Effect of hydroxy groups on X-ray-induced reactions of azo benzene derivatives†
Received
1st January 2025
, Accepted 11th March 2025
First published on 21st March 2025
Abstract
Caged compounds whose chemical bonds are cleavable by specific stimuli are useful tools for life science research because they facilitate control of various biological activities spatiotemporally. Although caged compounds activatable by hard X-rays can be employed for control in deep tissue owing to the high bio-permeability of X-rays, chemical bond cleavage by ionizing radiation has not been investigated adequately. Previously, we demonstrated that an azo bond tethered to a rhodamine scaffold can be efficiently cleaved by hydrated electrons, which is one of the radiolysis products of water, to release rhodamine. In this study, we synthesized novel azo benzene derivatives, AZO1–4, which can release 3-aminobenzamide (3-ABA), a poly (ADP-ribose) polymerase (PARP) inhibitor, and hydroxy groups or amino groups were introduced into them in order to assess the substituent effect on azo bond cleavage. While the amount of 3-ABA was nearly the same for all the azo compounds, decomposition of azo compounds increased according to the number of hydroxy groups. Furthermore, a methoxyl-radical-adding product was detected from AZO2. These results suggested that the hydroxy group accelerates not azo bond cleavage but the other decomposition pathway.
Introduction
Compounds activated by electromagnetic waves, such as caged compounds, are expected to be useful for life science research because they facilitate control of biological activities spatiotemporally. Although various caged compounds activatable by light have been developed,1–4 it is difficult to achieve control using these compounds in deep tissue owing to the low permeability of the body to light. To overcome this problem, we focused on hard X-rays as an external stimulus source. Because hard X-rays have high bio-permeability, X-ray-activatable compounds can potentially be used to manipulate biological activities deep inside the body. X-rays barely excite organic compounds compared to photoexcitation; however, they ionize water molecules to form several radical species. Among these radical species, the hydroxyl radical and hydrated electrons account for a large population. Although some groups have reported X-ray caged compounds based on the concept that compounds that can react with these radical species could be X-ray caged compounds,5–10 our understanding of X-ray-induced reactions remains inadequate.
Previously, we reported that an azo compound possessing a rhodamine structure can undergo azo bond cleavage to release aniline derivatives under X-ray irradiation (Fig. 1A).11 In these compounds, the azo bond was reduced to form a hydrazine intermediate by hydrated electrons, followed by N–N bond cleavage. Although it is considered that the rhodamine structure contributed toward increasing the reactivity with hydrated electrons by electrostatic interaction and lowering the LUMO level, the detailed structure–activity relationship of simpler azobenzene derivatives with X-ray irradiation has not been investigated. Furthermore, it was reported that hydrogen bonding between a hydroxy group and a nitrogen atom of the azo bond can contribute to the azo bond reduction by hydrated electrons (Fig. 1B).12 In this study, we synthesized some azo compounds possessing a hydroxy group or an amino group as a hydrogen bond donor (Fig. 1C, AZO1–4). These compounds were designed to release 3-aminobenzamide (3-ABA), which is an inhibitor of poly (ADP-ribose) polymerase,13 as a model drug compound. By comparing the release of 3-ABA and the decomposition of the azo compound, we investigated the substituent effect on X-ray-induced decomposition.
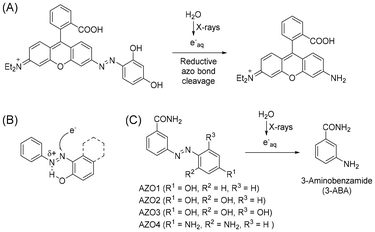 |
| | Fig. 1 (A) Previously reported azo compound bound to the rhodamine structure. (B) A plausible effect of the hydroxy group on enhancing reduction by hydrated electrons. (C) Design of azo compounds to release 3-ABA by X-ray irradiation. | |
Results and discussion
Synthesis
AZO1–4 were synthesized by azo coupling between 3-ABA and the corresponding reagents: phenol, resorcinol, phloroglucinol, and m-phenylenediamine (Scheme 1). The structure was confirmed by 1H-NMR, 13C-NMR, HRMS, and elemental analysis.
 |
| | Scheme 1 Synthetic scheme of AZO1–4. All the compounds were obtained by azo coupling reactions. | |
Experiment
Each AZO1–4 solution (25 μM) was prepared via dissolution in 1 mM phosphate buffer containing 40% methanol (total 2 mL). Under these conditions, methanol could act not only as a cosolvent to dissolve the azo compounds but also as a hydroxyl radical scavenger14,15 that can minimize the effect of the hydroxyl radical. Consequently, the effects of the solvated electrons could be evaluated under these conditions. The concentration of the solvated electrons (M Gy−1) can be calculated according to the following equation (eqn (1))| | | [Solvated electron] = (Ge × dsolvent)/(100e × NA). | (1) |
As an indicator of the number of species generated by X-rays, the G-value (Ge) is used. It is defined as the number of radical species generated by the absorption of 100 eV ionizing radiation. dsolvent is the density of the solvent, e is the elementary charge (1.60 × 10−19 C), and NA is Avogadro's number (6.02 × 1023 mol−1). For methanol and water, the G-values of the solvated electrons are reported to be 1.116 and 2.6,17 respectively. Thus, the G-value of the solvated electrons in the mixed solvent is assumed to be lower than that in water.
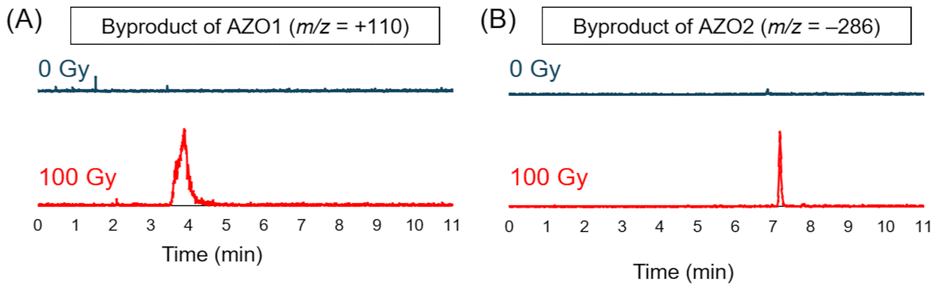
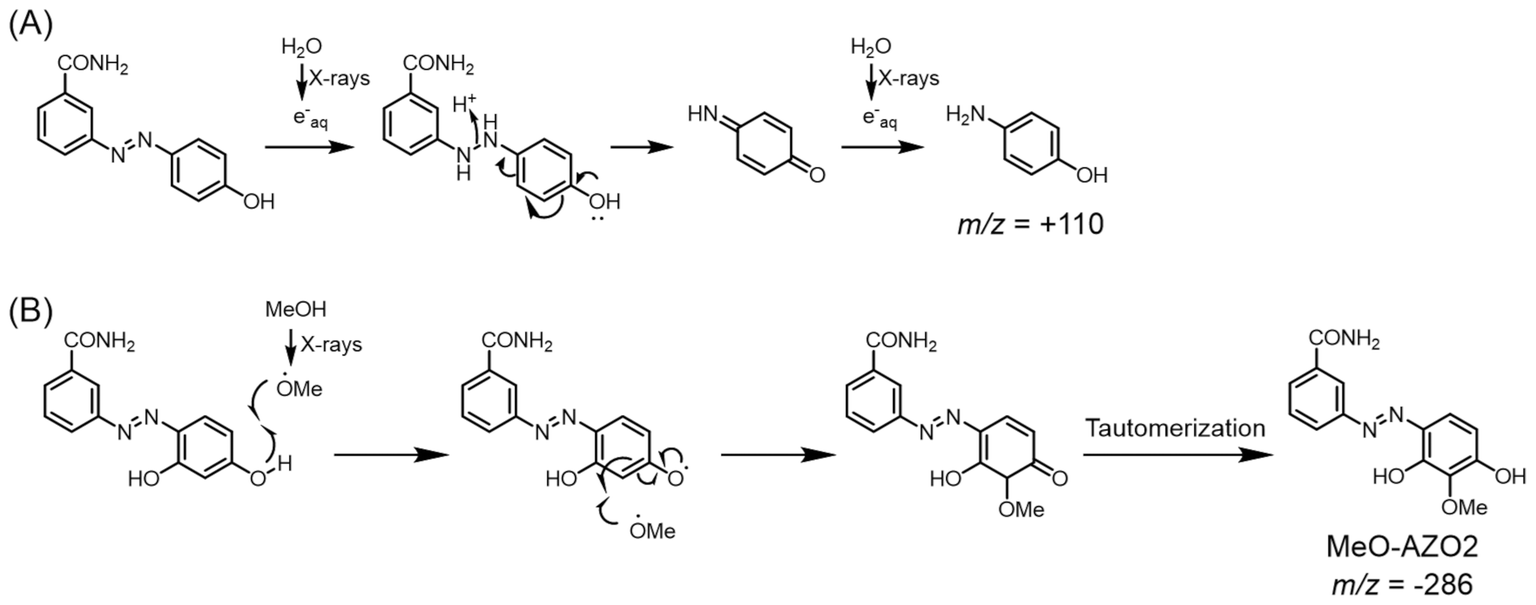
After deaerating the solution with argon gas for 10 min, 100 Gy X-rays were used for irradiating the solutions in an ice bath (irradiation dose rate: 4.7 Gy min−1). Under these conditions, the solvated electrons could be generated with a concentration of 27 μM at most, which is close to the concentration of AZO1–4. Then, the irradiated solution was analyzed by LC-MS. As shown in Fig. 2, while the peak of the azo compound decreased, the peak of 3-ABA was detected after X-ray irradiation for all the irradiated solutions. From the results of product analysis by LC-MS, the mass-to-charge ratio (m/z) of the decomposition products (+110 and −286) other than that of 3-ABA (+137) was obtained from a solution of AZO1 and AZO2, respectively (Fig. 3). While a byproduct of AZO1 (m/z +110) could be derived from the reductive azo bond cleavage as shown in Scheme 2A,11 that of AZO2 (m/z −286) could correspond to methoxy-group-substituted AZO2 (MeO-AZO2, Scheme 2B). The amounts of the reacted azo compounds were quantified as 5.9 μM (AZO1), 9.8 μM (AZO2), 16 μM (AZO3), and 7.4 μM (AZO4). Meanwhile, the amounts of released 3-ABA for all the compounds were quantified to be around 2.4–3.0 μM without a significant difference (Fig. 4).
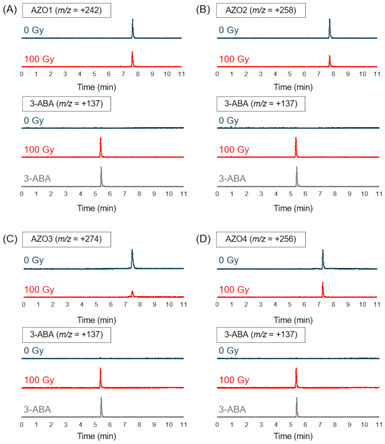 |
| | Fig. 2 LC-MS chromatograms of each azo compound solution before (blue, 0 Gy) and after (red, 100 Gy) X-ray irradiation to track the decomposition of the azo compound and release of 3-ABA for (A) AZO1, (B) AZO2, (C), AZO3, and (D) AZO4. A chromatogram of a standard solution of 3-ABA (gray) is shown at the bottom of each panel (Fig. S1†). | |
 |
| | Fig. 3 LC-MS chromatograms of byproducts detected from AZO1 and AZO2 solutions before (blue, 0 Gy) and after (red, 100 Gy) X-ray irradiation. (A) The byproduct with m/z = +110 was found in AZO1 solution and (B) the byproduct with m/z = –286 was found in AZO2 solution (Fig. S2†). | |
 |
| | Scheme 2 Plausible generation mechanisms of byproducts from (A) AZO1 and (B) AZO2. | |
 |
| | Fig. 4 Percentage of reacted azo compound after X-ray irradiation (A). Amount of released 3-ABA (B). The error bars represent the SEM: n = 3, Tukey–Kramer test, ***P < 0.001, **P < 0.01, and *P < 0.05. | |
Cyclic voltammetry (CV) was performed to evaluate the redox reactivity of each compound. The cyclic voltammograms of AZO1 and AZO3 showed reversible redox peaks, with the redox potentials calculated as −1.13 V for AZO1 and −0.938 V for AZO3 vs. a saturated calomel electrode (SCE). The cyclic voltammogram of AZO2 exhibited an irreversible reduction, and the redox potential was calculated as −0.933 V. The redox potential of AZO4 was not observed from −2 to 0 V (Fig. 5).
 |
| | Fig. 5 Cyclic voltammograms of (A) AZO1, (B) AZO2, (C) AZO3, and (D) AZO4. All the data were collected in DMF containing 0.1 M TBAP. The current flowed from the high potential side as indicated by the arrows on each graph (concentration of AZO1–3: 2 mM, AZO4: 4 mM, scan rate: 0.05 V s−1). | |
Discussion
From the quantification results, the amounts of the reacted azo compounds were in the following order: AZO3 > AZO2 > AZO1 = AZO4. Similarly, the redox potentials of AZO1–4 were in the following order: AZO3 > AZO2 > AZO1 > AZO4. Both the orders correspond to the number of hydroxy groups on the benzene moiety. Meanwhile, the amounts of 3-ABA, which is an azo bond cleavage product, were nearly the same for AZO1–4. Azo bond reduction mediated by a hydroxy group might not be attributed to the release of 3-ABA. A possible reason is that the hydroxy group supported tautomerization of the azo form to ortho-quinoid (as described previously by L. Das et al.),12 giving a different reduced product that decomposed immediately (Scheme 3). Moreover, from the result of the product analysis for AZO2, MeO-AZO2 was detected in the irradiated solution (Fig. 2B). A previous report suggested that the methoxyl radical can be generated by the radiolysis of methanol18 or its hydrogen ablation with the hydroxyl radical.15 Furthermore, it has also been reported that the alkoxyl radical can react with the hydroxy group of phenol.19 Therefore, the methoxyl radical should react with AZO2 to form MeO-AZO2 (Scheme 2B). However, a methoxy-group-substituted byproduct of AZO3 (MeO-AZO3) could not be detected in contrast to the case of AZO2. This is probably because AZO3 has more hydroxy groups than AZO2, which induced further decomposition.20–22 Hence, excessive hydroxy groups could cause an undesired reaction. Although both the amino group and the hydroxy group can act as electron-donating groups, AZO4 was less decomposed than AZO2. This result indicates that the hydroxy groups enhanced the reaction with the methoxyl radical much more than the amino groups.
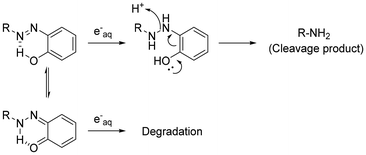 |
| | Scheme 3 Hypothesized degradation assisted by the hydroxy group. | |
Conclusions
In this study, we synthesized azo compounds AZO1–4 possessing hydroxy groups or amino groups and evaluated their X-ray-induced decomposition. By introducing hydroxy groups into the azo compound, the decomposition efficiency was increased. From the results of the product analysis, a decomposition product generated by the reaction with the methoxyl radical was detected. Thus, it was concluded that the hydroxy group in the azo compound might enhance the azo bond reduction efficiency, whereas introducing too many hydroxy groups could cause decomposition other than azo bond cleavage. To design X-ray-triggered caged compounds based on azo bond cleavage, not only the reductive cleavage of the azo bond but also undesired decomposition pathways should be considered. We believe that the structure–efficiency relationship of the hydroxy groups investigated in this study could act as a valuable guide in the further design of novel X-ray-triggered caged compounds for biological applications.
Experimental
Reagents and general information
AZO1–4 were all synthesized by azo coupling of 3-ABA with the corresponding reagents: phenol, resorcinol, phloroglucinol, and m-phenylenediamine. All the reagents used in the synthesis were purchased from FUJIFILM Wako Pure Chemicals Co. Ltd (Tokyo, Japan) or Kanto Chemical Co., Inc. (Tokyo, Japan), and they were used without further purification. 1H-NMR and 13C-NMR spectra were recorded on a JNM-ECX400P and a JNM-ECS400 (JEOL) at 400 MHz for 1H-NMR and 100 MHz for 13C-NMR. Chemical shifts were referred to a heavy solvent peak as a standard and expressed in ppm. Liquid chromatography–mass chromatography (LC-MS) analyses were conducted on an LC-MS system (Shimadzu Corporation, Kyoto, Japan) equipped with reverse-phase columns, Inertsil ODS-3 (2.1 mm × 50 mm) (GL Sciences Inc., Tokyo, Japan), using eluent A (0.1 M ammonium formate (HCOONH4) aqueous solution) and eluent B (99% CH3CN containing 1% H2O) with a set flow of 0.1 mL min−1 and eluent B conc. 1% (0 min) → 1% (3 min) → 80% (9 min) → 80% (11 min). X-rays were generated using a CLINAC (Varian Medical Systems, Palo Alto, California, USA) machine. Elemental analyses were performed using a CHN analyzer CE440 (Exeter Analytical Inc. Massachusetts, USA), and high-resolution mass spectroscopy (HRMS) was conducted using an Exactive Plus (Thermo Fisher Scientific Inc. Massachusetts, USA). Elemental analyses and HRMS were conducted by the Global Facility Center in Hokkaido University.
Preparation of AZO1
To a solution of 3-ABA (350 mg, 2.57 mmol) in a mixture of 1 M HCl aq (10 mL) and water (30 mL), a solution of NaNO2 (228 mg, 3.30 mmol) in water (1 mL) was gradually added at 0 °C. After stirring for 20 min at 0 °C, a solution of phenol (331 mg, 3.52 mmol) in ethanol (2 mL) was added to the reaction mixture. After stirring overnight while gradually reaching room temperature, the precipitate was collected by filtration and recrystallized with water and a small amount of EtOH to obtain AZO1 (200 mg, 0.829 mmol, 36%) as a yellow solid. 1H-NMR (400 MHz, CD3OD): δ 8.15 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.80 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.45 (dd, J = 7.8 Hz, 7.8 Hz, 1H), 6.81 (d, J = 8.0 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 167.82, 161.76, 152.57, 145.71, 136.08, 129.85, 129.72, 125.54, 125.06, 121.80, 116.54; HRMS (ESI) C13H11N3O2: [M − H]− calcd = 240.0779, found = 240.0785; anal. calcd for C13H11N3O2·1/10H2O: C, 64.36; H, 4.57; N, 16.94, found C, 64.24; H, 4.64; N, 17.29.
Preparation of AZO2
To a solution of 3-ABA (497 mg, 3.65 mmol) in a mixture of water (4 mL), 1 M HCl aq. (12 mL), and ethanol (4 mL), a solution of NaNO2 (280 mg, 4.06 mmol) in water (4 mL) was gradually added at 0 °C. After stirring at 0 °C for 20 min, a solution of resorcinol (444 mg, 4.03 mmol) in ethanol (4 mL) was added to the reaction mixture. After stirring overnight while gradually reaching room temperature, the precipitate was collected by filtration and recrystallized with water and a small amount of ethanol to obtain AZO2 (504 mg, 1.96 mmol, 54%) as a yellow solid. 1H-NMR (400 MHz, CD3OD): δ 8.31 (s, 1H), 7.99–7.92 (m, 2H), 7.72 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.62 (dd, J = 7.8 Hz, 7.8 Hz, 1H), 6.54 (d, J = 9.2 Hz, 1H), 6.32 (s, 1H); 13C-NMR (100 MHz, DMSO-d6): δ 167.76, 163.87, 157.25, 151.29, 136.08, 132.89, 129.92, 129.67, 129.33, 124.69, 121.32, 109.78, 103.52; HRMS (ESI) C13H11N3O3: [M − H]− calcd = 256.0728, found = 256.0729; anal. calcd for C13H11N3O3·1/4H2O: C, 58.54; H, 4.09; N, 15.85, found C, 58.62; H, 4.26; N, 15.78.
Preparation of AZO3
To a solution of 3-ABA (2.01 g, 14.7 mmol) in a mixture of water (16 mL), 1 M HCl aq (48 mL), and ethanol (10 mL), a solution of NaNO2 (1.08 g, 15.7 mmol) in water (16 mL) was gradually added at 0 °C. After stirring at 0 °C for 20 min, a solution of phloroglucinol (2.08 g, 16.5 mmol) in ethanol (16 mL) was added to the reaction mixture. After stirring overnight while gradually allowing it to reach room temperature, the precipitation was collected by filtration. The precipitates were recrystallized with water and a small amount of ethanol to obtain AZO3 (1.80 g, 6.59 mmol, 45%) as a yellow solid. 1H-NMR (400 MHz, CD3OD): δ 8.42 (s, 1H), 8.06 (d, J = 7.7 Hz, 1H), 8.04 (d, J = 7.7 Hz, 1H), 7.64 (dd, J = 7.7 Hz, 7.7 Hz, 1H), 5.95 (s, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 167.78, 166.38, 159.29, 150.25, 136.00, 129.90, 128.62, 124.28, 123.47, 120.70, 95.57; HRMS (ESI) C13H11N3O4: [M − H]− calcd = 272.0677, found = 272.0676; anal. calcd for C13H11N3O4·1/4H2O: C, 55.96; H, 3.84; N, 15.35, found C, 56.22; H, 4.17; N, 15.13.
Preparation of AZO4
To a solution of 3-ABA (497 mg, 3.65 mmol) in a mixture of H2O (4 mL), 1 M HCl aq. (2 mL), and EtOH (4 mL), a solution of NaNO2 (279 mg, 4.04 mmol) in H2O (4 mL) was added at 0 °C. After stirring at 0 °C, a solution of m-phenylenediamine (448 mg, 4.14 mmol) in EtOH (6 mL) was added to the reaction mixture. After stirring overnight while gradually reaching room temperature, the precipitate was collected by filtration and recrystallized with water and a small amount of EtOH to obtain AZO4 (882 mg, 3.18 mmol, 95%) as a yellow solid. 1H-NMR (400 MHz, CD3OD): δ 8.21 (s, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 6.6 Hz, 1H), 7.54–7.49 (m, 2H), 6.13 (d, J = 8.8 Hz, 1H), 5.98 (s, 1H); 13C-NMR (100 MHz, DMSO-d6): δ 168.30, 154.14, 153.65, 135.68, 129.89, 129.42, 126.80, 124.01, 120.36, 106.44, 96.93; HRMS (ESI) C13H13N5O: [M + H]+ calcd = 256.1154, found = 256.1191, [M + Na]+ calcd = 278.1012, found = 278.1009; anal. calcd for C13H13N5O·2/3H2O: C, 58.55; H, 5.12; N, 26.24, found C, 58.42; H, 5.42; N, 26.20.
Azo bond cleavage by X-ray irradiation
X-ray irradiation was conducted using 40% methanol because of the low solubility of AZO1–4. Because methanol can work as a hydroxyl radical scavenger, the reaction caused by hydrated electrons should be evaluated under these conditions. Each solution of AZO1–4 (25 μM) was prepared via dissolution in a mixture of 1 mM phosphate buffer (pH 7.4) and methanol (phosphate buffer/methanol = 6/4, 2 mL). After the solution was deaerated with argon gas through the spectrum cap of the sealed vial for 10 min, it was irradiated with X-rays (100 Gy) using a CLINAC (dose rate: 4.7 Gy min−1, tube voltage: 6 MeV). Each irradiated solution was mixed with 1 mM phosphate buffer (pH = 7.4) containing 10 μM fluorescein of the same volume and analysed by LC-MS.
Cyclic voltammetry
Cyclic voltammetry (CV) was conducted to measure the redox potentials using tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte. AZO1–3 (2 mM) or AZO4 (4 mM) was dissolved in DMF, which contains 0.1 M TBAP, and bubbled with argon gas. The working electrode, which had a Pt disc with a surface area of 0.020 cm2, the counter electrode made of a Pt wire, and the Ag/AgCl reference electrode were soaked in DMF. Voltage was applied at a scan rate of 0.05 V s−1. All the reduction potentials are shown vs. a SCE and by standardizing with ferrocene at +0.403 V.23
Author contributions
Conceptualization: M. O., data curation: K. O., funding acquisition: M. O., investigation: K. O., N. I., H. T., K. H., and O. I., methodology: K. O., H. T. and O. I., supervision: N. I., H. T., K. N., A. M., M. H., and M. O., visualization: K. O., N. I., and H. T., writing – original draft: K. O., and writing – review & editing: N. I., M. O.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Conflicts of interest
Dr Ogawa has received research grants from CREST. The funder was not involved in the study design, analysis, or interpretation of the data.
Acknowledgements
This work was partly supported by JST-CREST, Grant Number JPMJCR1902; a MEXT Project for Promoting Public Utilization of Advanced Research Infrastructure (Program for Supporting Introduction of the New Sharing System), Grant Number JPMXS0420100120; a Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED); and a Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP19am0101093. This work was also partly supported by Hokkaido University, Global Facility Center (GFC), Pharma Science Open Unit (PSOU), funded by MEXT under “Support Program for Implementation of New Equipment Sharing System”. In addition, this work was supported by JST SPRING, Grant Number JPMJSP2119.
References
- G. C. R. Ellis-Davies, Nat. Methods, 2007, 4, 619–628 CrossRef CAS PubMed.
- N. Ieda, Y. Hotta, N. Miyata, K. Kimura and H. Nakagawa, J. Am. Chem. Soc., 2014, 136, 7085–7091 CrossRef CAS PubMed.
- F. Bley, K. Schaper and H. Gö, Photochem. Photobiol., 2008, 84, 162–171 CrossRef CAS PubMed.
- Z. Zhao, J. Li, W. Yuan, D. Cheng, S. Ma, Y. F. Li, Z. J. Shi and K. Hu, J. Am. Chem. Soc., 2024, 146, 1364–1373 CrossRef CAS PubMed.
- Q. Fu, H. Li, D. Duan, C. Wang, S. Shen, H. Ma and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 21546–21552 CrossRef CAS PubMed.
- Z. Guo, H. Hong, Y. Zheng, Z. Wang, Z. Ding, Q. Fu and Z. Liu, Angew. Chem., Int. Ed., 2022, 61, 34 Search PubMed.
- J. Geng, Y. Zhang, Q. Gao, K. Neumann, H. Dong, H. Porter, M. Potter, H. Ren, D. Argyle and M. Bradley, Nat. Chem., 2021, 13, 805–810 CrossRef CAS PubMed.
- Q. Fu, Z. Gu, S. Shen, Y. Bai, X. Wang, M. Xu, P. Sun, J. Chen, D. Li and Z. Liu, Nat. Chem., 2024, 16, 1–9 CrossRef PubMed.
- Z. Ding, Z. Guo, Y. Zheng, Z. Wang, Q. Fu and Z. Liu, J. Am. Chem. Soc., 2022, 144, 9458–9464 CrossRef CAS PubMed.
- K. Tanabe, J. Ishizaki, Y. Ando, T. Ito and S. I. Nishimoto, Bioorg. Med. Chem. Lett., 2012, 22, 1682–1685 CrossRef CAS PubMed.
- K. Ogawara, O. Inanami, H. Takakura, K. Saita, K. Nakajima, S. Kumar, N. Ieda, M. Kobayashi, T. Taketsugu and M. Ogawa, Adv. Sci., 2024, 11(27), 2306586 CrossRef CAS PubMed.
- L. Das, S. Chatterjee, D. B. Naik and S. Adhikari, J. Hazard. Mater., 2015, 298, 19–27 CrossRef CAS PubMed.
- N. Makogon, T. Voznesenskaya, T. Bryzgina, V. Sukhina, N. Grushka and I. Alexeyeva, Reprod. Biol., 2010, 10, 215–226 CrossRef PubMed.
- L. Ding, Y. Hou, H. Liu, J. Peng, Z. Cao, Y. Zhang, B. Wang, X. Cao, Y. Chang, T. Wang and G. Liu, ACS ES & T Water, 2023, 3, 3534–3543 Search PubMed.
- L. G. Devi, S. G. Kumar, K. S. A. Raju and K. E. Rajashekhar, Chem. Pap., 2010, 64, 378–385 CAS.
- M. C. Sauer, S. Arai and L. M. Dorfman, J. Chem. Phys., 1965, 42, 708–712 CrossRef CAS.
- I. G. Draganic, T. Nenadovic and Z. D. Draganic, J. Phys. Chem., 1969, 73, 2564–2571 CrossRef CAS.
- J. Lind, A. Jowko and T. E. Eriksen, Radiat. Phys. Chem., 1979, 13, 159–163 CrossRef CAS.
- L. K. Folkes, S. Bartesaghi, M. Trujillo, R. Radi and P. Wardman, Free Radical Res., 2012, 46, 1150–1156 CrossRef CAS PubMed.
- J. Yu, H. X. Sheng, S. W. Wang, Z. H. Xu, S. Tang and S. L. Chen, Chem. Commun., 2019, 55, 4578–4581 RSC.
- S. Tang, Y. L. Deng, J. Li, W. X. Wang, Y. C. Wang, Z. Z. Li, L. Yuan, S. L. Chen and R. L. Sheng, Chem. Commun., 2016, 52, 4470–4473 RSC.
- S. Tang, D. Zhou, Y. Deng, Z. Li, Y. Yang, J. He and Y. Wang, Sci. China: Chem., 2015, 58, 684–688 CrossRef CAS.
- A. Paul, R. Borrelli, H. Bouyanfif, S. Gottis and F. Sauvage, ACS Omega, 2019, 4, 14780–14789 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Hideo
Takakura
a,
Hideo
Takakura