
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHetero-Diels–Alder reactions of (isobenzo)furans
Christopher J.
DeAngelis
and
Christopher G.
Newton
 *
*
Department of Chemistry, University of Georgia, 302 East Campus Road, Athens, GA 30602, USA. E-mail: chirs.newton@uga.edu
First published on 14th February 2025
Abstract
Hetero-Diels–Alder reactions of furans and isobenzofurans provide convergent redox-neutral access to hetero-oxanorbornene derivatives. These versatile intermediates serve as precursors to a variety of heterocycles, polymers, and complex natural products. Herein we comprehensively review this area of research and speculate on what developments are required to advance the field.
1. Introduction
Furans enjoy a rich and storied history within the context of the Diels–Alder (DA) reaction. Notable examples include Stork's 1951 total synthesis of cantharidin,1 generally acknowledged as the first planned stereospecific synthesis,2 and Dewar's fundamental mechanistic studies concerning the asynchronicity of DA cycloadditions.3 Today, furyl [4 + 2] cycloadditions regularly feature within the synthesis of natural products and polymers, while the reversibility of these processes lends itself to bioconjugation, drug delivery, and responsive material settings.4,5 Notably, the majority of these applications utilize carbo-dienophiles. In contrast, hetero-DA reactions of (isobenzo)furans remain comparatively exploratory in nature.6,7 In an effort to highlight this area of research, while also delineating what we believe to be some of the key unmet challenges, herein we provide the first comprehensive review of furan and isobenzofuran hetero-DA reactions and their applications. Our only exception pertains to the employment of singlet oxygen (1O2) as a dienophile, where instead we limit discussion to strategic considerations in natural product synthesis. This review is primarily organized according to the class of hetero-dienophile, as per the categorization and sequencing provided in Scheme 1. Reactions that proceed via a stepwise (formal) hetero-DA reaction are not discussed, although in cases where the mechanism is ambiguous or debated, we err on the side of caution, and such examples are included.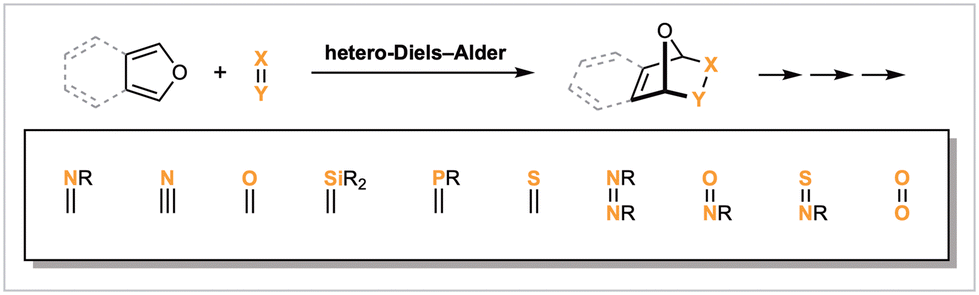 | ||
| Scheme 1 Scope and structure of this review. | ||
2. Diels–alder reactivity of (isobenzo)furans
Furans are widely described as poor DA dienes.5,8 This can largely be attributed to furan aromaticity, often resulting in a significant diene–dienophile HOMO–LUMO gap. This is further compounded by the generation of ring-strain within their oxanorbornene DA adducts.9 The combination of these two effects can manifest in facile retro-DA processes, which while desirable in some contexts is typically unwelcome. In contrast, isobenzofurans tend to be highly reactive as DA dienes while also benefiting from aromatic stabilization of their [4 + 2] cycloadducts, thus reducing the favorability of retro-DA processes.10,11 However, isobenzofurans themselves tend to exhibit poor stability profiles,11,12 with one notable exception: 1,3-diphenylisobenzofuran. This commercially available crystalline solid, which has been employed extensively within the context of this review, is kinetically stable while also highly reactive in DA settings. Although this makes it an excellent tool in demonstrating DA reactivity, its diphenyl substituents limit applicability in target-oriented settings.3. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) N bonds as dienophiles
N bonds as dienophiles
Acyclic dienophiles
Prior to 2025, only five examples of a furan engaging in an aza-DA reaction with an acyclic CN double bond had been reported, each within a wider study introducing a new class of aza-dienophile (Scheme 2a). The earliest example, from 1972, described the reactions of a highly activated triester imine with a small collection of dienes.13 While the cycloaddition with furan proceeded in good yield to generate cycloadduct 1, high-temperature and high-pressure were both necessary for reactivity. Several years later, Krolevets reported that O-silylated oximes underwent [4 + 2] cycloadditions with either furan or cyclopentadiene, the former generating aza-heterocycles of general structure 2.14 However, there may be grounds to question the accuracy of this report: only physical properties (e.g., boiling point, density) and in one case an infrared spectrum were reported for each of the cycloadducts. Perhaps more telling, O-silylated oximes have never again been employed as a dienophile in an aza-DA reaction. In 1988, Knunyants and coworkers explored the DA chemistry of various perfluorinated dienophiles.15 Within this study, the authors demonstrated that compound 3 could be accessed from the corresponding imine upon prolonged heating at 160 °C, however the yield was very low.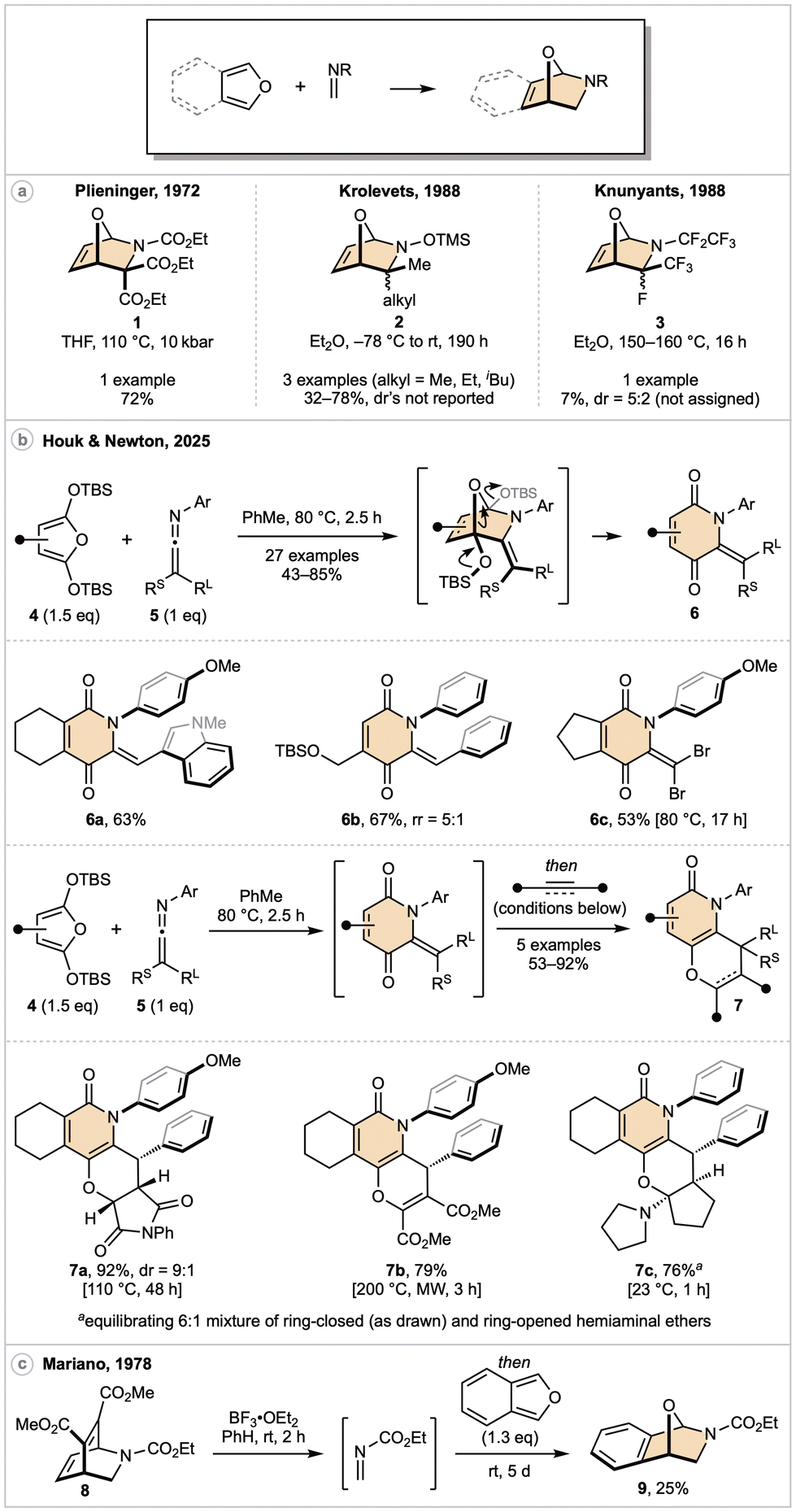 | ||
Scheme 2 Acyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds as dienophiles. N bonds as dienophiles. | ||
More recently, the authors of this review and their colleagues demonstrated that N-aryl ketenimines are highly reactive aza-dienophiles (Scheme 2b).16 Drawing upon mechanistic studies concerning the reactivity of ketenes as oxa-dienophiles,17 we hypothesized that ketenimine cycloadducts should be less prone to [3,3]-sigmatropic rearrangement processes that befall their oxa-analogues. Indeed, warming a mixture of our recently reported18 family of 2,5-bis(tert-butyldimethylsilyloxy)furans 4 with ketenimines 5 yielded metastable cycloadducts, typically with excellent regio- and diastereo-control. Fragmentation of the bridging ether occurred upon prolonged heating, providing oxygenated pyridone derivatives 6 in up to 85% yield. A wide reaction scope was demonstrated, including heteroaromatic substituted ketenimines (6a), unsymmetrically functionalized furans (6b), and fully substituted ketenimines (6c). Moreover, this DA/ring-opening process could be telescoped with an oxa-DA reaction to generate fused pyridone/pyran systems 7 in one-pot through the combination of three discrete molecules. With respect to oxa-DA scope, electron-poor olefins (7a), electron-poor alkynes (7b), and electron-rich enamines (7c), were all competent dienophiles. Finally, benchmarking studies were employed to demonstrate N-aryl ketenimines are significantly more reactive than related imines, an effect that was computationally ascribed to lower levels of diene distortion during ketenimine cycloaddition.
Given the highly forcing conditions required for the synthesis of 1 and 3, it is surprising that only a single example employing an isobenzofuran has been reported (Scheme 2c).19 While exploring the DA reactivity of various pyridine derivatives, Mariano observed that treatment of cycloadduct 8 with catalytic BF3·OEt2 promoted a retro-DA process at ambient temperature to generate an electron-poor formaldimine. The authors successfully trapped this intermediate with isobenzofuran, generating cycloadduct 9 in 25% yield.
Cyclic dienophiles: three-membered rings
The majority of reactions employing azirines as aza-dienophiles have utilized 1,3-diphenylisobenzofuran as coupling partner (Scheme 3a).20–22 With this highly reactive diene, electron-withdrawing groups on the azirine are not critical for reactivity. Rather, ring strain is sufficiently activating, provided the cycloaddition is run at elevated temperature (10–14). With the exception of spirocycle 12, each of these cycloadducts was isolated as a single diastereoisomer arising from cycloaddition through the exo-mode (defined with respect to the acyclic imine substituent) and on the less-sterically encumbered π-diastereoface of the azirine. In contrast, electron-poor azirines react with isobenzofurans at ambient temperature, albeit with poor diastereoselectivity. For example, cycloadduct 15 was isolated as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of exo- and endo-diastereomers.22 The authors note this reaction was highly reversible: repeating the procedure in refluxing THF exclusively yielded exo-15, while an NMR sample of endo-15 was observed to slowly convert into its exo-analog. A similar interconversion was also observed for 16. It is unclear if exo-10, exo-11, exo-13, and exo-14 also represent a thermodynamic sink or if facile reversibility is limited to electron-poor azirines.
:1 mixture of exo- and endo-diastereomers.22 The authors note this reaction was highly reversible: repeating the procedure in refluxing THF exclusively yielded exo-15, while an NMR sample of endo-15 was observed to slowly convert into its exo-analog. A similar interconversion was also observed for 16. It is unclear if exo-10, exo-11, exo-13, and exo-14 also represent a thermodynamic sink or if facile reversibility is limited to electron-poor azirines.
 | ||
| Scheme 3 Cyclic CN bonds as dienophiles: three-membered rings. | ||
Introduction of a conjugated ester on the azirine motif enables DA reactions with less reactive furan derivatives, as demonstrated by Gilchrist and Alves during their diastereoselective preparation of exo-17 and exo-18.23,24 The same authors reported additional (isobenzo)furan/azirine DA reactions, although instability of the presumed cycloadducts prevented their complete characterization.
Many (if not all) of the cycloadducts in Scheme 3 exhibit a poor stability profile as a result (at least in part) of their hemiaminal functionality. For example, Hassner and Anderson observed that subjecting crude exo-11a to chromatography with neutral alumina resulted in a ring expansion process, initiated by C–O bond cleavage, ultimately generating epoxide 19 in 81% yield (Scheme 3b).21 Notably, the regioselectivity of this type of process was reversed when exo-17a was treated with a Brønsted acid (Scheme 2c).24 In this case, C–N bond cleavage followed by unselective trapping of the oxocarbenium with methanol delivered 20a and 20b.
Cyclic dienophiles: four-membered rings
Four-membered aza-dienophiles remain relatively unexplored (Scheme 4). In seminal studies, Rees and Storr achieved the first ever synthesis of an aza-cyclobutadiene derivative via flash vacuum pyrolysis of 1,2,3-benzotriazines 21.25 Depending on the precise substitution pattern, these formally anti-aromatic compounds dimerize at temperatures as low as −40 °C. Despite this, the authors successfully demonstrated aza-cyclobutadienes can be trapped in DA reactions upon low-temperature addition of 1,3-diphenylisobenzofuran to deliver cycloadducts 22. While the overall yields of this process range from modest to poor, it is not clear if dienophile generation and/or cycloaddition is low yielding, nor is it clear if less reactive furan derivatives can outcompete dienophile dimerization. | ||
| Scheme 4 Cyclic CN bonds as dienophiles: four-membered rings. | ||
Cyclic dienophiles: five-membered rings
The Gaviña laboratory introduced a suite of five-membered aza-dienophiles, several of which were trapped in DA reactions with polymer-bound furoate 23 (Scheme 5).26 A representative example, utilizing ketal 24 as masked dienophile, proceeded via acid-promoted deprotection in refluxing dioxane. An aza-DA reaction to generate 25 was followed by basic hydrolysis to yield heteroaromatic 26 in 23% yield over the two steps. Hydrolysis products 27–30 were also accessed via the same general approach. | ||
| Scheme 5 Cyclic CN bonds as dienophiles: five-membered rings. | ||
4. C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N bonds as dienophiles
N bonds as dienophiles
A DA reaction of an (isobenzo)furan across a CN triple bond has yet to be unequivocally demonstrated. In general, nitriles are rarely used as dienophiles.7 Of the few reported examples, most require a highly reactive diene in conjunction with an electron-poor nitrile (e.g., tosyl cyanide).27 In the specific context of isobenzofurans, mechanistic studies from the Rickborn laboratory demonstrate their benzonitrile (formal) DA adducts undergo cycloreversion upon heating at 40 °C.28 As a consequence, Rickborn brings into question7 the accuracy of Babayan's 1974 report detailing the intramolecular DA reactivity of furyl-tethered nitriles (Scheme 6a).29 Here, the authors report a high-yielding cycloaddition occurs upon heating ammonium salts 31 or 33 under aqueous conditions. Cycloadducts 32 and 34 were only characterized using infrared spectroscopy and combustion analysis.
 | ||
Scheme 6 C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonds as dienophiles. N bonds as dienophiles. | ||
A more recent report also describes a potential intramolecular DA reaction with a nitrile as dienophile, although here the ambiguity lies with respect to the precise mechanism.30 The overall transformation involves heating 2-alkynylbenzoyl derivatives 35 with β-cyanocarbene complexes 36, providing access to hetero-functionalized phenanthridines in moderate to low yields (e.g., 37a–d). The authors propose multiple mechanistic possibilities, including chromium η5-complexed isobenzofuran derivatives acting as dienes in a reversible cycloaddition, followed by a deoxygenation/aromatization process.
5. CO bonds as dienophiles
Furyl oxa-DA reactions are remarkably rare. The earliest example, from Babayan's aforementioned 1974 report, detailed an intramolecular oxa-DA of methyl ester 38—however the resulting cycloadduct 39 was only characterized using infrared spectroscopy and elemental analysis (Scheme 7a).29
 | ||
| Scheme 7 CO bonds as dienophiles. | ||
In 2000, Wong et al. introduced 5,6-bis(trimethylsilyl)isobenzofuran as a new highly reactive DA diene (Scheme 7b).31 Proof of principle applications primarily focused on the synthesis of linear polyaromatic compounds using carbodienophiles (e.g., arynes and para-quinones), although a single oxa-DA reaction was also reported. Diene synthesis was achieved via extension of Warrener's approach to isobenzofuran:32 mixing furyl-DA adduct 40, 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine, and acetaldehyde at ambient temperature resulted in an inverse electron-demand DA/double retro-DA sequence to furnish the desired silylated isobenzofuran. This was proceeded by an in situ oxa-DA reaction with acetaldehyde to yield acetal 41 in 24% yield as a 1:1 mixture of diastereoisomers. Within the same study several carbo-DA examples proceeded in excellent yield, suggesting the oxa-DA reaction is the low yielding step in this transformation.
The most recent report, from Aoshima and coworkers, utilized a Lewis acid-catalyzed cationic furyl oxa-DA reaction for the copolymerization of furfural and 2-acetoxyethyl vinyl ether.33 A model study designed to probe the mechanism is presented in Scheme 7c, maintaining furfural as diene but introducing ether 42 as a replacement for the vinyl ether propagating species. The authors propose the reaction proceeds via GaCl3-catalyzed formation of a cationic alkylated furfural derivative, which undergoes an oxa-DA reaction across the carbonyl of furfural followed by a LiBH4 quench to generate 43.
6. CSi bonds as dienophiles
In the late 1970′s, the Brook laboratory demonstrated CSi bonds can engage as dienophiles in [4 + 2] reactions with simple 1,3-butadienes,34 later demonstrating such processes do not appear to proceed via a radical pathway.35 With respect to (isobenzo)furans as dienes, only three reactions have been disclosed, all within a single report from Auner and co-worker (Scheme 8).36 In this study, an electron-poor dienophile was generated at low temperature via a stepwise SN2′-type reaction of trichlorovinylsilane and tert-butyllithium. The transient sila-olefin intermediate was then treated with an excess of 2-methylfuran, affording silabicycle 44 as a single regioisomer in 70% yield, but with minimal diastereocontrol. The authors successfully reacted the same dienophile with furan and 2,5-dimethylfuran, although yields suffered in both cases. At ambient temperature these cycloadducts were observed to slowly revert to a mixture of their respective furan precursors and [2 + 2] dimers of the dienophile. Whether this proceeds via a retro-DA process remains unclear as attempts at generating crossed [2 + 2] adducts were unsuccessful. Unexpectedly, heating the same furyl cycloadducts for several days under vacuum at 170 °C resulted in a high yielding rearrangement to a dihydro-1,2-oxasilepine (shown for the conversion of 44 to 45). The authors propose this reaction proceeds via cleavage of the oxa-bridge to generate a stabilized allylic carbocation that also benefits from the β-silicon effect, followed by an anionic Brook-type rearrangement to furnish the ring-expanded product.
 | ||
| Scheme 8 CSi bonds as dienophiles. | ||
7. CP bonds as dienophiles
It wasn't until the late 1970s that synthetic routes to isolable phosphaalkenes and phosphaalkynes bearing localized CP and CP bonds were reported,37 paving the way for their application in DA settings. Examples employing phosphaalkenes as dienophiles began to appear in the early 1980s,38 followed shortly thereafter by the analogous reactions of phosphaalkynes.39 The earliest report of a phosphaalkene engaging in a DA reaction with an (isobenzo)furan was reported in 1985 by Grobe and coworker (Scheme 9a).40 In this example, 2-methylfuran was heated at 70 °C in the presence of stannyl phosphane 46. Thermolysis of the latter species generated a reactive perfluorinated phosphaalkene that underwent an unselective DA reaction to generate isomers 47a–d in a combined yield of 93%. This reaction is highly reversible and heating the cycloadducts at 70 °C in the presence of 2,3-dimethyl-1,3-butadiene yields the corresponding cross-DA adducts alongside 2-methylfuran. In a series of follow up studies, the Grobe laboratory trapped the same perfluorinated dienophile with furan40 and also introduced a related perfluorinated phosphaalkene was trapped with 2-methylfuran.40
 | ||
| Scheme 9 CP bonds as dienophiles. | ||
In 1987, Quin et al. expanded the scope of phosphaalkene dienophiles to include non-perfluorinated derivatives (Scheme 9b).41 Mild heating of phosphine 48 invoked a retro-DA process to generate 2-phosphapropene (and dimethyl phthalate as byproduct). In the absence of a trapping reagent, 2-phosphapropene decomposed via an unspecified intermolecular process. However, conducting the same reaction with an excess of 1,3-diphenylisobenzofuran yielded DA cycloadducts 49 as a 1:1 mixture of diastereomers. The instability of 49, and the analogous cycloadduct derived from furan as diene, precluded full characterization, and only 31P NMR data was acquired.
Over a series of papers, the first disclosed in 1985, Mathey and coworkers demonstrated that 1-chlorophosphaethene can be stabilized through π-complexation with a tungsten carbonyl complex.42,43 While not stable enough to be isolated in pure form, in situ characterization was feasible, and solutions could be heated to 65 °C without decomposition. Several years later, the same group described reactions of this dienophile with (isobenzo)furans as a convergent means to prepare phosphorus heterocycles (Scheme 9c).44 From a mechanistic perspective, the authors propose that heating phosphine 50 generates a tungsten-bound phosphinidene complex that undergoes a rapid CuCl-promoted rearrangement to generate the active dienophile.43 This intermediate was successfully trapped with a variety of simple furans, and representative cycloadducts include 51a–d. In each case, a mixture of isomers was obtained, favoring the regioisomer depicted (when relevant) and preferentially reacting via the exo-mode with respect to the bulky tungsten substituent. Cycloadduct derivatization by either aromatization or ring-expansion was demonstrated. In the case of exo-51a, treatment with stoichiometric boron tribromide followed by triethylamine yielded tungsten-complexed 2-hydroxyl phosphinine 52 in 46% yield. The reaction appears to proceed via an SN2′-type process to generate a boron-bound alkoxide, followed by a series of elimination events. Alternatively, if the alkoxide was quenched with water then bromohydrin 53 was isolated. A ring-expansion process, reminiscent of the work described by Auner in 1992 (Scheme 8), was achieved by the addition of TIPSCl and imidazole, ultimately leading to oxaphosphepine derivative 54 in 82% yield over the two steps. The Mathey laboratory has also demonstrated analogous DA/aromatization reactivity with 1,3-diphenylisobenzofuran for the synthesis of a phosphanaphthalene.45
8. CS bonds as dienophiles
In 1961, Middleton and coworkers synthesized a small library of perfluorinated thiocarbonyls via the reaction of bis(perfluoroalkyl)mercury salts with boiling sulfur.46 While the most reactive derivative, hexafluorothioacetone, is not sensitive toward water or oxygen, it dimerizes upon standing within several hours.47 Despite this, the authors went on to highlight its exceptional dienophilicity (“reacts instantaneously with butadiene at −78 °C to give a quantitative yield of the cyclic adduct”). Four years later, Middleton explored the DA reactivity of these perfluorinated thiocarbonyls in more detail, including the reaction of furan and hexafluorothioacetone for the synthesis of 55 (Scheme 10a). The authors noted this cycloadduct polymerizes on the order of hours when held neat at ambient temperature.48
 | ||
| Scheme 10 CS bonds as dienophiles. | ||
In 1978, Raasch prepared the thioketene derivative of hexafluorothioacetone [i.e., bis(trifluoromethyl)thioketene], observing that it can be stored at ambient temperature for months with minimal dimerization. A slight excess of this thioketene was mixed with 1,3-diphenylisobenzofuran at 0 °C, resulting in the formation of cycloadduct 56 in 83% yield. The DA reaction appears highly (if not completely) chemoselective for the CS over CC bond. Cycloadduct 56 was described as unstable to heat, decomposing by means of a retro-DA reaction. Eight years later, Eguchi et al. explored the DA reactivity of adamantanethione, a bench stable thioketone derivative.49 Its reaction with isobenzofuran proceeded in 24 hours at 100 °C to afford spirocycle 57 in 79% yield. Attempts to employ either furan or 1,3-diphenylisobenzofuran as dienes were unsuccessful, only yielding unreacted dienophile.
More recently, Abdel-Megeid and coworkers reported a thio-DA reaction of furan and an α-oxo thioketone (Scheme 10b).50 Only two dienophiles were reported, including one derived from cyclohexyl-substituted sulfenyl chloride 58. In this example, addition of potassium iodide generated the reactive α-oxo thioketone, leading to spirocycle 59 in 18% yield, alongside 33% of a dienophile [4 + 2] dimer.
The low reactivity of furans with CS dienophiles was well demonstrated within a study concerning the development of thermoreversible ligation systems (Scheme 10c).51 A small library of dienes were successfully reacted across the CS bond of symmetric thio-dienophile 60, and their respective rate constants are provided. Notably, ethyl sorbate, an acyclic electronically deactivated diene, proved to be competent in this reaction. However, no cycloaddition was observed with furan, and ab initio calculations indicate the equilibrium strongly favors the reactants in this case.
9 NN bonds as dienophiles
Acyclic dienophiles
The first furyl azo-DA reaction employed diethyl azodicarboxylate as dienophile for the synthesis of cycloadduct 63 (Scheme 11a).52 Over the next sixty years, several closely related reports describing the reaction of furans with acyclic dialkyl azodicarboxylates have been disclosed.53–56,57,58–61 While most examples employ minimally functionalized furans, a small number of more complex cycloadducts have also been accessed (e.g., 64 and 65).56 Typically, DA reactions of this nature tend to be high-yielding processes that proceed under mild conditions (e.g., ambient or slightly elevated temperatures). | ||
| Scheme 11 Acyclic NN bonds as dienophiles. | ||
Despite their ease of preparation, few furyl azo-DA cycloadducts have been applied in further settings, perhaps in part due to their modest stability profiles.58 Indeed, failed attempts to use them as antitumor agents,59 or as precursors to insecticides,54 oxadiazoles,58 pyridazines,53 or in ring-rearrangement metathesis60 have all been reported. Nevertheless, several research groups have productively channeled aza-cycloadduct reactivity/instability (Scheme 11b–e). For example, Sepúlveda-Arques and coworkers observed that heating furans 66 with diethyl azodicarboxylate unexpectedly yielded ring-expanded products 67 (Scheme 11b).62 Mechanistic studies suggest a three-step process beginning with an azo-DA reaction, followed by a strain-release promoted ring-opening before re-closure via the pendant aldehyde/ketone with concurrent formation of the bridging ether.
A proof-of-principle study introducing degradable polymers derived from furyl azo-DA adducts was reported in 2024 (Scheme 11c).61 Density functional theory calculations indicated cycloadducts 68, which were prepared neat on up to 100 g scale, exhibit greater ring-strain (22.8 kcal mol−1) than established degradable ring-opening metathesis polymerization monomers, prompting the authors to explore their suitability in living polymerization reactions. Treatment with Grubbs third generation catalyst provided efficient access to homopolymers 69, as well as various copolymers upon the inclusion of strained alkene comonomers (not shown). Finally, controlled polymer degradation, including biodegradation, was effectively demonstrated.
Two conceptually related but mechanistically distinct approaches to pyridazines (or their salts) from furyl azo-DA adducts have been reported (Scheme 11d and e). Not deterred by earlier unsuccessful efforts to access pyridazines via hydrolysis of furyl azo-DA cycloadducts,53 Bezhan and coworkers recognized that, while hydrolysis cleaves both CN bonds, this does not represent a dead end. Thus, cycloadducts 70 were treated with sulfuric acid to generate the corresponding 1,4-dicarbonyls, which, following addition of hydrazine, led to pyridazines 71 in up to 67% yield (Scheme 11d).57 In addition, one example employing 1,3-diphenylisobenzofuran as diene was reported, leading to derived phthalazine 72 in 91% yield. Strategically speaking, here the azo-dienophile ultimately serves as oxidant through conversion of furans into their corresponding dicarbonyl over the two-step cycloaddition/hydrolysis sequence.
In 2014, Heinrich demonstrated that mixing furans 73 and carboxylate salts 74 in the presence of a strong acid promoted decarboxylation to aryldiazenes (Scheme 11e).63 These unusual dienophiles were initially trapped as their furyl cycloadducts, followed by spontaneous aromatization to yield pyridazinium salts 75. In general, yields improved as electron-donating substituents were introduced onto the furan backbone (75a–d). Several mechanistic experiments were used to discount an alternate furan oxidation/hydrazine recombination mechanism (analogous to that depicted in Scheme 11). A follow-up study from the same group demonstrated aryldiazenes could also be prepared via aryl hydrazine oxidation, followed again by trapping in [4 + 2] cycloadditions with furans.64
Cyclic dienophiles
Phenyl-1,2,4-triazoline-3,5-dione (PTAD) is a highly reactive cyclic azo-dienophile that has been applied within a wide variety of DA reactions, often to demonstrate DA competency rather than looking toward specific applications.65 With respect to (isobenzo)furans as dienes,66–69 representative PTAD cycloadducts include ethoxy derivatives 76,67 dichloropyridine 77,68 and diynes 7869 (Scheme 12a). | ||
| Scheme 12 Cyclic NN bonds as dienophiles. | ||
The reversibility of such DA reactions was leveraged by Singha and coworkers for the preparation of self-healing polymers (Scheme 12b).70 Inspired by related approaches using furfuryl/maleimide DA reactions,71 the Singha laboratory demonstrated that polymer-bound furans 79 undergo a double DA reaction with bis-PTAD derivative 80 upon mixing at ambient temperature, generating crosslinked polymers of general structure 81. Differential scanning calorimetry and solubility studies were employed to establish the DA reaction is reversible upon heating in DMF at 130 °C, endowing the crosslinked polymers with >80% healing efficiency.
Moving away from PTAD and its derivatives, a small number of studies concerned with the development of new electron-deficient azo-compounds have leveraged isobenzofuran DA reactions to indirectly confirm their generation and/or probe their reactivity (Scheme 12c and d).72–74 With this goal in mind and building upon related exploratory work,72 the Amarasekara laboratory reported the synthesis and DA reactivity of 1,4-phthalazinedione (Scheme 12d).74 Upon mixing furans 85 with phthalhydrazide in the presence of two equivalents of lead tetraacetate, ring-contracted cycloadducts 86 were isolated in up to 64% yield. The authors proposed the reaction proceeded via oxidation of phthalhydrazide to 1,4-phthalazinedione, which was immediately trapped in an azo-DA reaction. A ring-opening/ring-closing sequence resembling Sepúlveda-Arques’ aforementioned ring-expansion methodology (Scheme 11b) was followed by an oxidative carbonyl-cleavage event to generate the final products.
A recent application that takes advantage of facile retro-furyl azo-DA processes was reported by Yoshino and Matsunaga (Scheme 12e).75 In the interests of developing new dynamic covalent bonding systems, the authors introduced a small family of para-urazine-derived azo-dienophiles. Dienophile design was guided by the hypothesis that attenuation of PTAD dienophilicty may result in a more facile retro-DA process. Proof of principle studies involved mixing 2,5-dimethylfuran with symmetric dienophiles of general structure 87, all of which reached thermodynamic equilibrium with cycloadducts 88 within ca. 10 minutes at ambient temperature. Following addition of sorbic alcohol as a more reactive diene, the majority of the azo-dienophile ultimately funneled to cycloadducts 91 within 90 minutes via a retro-DA/cross DA sequence. The authors explored various dienophile substituent effects, confirming the superiority of this new system relative to PTAD, before successfully applying it in a mild and reversible sol–gel transition.
10. NO bonds as dienophiles
In 1947, during attempts at a cheletropic cycloaddition, Wichterle unknowingly achieved what appears to be the first documented nitroso-DA reaction by mixing of 1,3-butadiene with nitrosobenzene.76 This cycloadduct was correctly assigned the following year,77 and shortly thereafter Mustafa et al. disclosed the first nitroso-DA reaction employing an (isobenzo)furan as diene (Scheme 13a).78 In this report, 1,3-diphenylisobenzofuran was reacted with either nitrosobenzene or its para-dimethylamino derivative to generate (the correctly assigned) cycloadducts 92 in undisclosed yields. Some years later, Taylor introduced two pyridyl derivatives for the synthesis of 93,79 and Moinet in 1993 described an electrochemical synthesis of 2-nitrosobenzamide, which was also trapped with 1,3-diphenylisobenzofuran to generate 94.80
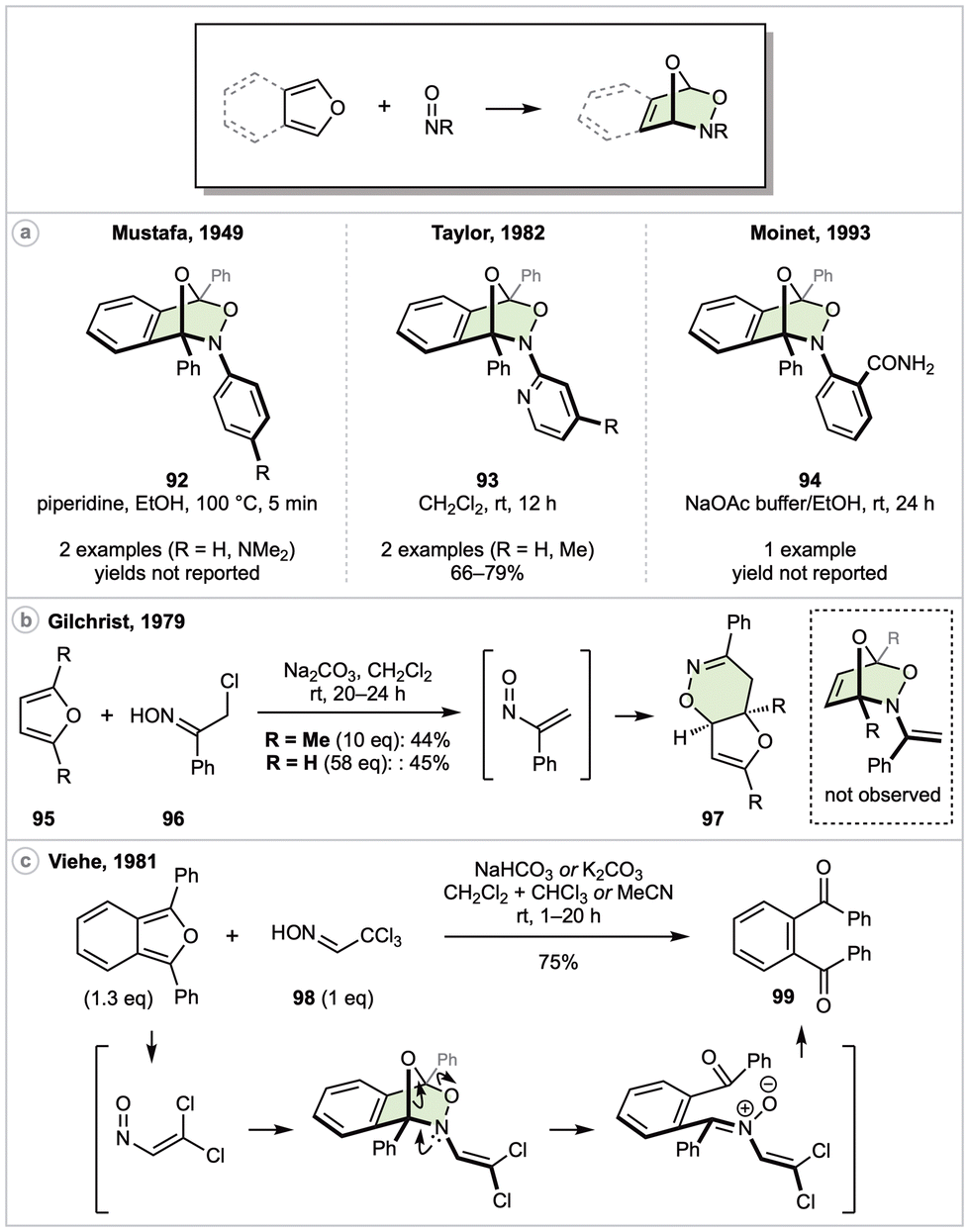 | ||
| Scheme 13 NO bonds as dienophiles. | ||
In 1979, Gilchrist and coworker explored the reactions of α-nitrosostyrene with various DA dienes (Scheme 13b).81 In all cases the nitroso component behaved as hetero-diene rather than hetero-dienophile. With respect to furans, mixing 95 with oxime 96 in the presence of a weak base led to DA adducts 97 irrespective of substitution at the 2- and 5-positions of furan. The authors note that 97 may alternatively form via a [4 + 2] cycloaddition across the α-nitrosostyrene CC bond, followed by a [3,3]-sigmatropic rearrangement.
In 1981, Viehe and coworkers also explored the reactions of alkenyl nitroso compounds with a variety of dienes, observing that peri- and chemo-selectivity (i.e., reacting as a hetero-diene versus hetero-dienophile verses carbo-dienophile) was associated with degree of alkene substitution.82 In the case of 1,3-diphenylisobenzofuran and nitroso precursor 98, 1,2-dibenzoylbenzene (99) was isolated in 75% yield, with the same reaction outcome also observed with 2,5-dimethyl furan (Scheme 13c). Although a precise mechanism was not proposed, presumably the reaction proceeds via elimination of HCl to generate a dichloroalkene that is trapped in a nitroso-DA reaction. C–O bond cleavage to the corresponding nitrone, followed by hydrolysis, would then yield the observed product. Overall, and in analogy to the aforementioned work of Bezhan (Scheme 11), here the hetero-dienophile ultimately serves as an oxidant.
11. NS bonds as dienophiles
While a number of DA reactions involving 1,3-butadiene derivatives across NS bonds have been reported, including sulfur diimides (RNSNR)83 and thionitroso compounds (RNS),84 sulfinylamines (RNSO) remain the most commonly explored.85 With respect to (isobenzo)furans as dienes, only two reactions have been described, although perhaps this will change given a renewed interest in this functionality.86 The first example, reported by Cava and coworker in 1963, described a Lewis acid-promoted DA reaction between 1,3-diphenylisobenzofuran and N-sulfinylaniline (100), leading to isoindole 101 in 78% yield (Scheme 14a).87 While no mechanistic experiments were conducted, the authors proposed the reaction proceeds first via a DA reaction across the NS bond of the sulfinylamine, followed by rearrangement to a cyclic sultam. A well-precedented extrusion of sulfur dioxide would then deliver the final product.88
 | ||
| Scheme 14 NS Bonds as dienophiles. | ||
In 2000, Hemming and coworkers reported the synthesis of a sulfinylamine derivative bearing a pendant azide, followed by in situ trapping with 1,3-butadienes.89 These DA adducts were then applied in a five-step sequence incorporating an aza-Wittig reaction for the synthesis of various benzothiadiazepines. Four years later, the chemistry of an isobenzofuran DA adduct was described, although no details regarding its preparation were provided.90 Drawing analogy to their first study, its synthesis presumably proceeds via treatment of azide 102 with thionyl chloride followed by addition of 1,3-diphenylisobenzofuran to generate cycloadduct 103 (Scheme 14b). A Staudinger reaction with triphenylphosphine would then yield iminophosphorane 104. The authors reported that heating this compound in anhydrous toluene yielded triphenylphosphine oxide (89%), 1,3-diphenylisobenzofuran (70%), and aniline 105 (59%). Based on a brief mechanistic study, the authors propose this reaction proceeds via an intramolecular aza-Wittig, followed by retro-DA reaction to generate a cyclic sulfur diimide, which finally hydrolyzes upon purification.
12. OO bonds as dienophiles
OO synthesis and general reactivity
Cycloadditions of (isobenzo)furans and 1O2 have been widely studied and are the subject of several excellent reviews.91–93 To distinguish our treatment, we have elected to focus exclusively on applications in complex natural product synthesis, organized primarily by strategic considerations.
1O2 is the lowest energy excited state of molecular oxygen and can be photochemically generated upon UV irradiation of ground state triplet oxygen in the presence of a suitable photosensitizer, such as rose bengal (RB), methylene blue (MB), or tetraphenylporphyrin (TPP) (Scheme 15).93 Operationally, this process is usually achieved at low temperatures and in the same flask as the reactant(s), although flow techniques have enabled alternate setups in recent years. Despite being a strong oxidant, 1O2 exhibits a great degree of chemoselectivity, typically leaving many oxidatively sensitive functional groups untouched (e.g., alcohols, carbonyls). With respect to pericyclic reactivity, 1O2 most commonly reacts via either: (i) a [4 + 2] cycloaddition with a 1,3-butadiene to generate an endo-peroxide, (ii) a Schenk-Ene reaction with an olefin bearing an allylic hydrogen, or (iii) via a [2 + 2] cycloaddition with an electron-rich olefin, and many of the factors that govern chemo- and peri-selectivity have been elucidated.92 It is worth highlighting early that Nature often takes advantage of 1O2 [4 + 2] cycloadditions,93 and as will become apparent, many syntheses discussed herein mirror current understanding of biosynthetic pathways (be it intentionally or otherwise).
 | ||
| Scheme 15 OO as dienophile: generation of 1O2. | ||
Synthesis of γ-hydroxybutenolides
The late-stage conversion of furans into γ-hydroxybutenolides via the intermediacy of 1O2 cycloadducts is a popular strategy in natural product synthesis (Scheme 16, boxed). One of the most common tactics involves employment of a base to effect a Kornblum–DeLaMare rearrangement, as well demonstrated within Trauner's enantioselective total synthesis of sandresolide B (Scheme 16a).94 Diastereoselective addition of methylmagnesium bromide to ketone 106 yielded unstable alcohol 107, which was subjected crude to a one-pot diastereoselective 1O2 cycloaddition/Kornblum–DeLaMare rearrangement to provide the natural product in 51% yield from 106. | ||
| Scheme 16 OO as dienophile: synthesis of γ-hydroxybutenolides. | ||
γ-Hydroxybutenolides can be readily reduced to butenolides, another motif commonly found in natural products. For example, in 2001 Metz described the conversion of furan 108 into γ-hydroxybutenolide 109, achieved using MB as photosensitizer in conjunction with a sterically encumbered base to engender chemoselective deprotonation (Scheme 16b).95 Reduction with sodium borohydride yielded (+)-ricciocarpin B in 46% yield over two steps.
In 1985, Katsumura, Isoe, and coworkers demonstrated 2-trimethylsilyl substituted furans react faster with 1O2 relative to their non-silylated congeners.96 Moreover, conducting the cycloaddition in methanol resulted in spontaneous and regiospecific conversion into the γ-hydroxybutenolide motif (Scheme 16, boxed). Mechanistically it has been proposed this transformation proceeds via a concerted 1,2-silyl migration/O–O cleavage event followed by a desilylative ring-closure upon addition of a weak acid.97 Strategically this tactic serves as a useful alternative to Kornblum–DeLaMare-based approaches that fail to yield the desired γ-hydroxybutenolide regioisomer,96 or if base-sensitive substrates are being considered. In addition, silyl incorporation has been observed to increase cycloaddition yields.98 An elegant application of this chemistry was described within Yang and co-workers’ synthesis of (−)-spirochensilide A (Scheme 16).99 Treatment of furan 110 with 1O2 was followed by addition of chloroacetic acid, leading to spirocycle 111 in 88% yield. Presumably, the reaction proceeds via an γ-hydroxybutenolide intermediate (not shown) that, upon elimination of water, undergoes a diastereoselective 6-exo-trig spirocyclization.
Alternative functionalization of the silylated, ring-opened intermediate is also feasible, as illustrated within Miyashita's synthesis of the complex natural product (−)-norzoanthamine (Scheme 16d).100 In this case, furan 112 was converted into the corresponding Z-configured unsaturated silyl ester upon reaction with 1O2. The crude product was then alkylated via treatment with tetra-n-butylammonium fluoride and methyl iodide, generating doubly activated carbo-dienophile 113 for a subsequent intramolecular DA reaction en route to the natural product.
Synthesis of γ-alkoxybutenolides
Alcoholic solvents (typically methanol), will add to furan-derived endoperoxides at temperatures approaching 0 °C, resulting in formation of a hydroperoxide (Scheme 17, boxed). Treatment with an acylating reagent then promotes a Kornblum–DeLaMare rearrangement for the synthesis of γ-alkoxybutenolides. The regioselectivity of alcohol addition is typically governed by stabilization of the incipient positive charge (i.e., addition to the more substituted position),101 although the opposite regioselectivity has also been observed (e.g., Scheme 18c). An elegant two-directional synthesis proceeding through γ-alkoxybutenolide intermediates was disclosed by Robertson et al. in 2014 (Scheme 17).102 Tethered bis-furan 114 was subjected to a double 1O2 cycloaddition. Employing methanol as solvent led to a bis-hydroperoxide intermediate, which following in situ acetylation/elimination yielded bis-methoxybutenolide 115 as a single (undefined) diastereoisomer. Removal of the Boc group was achieved with bromotrimethylsilane, providing secondary amine 116 in 75% yield over two steps. The synthesis was completed via a presumably biomimetic sequence of reactions. Mixing 116 in a biphasic mixture of sulfuric acid and dichloromethane promoted methoxide ionization, which was followed by a 6-exo-trig cyclization of the pendant amine to generate pandamarilcaone-1 in 12% yield. Competitive deprotonation of the same ionized intermediate, followed by a 5-exo-trig aza-Michael cyclization, generated pandamarilactonine A–D in a combined yield of 48%. | ||
| Scheme 17 OO as dienophile: synthesis of γ-alkoxybutenolides. | ||
 | ||
| Scheme 18 OO as dienophile: synthesis of α,β-unsaturated 1,4-dicarbonyls. | ||
Synthesis of α,β-unsaturated 1,4-dicarbonyls
α,β-Unsaturated 1,4-dicarbonyls at the dialdehyde oxidation level can be accessed directly from furyl 1O2 cycloadducts via reduction with triphenylphosphine or dimethyl sulfide (Scheme 18, boxed). For example, in efforts toward (±)-bielschowskysin, the Sarlah group demonstrated that macrocyclic furan 117 could be converted into the corresponding α,β-unsaturated diketone 118 under standard conditions (Scheme 18a). However, this intermediate was found to be sensitive toward E/Z isomerization, alongside other undetermined decomposition pathways, ultimately thwarting downstream attempts at a late-stage Norrish–Yang cyclization.In situ trapping of the reduction products has been leveraged in a variety of contexts. One such example in the context of natural product total synthesis was disclosed by Sammes and coworker in 1985 (Scheme 18b).103 Attempts at an Achmatowicz reaction for the conversion of furan 119 into pyran 120 under standard conditions (e.g., bromine and methanol or meta-chloroperoxybenzoic) proceeded with undesired oxidation of the 1,1-disubstituted olefin. Transitioning to a 1O2 cycloaddition/reduction sequence proved highly chemoselective and was accompanied by a 6-exo-trig cyclization of the pendant alcohol to generate 120 in 83% yield. Several steps were then necessary to access (±)-cryptofauronol.
α,β-Unsaturated 1,4-dicarbonyls can also be accessed via reduction of the hydroperoxide intermediates introduced in Scheme 17 by treatment with either triphenylphosphine or dimethyl sulfide (Scheme 18, boxed). As demonstrated within Vassilikogiannakis and Strataki's synthesis of (±)-litseaverticillol A, reacting furan 121 with 1O2 in the presence of MeOH led directly to hydroperoxide 122 in near quantitative yield (Scheme 18c).104 Addition of dimethyl sulfide prompted reductive cleavage to generate an α,β-unsaturated 1,4-dicarbonyl, which following addition of Hünig's base underwent an intramolecular aldol reaction to generate the natural product in 55% yield and with 19:1 diastereoselectivity. This overall transformation has since been applied to a handful of natural products, including Magauer's conversion of (−)-leucosceptroid A to C (Scheme 18d).105
13. Conclusion and future outlook
Several trends have become apparent throughout the preparation of this review. First and foremost, many (if not all) furyl hetero-DA cycloadducts exhibit poor stability profiles. This is to be expected given at least one sensitive oxygen–carbon–heteroatom bond is generated upon cycloaddition (e.g., ketal or hemiaminal functionality). This is further exacerbated if a weak heteroatom–heteroatom bond is also formed (e.g., nitroso- and azo-dienophiles) or if significant ring strain is generated (e.g., see the discussion surrounding Scheme 11c). These structural features can culminate in facile heteroatom bond cleavage events or mild retro-DA processes, ultimately complicating potential applications. However, if chemoselective cycloadduct functionalization can be achieved, hetero-DA reactions of (isobenzo)furans can become very empowering, as has been well demonstrated within the context of 1O2 as dienophile. Unfortunately, the rest of the field lags significantly behind. Perhaps most telling, with the exception of 1O2 as dienophile, a furyl hetero-DA reaction has never been successfully applied in the synthesis of a natural product. That being said, a small number of isolated applications concerning the synthesis of various heterocycle families are highly encouraging (e.g., oxygenated pyridones in Scheme 2b, oxaphosphepines in Scheme 9c, pyridazinium salts in Scheme 11e, and biodegradable polymers in Scheme 11c).Shifting focus from cycloadduct stability to hetero-DA reactivity, many of the dienophile classes discussed herein appear to be only reactive with isobenzofurans. A general tactic for addressing low DA reactivity (or engendering stereo- or regio-selectivity) involves the addition of a Lewis- or Brønsted-acid. While such additives are present in a small number of the hetero-DA reactions reviewed (e.g.Schemes 7c, 9c and 14a), it is not clear if they play a role in the cycloaddition itself. Rather, they appear to primarily (or perhaps exclusively) facilitate diene/dienophile generation or post-cycloaddition reactivity. Moreover, given Lewis- or Brønsted-acids can readily decompose hetero-oxanorbornene cycloadducts (e.g., Scheme 3b and c) and likely many of the dienes and dienophiles discussed throughout, it is perhaps not surprising a catalytic enantioselective (isobenzo)furan hetero-DA reaction has yet to be reported. Attempts to move away from frontier molecular orbital control in (isobenzo)furan hetero-DA reactions is also underdeveloped. While introduction of dienophile strain (e.g., arynes, cyclopropenes) is a well-established tactic for achieving DA reactivity with less reactive dienes, methods to employ hetero-derivatives lag significantly behind their carbo-analogs. Thus, with the exception of ketenimines (Scheme 2b), azirines (Scheme 3), and benzannulated azetes (Scheme 4), strain-promoted reactivity is exceptionally rare. Alternatively, enhancing furan reactivity through introduction of a cleavable functionality may also represent a viable approach.
Given the inherent challenges identified, we firmly believe that (isobenzo)furan hetero-DA reactions provide an excellent testing ground for the development of new, highly reactive hetero-dienophiles. We hope this review serves to inspire future practitioners to tackle challenges in this area, ultimately enabling the convergent synthesis of heteroatom-rich motifs.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Science Foundation (CHE-2340210 to C. G. N.) and the University of Georgia is gratefully acknowledged.References
- G. Stork, E. E. van Tamelen, L. J. Friedman and A. W. Burgstahler, J. Am. Chem. Soc., 1951, 73, 4501–4501 CrossRef; G. Stork, E. E. van Tamelen, L. J. Friedman and A. W. Burgstahler, J. Am. Chem. Soc., 1953, 75, 384–392 CrossRef.
- G. Stork, Oral history interview with Gilbert J. Stork Science History Institute, August 6, 1991, https://digital.sciencehistory.org/works/fb4949591 CrossRef PubMed; E. Nakamura, J. D. Winkler and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 57, 36–36 CrossRef PubMed.
- M. J. S. Dewar and A. B. Pierini, J. Am. Chem. Soc., 1984, 106, 203–208 CrossRef.
- M. V. Sargent and F. M. Dean, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon, 1984, p. 599–656 CrossRef; B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef; R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Angew. Chem., Int. Ed., 2022, 61, e202114720 CrossRef PubMed.
- C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179–14233 CrossRef.
- S. M. Weinreb and R. R. Staib, Tetrahedron, 1982, 38, 3087–3128 CrossRef.
- B. Rickborn, in Organic Reactions, ed. L. A. Paquette, John Wiley & Sons, Inc., New York, 1998, vol. 53, ch. 2, pp. 223–629 Search PubMed.
- K. Ando, N. Akadegawa and H. Takayama, J. Chem. Soc., Perkin Trans. 1, 1993, 2263–2268 RSC; P. A. Grieco, J. J. Nunes and M. D. Gaul, J. Am. Chem. Soc., 2002, 112, 4595–4596 CrossRef; T. Salavati-fard, S. Caratzoulas, R. F. Lobo and D. J. Doren, ACS Catal., 2017, 7, 2240–2246 CrossRef.
- Z. Liu, J. Vinskus, Y. Fu, P. Liu, K. Noonan and O. Isayev, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-dtq6q.
- D. Tobia and B. Rickborn, J. Org. Chem., 1987, 52, 2611–2615 CrossRef.
- R. Rodrigo, Tetrahedron, 1988, 44, 2093–2135 CrossRef.
- L. F. Fieser and M. J. Haddadin, J. Am. Chem. Soc., 2002, 86, 2081–2082 CrossRef.
- D. vor der Brück, R. Bühler and H. Plieninger, Tetrahedron, 1972, 28, 791–795 CrossRef.
- A. A. Krolevets, A. V. Adamov, A. G. Popov and I. V. Martynov, Russ. Chem. Bull., 1988, 37, 1737–1737 CrossRef CAS; A. A. Krolevets, A. G. Popov, A. V. Adamov and I. V. Martynov, Izv. Akad. Nauk, Ser. Khim., 1988, 1938–1939 Search PubMed; A. A. Krolevets, A. V. Adamov, A. G. Popov and I. V. Martynov, Dokl. Akad. Nauk SSSR, 1988, 303, 876–878 Search PubMed.
- V. A. Al'bekov, A. F. Benda, A. F. Gontar, G. A. Sokol'skii and I. L. Knunyants, Russ. Chem. Bull., 1988, 37, 777–780 CrossRef; V. A. Al'bekov, A. F. Benda, A. F. Gontar, G. A. Sokol'skii and I. L. Knunyants, Izv. Akad. Nauk, Ser. Khim., 1988, 897–900 Search PubMed.
- C. J. DeAngelis, G. Goyal, M. J. Liss, J. E. Budwitz, M. S. Herlihy, A. V. Conner, S. E. Wheeler, P. Ma, M. Li, K. N. Houk and C. G. Newton, J. Am. Chem. Soc., 2025 DOI:10.1021/jacs.4c17174.
- S. Yamabe, T. Dai, T. Minato, T. Machiguchi and T. Hasegawa, J. Am. Chem. Soc., 1996, 118, 6518–6519 CrossRef; T. Machiguchi, T. Hasegawa, A. Ishiwata, S. Terashima, S. Yamabe and T. Minato, J. Am. Chem. Soc., 1999, 121, 4771–4786 CrossRef; B. R. Ussing, C. Hang and D. A. Singleton, J. Am. Chem. Soc., 2006, 128, 7594–7607 CrossRef PubMed; E. G. Mackay and C. G. Newton, Aust. J. Chem., 2016, 69, 1365–1374 CrossRef.
- I. Dissanayake, J. D. Hart, E. C. Becroft, C. J. Sumby and C. G. Newton, J. Am. Chem. Soc., 2020, 142, 13328–13333 CrossRef PubMed; C. G. Newton and J. E. Budwitz, Synlett, 2022, 1473–1480 CrossRef; C. G. Newton and J. E. Budwitz, Org. Synth., 2023, 100, 159–185 CrossRef.
- P. S. Mariano, M. E. Osborn, D. Dunaway-Mariano, B. C. Gunn and R. C. Pettersen, J. Org. Chem., 1977, 42, 2903–2910 CrossRef CAS; P. S. Mariano, P. L. Huesmann, R. L. Beamer and D. Dunaway-Mariano, Tetrahedron, 1978, 34, 2617–2626 CrossRef.
- V. Nair, J. Org. Chem., 1972, 37, 2508–2510 CrossRef CAS; O. Tsuge, T. Ohnishi and H. Watanabe, Heterocycles, 1981, 16, 2085–2090 CrossRef; M. J. Alves A. G. Fortes, Américo Lemos and C. Martins, Synthesis, 2005, 555–558 Search PubMed.
- A. Hassner and D. J. Anderson, J. Org. Chem., 1974, 39, 2031–2036 CrossRef.
- M. J. Alves, N. G. Azoia and A. G. Fortes, Heterocycles, 2005, 65, 1329–1345 CrossRef.
- M. J. Alves and T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 1998, 299–304 RSC.
- M. J. Alves, N. G. Azoia, J. F. Bickley, A. G. Fortes, T. L. Gilchrist and R. Mendonça, J. Chem. Soc., Perkin Trans. 1, 2001, 2969–2976 RSC.
- B. M. Adger, M. Keating, C. W. Rees and R. C. Storr, J. Chem. Soc., Chem. Commun., 1973, 19–20 RSC; B. M. Adger, C. W. Rees and R. C. Storr, J. Chem. Soc., Perkin Trans. 1, 1975, 45–52 RSC.
- F. Gavina, A. M. Costero, M. R. Andreu, M. Carda and S. V. Luis, J. Am. Chem. Soc., 1988, 110, 4017–4018 CrossRef; F. Gavina, A. M. Costero, M. R. Andreu and S. V. Luis, J. Org. Chem., 1988, 53, 6112–6113 CrossRef; F. Gavina, A. M. Costero and M. R. Andreu, J. Org. Chem., 1990, 55, 434–437 CrossRef; F. Gavina, A. M. Costero, M. R. Andreu and M. D. Ayet, J. Org. Chem., 1991, 56, 5417–5421 CrossRef.
- J. C. Jagt and A. M. Van Leusen, J. Org. Chem., 1974, 39, 564–566 CrossRef; C. K. McClure and J. S. Link, J. Org. Chem., 2003, 68, 8256–8257 CrossRef PubMed; M. Sasaki, P. J. Hamzik, H. Ikemoto, S. G. Bartko and R. L. Danheiser, Org. Lett., 2018, 20, 6244–6249 CrossRef; S. G. Bartko, P. J. Hamzik, L. Espindola, C. Gomez and R. L. Danheiser, J. Org. Chem., 2020, 85, 548–563 CrossRef.
- S. E. Whitney, M. Winters and B. Rickborn, J. Org. Chem., 1990, 55, 929–935 CrossRef.
- K. T. Tagmazyan, R. S. Mkrtchyan and A. T. Babayan, Russ. J. Org. Chem., 1974, 1657–1662 Search PubMed; K. T. Tagmazyan, R. S. Mkrtchyan and A. T. Babayan, Zh. Org. Khim., 1974, 10, 1642–1648 Search PubMed.
- B. K. Ghorai, D. Jiang and J. W. Herndon, Org. Lett., 2003, 5, 4261–4263 CrossRef PubMed; J. Herndon, B. Ghorai, S. Duan and D. Jiang, Synthesis, 2006, 3661–3669 CrossRef.
- C.-Y. Yick, S.-H. Chan and H. N. C. Wong, Tetrahedron Lett., 2000, 41, 5957–5961 CrossRef.
- R. N. Warrener, J. Am. Chem. Soc., 1971, 93, 2346–2348 CrossRef.
- S. Matsumoto, A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2017, 139, 7713–7716 CrossRef.
- A. G. Brook, J. W. Harris, J. Lennon and M. El Sheikh, J. Am. Chem. Soc., 1979, 101, 83–95 CrossRef.
- A. G. Brook, K. Vorspohl, R. R. Ford, M. Hesse and W. J. Chatterton, Organometallics, 1987, 6, 2128–2137 CrossRef.
- N. Auner and A. Wolff, Chem. Ber., 1993, 126, 575–580 CrossRef.
- R. Appel, F. Knoll and I. Ruppert, Angew. Chem., Int. Ed. Engl., 1981, 20, 731–744 CrossRef.
- R. Appel, S. Korte, M. Halstenberg and F. Knoch, Chem. Ber., 1982, 115, 3610–3617 CrossRef CAS; J. Heinicke and A. Tzschach, Tetrahedron Lett., 1983, 24, 5481–5484 CrossRef; R. Appel and R. Zimmermann, Tetrahedron Lett., 1983, 24, 3591–3594 CrossRef; A. Meriem, J.-P. Majoral, M. Revel and J. Navech, Tetrahedron Lett., 1983, 24, 1975–1978 CrossRef.
- W. Rösch and M. Regitz, Z. Naturforsch., B: J. Chem. Sci., 1986, 41, 931–933 CrossRef CAS; E. P. O. Fuchs, W. Rösch and M. Regitz, Angew. Chem., Int. Ed. Engl., 1987, 26, 1011–1012 CrossRef; M. Regitz, Chem. Rev., 1990, 90, 191–213 CrossRef.
- J. Grobe and D. Le Van, Tetrahedron Lett., 1985, 26, 3681–3684 CrossRef CAS.
- L. D. Quin, A. N. Hughes and B. Pete, Tetrahedron Lett., 1987, 28, 5783–5786 CrossRef CAS.
- B. Deschamps and F. Mathey, J. Chem. Soc., Chem. Commun., 1985, 1010–1012 RSC.
- B. Deschamps and F. Mathey, J. Organomet. Chem., 1988, 354, 83–90 CrossRef CAS.
- Y. Mao and F. Mathey, Org. Lett., 2010, 12, 3384–3385 CrossRef CAS PubMed; Y. Mao and F. Mathey, Chem. – Eur. J., 2011, 17, 10745–10751 CrossRef PubMed.
- Y. Mao, K. M. H. Lim, R. Ganguly and F. Mathey, Org. Lett., 2012, 14, 4974–4975 CrossRef CAS PubMed.
- W. J. Middleton, E. G. Howard and W. H. Sharkey, J. Am. Chem. Soc., 1961, 83, 2589–2590 CrossRef CAS.
- W. J. Middleton, E. G. Howard and W. H. Sharkey, J. Org. Chem., 1965, 30, 1375–1384 CrossRef CAS.
- W. J. Middleton, J. Org. Chem., 1965, 30, 1390–1394 CrossRef.
- T. Katada, S. Eguchi and T. Sasaki, J. Org. Chem., 1986, 51, 314–320 CrossRef CAS.
- M. I. Hegab, A. E.-G. A. Amr and F. M. E. Abdel-Megeid, Z. Naturforsch., B: J. Chem. Sci., 2002, 57, 922–927 CrossRef CAS.
- K. Pahnke, N. L. Haworth, J. Brandt, U. Paulmann, C. Richter, F. G. Schmidt, A. Lederer, M. L. Coote and C. Barner-Kowollik, Polym. Chem., 2016, 7, 3244–3250 RSC.
- K. Alder and H. Niklas, Justus Liebigs Ann. Chem., 1954, 585, 81–96 CrossRef CAS.
- J. Levisalles and R. Baranger, Bull. Soc. Chim. Fr., 1957, 704–708 Search PubMed.
- J. G. Kuderna, J. W. Sims, J. F. Wikstrom and S. B. Soloway, J. Am. Chem. Soc., 1959, 81, 382–386 CrossRef CAS.
- Y. K. Yur'ev and N. S. Zefirov, Zh. Obshch. Khim., 1959, 29, 2954–2960 Search PubMed.
- M. Häring, Helv. Chim. Acta, 1960, 43, 556–561 CrossRef CAS; A. K. Mahida, S. B. Kale and U. Das, Eur. J. Org. Chem., 2017, 6427–6433 CrossRef.
- K. N. Zelenin and I. P. Bezhan, Dokl. Akad. Nauk SSSR, 1966, 2, 1524–1525 CAS; K. N. Zelenin and I. P. Bezhan, Russ. J. Org. Chem., 1966, 1505–1505 Search PubMed; K. N. Zelenin and I. P. Bezhan, Dokl. Akad. Nauk SSSR, 1970, 191, 1292–1294 Search PubMed.
- B. K. Bandlish, J. N. Brown, J. W. Timberlake and L. M. Trefonas, J. Org. Chem., 1973, 38, 1102–1105 CrossRef CAS.
- W. K. Anderson, R. H. Dewey and B. Mulumba, J. Med. Chem., 1979, 22, 1270–1272 CrossRef CAS PubMed.
- H. Clavier, J. Broggi and S. P. Nolan, Eur. J. Org. Chem., 2010, 937–943 CrossRef CAS.
- X. Wang, Y. Wen, Y. Wang, W. Li, X. Lu and W. You, CCS Chem., 2024, 6, 2305–2317 CrossRef CAS.
- E. Zaballos-García, M. E. González-Rosende, J. M. Jorda-Gregori, J. Sepúlveda-Arques, W. B. Jennings, D. O'Leary and S. Twomey, Tetrahedron, 1997, 53, 9313–9322 CrossRef; M. E. González-Rosende, J. Sepúlveda-Arques, E. Zaballos-Garcia, L. R. Domingo, R. J. Zaragozá, W. B. Jennings, S. E. Lawrence and D. O'Leary, J. Chem. Soc., Perkin Trans. 2, 1999, 73–80 RSC.
- S. K. Fehler, G. Pratsch and M. R. Heinrich, Angew. Chem., Int. Ed., 2014, 53, 11361–11365 CrossRef CAS PubMed.
- S. Gradl, J. Köckenberger, J. Oppl, M. Schiller and M. R. Heinrich, J. Org. Chem., 2021, 86, 6228–6238 CrossRef CAS.
- I. K. Korobitsyna, A. V. Khalikova, L. L. Rodina and N. P. Shusherina, Chem. Heterocycl. Compd., 1983, 19, 117–136 CrossRef.
- D. C. Oniciu, I. Ghiviriga, F. Iordache, D. Istrate and I. G. Dinulescu, Rev. Roum. Chim., 1992, 37, 1285–1290 CAS.
- R. W. Saalfrank, W. Hafner, J. Markmann, A. Welch, K. Peters and H. G. v. Schnering, Z. Naturforsch., B: J. Chem. Sci., 1994, 49, 389–406 CrossRef CAS.
- T. K. Sarkar, S. K. Ghosh and T. J. Chow, J. Org. Chem., 2000, 65, 3111–3115 CrossRef CAS.
- D. Song, Y. Tian, S. Huang, B. Li, Y. Yuan and A. Hu, J. Mater. Chem. B, 2015, 3, 8584–8588 RSC.
- P. Mondal, G. Jana, T. S. Pal, P. K. Chattaraj and N. K. Singha, Polym. Chem., 2021, 12, 6283–6290 RSC.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145 RSC.
- T. J. Kealy, J. Am. Chem. Soc., 1962, 84, 966–973 CrossRef CAS; L. G. Rashidyan, G. B. Oganesyan and G. T. Tatevosyan, Arm. Khim. Zh., 1974, 27, 1000–1003 Search PubMed.
- R. W. Hoffmann, G. E. Vargas-Núñez, G. Guhn and W. Sieber, Chem. Ber., 1965, 98, 2074–2085 CrossRef CAS; S. P. Darwen and R. K. Smalley, Tetrahedron Lett., 1985, 26, 5707–5708 CrossRef; M. Squillacote and J. De Felippis, J. Org. Chem., 1994, 59, 3564–3571 CrossRef.
- A. S. Amarasekara and S. Chandrasekara, Org. Lett., 2002, 4, 773–775 CrossRef CAS PubMed.
- K. Kawai, K. Ikeda, A. Sato, A. Kabasawa, M. Kojima, K. Kokado, A. Kakugo, K. Sada, T. Yoshino and S. Matsunaga, J. Am. Chem. Soc., 2022, 144, 1370–1379 CrossRef CAS.
- O. Wichterle, Collect. Czech. Chem. Commun., 1947, 12, 292–304 CrossRef CAS.
- Y. A. Arbuzov, Dokl. Akad. Nauk SSSR, 1948, 60, 993–996 CAS.
- A. Adams, D. H. Hey, A. Mustafa, S. D. Nicholas, F. Smith, H. Burton and P. F. Hu, J. Chem. Soc., 1949, 255–258 RSC.
- E. C. Taylor, C. P. Tseng and J. B. Rampal, J. Org. Chem., 1982, 47, 552–555 CrossRef CAS.
- A. Guilbaud-Criqui and C. Moinet, Bull. Soc. Chim. Fr., 1993, 130, 101–110 CAS.
- R. Faragher and T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 1979, 249–257 RSC.
- E. Francotte, R. Merényi, B. Vandenbulcke-Coyette and H. G. Viehe, Helv. Chim. Acta, 1981, 64, 1208–1218 CrossRef CAS.
- E. Turos, M. Parvez, R. S. Garigipati and S. M. Weinreb, J. Org. Chem., 1988, 53, 1116–1118 CrossRef CAS; R. Lux and G. Kresze, Liebigs Ann., 1989, 1989, 605–606 CrossRef.
- M. Takahashi and R. Okazaki, J. Sulphur Chem., 1993, 13, 293–338 CAS.
- G. Kresze and W. Wucherpfennig, Angew. Chem., Int. Ed. Engl., 1967, 6, 149–167 CrossRef CAS PubMed; S. M. Weinreb, Acc. Chem. Res., 1988, 21, 313–318 CrossRef.
- T. Q. Davies and M. C. Willis, Chem. – Eur. J., 2021, 27, 8918–8927 CrossRef CAS PubMed.
- M. P. Cava and R. H. Schlessinger, J. Org. Chem., 1963, 28, 2464–2465 CrossRef CAS.
- B. Helferich, R. Dhein, K. Geist, H. Jünger and D. Wiehle, Justus Liebigs Ann. Chem., 1961, 646, 45–48 CrossRef CAS.
- B. Anwar, P. Grimsey, K. Hemming, M. Krajniewski and C. Loukou, Tetrahedron Lett., 2000, 41, 10107–10110 CrossRef CAS.
- K. Hemming, C. Loukou, S. Elkatip and R. K. Smalley, Synlett, 2004, 101–105 CrossRef CAS.
- B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 1987, 106, 469–488 CrossRef CAS; T. Montagnon, D. Noutsias, I. Alexopoulou, M. Tofi and G. Vassilikogiannakis, Org. Biomol. Chem., 2011, 9, 2031–2039 RSC; M. C. Kimber and D. S. Lee, Nat. Prod. Rep., 2024, 41, 813–833 RSC.
- T. Montagnon, D. Kalaitzakis, M. Triantafyllakis, M. Stratakis and G. Vassilikogiannakis, Chem. Commun., 2014, 50, 15480–15498 RSC.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS.
- I. T. Chen, I. Baitinger, L. Schreyer and D. Trauner, Org. Lett., 2014, 16, 166–169 CrossRef CAS.
- C. Held, R. Fröhlich and P. Metz, Angew. Chem., Int. Ed., 2001, 40, 1058–1060 CrossRef CAS.
- S. Katsumura, K. Hori, S. Fujiwara and S. Isoe, Tetrahedron Lett., 1985, 26, 4625–4628 CrossRef CAS.
- W. Adam and A. Rodriguez, Tetrahedron Lett., 1981, 22, 3505–3508 CrossRef CAS.
- R. Shiraki, A. Sumino, K.-i. Tadano and S. Ogawa, Tetrahedron Lett., 1995, 36, 5551–5554 CrossRef CAS.
- X.-T. Liang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2020, 142, 8116–8121 CrossRef CAS PubMed.
- M. Miyashita, M. Sasaki, I. Hattori, M. Sakai and K. Tanino, Science, 2004, 305, 495–499 CrossRef CAS; F. Yoshimura, M. Sasaki, I. Hattori, K. Komatsu, M. Sakai, K. Tanino and M. Miyashita, Chem. – Eur. J., 2009, 15, 6626–6644 CrossRef PubMed.
- C. S. Foote, M. T. Wuesthoff, S. Wexler, I. G. Burstain, R. Denny, G. O. Schenck and K. H. Schulte-Elte, Tetrahedron, 1967, 23, 2583–2599 CrossRef CAS.
- K. Y. Seah, S. J. Macnaughton, J. W. P. Dallimore and J. Robertson, Org. Lett., 2014, 16, 884–887 CrossRef CAS PubMed.
- P. G. Sammes and L. J. Street, J. Chem. Soc., Chem. Commun., 1983, 666–668 RSC.
- G. Vassilikogiannakis and M. Stratakis, Angew. Chem., Int. Ed., 2003, 42, 5465–5468 CrossRef CAS.
- C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807–3810 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |