
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic cyclization-rearrangement cascade reaction: asymmetric construction of γ-lactams†
Sheng-Feng
Wu
,
Zhi-Yuan
Wang
and
Xing-Wen
Sun
 *
*
Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: sunxingwen@fudan.edu.cn
First published on 31st March 2025
Abstract
We report a chiral thiourea-catalyzed Michael/ring-reorganization cyclization of 3-methyleneoxindoles with N-(p-toluenesulfonyl)-α-amino ketones, facilitating the asymmetric synthesis of γ-lactams. This method efficiently generates a range of optically pure γ-lactams, in yields ranging from 60% to 86% and excellent stereoselectivity (up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, >99% ee). The gram-scale experiments confirmed the scalability of the reaction without compromising the yield or stereoselectivity. Performed under mild conditions, this investigation showcases its potential applicability for synthesizing complex chiral γ-lactams with consecutive three chiral centers.
:1 dr, >99% ee). The gram-scale experiments confirmed the scalability of the reaction without compromising the yield or stereoselectivity. Performed under mild conditions, this investigation showcases its potential applicability for synthesizing complex chiral γ-lactams with consecutive three chiral centers.
Introduction
The γ-lactam framework constitutes a pivotal structural motif in natural products and bioactive molecules, underpinning their diverse pharmacological activities including antibacterial, anticancer, and antiviral effects.1–3 Despite its pharmaceutical relevance, the synthesis of γ-lactams bearing multiple stereocenters presents formidable challenges in stereochemical control, a critical determinant of drug efficacy and specificity.4 This challenge highlights the central role of asymmetric synthesis in modern organic synthesis, particularly in pharmaceutical and agrochemical development.Organocatalytic asymmetric synthesis has emerged as a vital strategy for constructing chiral γ-lactams, where the strategic selection of synthetic building blocks dictates the efficiency of stereochemical control.5–12 Among commonly used precursors, Ts-protected α-amino ketones and α-amino esters exhibit unique reactivity profiles due to the dual activation effects of the p-toluenesulfonyl group.13–20 This protecting group not only elevates the NH acidity (pKa 10–16) to enable facile deprotonation under mild basic conditions but also creates a chemoselectivity dilemma by activating both α-carbon and nitrogen as potential nucleophilic sites, a phenomenon first systematically elucidated by Uria et al.21 Uria's seminal work revealed substrate-dependent chemoselectivity through comparative studies (Scheme 1a). They investigated the reactions of Ts-protected α-amino ketones with α,β-unsaturated aldehydes and observed two distinct outcomes. Specifically, when Ts-protected α-aminoacetophenone was used, the product predominantly resulted from a carbon nucleophilic attack. Conversely, when Ts-protected α-aminopropiophenone reacted with α,β-unsaturated aldehydes, the product mainly arose from a nitrogen nucleophilic attack.
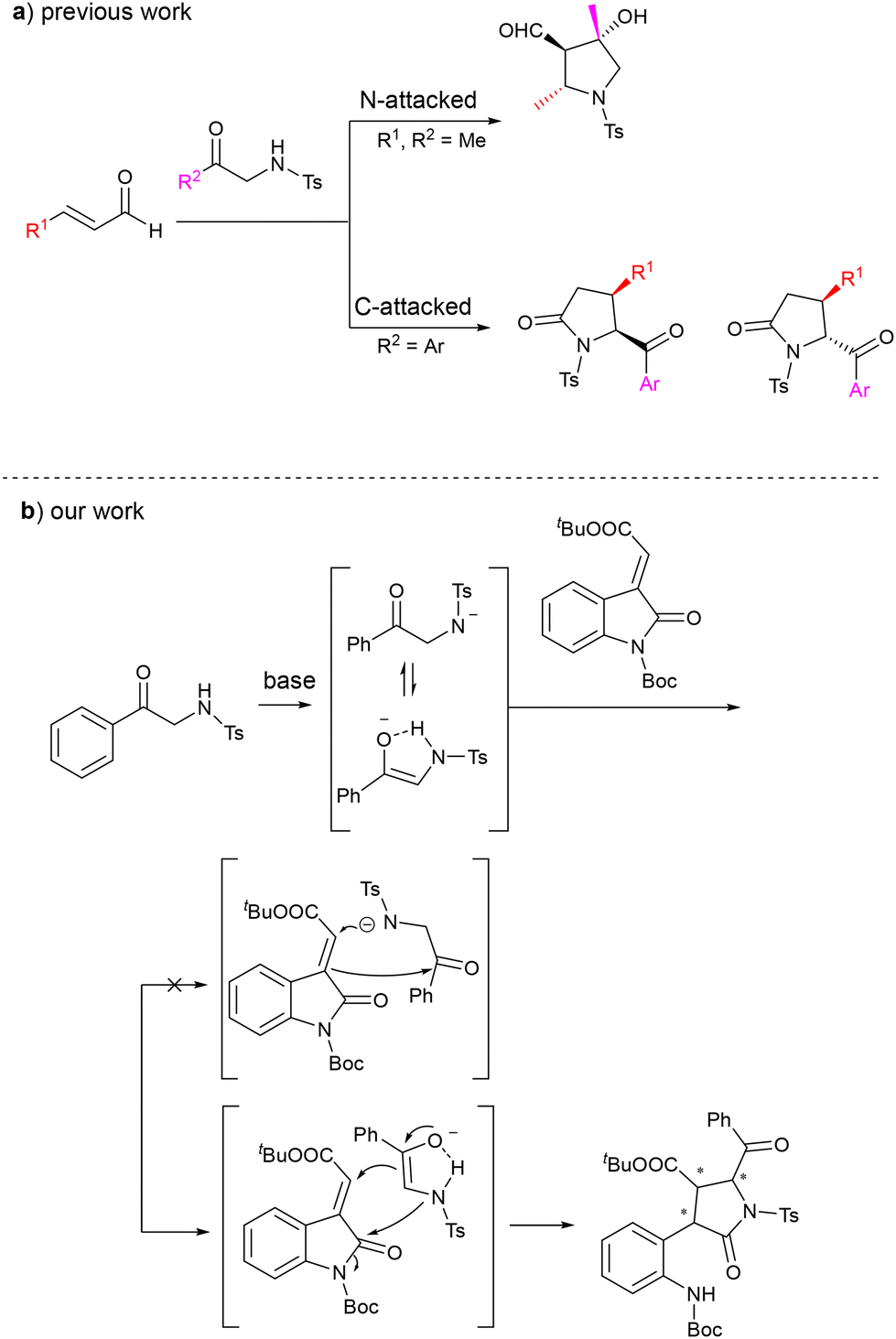 | ||
| Scheme 1 α-Aminophenone for construction of nitrogen-containing heterocyclic units. | ||
Based on our previous work regarding the construction of azaspirocyclic oxindoles through aza-Michael/annulation cascades of 3-methyleneoxindoles,22–24 we sought to extend this strategy to extend this strategy to the asymmetric synthesis of nitrogen-containing heterocycles. As depicted in Scheme 1b, Ts-protected α-amino ketones can adopt two distinct tautomeric forms under basic conditions, potentially displaying different reactivities towards 3-methyleneoxindole. If the nitrogen atom initiates the reaction, an aza-Michael/aldol cascade may ensue; conversely, if the α-carbon is the initial point of attack, a Michael addition could be followed by a possible ring-opening event. Empirical data have consistently demonstrated that the latter pathway predominates in these reactions.
Results and discussion
To further optimize the conditions, we used 1a and 2a as substrates for screening (Table 1). Initially, we screened various catalysts (Table 1). We began by evaluating bifunctional cinchona alkaloid catalysts. As shown in Table 1, all cinchona alkaloid-based catalysts yielded the desired products with excellent stereoselectivity (99% ee, 20:1 dr), with the main differences being in yield. Thiourea-based catalysts performed better in terms of yield compared to squaramide-based catalysts (entry 3 versus entry 5). The best cinchona alkaloid catalyst was the thiourea catalyst 3e, achieving a yield of 71%.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ratio | Cat. | Solvent | drb | Yield (%) | eec (%) |
| a Typical reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), cat. 3 (0.02 mmol) in solvent (0.5 mL), at 25 °C for 36 h. b Determined by 1H NMR analysis of the crude reaction mixture before purification. c Determined by HPLC analysis. d Prolong the reaction time to 72 h. | ||||||
| 1 | 1:1:0.1 |
3a | DCM | 20:1 |
54 | 99 |
| 2 | 1:1:0.1 |
3b | DCM | 20:1 |
65 | 99 |
| 3 | 1:1:0.1 |
3c | DCM | 20:1 |
56 | 99 |
| 4 | 1:1:0.1 |
3d | DCM | 20:1 |
60 | −99 |
| 5 | 1:1:0.1 |
3e | DCM | 20:1 |
71 | 99 |
| 6 | 1:1:0.1 |
3f | DCM | 8:1 |
62 | 99 |
| 7 | 1:1:0.1 |
3g | DCM | 7:1 |
45 | −97 |
| 8 | 1:1:0.1 |
3h | DCM | — | Trace | — |
| 9 | 1:1:0.1 |
3i | DCM | 3:1 |
35 | 90 |
| 10 | 1:1:0.1 |
3j | DCM | 4:1 |
40 | 97 |
| 11 | 1:1:0.1 |
3e | CHCl3 | 20:1 |
72 | 99 |
| 12 | 1:1:0.1 |
3e | Toluene | 20:1 |
70 | 99 |
| 13 | 1:1:0.1 |
3e | EtOAc | 20:1 |
76 | 99 |
| 14 | 1:1:0.1 |
3e | THF | 20:1 |
42 | 99 |
| 15 | 1:1:0.05 |
3e | EtOAc | 20:1 |
61 | 99 |
| 16 | 1:1:0.02 |
3e | EtOAc | 20:1 |
36 | 99 |
| 17d | 1:1:0.05 |
3e | EtOAc | 20:1 |
64 | 99 |

Next, cyclohexanediamine-based catalysts were screened to further optimize the conditions. Unfortunately, cyclohexanediamine-based catalysts generally demonstrated inferior stereoselectivity compared to cinchona alkaloid catalysts. The best cyclohexanediamine-based catalyst was 3f, yielding the desired product with 62% yield, 8:1 diastereoselectivity, and 99% enantioselectivity (entry 6). Additionally, increasing the steric hindrance of cyclohexanediamine led to decreased stereoselectivity and yield, prompting us to discontinue further screening of these catalysts. Ultimately, we selected 3e as the optimal catalyst.
After identifying the optimal catalyst, we screened for the best reaction solvent. The results indicated no differences in stereoselectivity among the solvents tested, with ethyl acetate giving the highest yield of 76%, making it the chosen solvent (entry 13). To further optimize the conditions, we examined the catalyst loading. Reducing the catalyst loading to 5 mol% and 2 mol% (entries 15 and 16) did not affect stereoselectivity but resulted in lower yields. We hypothesized that the lower catalyst concentration slowed the reaction rate. Extending the reaction time to 72 hours with a 5 mol% catalyst loading (entry 17) resulted in only a slight increase in yield by 3%. Considering all factors, we determined the conditions in entry 13 to be the optimal reaction conditions.
Following the determination of optimal reaction conditions, we expanded our investigation to explore the substrate scope, as delineated in Scheme 2. Initially, we focused on the impact of various substituents on the aryl ring of 3-methyleneoxindole. Overall, these substituents exhibited good stereoselectivity. However, the nitro-substituted derivative 4h showed decreased diastereoselectivity and yield, with only a 10:1 dr and a 65% yield. This could be attributed to the strong hydrogen-bonding ability of the nitro group, which might interfere with the catalyst's efficiency. Similarly, the 7-methyl-substituted oxindole substrate 4k also exhibited reduced performance. The hydrogen-bonding sites of the catalyst are typically located on the carbonyl oxygen of the protecting group Boc and the 2-oxo group of the oxindole. The 7-methyl substituent may hinder the hydrogen bonding, resulting in less efficient catalysis. The reaction with substrate 1, containing a methyl group at the terminal position of the Michael acceptor site, was attempted but proved unsuccessful. This result highlights a clear limitation of the reaction scope, indicating that only highly activated alkylideneoxindoles with a terminal electron-withdrawing group are suitable substrates for this transformation.
 | ||
| Scheme 2 Substrate applicability evaluation. Typical reaction conditions: 1 (0.2 mmol), 2 (0.2 mmol), 3e (0.02 mmol) in EtOAc (0.5 mL), at 25 °C for 36h. aIsolated yield. bDetermined by 1H NMR analysis of the crude reaction mixture before purification. cDetermined by HPLC analysis. dGram-scale experiment. | ||
Next, we examined α-amino ketone compounds. The results showed that only the ortho-substituted derivative 4r experienced reduced stereoselectivity, with a 10:1 dr. The other derivatives maintained excellent stereoselectivity (20:1 dr, 99% ee) and yielded the desired products in moderate to good yields. We also attempted the reaction with a heterocyclic α-amino ketone, 4s, which afforded the product with a yield of 65%, a diastereoselectivity of 20:1, and an enantioselectivity of 99%.
To assess the practicality of the method, we conducted a gram-scale experiment, as illustrated in Scheme 2. Employing the standard conditions, we utilized the template substrates 1a and 2a in the reaction, yielding the product 4a with a 70% yield, a diastereoselectivity greater than 20:1, and an enantioselectivity exceeding 99%. Although the isolated yield was slightly lower compared to the small-scale experiments, the excellent stereoselectivity was maintained, indicating the promising applicability of the approach. Some synthetic elaborations of 4a were made to explore the derivatization of the compounds, including Boc removal, hydrolysis of the tert-butyl ester, and exhaustive reduction of the lactam to the pyrrolidine, as well as complete hydrolysis to the open-chain amino acid derivative. But unfortunately, these attempts were unsuccessful, likely due to the high sensitivity of the substrates to acidic or basic conditions.
Based on the absolute configuration of the final product (CCDC 2363697†) (Scheme 3) and the catalytic mode of the bifunctional catalyst,25–28 we propose a plausible transition state model for this cascade reaction. Our previous research has identified that the carbonyl oxygen of the Boc group and the carbonyl oxygen of the oxindole are excellent hydrogen-bonding sites for thiourea. These interactions not only create a chiral environment but also enhance the electron-withdrawing effect of the double carbonyls, facilitating the nucleophilic addition to the double bond of the oxindole (Scheme 3). The tertiary amine of the cinchona alkaloid catalyst polarizes the enolate's H–O bond, enabling a more favorable Re-face attack on the double bond of the oxindole via Michael addition. Upon completion of the addition, the Ts-protected amine undergoes a nucleophilic reaction with the carbonyl group of the lactam, leading to the cleavage of the C–N bond, ring-opening, and the formation of a new lactam ring, ultimately yielding the desired product 4i.
 | ||
| Scheme 3 Proposed mechanism and transition state for the reaction. | ||
Experimental
General information
All reagents and all solvents were obtained from commercial suppliers and used without further purification except as indicated below. The silica gel (300–400 mesh) was used for column chromatography and TLC inspections were on silica gel GF 254 plates (0.25 mm layer thickness). NMR spectra were all recorded on a Bruker AM400 (400 MHz) spectrometer. Chemical shifts are reported in δ ppm referenced to an internal SiMe4 standard for 1H NMR and chloroform-d (δ 77.16) for 13C NMR. Enantioselectivities were determined by high-performance liquid chromatography (HPLC) with an Aglilent-1260 intelligent uv/vis detector (λ = 214 nm, 220 nm or 254 nm) and a Daicel IA or Daicel AD-H column. Optical rotations were measured in CHCl3 on a Pekin-Elmer 241MC automatic polarimeter. HRESIMS were recorded on an Agilent 6210 TOF LC/MS equipped with an electrospray ionization (ESI) probe operating in positive or negative ion mode.Typical procedure and analytical data
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 16.1 min, ee > 99%. [α]20D 5.2 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.15 (d, J = 7.8 Hz, 2H), 7.79–7.53 (m, 6H), 7.44 (s, 1H), 7.25 (dd, J = 11.2, 6.7 Hz, 3H), 7.11 (d, J = 8.2 Hz, 2H), 6.27 (d, J = 8.9 Hz, 1H), 4.56 (d, J = 11.4 Hz, 1H), 3.78–3.58 (m, 1H), 2.40 (s, 3H), 1.53 (s, 9H), 0.94 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 172.3, 167.8, 153.7, 145.7, 137.0, 135.6, 134.8, 133.9, 129.5, 129.2, 129.2, 129.1, 129.0, 128.9, 128.8, 128.6, 125.0, 84.1, 80.2, 56.9, 50.8, 45.8, 28.3, 27.2, 21.7. HRMS (ESI) m/z calcd for C34H38N2O8S [M + Na]+ 657.2246, found 657.2246.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 14.6 min, ee > 99%. [α]20D 12.0 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.21–8.10 (m, 2H), 7.74–7.68 (m, 3H), 7.59 (t, J = 7.7 Hz, 2H), 7.49 (s, 1H), 7.27–7.21 (m, 2H), 7.20–7.12 (m, 1H), 7.08 (dd, J = 8.4, 2.0 Hz, 1H), 6.87 (d, J = 2.0 Hz, 1H), 6.26 (d, J = 9.0 Hz, 1H), 4.52 (d, J = 11.4 Hz, 1H), 3.68 (dd, J = 11.4, 8.9 Hz, 1H), 2.41 (s, 3H), 2.28 (s, 3H), 1.52 (s, 9H), 0.95 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 172.4, 167.8, 153.9, 145.7, 135.7, 134.8, 134.7, 134.3, 133.9, 129.4, 129.4, 129.2, 129.1, 129.1, 129.0, 128.2, 125.5, 83.9, 80.0, 57.0, 50.8, 46.0, 28.3, 27.2, 21.7, 20.9. HRMS (ESI) m/z calcd for C35H40N2O8S [M + Na]+ 671.2403, found 671.2402.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel AD-H column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 18.5.0 min, tminor = 13.0 min, ee > 99%. [α]20D 9.7 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.22–8.09 (m, 2H), 7.73 (d, J = 8.1 Hz, 3H), 7.61 (t, J = 7.7 Hz, 3H), 7.33–7.17 (m, 3H), 7.06–6.94 (m, 1H), 6.83 (dd, J = 9.1, 2.9 Hz, 1H), 6.27 (d, J = 8.9 Hz, 1H), 4.58 (d, J = 11.5 Hz, 1H), 3.70 (dd, J = 11.5, 8.8 Hz, 1H), 2.43 (s, 3H), 1.55 (s, 9H), 0.98 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 171.8, 167.5, 161.1, 158.6, 154.0, 145.9, 135.6, 134.9, 133.9, 133.0, 131.6, 129.5, 129.3, 129.1, 127.8, 115.7, 115.5, 84.2, 80.4, 57.0, 50.3, 46.3, 28.3, 27.2, 21.7. 19F NMR (376 MHz, CDCl3) δ −116.5. HRMS (ESI) m/z calcd for C34H37FN2O8S [M + Na]+ 675.2152, found 675.2152.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 27.5 min, ee > 99%. [α]20D 10.6 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.21–8.10 (m, 2H), 7.73 (d, J = 8.0 Hz, 3H), 7.63 (q, J = 8.9, 7.6 Hz, 3H), 7.46 (s, 1H), 7.26 (dd, J = 10.5, 8.2 Hz, 3H), 7.04 (d, J = 2.4 Hz, 1H), 6.28 (d, J = 8.9 Hz, 1H), 4.56 (d, J = 11.6 Hz, 1H), 3.68 (dd, J = 11.6, 8.9 Hz, 1H), 2.44 (s, 3H), 1.54 (s, 9H), 0.99 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 171.6, 167.6, 153.6, 145.9, 135.9, 135.6, 134.9, 133.8, 130.3, 130.0, 129.5, 129.4, 129.2, 129.1, 128.8, 128.7, 126.4, 84.4, 80.6, 56.9, 50.7, 45.7, 28.3, 27.2, 21.7. HRMS (ESI) m/z calcd for C34H37ClN2O8S [M + Na]+ 691.1857, found 691.1857.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 14.0 min, tminor = 12.6 min, ee > 99%. [α]20D 14.7 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.14 (d, J = 7.7 Hz, 2H), 7.71 (d, J = 7.8 Hz, 3H), 7.59 (t, J = 7.6 Hz, 3H), 7.49 (s, 1H), 7.42–7.32 (m, 1H), 7.26 (d, J = 7.8 Hz, 2H), 7.15 (s, 1H), 6.26 (d, J = 8.8 Hz, 1H), 4.54 (d, J = 11.5 Hz, 1H), 3.72–3.56 (m, 1H), 2.41 (s, 3H), 1.51 (s, 9H), 0.96 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 171.6, 167.6, 153.4, 145.9, 136.4, 135.5, 134.9, 133.7, 131.6, 131.4, 130.4, 129.5, 129.3, 129.1, 129.1, 126.4, 117.5, 84.4, 80.6, 56.8, 50.7, 45.4, 28.2, 27.2, 21.7. HRMS (ESI) m/z calcd for C34H37BrN2O8S [M + Na]+ 735.1352, found 735.1348.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 20.8 min, tminor = 31.2 min, ee = 99%. [α]20D 11.2 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.20–8.07 (m, 2H), 7.73–7.68 (m, 3H), 7.58 (t, J = 7.7 Hz, 2H), 7.25 (dd, J = 9.2, 3.0 Hz, 2H), 7.17 (dt, J = 8.0, 2.5 Hz, 1H), 6.82 (dd, J = 8.8, 2.9 Hz, 1H), 6.65 (d, J = 2.9 Hz, 1H), 6.23 (d, J = 8.9 Hz, 1H), 4.49 (t, J = 11.0 Hz, 1H), 3.75 (s, 4H), 2.41 (s, 3H), 1.54 (s, 9H), 0.96 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 172.3, 167.5, 157.4, 154.5, 145.7, 135.7, 134.7, 134.1, 129.7, 129.4, 129.3, 129.1, 129.0, 128.4, 127.3, 115.0, 114.1, 83.8, 80.2, 57.2, 55.5, 50.1, 47.0, 28.3, 27.3, 21.7. HRMS (ESI) m/z calcd for C35H40N2O9S [M + Na]+ 687.2352, found 687.2350.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 5/95, 1.0 ml min−1, 220 nm), retention time: tmajor = 17.6 min, ee > 99%. [α]20D 16.3 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.23–8.08 (m, 2H), 7.74 (dt, J = 7.5, 2.5 Hz, 4H), 7.61 (t, J = 7.7 Hz, 2H), 7.46 (s, 1H), 7.35–7.23 (m, 2H), 7.16 (dt, J = 9.0, 1.9 Hz, 1H), 6.92 (d, J = 2.8 Hz, 1H), 6.28 (d, J = 8.9 Hz, 1H), 4.59 (d, J = 11.6 Hz, 1H), 3.69 (dd, J = 11.6, 8.9 Hz, 1H), 2.44 (s, 3H), 1.55 (s, 9H), 0.99 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.6, 171.6, 167.4, 153.7, 146.0, 145.8, 135.9, 135.6, 134.9, 133.9, 130.5, 129.5, 129.4, 129.1, 126.7, 121.7, 121.5, 121.3, 119.1, 84.4, 80.7, 57.0, 50.2, 46.1, 28.3, 27.2, 21.7. 19F NMR (376 MHz, CDCl3) δ −58.0. HRMS (ESI) m/z calcd for C35H37F3N2O9S [M + Na]+ 741.2070, found 741.2072.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 27.0 min, tminor = 16.5 min, ee = 98%. [α]20D 11.8 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.23–8.15 (m, 3H), 8.15–8.06 (m, 2H), 7.88 (d, J = 2.3 Hz, 1H), 7.74 (dd, J = 7.8, 5.7 Hz, 3H), 7.61 (t, J = 7.7 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 6.31 (d, J = 8.8 Hz, 1H), 4.63 (d, J = 11.7 Hz, 1H), 3.75 (dd, J = 11.6, 8.8 Hz, 1H), 2.43 (s, 3H), 1.53 (s, 9H), 0.97 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 171.2, 167.8, 152.6, 146.2, 144.1, 143.3, 135.4, 135.1, 133.7, 129.6, 129.5, 129.3, 129.2, 129.1, 127.0, 124.1, 122.6, 85.0, 81.5, 56.8, 50.5, 45.1, 28.2, 27.2, 21.8. HRMS (ESI) m/z calcd for C34H37N3O10S [M + Na]+ 702.2097, found 702.2098.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 17.1 min, tminor = 14.5 min, ee > 99%. [α]20D 15.7 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.23–8.08 (m, 2H), 7.81 (s, 1H), 7.78–7.57 (m, 6H), 7.26 (dt, J = 8.4, 2.9 Hz, 2H), 7.10 (dq, J = 8.4, 2.2 Hz, 1H), 7.01 (dd, J = 8.5, 4.0 Hz, 1H), 6.28 (dd, J = 8.8, 4.0 Hz, 1H), 4.54 (dd, J = 11.7, 4.0 Hz, 1H), 3.63 (ddd, J = 11.2, 8.7, 4.1 Hz, 1H), 2.43 (s, 3H), 1.54 (s, 9H), 0.97 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 171.9, 167.9, 153.2, 145.9, 138.5, 135.6, 134.9, 134.3, 133.8, 129.8, 129.5, 129.3, 129.1, 129.1, 126.3, 124.8, 124.2, 84.5, 80.7, 56.8, 51.0, 45.2, 28.3, 27.2, 21.7. HRMS (ESI) m/z calcd for C34H37ClN2O8S [M + Na]+ 691.1857, found 691.1857.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 220 nm), retention time: tmajor = 18.2 min, tminor = 15.0 min, ee = 99%. [α]20D 13.6 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.18–8.10 (m, 2H), 7.98–7.89 (m, 1H), 7.76–7.66 (m, 3H), 7.65–7.54 (m, 3H), 7.26–7.19 (m, 3H), 6.93 (d, J = 8.4 Hz, 1H), 6.26 (d, J = 8.9 Hz, 1H), 4.51 (d, J = 11.5 Hz, 1H), 3.60 (dd, J = 11.5, 8.9 Hz, 1H), 2.41 (s, 3H), 1.52 (s, 9H), 0.95 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 171.8, 167.9, 153.2, 145.9, 138.6, 135.6, 134.9, 133.8, 130.0, 129.5, 129.3, 129.1, 129.1, 127.8, 127.2, 127.0, 122.3, 84.5, 80.7, 56.8, 50.9, 45.3, 28.3, 27.2, 21.7. HRMS (ESI) m/z calcd for C34H37BrN2O8S [M + Na]+ 735.1352, found 735.1348.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel AD-H column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 10.0 min, tminor = 8.5 min, ee = 98%. [α]20D 15.1 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.11 (d, J = 7.7 Hz, 2H), 7.71 (dt, J = 9.8, 5.0 Hz, 3H), 7.58 (t, J = 7.7 Hz, 2H), 7.30–7.19 (m, 3H), 7.15 (q, J = 7.0, 6.5 Hz, 2H), 6.20 (d, J = 9.2 Hz, 2H), 4.47 (d, J = 11.1 Hz, 1H), 3.86 (t, J = 10.3 Hz, 1H), 2.42 (s, 3H), 2.28 (s, 3H), 1.64 (s, 9H), 0.98 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.9, 173.1, 167.4, 154.5, 145.5, 138.7, 135.9, 134.5, 134.4, 130.9, 129.3, 129.2, 129.1, 129.0, 129.0, 127.7, 83.2, 80.3, 57.4, 49.2, 28.2, 27.3, 21.7, 18.0. HRMS (ESI) m/z calcd for C35H40N2O8S [M + Na]+ 671.2403, found 671.2402.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IB column, i-PrOH/n-hexane = 5/95, 1.0 ml min−1, 254 nm), retention time: tmajor = 19.4 min, ee > 99%. [α]20D 4.4 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.19 (dd, J = 8.6, 5.2 Hz, 2H), 7.67 (dd, J = 19.7, 8.0 Hz, 3H), 7.37 (s, 1H), 7.27 (dt, J = 12.3, 5.7 Hz, 5H), 7.17–7.01 (m, 2H), 6.21 (d, J = 8.9 Hz, 1H), 4.56 (d, J = 11.4 Hz, 1H), 3.69 (dd, J = 11.4, 8.8 Hz, 1H), 2.41 (s, 3H), 1.52 (s, 9H), 0.99 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 194.4, 172.2, 168.0, 167.8, 165.4, 153.7, 145.8, 137.0, 133.8, 132.3, 132.2, 129.3, 129.0, 128.9, 128.7, 125.1, 116.4, 116.2, 84.1, 80.2, 56.8, 50.7, 45.9, 28.3, 27.3, 21.7. 19F NMR (376 MHz, CDCl3) δ −101.6. HRMS (ESI) m/z calcd for C34H37FN2O8S [M + Na]+ 675.2152, found 675.2152.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 21.6 min, ee > 99%. [α]20D 6.3 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.09 (d, J = 8.2 Hz, 2H), 7.76–7.51 (m, 5H), 7.34 (s, 1H), 7.26 (t, J = 8.2 Hz, 3H), 7.17–7.02 (m, 2H), 6.19 (d, J = 8.9 Hz, 1H), 4.55 (d, J = 11.3 Hz, 1H), 3.76–3.62 (m, 1H), 2.41 (s, 3H), 1.53 (s, 9H), 1.00 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.0, 172.2, 167.8, 153.8, 145.8, 141.4, 137.0, 134.1, 133.8, 130.7, 129.4, 129.3, 129.0, 128.9, 128.7, 125.4, 125.2, 84.2, 80.3, 56.8, 50.6, 45.9, 28.3, 27.3, 21.7. HRMS (ESI) m/z calcd for C34H37ClN2O8S [M + Na]+ 691.1857, found 691.1856.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 22.7 min, ee > 99%. [α]20D 7.7 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.01 (d, J = 8.2 Hz, 2H), 7.69 (td, J = 20.9, 20.4, 7.7 Hz, 5H), 7.34 (s, 1H), 7.26 (t, J = 8.0 Hz, 3H), 7.10 (dt, J = 14.9, 7.7 Hz, 2H), 6.18 (d, J = 8.9 Hz, 1H), 4.55 (d, J = 11.4 Hz, 1H), 3.78–3.60 (m, 1H), 2.41 (s, 3H), 1.52 (s, 9H), 1.00 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.3, 172.2, 167.8, 153.8, 145.9, 137.0, 134.5, 133.8, 132.4, 130.8, 130.3, 129.3, 129.0, 128.9, 128.7, 125.4, 125.2, 84.2, 80.3, 56.8, 50.6, 45.9, 28.3, 27.3, 21.7. HRMS (ESI) m/z calcd for C34H37BrN2O8S [M + Na]+ 735.1352, found 735.1348.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 9.4 min, ee > 99%. [α]20D 3.3 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.04 (d, J = 7.8 Hz, 2H), 7.67 (t, J = 9.0 Hz, 3H), 7.47 (s, 1H), 7.38 (d, J = 7.9 Hz, 2H), 7.25 (dd, J = 15.2, 5.8 Hz, 3H), 7.11 (d, J = 7.4 Hz, 2H), 6.23 (d, J = 8.8 Hz, 1H), 4.57 (d, J = 11.3 Hz, 1H), 3.67 (dd, J = 11.4, 8.8 Hz, 1H), 2.48 (s, 3H), 2.40 (s, 3H), 1.52 (s, 9H), 0.96 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.2, 172.4, 167.8, 153.7, 146.0, 145.7, 137.1, 133.9, 133.3, 129.7, 129.6, 129.2, 129.1, 128.9, 128.9, 128.5, 125.0, 84.0, 80.2, 56.8, 50.9, 45.8, 28.3, 27.2, 21.9, 21.7. HRMS (ESI) m/z calcd for C35H40N2O8S [M + Na]+ 671.2403, found 671.2402.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IB column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 10.2 min, tminor = 8.9 min, ee > 99%. [α]20D 8.3 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 7.99–7.87 (m, 2H), 7.69 (dd, J = 23.7, 8.1 Hz, 3H), 7.48 (dt, J = 19.9, 6.0 Hz, 3H), 7.25 (t, J = 6.8 Hz, 3H), 7.10 (q, J = 8.6, 8.1 Hz, 2H), 6.26 (d, J = 8.9 Hz, 1H), 4.55 (d, J = 11.5 Hz, 1H), 3.73–3.58 (m, 1H), 2.48 (s, 3H), 2.41 (s, 3H), 1.52 (s, 9H), 0.95 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.7, 172.3, 167.8, 153.7, 145.7, 138.9, 137.1, 135.6, 135.5, 133.9, 129.9, 129.2, 129.2, 128.9, 128.8, 128.7, 128.6, 126.7, 125.1, 125.0, 84.0, 80.2, 57.1, 50.8, 45.8, 28.3, 27.1, 21.7, 21.4. HRMS (ESI) m/z calcd for C35H40N2O8S [M + Na]+ 671.2403, found 671.2405.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IB column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 13.0 min, tminor = 10.6 min, ee = 99%. [α]20D 5.6 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 7.85–7.56 (m, 5H), 7.54–7.36 (m, 2H), 7.25 (t, J = 7.0 Hz, 4H), 7.15–7.01 (m, 2H), 6.23 (d, J = 8.9 Hz, 1H), 4.55 (d, J = 11.4 Hz, 1H), 3.89 (s, 3H), 3.69 (dd, J = 11.5, 8.9 Hz, 1H), 2.40 (s, 3H), 1.52 (s, 9H), 0.97 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.5, 172.3, 167.8, 160.1, 153.7, 145.7, 137.0, 136.9, 133.9, 130.0, 129.2, 129.1, 128.9, 128.8, 128.6, 125.2, 125.0, 122.2, 121.3, 113.4, 84.1, 80.2, 57.2, 55.6, 50.7, 45.8, 28.3, 27.2, 21.7. HRMS (ESI) m/z calcd for C35H40N2O9S [M + Na]+ 687.2352, found 687.2350.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 240 nm), retention time: tmajor = 25.8 min, tminor = 21.5 min, ee = 98%. [α]20D 6.3 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.11 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.43 (t, J = 7.5 Hz, 2H), 7.37–7.21 (m, 5H), 7.15–7.03 (m, 2H), 6.23 (d, J = 8.7 Hz, 1H), 4.52 (d, J = 11.9 Hz, 1H), 3.62 (dd, J = 12.0, 8.6 Hz, 1H), 2.51 (s, 3H), 2.42 (s, 3H), 1.51 (s, 9H), 0.93 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 196.5, 172.0, 167.9, 153.6, 145.7, 141.7, 137.1, 134.5, 134.0, 133.5, 132.8, 131.0, 129.3, 129.2, 128.8, 128.7, 128.6, 128.6, 126.1, 124.9, 83.8, 80.1, 67.1, 59.2, 50.9, 45.4, 28.3, 27.1, 21.8, 21.7. HRMS (ESI) m/z calcd for C35H40N2O8S [M + Na]+ 671.2403, found 671.2400.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IB column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 254 nm), retention time: tmajor = 16.9 min, tminor = 14.3 min, ee = 99%. [α]20D 14.4 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.07 (d, J = 3.9 Hz, 1H), 7.87 (d, J = 4.9 Hz, 1H), 7.68 (dd, J = 16.4, 7.9 Hz, 3H), 7.49 (s, 1H), 7.27 (dt, J = 20.0, 6.1 Hz, 4H), 7.17–7.03 (m, 2H), 6.01 (d, J = 8.8 Hz, 1H), 4.59 (d, J = 11.4 Hz, 1H), 3.68 (dd, J = 11.4, 8.7 Hz, 1H), 2.40 (s, 3H), 1.52 (s, 9H), 1.05 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 188.1, 172.2, 167.8, 153.7, 145.8, 142.9, 137.0, 136.9, 135.3, 133.9, 129.3, 129.1, 129.0, 128.9, 128.7, 128.6, 125.1, 125.1, 84.0, 80.2, 58.7, 50.9, 45.8, 28.3, 27.2, 21.7. HRMS (ESI) m/z calcd for C32H36N2O8S2 [M + H]+ 641.1991, found 641.1994.
:1. HPLC: the ee value was determined by HPLC analysis (Chiralcel IA column, i-PrOH/n-hexane = 10/90, 1.0 ml min−1, 240 nm), retention time: tmajor = 11.5 min, ee > 99%. [α]20D 15.3 (c 1.0, CHCl3). 1H NMR (400 MHz, chloroform-d) δ 8.74 (s, 1H), 8.09 (dd, J = 15.1, 8.4 Hz, 2H), 7.97 (dd, J = 22.5, 8.4 Hz, 2H), 7.67 (dt, J = 24.2, 7.7 Hz, 5H), 7.47 (s, 1H), 7.31–7.18 (m, 3H), 7.17–7.07 (m, 2H), 6.45 (d, J = 8.9 Hz, 1H), 4.62 (d, J = 11.4 Hz, 1H), 3.75 (dd, J = 11.4, 8.8 Hz, 1H), 2.40 (s, 3H), 1.54 (s, 9H), 0.85 (s, 9H). 13C NMR (101 MHz, chloroform-d) δ 195.5, 172.3, 167.9, 153.7, 145.7, 137.1, 136.2, 133.9, 133.0, 132.4, 132.1, 130.0, 129.6, 129.2, 129.1, 129.0, 128.9, 128.8, 128.8, 128.6, 127.9, 127.3, 125.0, 124.0, 84.0, 80.2, 57.1, 50.9, 45.9, 28.3, 27.1, 21.7. HRMS (ESI) m/z calcd for C38H40N2O8S [M + Na]+ 707.2403, found 707.2405.
Conclusions
In this study, we have successfully catalyzed a Michael/ring-reorganization cyclization reaction between 3-methyleneoxindoles and N-(p-toluenesulfonyl)-α-amino ketones using a thiourea-based bifunctional catalyst. This approach has yielded highly functionalized γ-lactams with good yields (up to 86%) and excellent stereoselectivity (up to 20:1 dr, >99% ee). The gram-scale experiments demonstrated that the scalability of the reaction does not compromise yield or stereoselectivity, indicating a promising potential for application. The reaction conditions are mild, and the substrate scope is broad, further highlighting the versatility and robustness of this catalytic system.
Data availability
The data that support the findings of this study are available on request from the corresponding author. For this article, crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number 2363697.† The 1H and 13C NMR spectra for all new compounds are included as part of the ESI.† Copies of the spectra can also be obtained from the authors upon request.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (21472019).References
- J. Li, S. Liu, S. Niu, W. Zhuang and Y. Che, Pyrrolidinones from the Ascomycete Fungus Albonectria rigidiuscula, J. Nat. Prod., 2009, 72, 2184–2187 CrossRef CAS.
- J. Caruano, G. G. Muccioli and R. Robiette, Biologically active γ-lactams: synthesis and natural sources, Org. Biomol. Chem., 2016, 14, 10134–10156 Search PubMed.
- F. I. Saldívar-González, E. Lenci, A. Trabocchi and J. L. Medina-Franco, Exploring the chemical space and the bioactivity profile of lactams: a chemoinformatic study, RSC Adv., 2019, 9, 27105–27116 Search PubMed.
- F. Rivas and T. Ling, Advances toward the Synthesis of Functionalized γ-Lactams, Org. Prep. Proced. Int., 2016, 48, 254–295 Search PubMed.
- X.-Y. Chen, Z.-H. Gao, C.-Y. Song, C.-L. Zhang, Z.-X. Wang and S. Ye, N-Heterocyclic, carbene catalyzed cyclocondensation of α,β-unsaturated carboxylic acids: Enantioselective synthesis of pyrrolidinone and dihydropyridinone derivatives, Angew. Chem., Int. Ed., 2014, 53, 11611–11615 Search PubMed.
- J. Caruano, G. G. Muccioli and R. Robiette, Biologically active γ-lactams: synthesis and natural sources, Org. Biomol. Chem., 2016, 14, 10134–10156 Search PubMed.
- F. Rivas and T. T. Ling, Advances toward the Synthesis of Functionalized γ-Lactams, Org. Prep. Proced. Int., 2016, 48, 254–295 CrossRef CAS.
- X. del Corte, A. Maestro, J. Vicario, E. M. Marigorta and F. Palacios, Brönsted-Acid-Catalyzed Asymmetric Three-Component Reaction of Amines, Aldehydes, and Pyruvate Derivatives. Enantioselective Synthesis of Highly Functionalized γ-Lactam Derivatives, Org. Lett., 2018, 20, 317–320 Search PubMed.
- Z. Hu, Y. Zhu, Z. Fu and W. Huang, Asymmetric synthesis of enantioenriched 6-hydroxyl butyrolactams promoted by N-heterocyclic carbene, J. Org. Chem., 2019, 84, 10328–10337 Search PubMed.
- X. del Corte, A. López-Francés, A. Maestro, E. M. Marigorta, F. Palacios and J. Vicario, Brönsted Acid Catalyzed Multicomponent Synthesis of Phosphorus and Fluorine-Derived γ-Lactam Derivatives, J. Org. Chem., 2020, 85, 14369–14383 CAS.
- S.-J. Liu, Q. Mao, G. Zhan, R. Qin, B.-H. Chen, J. Xue, M.-L. Luo, Q. Zhao and B. Han, Stereoselective synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam through the organocatalytic cascade reaction of 3-((2,2,2-trifluoroethyl)amino)indolin-2-one, Org. Biomol. Chem., 2021, 19, 467–475 RSC.
- Y. Pu, M. Wang, W. Tian, X. Ge, D. Zhu, C. Wang, Y. Zeng, F. Tao, Y. Deng and J. Lu, N-heterocyclic carbene catalyzed [2 + 3] annulation reaction for the synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam, RSC Adv., 2024, 14, 18453–18458 CAS.
- J.-W. Ren, Q.-L. Zhao, J.-A. Xiao, P.-J. Xia, H.-Y. Xiang, X.-Q. Chen and H. Yang, A One-Pot Ring-Opening/Ring-Closure Sequence for the Synthesis of Polycyclic Spirooxindoles, Chem. – Eur. J., 2019, 25, 4673–4677 Search PubMed.
- J.-W. Ren, Z.-Z. Xie, L. Zheng, Z.-P. Ye, Z.-X. Deng, Q.-L. Zhao, J.-A. Xiao, K. Chen, H.-Y. Xiang, X.-Q. Chen and H. Yang, An organocatalytic enantioselective ring-reorganization domino sequence of methyleneindolinones with 2-aminomalonates, Org. Chem. Front., 2021, 8, 778–783 Search PubMed.
- L. V. Frolova, N. M. Evdokimov, K. Hayden, I. Malik, S. Rogelj, A. Kornienko and I. V. Magedov, One-Pot Multicomponent Synthesis of Diversely Substituted 2-Aminopyrroles. A Short General Synthesis of Rigidins A, B, C, and D, Org. Lett., 2011, 13, 1118–1121 Search PubMed.
- R. O. Torres-Ochoa, T. Buyck, Q. Wang and J. Zhu, Heteroannulation of Arynes with α-Amino Imides: Synthesis of 2,2-Disubstituted Indolin-3-ones and Application to the Enantioselective Total Synthesis of (+)-Hinckdentine A, Angew. Chem., Int. Ed., 2018, 57, 5679–5683 Search PubMed.
- J.-W. Ren, L. Zheng, Z.-P. Ye, Z.-X. Deng, Z.-Z. Xie, J.-A. Xiao, F.-W. Zhu, H.-Y. Xiang, X.-Q. Chen and H. Yang, Organocatalytic, Enantioselective, Polarity-Matched Ring-Reorganization Domino Sequence Based on the 3-Oxindole Scaffold, Org. Lett., 2019, 21, 2166–2170 Search PubMed.
- X.-C. Yang, M.-M. Liu, F. Mathey, H. Yang, Y.-Z. Hua and M.-C. Wang, Access to Chiral 2,5-Pyrrolidinyl Dispirooxindoles via Dinuclear Zinc-Catalyzed Asymmetric Cascade Reactions, J. Org. Chem., 2019, 84, 7762–7775 Search PubMed.
- R. Shrestha, H. D. Khanal, W.-G. Yang, S. H. Kim, J.-J. Shim and Y. R. Lee, Metal-Free N-Annulation of 3-Formylchromones with α-Amino Ketones for the Construction of Diverse N-Functionalized Pyrroles, Asian J. Org. Chem., 2020, 9, 1070–1075 CAS.
- Y. Zhang, Y. Zhang, J. Guo, J. Han, X. Zhou and Z. Fu, Org. Chem. Front., 2021, 8, 5087–5091 Search PubMed.
- G. Talavera, E. Reyes, J. L. Vicario, L. Carrillo and U. Uria, Using Conveniently Designed α-Amino Ketones in Michael Reactions under Iminium Catalysis: Enantioselective Synthesis of γ-Lactams and γ-Amino-δ-keto Esters, Adv. Synth. Catal., 2013, 355, 653–658 Search PubMed.
- Q.-G. Tang, S.-L. Cai, C.-C. Wang, G.-Q. Lin and X.-W. Sun, Organocatalytic aza-Michael/Michael cyclization cascade reaction: Enantioselective synthesis of spiro-oxindole piperidin-2-one derivatives, Org. Lett., 2020, 22, 3351–3355 CAS.
- X.-T. Li, H.-Z. Tian and X.-W. Sun, Organocatalytic aza-Michael/Mannich cascade reaction: Synthesis of enantioenriched 3,3′-spirooxindole γ-lactams, J. Org. Chem., 2022, 87, 12345–12356 Search PubMed.
- H.-Z. Tian, Q.-G. Tang, G.-Q. Lin and X.-W. Sun, Asymmetric synthesis of chiral 1,2-oxazinane and hexahydropyridazin spirocyclic scaffolds by organocatalytic [4 + 2] cycloaddition, RSC Adv., 2022, 12, 15713–15717 CAS.
- C. M. R. Volla, I. Atodiresei and M. Rueping, Catalytic C–C Bond-Forming Multi-Component Cascade or Domino Reactions: Pushing the Boundaries of Complexity in Asymmetric Organocatalysis, Chem. Rev., 2014, 114, 2390–2431 CAS.
- N. Bania and S. C. Pan, Organocatalytic asymmetric Michael/hemiketalization/acyl transfer reaction of 1,3-propanediones with (E,)-2-(2-nitrovinyl)phenols, Org. Biomol. Chem., 2019, 17, 1718–1721 RSC.
- Y. Mo, S. Liu, Y. Liu, L. Ye, Z. Shi, Z. Zhao and X. Li, Highly stereoselective synthesis of 2,3-dihydrofurans via a cascade Michael addition-alkylation process: a nitro group as the leaving group, Chem. Commun., 2019, 55, 6285–6288 RSC.
- N. Saini, C. Khajuria, R. G. Biswas and V. K. Singh, Organocatalytic Asymmetric Cascade Michael-Acyl Transfer Reaction between 2-Fluoro-1,3-diketones and 2-Hydroxynitrostyrenes, J. Org. Chem., 2024, 89, 1264–1274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2363697. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00010f |
| This journal is © The Royal Society of Chemistry 2025 |