogen atmosphere was abandoned for these shake-flask cultures. Some loss of deuterium is likely to have occurred because of exchange between the medium and atmospheric moisture in the headspace above the medium, which was free to diffuse through a breathable culture plug. In none of the 2H or 13C NMR analyses of the biosynthesised (S)-(2-2H1)5 from the experiments in Table 1 did we observe labelling in any other than the 2-position, regardless of the pH of the reaction medium.To address the issue of protium being introduced using peptone and yeast extract we repeated the reaction but did not use the HS culture directly, instead isolating the cells by centrifugation and resuspending them in a deficient HS medium without peptone and yeast extract but containing the same 20 g L−1 of glycerol-d8 (entry 11). After one day in this deficient medium, 1 was added and after 5 days shaking (S)-(2-2H1)5 was isolated and found to have an isotopic purity of 94%-d and an enantiomeric excess of 93% (by formation of the Mosher ester after transformation of (S)-(2-2H1)5 into the target compound, (S)-(2-2H1)2. This synthesis was carried out by installation of the acetonide to give (S)-(2-2H1)6, followed by cleavage of the benzoate ester. The absolute configuration (AC) of the major isomer produced was the same as for the baker's yeast strain from the supermarket (previously established as (S)-5, leading to (R)-2via the transformations in Scheme 3).
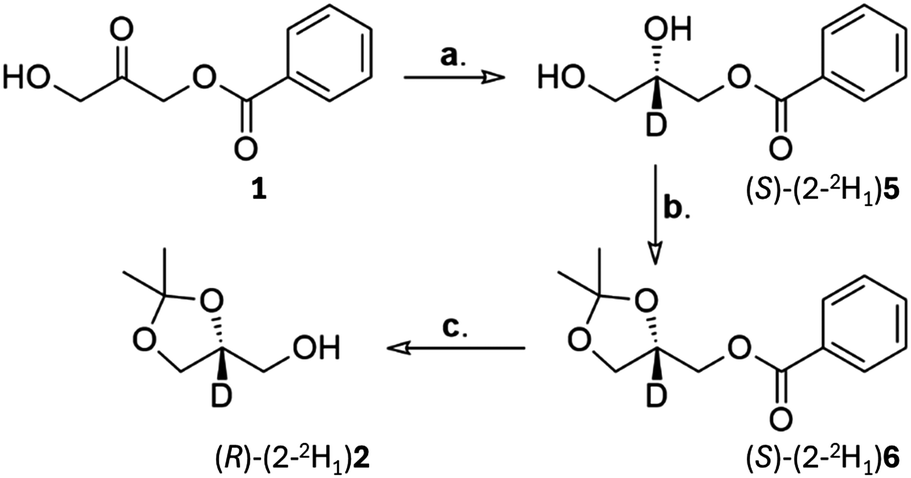 | ||
| Scheme 3 Synthesis of (R)-(2-2H1)2. Reagents and conditions: (a) MBG5177, glycerol-d8; defined HS medium; 30 °C; 5 days; 93%, 94%-d, 93% ee by 19F NMR analysis of the Mosher ester of (R)-(2-2H1)2. (b) Me2C(OMe)2, TsOH; CH2Cl2; rt; 16 h; 91%. (c) NaOH; EtOH; 50 °C; 5 h; 66%. | ||
The (L)-cysteine additive employed with baker's yeast (in deference to the literature protocol) has a profound effect on reaction outcome. Replacing it with ethanolamine was reported to reduce the enantioselectivity from >97% to 86% and the yield from 80 to 76%.26 Although we employed it in supermarket-bought baker's yeast reductions (entries 3–9, Table 1), we did not employ it with MBG5177 since we did not wish to introduce further variables but rather to focus on reducing the cost of reagents while maintaining high levels of deuterium incorporation. When compared to the literature reduction of 1 by supermarket-bought baker's yeast, the enantiomeric excess from the MBG5177 reduction is superior to the reported performance of baker's yeast without (L)-cysteine (entry 2). In general, the addition of reducing reagents like cysteine, exclusion of air and the use of potentially reducing dimethylsulfoxide as a solvent for adding substrates to baker's yeast reductions are all expected to affect the course of microbial reduction by changing the redox environment of the medium.
We established the enantiomeric excess of all the solketal isotopologues produced in this work by Mosher ester analysis. This choice of method was motivated by the presence of traces of biosynthesised impurities in the reduction product, which potentially interfere with HPLC analysis, as well as traces of the 2-O-benzoylglycerol isomer. The use of optical rotation as a tool for quantitation of enantioselectivity was also judged unlikely to be reliable given the small reported optical rotation values and existing confusion in the literature about the sign of rotation of the two enantiomers.25 Equally, we were unsuccessful in developing a method for analysis of (S)-(2-2H1)5 using NMR chiral shift reagents. We found that analysis of the Mosher ester of (R)-(2-2H1)2 was best carried by 19F NMR, with acetone-d6 as the solvent that gave the most disperse signals. Incidentally, this permits analysis of variously deuterated isotopologues of 2 since some or all the 1H NMR signals would be absent in more highly deuterated variants. Our next task was to include the other component of our intended method, viz. the introduction of deuterium atoms by H/D exchange of α-hydrogen atoms.
5. Synthesis of (1,1,3,3-2H4)solketal
Our next target was (1,1,3,3-2H4)2, to be produced by protiative reduction of (1,1,3,3-2H4)1 (97%-d, Scheme 4) using baker's yeast. Compound (1,1,3,3-2H4)1 was prepared from 1,3-dihydroxy(1,1,3,3-2H4)acetone (itself produced by H/D exchange according to a literature protocol)29 and subjected to the standard reaction conditions from Table 1. | ||
| Scheme 4 Synthesis of (R)-(1,1,3,3-2H4)2. Reagents and conditions: (a) baker's yeast, glucose, (L)-cysteine; 0.15 M H2O pH 5 phosphate buffer; 30 °C; 5 days; 89%, 97%-d, 95% ee by Mosher ester analysis of (R)-(1,1,3,3-2H4)2. (b) Me2C(OMe)2, TsOH; CH2Cl2; rt; 16 h; 90%. (c) NaOH; EtOH; 50 °C; 5 h; 85%. See also Table 2, entry 3. | ||
In conformance to the results for the synthesis of (2-2H1)5, (where deuterium incorporation from the labelled medium into the exchangeable α-positions was not observed), we observed retention of the 97%-d labelling from the starting material (cf: entries 1, 2 with 3, Table 2). The reduction proceeded in high yield (89%) with 95% enantiomeric excess. Compound (1,1,3,3-2H4)5 is cheap and simple to produce as it employs baker's yeast in a protiated medium.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Yeast species and conditions | Carbon source | Yield | %-d | ee (%) |
| 1 | H | 20 h reaction with 130 mg mL−1 dried baker's yeast in pH 7 H2O buffer, N2 atmosphere, addition of reactant in EtOH/DMSO, (L)-cysteine as additive.26 | Glucose (100g L−1) | 80% | N/A | >97% |
| 2 | H | 4 h reaction with 130 mg mL−1 dried baker's yeast in pH 7 H2O buffer, N2 atmosphere, addition of reactant in EtOH, no additive.26 | Glucose (100 g L−1) | 56% | N/A | 85% |
| 3 | D | 16 h reaction with 50 mg mL−1 dried baker's yeast in pH 7 H2O buffer, N2 atmosphere, addition of neat reactant, (L)-cysteine as additive. | Glucose (36 g L−1) | 89% | 97% | 95% |
| 4 | H | 4 d reaction with X-33, air atmosphere, YP medium (5 g L−1 yeast extract, 10 g L−1 peptone) | Glycerol (20 g L−1) | 87% | N/A | 93% |
| 5 | H | 4 d reaction with X-33, air atmosphere, YP medium (10 g L−1 yeast extract, 20 g L−1 peptone) | Methanol (20 g L−1) | 79% | N/A | 91% |
| 6 | D | 4 d reaction with X-33, air atmosphere, YP medium (10 g L−1 yeast extract, 20 g L−1 peptone) | Glycerol (20 g L−1) | 88% | 97% | 91% |

Despite the requirement to synthesise (1,1,3,3-2H4)1 from deuterated 1,3-dihydroxyacetone, this reduction offers a means to prepare asymmetrically substituted glycerol derivatives with nearly 80% (4/5 hydrogen atoms) glycerol backbone deuteration. This material is ideal for the construction of phospholipids for neutron studies since the enantiomeric excess is high and the estimated contrast match point for the corresponding glycerol C3H1D7O3 (i.e. after exchange of the oxygen-bound hydrogen atoms with D2O) is very close to pure deuterium oxide.‡
Lipids synthesised from (1,1,3,3-2H4)2 would not, however, be ideal mass spectrometry standards since complex lipids contain enough carbon-13 to give some signal overlap with such a standard. For instance, the common phospholipid POPC (m/z = 760) contains 0.36% of m/z = 764 from the native isotopic distribution. Compound (1,1,2,3,3-2H5)2 would be preferred as the starting material for synthesising POPC-d5 – a mass spectrometry standard with negligible overlap of analyte signal. Prior to embarking on the synthesis of (1,1,2,3,3-2H5)2 by deuterative reduction we desired to explore means to increase the extent of deuteration (from 94%-d) achieved in the synthesis of (2-2H1)2 and to further reduce the material costs by using a yet cheaper deuterated carbon source than glycerol-d8, namely methanol-d4. To do this, we employed the methylotropic yeast Pichia pastoris (Komagataella pastoris) wild type strain X-33 (Invitrogen), which is commonly used for recombinant protein expression.30 X-33 can grow to high cell density in simple and defined media, a useful quality since we wished to reserve the option to enhance the deuteration level by restricting the addition of protiated components to the culture. The use of minimal media may enhance the resistance of the culture to contamination by providing a less hospitable environment for bacteria. We employed a biohazard cabinet for culture manipulation and used sterile media but did not add antibiotics to the medium. Using these precautions we have not thus far observed contamination events in our work – but it was a concern given the 1-week reaction times. In the event, a glycerol-fed culture of X-33 (Table 2, entry 4) was found to reduce 1 to 5 in 87% yield and 93% enantiomeric excess in a 4-day reaction. To the best of our knowledge this is the first report of P. pastoris used to perform a microbial reduction of an added small molecule.
The AC of the major enantiomer produced by strain X-33 was again the same as for the baker's yeast strain from the supermarket, established by Mosher ester analysis. The enantioselectivity was slightly less than the reduction by baker's yeast in the presence of (L)-cysteine (entry 1), but higher than without this additive (entry 2). An enantiomeric excess of >90% was retained both when the carbon source was changed to methanol (entry 5) and when (1,1,3,3-2H4)1 was reduced (entry 6). The products of the reductions in Table 2 were transformed into 2 and the enantiomeric excess obtained by Mosher ester analysis.
For the experiments in Table 2 we added the substrate as the neat solid to the culture and continued this practice throughout the remainder of the work. This change avoided the introduction of protiated organic solvent or the expense of using deuterated solvents. Methanol was dosed gradually with a target concentration <0.5% v/v to avoid toxicity to the organism, monitoring the dissolved oxygen tension. Strain X-33 fed with methanol-d4 seemed the most promising lead for minimising the cost of labelled reagents and was selected as the system for producing fully backbone-deuterated solketal, our final target isotopic variant of this substance.
6. Synthesis of (1,1,2,3,3-2H5)solketal
To produce the fully backbone-deuterated solketal would require the introduction of two stressors to the P. pastoris culture, being the transition to D2O growth media and the shift from glycerol to methanol feeding. In keeping with the aim of achieving a high level of deuteration with economical use of deuterated carbon source, the organism was adapted first to growth in D2O media containing 20 g L−1 protiated glycerol, followed by the introduction of protiated methanol and then methanol-d4 at 4 g L−1 with simultaneous stepwise deprivation of the glycerol, in a series of small shake flasks. The final shake flask in this series became the inoculum for a bioreactor culture, allowing for real-time monitoring during cultivation and the provision of real-time monitoring of biomass and substrate consumption. At the stationary phase, the bioreactor culture was divided back into shake flasks for the reduction reactions. Strain X-33, as cultured above, proved competent in production of the fully deuterated material (97% backbone deuteration, Scheme 5). The enantiomeric excess (92%) was higher than baker's yeast achieves without the (L)-cysteine additive, while the yield (85%) was similar to the highest yields found to be achievable with baker's yeast. | ||
| Scheme 5 Synthesis of (R)-(1,1,2,3,3-2H5)2. Reagents and conditions: (a) strain X-33, methanol-d4, YP medium; 30 °C; 4 days; 85%, 97%-d, 92% ee by Mosher ester analysis of (R)-(1,1,2,3,3-2H5)2. (b) Me2C(OMe)2, TsOH; CH2Cl2; rt; 16 h; 90%. (c) NaOH; EtOH; 50 °C; 5 h; 53%. | ||
All three organisms used in the microbial reduction of 1 demonstrated negligible back exchange at the α-positions prior to reduction, regardless of the pH of the reaction medium. One aspect of the biochemistry which is undesirable is the tendency towards formation of the optically inactive 2-O-benzoyl ester (generally, it could be completely removed chromatographically from 2 after being partially removed during purification of 6). With strain X-33 this impurity was always less than 3% of the product mixture by 1H NMR. This byproduct becomes problematic if the ester is shuttled between sn1, sn2 and sn3 positions in the product – this process would then be a racemisation. We saw no evidence that the racemisation process was significant, given that the enantiomeric excess remained high with P. pastoris X-33 and both strains of S. cerevisiae.
The typical amount of growth medium for shake flask cultures of strains X-33 and MBG5177 was 300 mL, containing 20 g L−1 of the carbon source (i.e. 6 g). At the time of writing this is about 550 USD of D2O. Using glycerol-d8 as the carbon source makes the cost of labelled materials for such a shake flask culture about 800 USD in total. The reactions in Schemes 4 and 5 used 100 mg of 1 to produce about 90 mg of 5, giving labelled material costs of about 9000 USD per gram of 5. Replacing glycerol-d8 with methanol-d4 reduces the labelled material costs for a 300 mL deuterative shake flask culture to about 6000 USD per gram of 5 (prices at time of writing). This is not cheap but does compare favourably with attempting to obtain mono-protected, backbone perdeuterated, chiral glycerols commercially, since, to the best of our knowledge, there are none available except perhaps by custom synthesis. In fact, even backbone perdeuterated D-glyceraldehyde is not available for less than 2600 USD per gram (250 mg scale). It is also expected that the cost of microbial, deuterative reduction could be improved significantly by recycling the deuterated yeast cell mass.
In our experiments with our first target substrate, selective deuterium labelling at the chiral, secondary alcohol was achieved in the synthesis of (S)-(2-2H1)2, with a high yield and enantiopurity (93% ee, 94%-d). Backbone perdeuterated material ((S)-(1,1,2,3,3-2H5)2) was also produced (92% ee, 97%-d) for application to the synthesis of lipid mass spectrometry standards while material with 4 deuterons ((S)-(1,1,3,3-2H4)2) was obtained (95% ee, 97%-d) from a protiated culture for application to neutron studies. We then turned our attention to our second target, 1,2-propanediol, 1-O-benzyl ether.
7. Synthesis of deuterium-labelled 1,2-propanediol
The second target presents an opportunity for the maximum level of deuterium incorporation into a three-carbon framework (6 deuterons) whilst still being able to produce a chiral, secondary alcohol via reduction. As for our first target, we began by studying the protiative reduction of protiated and deuterated starting materials. Compound (1,1,3,3,3-2H5)3 was produced with 98%-d by base-catalysed exchange of the α-hydrogens of 3 with deuterium oxide. Baker's yeast is known to reduce 3 (Table 3, entry 1) with high yield (76%) and moderately high enantiomeric excess (90% ee).31 The reported transformation uses a large quantity of cells (100 g of dried baker's yeast) to produce 0.35 g of 4. This scale would be unsuitable for screening deuterative reduction conditions given the expense of culturing such a large quantity of cells in a deuterated medium and the difficulty in achieving cell densities as high as the reported 100 mg mL−1. We therefore performed a test reduction with baker's yeast on a 200 mg scale in 100 mL of water to serve as a reference for achievable yield and stereoselectivity in a fully protiated system on a more practical scale (entry 2). We found that the enantiomeric excess (96%) was higher than the previous report and a yield of 57% was achieved after a 16-hour reaction, with chromatographic separation of remaining starting material found to be practical.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | Yeast species and conditions | Carbon source | Yield | %-d | ee (%) |
| a 33% recovery of starting material. b Measured by Mosher ester analysis. c Measured by HPLC. d Measured by MRR. Methanol was dosed gradually to keep the concentration below 4 g L−1 at any time point. | ||||||
| 1 | H | 6 d reaction with 100 mg mL−1 dried baker's yeast in unbuffered H2O, addition of reactant in EtOH over 8 h.31 | Sucrose (100 g L−1) | 76% | N/A | 90%a |
| 2 | H | 16 h reaction with 80 mg mL−1 dried baker's yeast in pH 5 H2O buffer, N2 atmosphere, addition of neat reactant. | Glucose (30 g L−1) | 57%a | N/A | 96%b,c |
| 3 | D | 40 h reaction with 60 mg mL−1 dried baker's yeast in pH 5 H2O buffer, N2 atmosphere, (L)-cysteine as additive (2 repeats). | Glucose (30 g L−1) | 84% + 88% | 98% + 98% | 96%c + 95%c |
| 4 | D | 40 h reaction with 48 mg mL−1 dried baker's yeast in pH 5 H2O buffer, air atmosphere, addition of neat reactant, (L)-cysteine as additive. | Glucose (2.4 g L−1) | 92% | 98% | 92%,c 91%d |
| 5 | H | 7 d reaction with X-33, air atmosphere, YP medium (10 g L−1 yeast extract, 20 g L−1 peptone) | Glycerol (20 g L−1) | 85% | N/A | 53%b |
| 6 | D | 4 d reaction with X-33, air atmosphere, YP medium (10 g L−1 yeast extract, 20 g L−1 peptone) | Glycerol (20 g L−1) | 86% | 98% | 52%b |
| 7 | H | 7 d reaction with X-33, air atmosphere, YP medium (10 g L−1 yeast extract, 20 g L−1 peptone) | Methanol (20 g L−1) | 12% | N/A | 5%b |

Increasing the reaction time to 40 hours, decreasing the concentration of carbon source and including (L)-cysteine as an additive gave (S)-(1,1,3,3,3-2H5)4 in high yield (84% and 88%; entry 3; duplicate reactions to check repeatability) and enantiomeric excess (96% and 95% ee). There was no detectable loss of labelling under any of the conditions studied in Table 3. Chiral HPLC was found to be a suitable method for determining enantiomeric excess, but HPLC analysis required treatment of the crude mixture with lithium hydroxide solution to cleave traces of lipids obtained from the culture, followed by chromatographic purification to achieve material of sufficient purity for chiral HPLC analysis (this is partly because of the weak UV response of the analyte). Optical rotation was again ruled out because of the presence of trace impurities. For this reason, we again turned to Mosher ester analysis to determine the enantiomeric excess of the product (see: ESI I,† pages 7 & 22–23). The enantiomeric excess of the baker's yeast reductions tended to be highest under a nitrogen atmosphere (96% ee, entries 2 and 3) and dropped to 92% when the reaction was performed under air (entry 4). The presence or absence of (L)-cysteine was not noted to influence the enantioselectivity, in contrast to reductions of 1. Reducing the quantity of cells and of the carbon source (entry 4) did not restrict completion of the baker's yeast reduction of (1,1,3,3,3-2H5)3 in a 40-hour time window.
We then moved to study the corresponding reduction by cultured P. pastoris X-33. Initially, we trialled the reduction of the protiated material (3) using glycerol as the carbon source (entry 5). The enantiomeric excess was a modest 53% although the yield was acceptable (85%). The reduction required a week for complete consumption of the starting material. The lower enantiomeric excess was not surprising. In microbial reductions tailoring of the steric and electronic properties of the protecting group are generally required for high enantioselectivity. For instance, replacing the benzoate ester of our first target substrate, 1, with a benzyl ether results in a drop in enantiomeric excess from >97% to 55% in a baker's yeast reduction.26 Since the current study is aimed at finding economical conditions for high deuterium incorporation without loss of the enantiomeric excess achieved in the protiative reduction, we considered this result to be a reasonable basis for further experiments on this substrate. Coproduced with 3, and tedious to separate from it by chromatography, was 2-phenylethanol. Since this material was known to be produced naturally by fermenting P. pastoris we initially did not consider it as being linked to the microbial reduction being studied.32
The protiative reduction of the deuterated starting material (entry 6) with strain X-33 occurred with the degree of deuteration and enantiomeric excess essentially unchanged. However, when methanol was used as the carbon source a dramatic reduction in yield (to 12%) for the week-long reaction was observed (entry 7). The production of 2-phenylethanol increased (in absolute terms and not just relative to the desired product) and it became the major product isolated. The enantiomeric excess dropped to 5%. Growing Pichia on methanol induces expression of the genes required for metabolising this carbon source, and the long reaction time and relatively low amount of methanol in the culture may have led to exhaustion of the carbon source and subsequent metabolism of (S)-(1,1,3,3,3-2H5)4 may have been a consequence.
To prevent loss of the reduction product the corresponding deuterative culture (with methanol-d4 as carbon source) was carried out over 4 days (Scheme 6). The yield was 56%, with some starting material observed but not recovered, while the enantiomeric excess (54% ee) matched the Pichia protiative cultures in Table 3. A high level of backbone product deuteration (98%-d) was measured. Residual 1H NMR analysis of the starting material and product (see: Scheme 6; see: ESI I,† pages 51 & 55 for spectra) indicates retention of the original extent of labelling (teal) and 97%-d for the deuteron introduced by microbial reduction (purple).
 | ||
| Scheme 6 Synthesis of (S)-(1,1,2,3,3,3-2H6)4. Reagents and conditions: X-33, methanol-d4; YP medium (10 g L−1 yeast extract, 20 g L−1 peptone); air atmosphere; 30 °C; 4 days; 56%, 98%-d, 54% ee by Mosher ester analysis of (S)-(1,1,2,3,3,3-2H6)4. | ||
Again, 2-phenylethanol was co-produced with (S)-(1,1,2,3,3,3-2H6)4. We were forced to revise our assessment that this material was irrelevant to the reductive processes being studied, as it was found to have an anomalous isotope distribution ((2H)7, Scheme 7) which indicated that it may have been derived not from de novo biosynthesis (despite the known capacity of this yeast species to do so) but was instead a product of the metabolism of (S)-(1,1,2,3,3,3-2H6)4. The mechanism(s) of this putative metabolic transformation are obscure, and the origin of the alcohol carbon (purple) is unknown while there is at least some deuteration of the benzylic carbon atom (teal). We cannot rule out biosynthesis from protiated phenylalanine supplied to the culture as part of the nitrogen source, but the increase in this byproduct occurring alongside the decrease in the expected product suggested transformation of 3 to 7. The production of 7 is representative of a general consideration in biosynthesis: supplying an organism with a material structurally-related to its own suite of biomolecules creates potential for interception of the reactant or product by undesired biosynthetic processes.
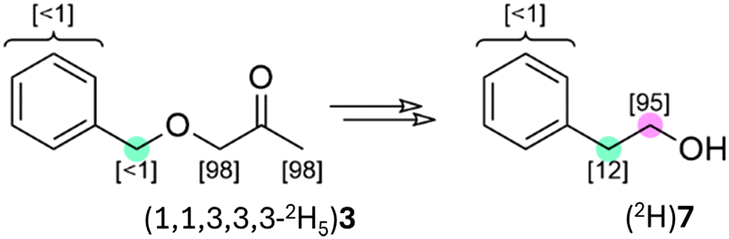 | ||
| Scheme 7 Site-specific deuteration levels of the (2H)7 byproduct of the deuterative reduction of (S)-(1,1,3,3,3-2H5)3 by X-33, measured by integration of the 13C{1H,2H} NMR. | ||
There was a more pronounced difference between the action of baker's yeast and X-33 in the reduction of the second substrate than in reductions to produce solketal. Despite the poor yield it had been demonstrated that high deuteration levels could be achieved without lowering the enantiomeric excess observed in the protiative reduction.
In fact, the most difficult aspect of performing this work was the determination of enantiomeric excess on a small scale. In the case of solketal, the product is sufficiently volatile to be problematic to isolate on a scale typical of test reactions and yields were accordingly affected. This was not a concern with the second, protected 1,2-propanediol substrate but in both cases traces of impurities from the microbial reduction made it challenging to ensure reliable quantification of enantiomeric excess by 19F NMR, generally requiring chromatography, with effort required to ensure the observed diastereopurity of the crude mix of Mosher esters was not affected by chromatography. 1H NMR of the Mosher esters was not suitable either because the substrates were variously deuterated and the low steric differentiation (viz. hydrogen versus methyl for the second substrate) led to low differences in the chemical shifts of corresponding positions on the Mosher diastereomers. Of the materials studied in this work, chiral HPLC was found to be practical for 4 only. To validate the accuracy of Mosher ester analysis and with the intent to expedite and enhance future studies, we turned to molecular rotational resonance (MRR) spectroscopy.
8. MRR analysis of (S)-1,2-propanediol, 1-O-benzyl ether
Molecular rotational resonance (MRR) spectroscopy has recently emerged as an analytical technique to provide site-specific, quantitative analysis of isotopic mixtures.33–36 MRR has exceptionally high resolution, making it possible to distinguish closely related analytes, including isotopic variants, without chromatographic separation. MRR spectra are directly related to the three-dimensional structure of the analyte; consequently, distinct isotopic variants have a unique MRR spectra that can be calculated with a high degree of accuracy using DFT calculations.37 For isotopic variants, a single equilibrium geometry is used to calculate all possible isotopic variants of an analyte. MRR spectra are identified by comparing experimental with calculated spectroscopic parameters, which enables identification of MRR spectra without the need for reference samples, which are not always readily available. The relative intensity of the identified spectra can be analyzed to provide quantitative data on the site-specific deuteration levels.36 Additionally, MRR can also be used to resolve enantiomers using a technique called chiral tagging. Chiral tagging resolves enantiomers, which are spectroscopically indistinguishable, through the non-covalent derivatization in the gas phase with a small chiral molecule referred to as a chiral tag.38,39 Similar to analysis of monomer species, the non-covalent complex spectra are identified using quantum chemistry calculations which enable AC determination without reference compounds.40 The relative intensity of the resulting diastereomeric complexes is used to determine the enantiomeric excess of the analyte and establish AC. This means that a single technique can be used to assess enantiopurity, configuration, the distribution of isotopologues and the overall deuteration level without full chromatographic purification of the product or derivatisation.Examination of the chiral HPLC chromatogram of biosynthetic (S)-(1,1,3,3,3-2H5)4 (Table 3, entry 4; see ESI I,† page 28) showed a trace amount of comparatively highly UV active starting material as an impurity. Although peak separation was perhaps sufficient to analyse the material, we nevertheless considered this an unsatisfactory analysis and therefore selected this material as a test case for MRR analysis.
MRR spectra are identified by comparing experimental and calculated spectroscopic parameters, meaning that for screening studies such as detailed in this study a single set of calculations can support analysis of a screen of biosynthetic conditions. In this instance the easy availability of the protiated building block 1,2-propanediol 1-O-benzyl ether allows for refinement of the calculated spectrum to facilitate prediction of the spectrum for the isotopic variants and therefore provide an analysis of the relative composition of the isotopologues.
The -2H0 sample was used to characterize the analyte by MRR. Multiple conformers of the analyte were observed; however, only the lowest energy conformer was used for the isotope analysis. A complete summary of the experimental information and spectroscopic assignments can be found in the ESI.† The dominant conformer established by calculation (Table 4), unsurprisingly places the largest groups on each of the two carbon atoms along the indicated (C-1 to C-2) bond in an anti-configuration (the A-value for a methyl group, 1.74 is greater than a hydroxyl with 0.6–1.04),41 and this conformation may furthermore permit a favourable H-bonding interaction.
|
|
|||
|---|---|---|---|
| Variant | Isotopologue | Sites of deuteration | Relative composition by MRR |
| 1 | -d5 | 1, 2, 3, 4, 5 | 87.0% |
| 2 | -d4 | 1, 2, 4, 5 | 9.2% |
| 1, 2, 3, 5 | |||
| 1, 2, 3, 4 | |||
| 3 | -d4 | 1, 3, 4, 5 | 3.7% |
| 2, 3, 4, 5 | |||

Six distinct spectra attributed to 2H5 and 2H4 isotopic variants were assigned (Table 4). They have been grouped into 3 variants to provide site-specific deuteration data – variant 1 being the fully deuterated material with variants 2 and 3 representing, respectively, an extra residual proton at the C-3 methyl group and the C-1 methylene. There was sufficient sensitivity in the spectrum to detect species at a level of 1% or higher relative to the main variant. None of the ten possible distinct 2H3 species possible from underdeuteration were observed. One erroneous assumption generally unremarked upon in the mass spectrometric measurement of deuterated materials is that the response factor is identical between isotopologues. Therefore, the use of multiple methods in parallel may allow for detection of erroneous claims arising from limitations of individual analytical methods.
Mass spectrometric analysis uncorrected for response factors showed that the studied material contained 86.3% of variant 1, 12.1% of variants 2 and 3 and 1.65% of trideuterated variants. Table 5 shows a comparison of site-specific deuteration data obtained by integration of the residual protons in 1H NMR, mass spectrometric analysis and MRR analysis (see: Scheme 6; see: ESI I,† pages 29 & 30; see: ESI II†). 13C{1H,2H} NMR was not employed due to the insufficient signal to noise ratio. It should also be noted that residual 1H NMR analysis is contingent in general on sufficient sample size and the absence of overlapping signals.
| Site | % backbone deuteration for 2Hn | ||
|---|---|---|---|
| Residual 1H NMR | Mass spectrometry | MRR | |
| C-1 (n = 2) | 98% | — | 98% |
| C-3 (n = 3) | 97% | — | 97% |
| Overall (n = 5) | 97% | 97% | 97% |
The overall deuteration level by mass spectrometry (calculated using the DGet! software package)42 was 97%-d for 2H5 as well as by calculation from the MRR data (which excludes the 2H3 variants but will only slightly overestimate the overall deuteration level at these relatively high overall deuteration levels). Although our study did not attempt to quantify the errors in the latter measurements, the MRR result conformed with the mass spectrometric result, which is the standard method used to quantify deuteration extent.
Having established that MRR provides data for overall and site-specific deuteration in conformance to two standard techniques (residual 1H NMR analysis and mass spectrometry) we turned to analysis of the enantiomeric excess of biosynthetic (S)-(1,1,3,3,3-2H5)4. This was performed by chiral tagging – resolving the MRR spectrum of non-covalent complexes of a chiral tag of known enantiopurity and the analyte with reference to complexes with the racemic tag. Agreement of the measured and the calculated spectra allowed determination of the absolute configuration of the analyte without a reference sample.
The enantiomeric excess of biosynthetic (S)-(1,1,3,3,3-2H5)4 had been found to be 92% by 19F NMR of the Mosher ester. Using racemic and (S)-1,1,1-trifluoro-2-propanol as the chiral tag, the enantiomeric excess was measured at 91.3 ± 1.4% (1σ error), with the (S)-enantiomer being predominant. This result is in close agreement with Mosher ester analysis and HPLC analysis. It is also in conformance with a previous report for reduction of the protiated material31 as well as in conformance with Prelog's rule. The requirement to fully purify the material and derivatise as the Mosher ester is far more time-consuming than MRR, which required only 2 mg of sample and took ca. 15 minutes to complete.
It is also notable that MRR was used to find the enantiomeric excess of variant 1 only (i.e. the 87% of the measured material with the desired 2H5 isotopic substitution; see: Table 4). With sufficient concentration the other isotopologues could be analysed this way also. The ability to measure the enantiomeric excess of compounds differing only in the pattern of deuteration within a mixture is a unique feature of MRR.
The two principal advantages of MRR with respect to chiral HPLC, therefore, is that it allows for routine chiral analysis in complex mixtures firstly without chromatographic separation and secondly without a racemic sample of analyte. These factors are advantageous when working with complex mixtures obtained via biocatalytic methods involving whole microbes and particularly advantageous when working with isotopically labelled compounds where racemic versions of the analyte would likely have to be synthesised.
9. Conclusion
It was found that ca. 50% deuterium incorporation by reduction was possible using deuterium oxide as the only deuterium source and pre-stirring of the yeast prior to addition of the material to be reduced. Higher levels require a deuterated carbon source. Given this constraint, the key factors in developing a reasonably economical deuterative microbial reduction were the use of cheaper carbon sources and lower cell densities, with yeast strains suitable for this purpose. In general, the use of cultured cells rather than dried baker's yeast and lower concentrations of the carbon source had no deleterious effects upon reaction outcomes. When cultured cells are used by inoculating a suitable growth medium, pre-stirring additionally becomes irrelevant as a factor. We recommend that, as a first resort when exploring microbial reductions (deuterated or otherwise), the material to be reduced is added neat to a low cell density culture (even if the material is water-insoluble like 1 and 3) and that no buffer is used. Less viscous media (cultured or formed by addition of moderate amounts of dried yeast) often gave higher yields of the target material and could be extracted directly without filtration, potentially allowing for reuse of the deuterated cells.Throughout this work we observed that the enantiomeric excess observed in a protiated culture was almost unchanged by switching to a deuterated medium. Furthermore, the enantioselectivity was almost unchanged when the α-hydrogen atoms of the two ketone starting materials were exchanged for deuterium. These deuterium atoms are not appreciably back exchanged in either buffered or unbuffered cultures, either. It is possible to achieve >97% backbone perdeuteration by the two-step protocol of α-exchange and deuterative microbial reduction. This protocol allows for three deuterated isotopologues each of 2 and 4 to be prepared. We synthesised 5 of these new isotopologues in enantioenriched form.
The use of cheaper carbon sources with a different strain of S. cerevisiae (MBG5177) as well as P. pastoris strain X-33 showed that even for a ‘best-in-class’ reduction the performance of other yeast strains and species may be comparable or superior to baker's yeast. This is despite the fact that these strains have no intentional, specific carbonyl-reducing properties. As for all microbial reductions, optimisation of the strain and substrate is necessary for industrial applications.
Under an air atmosphere MBG5177 gave a higher yield and enantioselectivity than baker's yeast in the synthesis of (2-2H1)solketal ((R)-(2-2H1)2). Strain X-33 was found to provide the desired >95% deuteration by microbial reduction when provided with inexpensive methanol-d4 as a carbon source. For the synthesis of (S)-1,2-propanediol, 1-O-benzyl ether ((S)-4) we observed a reduced enantioselectivity by P. pastoris X-33 with respect to baker's yeast as well as an unexpected biosynthesis of 2-phenylethanol (7). These results underline the significance of having a range of biochemically competent microbes available to assist not just the isotope-labelling chemist, but for research and industrial chemistry in general.
Additionally, application of the transformative tool molecular rotational resonance spectroscopy (MRR) to the evaluation of the studied biochemical processes showed close agreement to standard techniques for determining site-specific and overall deuteration levels, as well as enantiopurity, with considerably less labour required to obtain these data.
This work aimed to develop cost-effective methods for producing chiral building blocks with a high degree of isotopic labelling. While this goal was achieved, the substrate scope in the current work was restricted by the focus on comparing deuterated and natively isotopically-substituted systems. Comparing a larger range of yeast strains is essential for identifying those with optimal reactivity towards a given substrate; however, the need for species-specific conditions and feedstocks makes broad screening inefficient. A deeper understanding of each strain's inherent reducing behaviour with respect to various substrates would streamline this process. Systematic studies of different species of yeast acting upon a carefully selected set of prochiral ketones could reveal these inherent capabilities, ultimately enabling AI-enhanced design of experiments (DoE) for more efficient strain selection and subsequent protecting group and strain optimisation. Validating the reuse of the deuterated cell mass is another essential step towards larger-scale implementation of enantioselective microbial deuteration.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†This work describes studies in enantioselective microbial deuteration. In the course of the work several new compounds were generated. All novel compounds have been characterised by NMR (1H, 2H, 13C{1H} and 13C{1H,2H}), IR and mass spectrometry. Annotated copies of NMR spectra have been provided in ESI I,† along with experimental details.
MRR results are accompanied by ESI II,† containing DFT calculations, MRR spectra and enantiomer analysis.
Where enantioselectivity of a reaction has been quantified this claim is supported by HPLC analysis, MRR and/or NMR analysis of the corresponding Mosher esters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the contribution of Dr Michael Moir, particularly his insights on opportunities for the creation of deuterated standards. S. cerevisiae strain MBG5177 was kindly provided by MicroBioGen Pty Ltd. We acknowledge the Australian Government in supporting ANSTO's National Deuteration Facility, which is partly funded through the National Collaborative Research Infrastructure Strategy (NCRIS).References
- M. Lecomte, M. Lahboubi, P. Thilmany, A. El Bouzakhi and G. Evano, Chem. Sci., 2021, 12, 11157–11165 RSC.
- H. Li, M. Shabbir, W. Li and A. Lei, Chin. J. Chem., 2024, 42, 1145–1156 CrossRef CAS.
- G. Fronza, C. Fuganti, P. Grasselli, A. Mele, A. Sarra, G. Allegrone and M. Barbeni, Tetrahedron Lett., 1993, 34, 6467–6470 CrossRef CAS.
- E. Santaniello, P. Ferraboschi and P. Grisenti, in Encyclopedia of Reagents for Organic Synthesis, 2001, DOI:10.1002/047084289X.rb001.
- V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 CrossRef CAS.
- C. Andreu and M. del Olmo, Catalysts, 2024, 14, 767 CrossRef CAS.
- C. Geijer, R. Ledesma-Amaro and E. Tomás-Pejó, FEMS Yeast Res., 2022, 22, foab071 CrossRef CAS PubMed.
- Q. Hu, Y. Xu and Y. Nie, Bioresour. Technol., 2010, 101, 8502–8508 CrossRef CAS PubMed.
- S. Servi, Synthesis, 1990, 1–25 CrossRef CAS.
- K. Ushio, J. Hada, Y. Tanaka and K. Ebara, Enzyme Microb. Technol., 1993, 15, 222–228 CrossRef CAS.
- T. Tsujigami, T. Sugai and H. Ohta, Tetrahedron: Asymmetry, 2001, 12, 2543–2549 CrossRef CAS.
- M. Y. Wang, S. J. Cai, J. C. Lin, X. J. Ji and Z. G. Zhang, Molecules, 2023, 28, 1422 CrossRef CAS PubMed.
- J. S. Rowbotham, M. A. Ramirez, O. Lenz, H. A. Reeve and K. A. Vincent, Nat. Commun., 2020, 11, 1454 CrossRef CAS PubMed.
- C. H. Wong and G. M. Whitesides, J. Am. Chem. Soc., 1983, 105, 5012–5014 CrossRef CAS.
- K. Edegger, C. C. Gruber, T. M. Poessl, S. R. Wallner, I. Lavandera, K. Faber, F. Niehaus, J. Eck, R. Oehrlein, A. Hafner and W. Kroutil, Chem. Commun., 2006, 2402–2404, 10.1039/B602487D.
- J. S. Rowbotham, J. H. Nicholson, M. A. Ramirez, K. Urata, P. M. T. Todd, G. Karunanithy, L. Lauterbach, H. A. Reeve, A. J. Baldwin and K. A. Vincent, ChemRxiv, 2022 preprint, pp. 1–6, DOI:10.26434/chemrxiv-2022-82tz0.
- A. Yahashiri, A. Sen and A. Kohen, J. Labelled Compd. Radiopharm., 2009, 52, 463–466 CrossRef CAS PubMed.
- Z. Zhang, L. Chen, L. Liu, X. Su and J. D. Rabinowitz, J. Am. Chem. Soc., 2017, 139, 14368–14371 CrossRef CAS PubMed.
- E. Brenna, G. Fronza, C. Fuganti and F. G. Gatti, Eur. J. Org. Chem., 2010, 5077–5084 CrossRef CAS.
- E. Brenna, G. Fronza, C. Fuganti, F. G. Gatti, A. Manfredi, F. Parmeggiani and P. Ronchi, J. Mol. Catal. B: Enzym., 2012, 84, 94–101 CrossRef CAS.
- G. Fogliato, G. Fronza, C. Fuganti, S. Lanati, R. Rallo, R. Rigoni and S. Servi, Tetrahedron, 1995, 51, 10231–10240 CrossRef CAS.
- G. Fronza, C. Fuganti, A. Mele, G. Pedrocchi-Fantoni and S. Servi, J. Org. Chem., 1992, 57, 999–1002 CrossRef CAS.
- G. Fronza, C. Fuganti, M. Mendozza, R. Rigoni, S. Servia and G. Zucchi, Pure Appl. Chem., 1996, 68, 2065–2071 CrossRef CAS.
- G. Fronza, C. Fuganti and S. Serra, Eur. J. Org. Chem., 2009, 6160–6171 CrossRef CAS.
- S. Casati, P. Ciuffreda and E. Santaniello, Tetrahedron: Asymmetry, 2011, 22, 658–661 CrossRef CAS.
- J. Bálint, G. Egri, A. Kolbert, C. Dianóczky, E. Fogassy, L. Novák and L. Poppe, Tetrahedron: Asymmetry, 1999, 10, 4017–4028 CrossRef.
- C. Recsei, R. A. Russell, M. Cagnes and T. Darwish, Org. Biomol. Chem., 2023, 21, 6537–6548 RSC.
- J. Möller, C. Recsei, R. Russell and T. Darwish, RSC Adv., 2024, 14, 26002–26006 RSC.
- G. Fronza, C. Fuganti, A. Mele, G. Pedrocchi-Fantoni, A. Sarra and S. Servi, Gazz. Chim. Ital., 1995, 125, 305 CAS.
- D. A. Peña, B. Gasser, J. Zanghellini, M. G. Steiger and D. Mattanovich, Metab. Eng., 2018, 50, 2–15 CrossRef PubMed.
- A. Manzocchi, A. Fiecchi and E. Santaniello, Synthesis, 1987, 1007–1009 CrossRef CAS.
- S. Kong, H. Pan, X. Liu, X. Li and D. Guo, Enzyme Microb. Technol., 2020, 133, 109459 CrossRef CAS PubMed.
- J. D. Dabbs, C. C. Taylor, M. S. Holdren, S. E. Brewster, B. T. Quillin, A. Q. Meng, D. A. Dickie, B. H. Pate and W. D. Harman, Nat. Commun., 2024, 15, 8473 CrossRef CAS PubMed.
- S. E. Sloane, Z. P. Vang, G. Nelson, L. Qi, R. E. Sonstrom, I. Y. Alansari, K. T. Behlow, B. H. Pate, S. R. Neufeldt and J. R. Clark, JACS Au, 2023, 3, 1583–1589 CrossRef CAS PubMed.
- J. A. Smith, K. B. Wilson, R. E. Sonstrom, P. J. Kelleher, K. D. Welch, E. K. Pert, K. S. Westendorff, D. A. Dickie, X. Wang, B. H. Pate and W. D. Harman, Nature, 2020, 581, 288–293 CrossRef CAS PubMed.
- Z. P. Vang, A. Reyes, R. E. Sonstrom, M. S. Holdren, S. E. Sloane, I. Y. Alansari, J. L. Neill, B. H. Pate and J. R. Clark, J. Am. Chem. Soc., 2021, 143, 7707–7718 CrossRef CAS PubMed.
- S. Grimme and M. Steinmetz, Phys. Chem. Chem. Phys., 2013, 15, 16031–16042 RSC.
- B. H. Pate, L. Evangelisti, W. Caminati, Y. Xu, J. Thomas, D. Patterson, C. Perez and M. Schnell, in Chiral Analysis, ed. P. L. Polavarapu, Elsevier, 2nd edn, 2018, pp. 679–729, DOI:10.1016/B978-0-444-64027-7.00019-7.
- J. L. Neill, L. Evangelisti and B. H. Pate, Anal. Sci. Adv., 2023, 4, 204–219 CrossRef CAS PubMed.
- K. Mayer, C. West, F. E. Marshall, G. Sedo, G. S. Grubbs, L. Evangelisti and B. H. Pate, Phys. Chem. Chem. Phys., 2022, 24, 27705–27721 RSC.
- E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, 2006 Search PubMed.
- T. E. Lockwood and A. Angeloski, J. Cheminf., 2024, 16, 36 Search PubMed.
- P. Kienzle , Neutron activation and scattering calculator, https://www.ncnr.nist.gov/resources/activation/, (accessed 05/10/2024).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00072f |
| ‡ Approximating the density of glycerol-d7 as 1.34 g mL−1 since glycerol has a density of 1.26 g mL−1 and glycerol-d8 has 1.37 g mL−1 and by using 95%-d as the extent of labelling for -d4 gives an estimated scattering-length density equal to 99.9%-d deuterium oxide.43 |
| This journal is © The Royal Society of Chemistry 2025 |