
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformational properties of alkyl glucosyl sulfones in solution†
Carlos A.
Sanhueza
 ab,
Rosa L.
Dorta
a and
Jesús T.
Vázquez
*a
ab,
Rosa L.
Dorta
a and
Jesús T.
Vázquez
*a
aDepartamento de Química Orgánica, Instituto Universitario de Bio-Orgánica “Antonio González”, Universidad de La Laguna, Avda. Astrofísico Francisco Sánchez 2, La Laguna, Tenerife 38206, Spain. E-mail: jtruvaz@ull.edu.es
bDepartment of Pharmaceutical Sciences, St John's University, 8000 Utopia Parkway, Queens, NY 10439, USA
First published on 2nd July 2025
Abstract
The conformational properties of ester-protected alkyl glucosyl sulfones were studied by means of nuclear magnetic resonance (NMR) and circular dichroism (CD). The equilibrium about the C5–C6 rotational axis (hydroxymethyl group) in polar and apolar media is distributed between the gg and gt rotamers, and the equilibrium contributions of each rotamer are modulated by the steric properties of the aglycone, where an increment in the alkyl substituent's bulkiness leads to progressive increments in gt contributions at the expense of gg. The equilibrium about the glycosidic bond is also dependent on the aglycone's bulkiness and appears more sensitive to the media polarity. Glucosyl sulfones carrying small aglycones, e.g, those substituted with methyl and ethyl groups, show a ϕ torsion predominated by the g+ conformer in apolar media (C6D6) and distributed between g− and g+ conformers in a polar solvent (CD3CN). Compounds substituted with larger alkyl groups, such as iso-propyl and tert-butyl groups, show glycosidic bond conformations predominated by the g− conformer.
Introduction
Carbohydrates are intrinsically flexible molecules playing critical roles in living systems.1 The molecular recognition of cell-associated saccharides by protein receptors typically triggers a plethora of physiological and pathological functions including fertilization,2 immunity,3 infection,4 metastasis,5 and many others.6 To successfully elicit such functions, the carbohydrate ligand must fulfill geometrical and conformational requirements for an optimal interaction with the receptor. Hence, the study of the conformational properties of carbohydrates is vital to fully characterize and understand the molecular recognition mechanisms at the molecular level. Moreover, understanding the conformational patterns of sugars and the factors driving them could prove useful in future studies of carbohydrates and glycomimetics.The conformational flexibility of hexopyranoses, the prevalent monosaccharide configuration in mammalian glycans, lies on the glycosidic C1–O1 bond and the C5–C6 bond (hydroxymethyl group) (Fig. 1A). The torsional angles ϕ and ψ characterize the glycosidic bond's conformation. The free rotation about ϕ generates three staggered rotamers called exo–syn (or g−), exo–anti (or g+), and non–exo (or anti) (Fig. 1B) whose contributions to the glycosidic conformational equilibrium are determined by the regio- and stereochemistry of the glycosidic linkage, the media polarity, the anomeric effect, and the presence of intramolecular polar interactions. For example, in aryl S-β-glucosides, the aglycone's electronic properties appear to be a relevant conformational modulator about ϕ, where exo–syn is the predominant conformer in glucosides carrying electron-deficient aryl rings whereas those carrying electron-richer substituents exhibit larger exo–anti and non–exo contributions.7 The free rotation about ω (hydroxymethyl group) generates three rotamers called gauche–gauche (gg), gauche–trans (gt), and trans–gauche (tg) (Fig. 1C). The contributions of each rotamer to the conformational equilibrium about C5–C6 depend on several factors including the stereochemical configuration of C4, the nature of the C4 and C6 substituents, the polarity of the media, the anomeric configuration, and the chemical nature of the aglycone, including its stereochemistry. We have extensively studied the effect of the aglycone's nature on the hydroxymethyl conformation of alkyl O-,8S-,9 and C-10 glycosides as well as glycosyl sulfoxides,11 finding that the rotameric distribution about C5–C6 relates to the steric properties of the alkyl aglycone, where a progressive increment of its bulkiness leads to a correlating increment of gt percent contributions at the expense of the predominant gg conformer. We found a similar conformational behavior in aryl S-glycosides, where increased gt contributions correlate with glycosides carrying stronger electron-withdrawing aryl substituents.7 The elimination of the endocyclic oxygen atom leads to carbasugar derivatives showing no conformational dependence on the aglycone's nature.12 We envisage these structure–conformation relationships as an invaluable tool for upcoming studies of conformationally modulated carbohydrates and glycomimetics that could be useful in areas such as drug discovery and molecular probe design. In our pursuit to expand our knowledge on structure–conformation relationships, we centered our attention on the glycosidic and hydroxymethyl flexibility of alkyl glucosyl sulfones. Glycosyl sulfones are an underexplored, yet promising, class of glycosyl donors capable of generating cationic and radical glycosylating intermediates.
 | ||
| Fig. 1 Conformational properties of glycosides. (A) Flexible bonds in O-glycosides; (B) glycosidic bond rotamers; (C) hydroxymethyl group rotamers. | ||
Synthesis and structural characterization of alkyl glycosyl sulfones
We designed two series of model compounds namely acetates (4a–4f) and dibenzoates (9a–9f) suitable for performing the conformational analysis by nuclear magnetic resonance (NMR) and circular dichroism (CD) respectively (Scheme 1). We started the preparation of acetates 4a–4f from free glucose 1, which was converted into glucose per-acetylated by treatment with Ac2O in pyridine at room temperature. The anomeric acetate in 2 was then substituted by an alkyl thiyl group by treating the glycosyl donor with BF3·Et2O in DCM in the presence of the respective alkyl thiol.13 This procedure afforded alkyl S-glycosides 3b–3f in moderate to good yields.9 Since the volatility of methanethiol makes its direct glycosylation difficult to implement, methyl derivative 4a was prepared by the in situ methylation of a glucosyl thiouronium salt prepared by refluxing a MeCN solution of 2, thiourea, and BF3·Et2O.9,14 The subsequent oxidation of S-glycosides 3a–3f with per-acetic acid, generated in situ by reacting H2O2 with Ac2O over SiO2 as a catalyst,15 afforded the respective alkyl glycosyl sulfones 4a–4f in excellent yields (91–99%). To prepare dibenzoates 9a–9f, we subjected the C4 and C6 hydroxyl groups in 1 to regioselective protection as benzylidene acetal by treatment with benzaldehyde dimethoxy acetal and p-toluenesulphonic acid in DMF at 50 °C. The resulting benzylidene was subsequently per-acetylated with Ac2O in pyridine to afford 5 as an anomeric mixture. The C4/C6 diol 6 was obtained by dissolving 5 in an AcOH/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture and heating at 60 °C. Subsequent installation of the p-bromobenzoate chromophores over C4 and C6, required for CD analysis, was accomplished by reacting 6 with p-bromobenzoyl chloride and DMAP in pyridine at 60 °C. The resulting glycosyl donor 7 was dissolved in DCM and activated with BF3·Et2O in the presence of different alkyl thiols to afford the respective S-glycosides 8b–8f in good yields (40–55%).9 Methyl S-glycoside 8a was prepared in 42% yield via methylation of the respective glucosyl thiouronium salt.9 The oxidation of alkyl S-glycosides 8a–8f with H2O2 in the presence of Ac2O and silica gel in DCM afforded the target alkyl glycosyl sulfones 9a–9f in excellent yields (92–98%).
:1) mixture and heating at 60 °C. Subsequent installation of the p-bromobenzoate chromophores over C4 and C6, required for CD analysis, was accomplished by reacting 6 with p-bromobenzoyl chloride and DMAP in pyridine at 60 °C. The resulting glycosyl donor 7 was dissolved in DCM and activated with BF3·Et2O in the presence of different alkyl thiols to afford the respective S-glycosides 8b–8f in good yields (40–55%).9 Methyl S-glycoside 8a was prepared in 42% yield via methylation of the respective glucosyl thiouronium salt.9 The oxidation of alkyl S-glycosides 8a–8f with H2O2 in the presence of Ac2O and silica gel in DCM afforded the target alkyl glycosyl sulfones 9a–9f in excellent yields (92–98%).
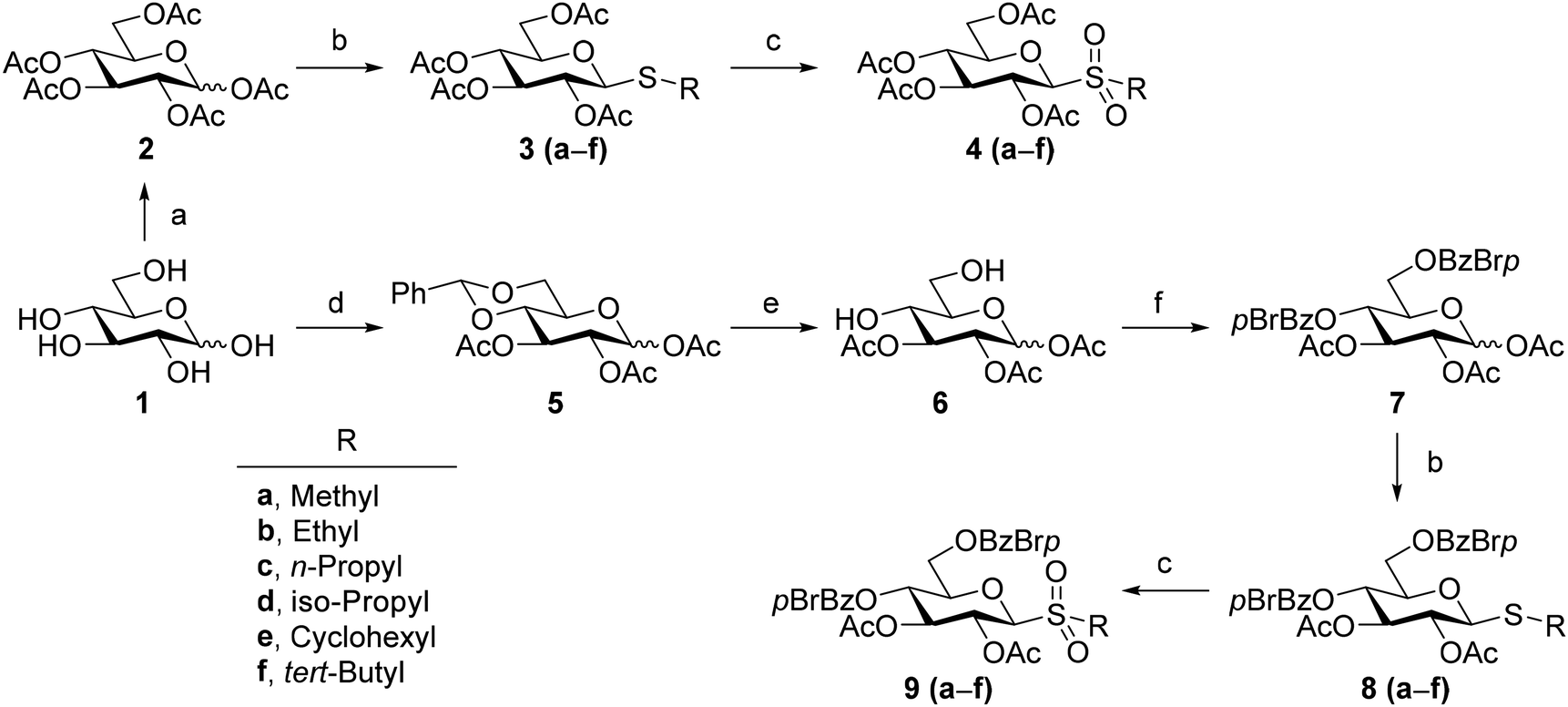 | ||
| Scheme 1 Synthesis of alkyl glucosyl sulfones. Conditions: (a) Ac2O, pyridine, rt; (b) for 4b–4f and 8b–8f: R-SH, BF3·OEt2, DCM, rt 4–12 h; for 4a and 8a: (i) (NH2)2CS, BF3·OEt2, MeCN, reflux; (ii) Et3N, CH3I, reflux; (c) H2O2, Ac2O, silica gel 60, DCM, rt; (d) (i) PhCH(OMe)2, p-TsOH, DMF, 50 °C; (ii) Ac2O, pyridine, rt; (e) AcOH/H2O (8:2), 60 °C; (f) p-BrBzCl, pyridine, DMAP, 60 °C. | ||
The characterization of the glycosyl sulfones was performed by means of 1D (1H, 13C{1H}, 1D-NOESY) and 2D (COSY, HSQC) NMR spectroscopy. The β anomeric configuration was determined from the 3JH1,H2 coupling constants which oscillated between 9.4 to 9.9 Hz among sulfones from both series. Moreover, the assigned anomeric stereochemistry was confirmed from the observed cross-peaks between the anomeric proton H1 with H3 and H5 in the selective 1D-NOESY experiments.
Results and discussion
The correct identification of the pro-chiral H6S and H6R protons is critical for the correct assessment of the conformational equilibrium about ω. We identified H6S and H6R following the protocol established by Ohrui et al.16 Briefly, in ester-protected D-glucopyranosides, H6R appears downfield with respect to H6S (δH6R > δH6S) and the 3JH5,H6R coupling constant values are higher than 3JH5,H6S regardless of the protective group patterns and media. All synthesized glycosyl sulfones showed well-isolated H6R and H6S peaks in their 1H NMR spectra, and the reported δH6R > δH6S relationship is fulfilled by the per-acetates 4a (R = Me), 4b (R = Et) and 4c (R = n-Pr). As the size of the alkyl aglycones increases, we observe progressive H6R deshielding and H6S shielding, resulting in sulfones 4d (R = i-Pr), 4e (R = Cy), and 4f (R = t-Bu) showing H6S downfield with respect to H6R (δH6S > δH6R). In the dibenzoate series, H6S appears downfield with respect to H6R in all derivatives. Because of this discrepancy, which probably originated from a combination of the anisotropic effect of the sulfinyl group and the conformational preferences about ϕ and ω, we assigned the proton identities based on the 3JH5,H6R and 3JH5,H6S values. Since in simple glucosides, the contributions of the tg rotamer to the C5–C6 conformational equilibrium are negligible, we expect low 3JH5,H6S and high 3JH5,H6R values, reflecting the predominance of the gg and gt rotamers typically observed on glucosides.Table 1 collects selected NMR data (CDCl3) for acetates 4a–4f and dibenzoates 9a–9f. In the acetates, we can observe an interesting trend in the chemical shifts of H6R and H6S with respect to the aglycone's structure. As the alkyl substituent's bulkiness increases, δH6R moves upfield whereas δH6S moves downfield. Thus, H6R progressively shields from 4.32 (4a, R = Me) to 4.13 ppm (4f, R = t-Bu) and H6S deshields from 4.20 (4a, R = Me) to 4.24 ppm (4f, R = t-Bu), with H6R being the proton exhibiting the largest Δδ of 0.19 ppm (vs. Δδ = 0.04 ppm for H6S). In contrast, both δH6R and δH6S in dibenzoates 9a–9f show a small upfield displacement (Δδ = 0.05 ppm) as the aglycone becomes bulkier. The chemical shifts of the anomeric proton (H1) and carbon (C1) also appear to depend on the aglycone's structure in both sulfone series, where δH1 moves downfield and δC1 upfield as the aglycone increases its volume. In the acetate series, the anomeric proton H1 deshields from 4.35 (4a, R = Me) to 4.84 ppm (4f, R = t-Bu) (Δδ = 0.49 ppm) and from 4.47 (9a, R = Me) to 4.95 ppm (9f, R = t-Bu) in the dibenzoates (Δδ = 0.48 ppm). In turn, the anomeric carbon shields from 88.4 (4a, R = Me) to 86.3 ppm (4f, R = t-Bu) in the acetates (Δδ = 2.1 ppm) and from 88.2 (9a, R = Me) to 86.4 ppm (9f, R = t-Bu) in the dibenzoates (Δδ = 1.8 ppm).
|
|
|||||
|---|---|---|---|---|---|
| Cmpd | R | δC1 | δH1 | δH6R | δH6S |
| Acetate series (R′ = Ac) | |||||
| 4a | Me | 88.4 | 4.35 | 4.32 | 4.20 |
| 4b | Et | 87.5 | 4.46 | 4.26 | 4.21 |
| 4c | n-Pr | 87.8 | 4.42 | 4.26 | 4.20 |
| 4d | i-Pr | 85.9 | 4.63 | 4.20 | 4.23 |
| 4e | Cy | 85.7 | 4.60 | 4.16 | 4.22 |
| 4f | t-Bu | 86.3 | 4.84 | 4.13 | 4.24 |
| Dibenzoate series (R′ = p-BrBz) | |||||
| 9a | Me | 88.2 | 4.47 | 4.44 | 4.62 |
| 9b | Et | 87.4 | 4.58 | 4.44 | 4.60 |
| 9c | n-Pr | 87.8 | 4.54 | 4.44 | 4.60 |
| 9d | i-Pr | 85.8 | 4.74 | 4.43 | 4.56 |
| 9e | Cy | 85.6 | 4.70 | 4.42 | 4.55 |
| 9f | t-Bu | 86.4 | 4.95 | 4.39 | 4.57 |

Conformational analysis of the hydroxymethyl group
We analyzed the acetates 4a–4f and dibenzoates 9a–9f in media with different polarities. The observed 3JH5,H6R and 3JH5,H6S values in each solvent are collected in Table 2 for acetates 4a–4f and in Table 3 for dibenzoates 9a–9f. In the acetate series, analyzed in CDCl3, we observe 3JH5,H6R values to vary between 4.7 and 6.5 Hz whereas 3JH5,H6S shows smaller fluctuations between 2.1 and 2.4 Hz. Moreover, we observe a 3JH5,H6R dependency on the aglycone's structure, namely 3JH5,H6R increases as the aglycone becomes bulkier. Thus, glycosides 4a (R = Me), 4b (R = Et), 4c (R = n-Pr) and 4d (R = i-Pr) show 3JH5,H6R values of ∼4.8 Hz which increase to 5.6 and 6.5 Hz in cyclohexyl (4e) and tert-butyl (4f) derivatives respectively. We observe the same trend for the dibenzoates in CDCl3. In this series, methyl derivative 9a shows a 3JH5,H6R value of 4.9 Hz which progressively increases to 6.7 Hz in tert-butyl sulfone 9f. The 3JH5,H6S values range from 2.1 to 2.4 Hz for this series in CDCl3. The analysis of the 3JH5,H6R and 3JH5,H6S data in the remaining solvents shows the same trends for both sulfone series. Regardless of the solvent's polarity, 3JH5,H6R exhibits larger values that increase as the aglycone's size increases whereas 3JH5,H6S shows smaller values with small fluctuations throughout each series.| Cmpd | R | C6D6 | CDCl3 | CD3OD | CD3CN | ||||
|---|---|---|---|---|---|---|---|---|---|
| 3 J H5,H6R | 3 J H5,H6S | 3 J H5,H6R | 3 J H5,H6S | 3 J H5,H6R | 3 J H5,H6S | 3 J H5,H6R | 3 J H5,H6S | ||
| 4a | Me | 4.7 | 2.0 | 4.8 | 2.2 | 4.9 | 2.3 | 5.1 | 2.4 |
| 4b | Et | 4.8 | 2.2 | 4.7 | 2.1 | 5.0 | 2.2 | 5.4 | 2.4 |
| 4c | n-Pr | 4.8 | 1.9 | 4.8 | 2.3 | 5.0 | 2.3 | 5.4 | 2.4 |
| 4d | i-Pr | 4.4 | 3.0 | 4.8 | 2.4 | 5.4 | 2.4 | 6.0 | 2.4 |
| 4e | Cy | 5.4 | 2.2 | 5.6 | 2.3 | 5.4 | 2.4 | 5.3 | 3.0 |
| 4f | t-Bu | 6.4 | 2.2 | 6.5 | 2.4 | 6.2 | 2.4 | 6.1 | 2.9 |
| Cmpd | R | CDCl3 | CD3CN | ||
|---|---|---|---|---|---|
| 3 J H5,H6R | 3 J H5,H6S | 3 J H5,H6R | 3 J H5,H6S | ||
| 9a | Me | 4.9 | 3.0 | 4.7 | 3.0 |
| 9b | Et | 5.3 | 3.0 | 5.0 | 3.0 |
| 9c | n-Pr | 5.4 | 3.0 | 5.1 | 3.0 |
| 9d | i-Pr | 5.8 | 2.7 | 5.5 | 2.9 |
| 9e | Cy | 5.9 | 2.4 | 5.7 | 2.8 |
| 9f | t-Bu | 6.7 | 2.3 | 6.3 | 2.1 |
Due to the small chemical shift differences between H6R and H6S in the 1H NMR spectra, ABX systems were obtained. To test how this could affect the values of 3JH5,H6R and 3JH5,H6S, we proceed to simulate the NMR regions containing H6R, H6S, H5 and H4 as an ABXY pattern for acetylated sulfones 4a–4f (CDCl3 and CD3CN) and dibenzoates 9a–9f (CDCl3) using the built-in algorithm (DAISY) in Bruker TopSpin 4.0.7. The overlaid spectral comparison between those obtained directly and those after simulation is in excellent agreement, either in chemical shifts or in coupling constants (ESI Tables S2–S4 and Fig. S26–S45†). Both analyses show the same general behaviour, an increase and a decrease in the 3JH5,H6R and 3JH5,H6S values, respectively, as the bulkiness of the alkyl group increases. The conformational information contained in the 3JH5,H6R and 3JH5,H6S constants was converted into percent contributions of gg, gt, and tg (Pgg, Pgt, and Ptg) using eqn (1)–(3), developed by Crich et al.17 These equations include optimized limiting coupling constants for the gg, gt, and tg rotamers allowing for accurate Pgg, Pgt, and Ptg calculations.
| 3JH5,H6R = 1.0 Pgg + 11.0 Pgt + 4.8 Ptg | (1) |
| 3JH5,H6S = 2.2 Pgg + 2.5 Pgt + 10.2 Ptg | (2) |
| 1 = Pgg + Pgt + Ptg | (3) |
Table 4 shows the calculated gg, gt, and tg rotamer populations for the acetyl derivatives 4a–4f and Table 5 presents the respective conformational data for dibenzoates 9a–9f obtained by using the experimental data contained in Tables 2–3.
| Cmpd | R | C6D6 | CDCl3 | CD3OD | CD3CN | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P gg | P gt | P tg | P gg | P gt | P tg | P gg | P gt | P tg | P gg | P gt | P tg | ||
| 4a | Me | 63 | 37 | 0 | 62 | 38 | 0 | 61 | 39 | 0 | 58 | 41 | 1 |
| 4b | Et | 62 | 38 | 0 | 63 | 37 | 0 | 60 | 40 | 0 | 56 | 44 | 0 |
| 4c | n-Pr | 62 | 38 | 0 | 62 | 38 | 0 | 60 | 40 | 0 | 56 | 44 | 0 |
| 4d | i-Pr | 61 | 31 | 8 | 61 | 38 | 1 | 56 | 44 | 0 | 50 | 50 | 0 |
| 4e | Cy | 56 | 44 | 0 | 54 | 46 | 0 | 56 | 44 | 0 | 52 | 40 | 8 |
| 4f | t-Bu | 46 | 54 | 0 | 45 | 55 | 0 | 48 | 52 | 0 | 45 | 48 | 7 |
| Cmpd | R | CDCl3 | CD3CN | ||||
|---|---|---|---|---|---|---|---|
| P gg | P gt | P tg | P gg | P gt | P tg | ||
| 9a | Me | 56 | 36 | 8 | 58 | 34 | 8 |
| 9b | Et | 52 | 40 | 8 | 55 | 37 | 8 |
| 9c | n-Pr | 51 | 41 | 8 | 54 | 38 | 8 |
| 9d | i-Pr | 49 | 46 | 5 | 51 | 42 | 7 |
| 9e | Cy | 51 | 49 | 0 | 49 | 45 | 6 |
| 9f | t-Bu | 44 | 46 | 0 | 48 | 52 | 0 |
In the per-acetylated glucosyl sulfones 4a–4f, the conformational equilibrium about ω distributes between the gg and gt rotamers with small to no contributions of tg in all solvents. Independent of the media polarity, the gg conformer predominates the equilibrium in most sulfones. However, the gg predominance clearly depends on the aglycone's nature since Pgg progressively declines as the size of the alkyl substituent increases, and this Pgg detriment correlates with a progressive increment in the gt contributions. In C6D6, the lowest polarity solvent used in this study, gg shows a 63% contribution in the methyl-substituted compound 4a, which falls to 46% in the tert-butyl sulfone 4f, whereas the gt contribution correspondingly increases from 37% (4a, R = Me) to 54% (4f, R = t-Bu). Similarly, in CDCl3, Pgg diminishes from 62% (4a, R = Me) to 45% (4f, R = t-Bu) and Pgt increases from 38% (4a) to 55% (4f). In CD3OD, the population fluctuations are Pgg: 61% (4a)/48% (4f) and Pgt: 39% (4a)/52% (4f) and in CD3CN, they are Pgg: 58% (4a)/45% (4f) and Pgt 41% (4a)/48% (4f).
Regarding the tg rotamer, three per-acetylated sulfones exhibit tg contributions with the iso-propyl derivative 4d being the only compound showing tg contributions in two different media with Ptg values of 8% and 1% in C6D6 and CDCl3, respectively. In CD3CN, methyl, cyclohexyl and tert-butyl sulfones 4a, 4e and 4f show small 1%, 8%, and 7% tg contributions, respectively.
The C5–C6 conformational dependency on the aglycone's substituent is also present in the dibenzoates. For compounds 9a–9f we observe an equilibrium predominated by gg and gt regardless of the media polarity. In CDCl3, methyl sulfone 9a shows a gg contribution of 56% which progressively diminishes to 44% in the tert-butyl derivative 9f, while the gt population increases from 36% (9a, R = Me) to 46% (9f, R = t-Bu). Likewise, in CD3CN, the gg population gradually decreases from 58% (9a, R = Me) to 48% (9f, R = t-Bu) and correspondingly, the gt population increases from 34% (9a) to 52% (9f). In both CDCl3 and CD3CN, the tg contributions remain about ∼8% for most sulfones. Dibenzoates show larger tg contributions compared with their parent acetates, probably by a favorable π–π interaction between the two aromatic rings, and independently of the media, the tg rotamer decreases its population from the methyl (9a, 8%) to the tert-butyl (9f, 0%) derivative, favoring the gt population.
Given the evident relationship between the hydroxymethyl's rotational populations and the aglycone's volume, we represented the calculated Pgg and Pgt data set versus the Charton values (ν) of the respective alkyl substituents Fig. 2.24 The Charton value is a steric descriptor derived from Taft's steric parameter24,25a–d which was developed to include electronic factors and is a practical choice for a broader understanding of steric trends across physicochemical phenomena.26 From the Pgg and Pgtversus ν plots shown in Fig. 2, the linear relationship between the rotational populations and the steric descriptor becomes clear. In both per-acetyl and dibenzoate series, we observe a positive linear correlation of gt with ν and a negative one for gg in all analysed solvents. As the Charton value increases, i.e., the aglycone's alkyl substituent becomes bulkier, the gt population linearly increases at the expense of gg until reaching predominance in sulfones 4f and 9f, both substituted with the bulkiest alkyl group examined in this study.
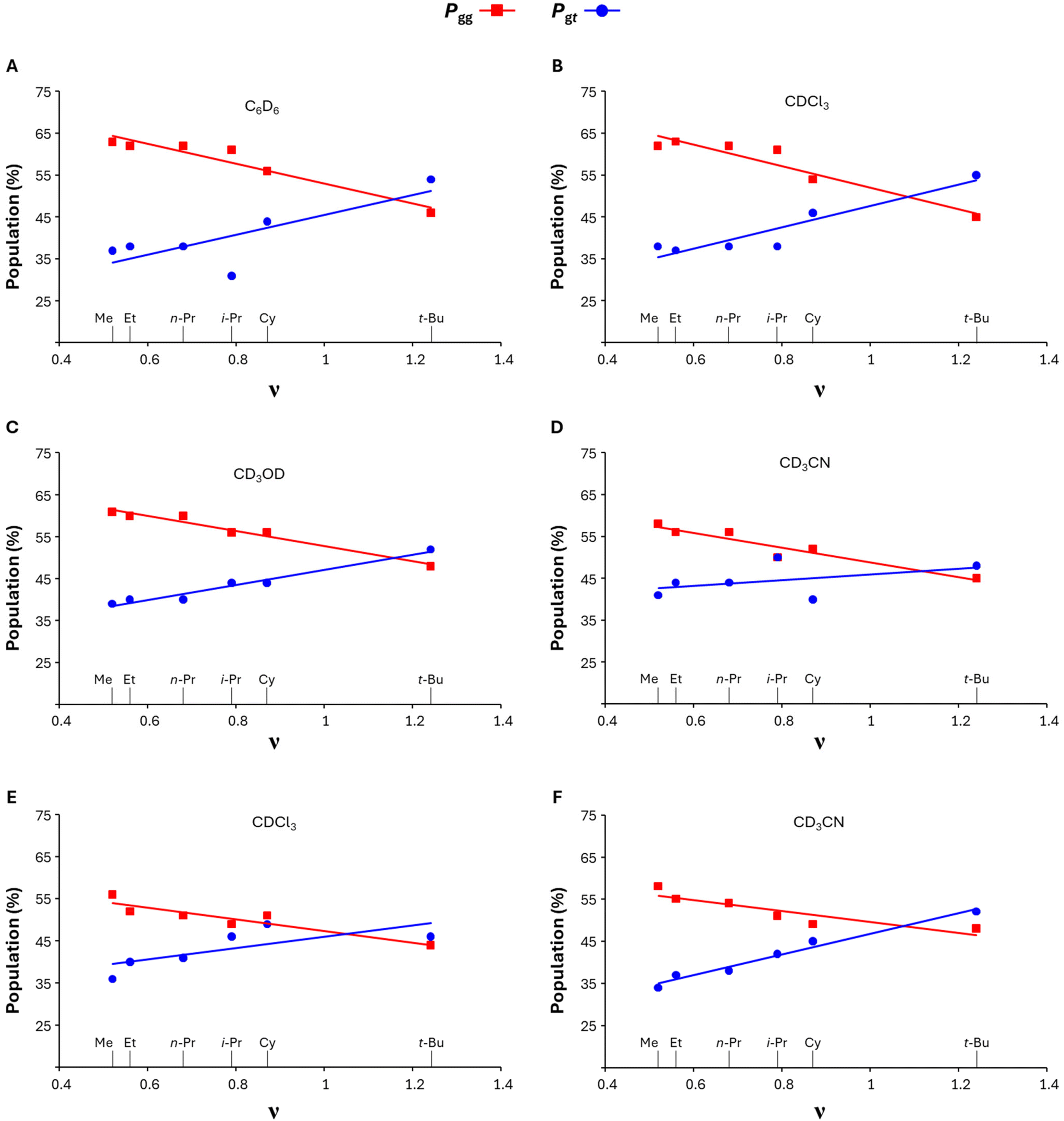 | ||
| Fig. 2 P gg (red color) and Pgt (blue color) versus alkyl's Charton values (ν) for per-acetylated glucosyl sulfones 4a–4f in: (A) C6D6;18 (B) CDCl3;19 (C) CD3OD;20 (D) CD3CN;21 and for dibenzoate glucosyl sulfones 9a–9f in: (E) CDCl3;22 (F) CD3CN.23 | ||
This relationship between aglycone's bulkiness and rotational populations was also observed in the respective P% vs. ν plots performed based on the calculated 3JH5,H6R and 3JH5,H6S values (algorithm DAISY, in Bruker TopSpin 4.0.7.), confirming the above-mentioned conclusion (see ESI Table S5 and Fig. S46–S48†).
Study of hydroxymethyl conformation by circular dichroism
Circular dichroism (CD) is a powerful technique that has been mainly used for the determination of absolute configurations of a great number of compounds of both natural and synthetic origin. Further developments came with the CD exciton chirality method,27 which allows the determination of absolute configurations in a non-empirical way. Applications of circular dichroism have also been widely applied to the conformational studies in a solution of small-to-medium sized molecules, as well as macromolecules, especially proteins.28The CD exciton chirality method is based on the through-space interaction of the electric transition moments of two chromophores that gives rise to an excited state split into two energy levels. Excitations to these levels lead to a CD spectrum with two Cotton effects of opposite signs, namely to a “split” CD curve. The chiral environment of the two chromophores determines the sign of the Cotton effects, with the sign of the exciton chirality being that of the first Cotton effect, the one at a longer wavelength (Fig. 3).
 | ||
| Fig. 3 Nondegenerate system in which chromophoric groups i and j undergo O → a transitions to give split levels separated by interaction energy 2Vij. Δε is the molar circular dichroism, Ri,j is the interchromophoric distance vector from i to j, and μioa and μjoa are electric transition dipole moments of excitation O → a. 1La is the transition dipole moment in p-substituted benzoate chromophores. | ||
In the belief that CD is an extremely sensitive spectroscopic technique to study the rotational population of the hydroxymethyl group in alkyl glucopyranoside sulfones, the CD spectra of compounds 9a–9f were recorded in CH3CN.
The CD curves of compounds 9a–9f (Fig. 4) showed a first Cotton effect around 251 nm and a second Cotton effect around 233 nm, centered around the UV λmax 245 nm (Table 6), in agreement with the exciton chirality method. In addition, a general decrease in the intensities of the first and second Cotton effects, or their A values, from the methyl derivative 9a (A value 21.5), to the ethyl derivative 9b (19.4), to the n-propyl 9c (18.7), to the isopropyl 9d (16.3), to the cyclohexyl 9e (14.2), and to the tert-butyl derivative 9f (11.2), can be observed.
 | ||
| Fig. 4 CD curves (CH3CN) for alkyl glucosyl sulfones 9a–9f at 25 °C. | ||
| Cmpd | R | First Cotton effect | Second Cotton effect | A value | ||
|---|---|---|---|---|---|---|
| λ (nm) | Δε | λ (nm) | Δε | |||
| 9a | Me | 251 | 14.9 | 233 | −6.6 | 21.5 |
| 9b | Et | 251 | 13.5 | 233 | −5.9 | 19.4 |
| 9c | n-Pr | 251 | 12.9 | 235 | −5.8 | 18.7 |
| 9d | i-Pr | 252 | 10.9 | 232 | −5.4 | 16.3 |
| 9e | Cy | 252 | 9.8 | 229 | −4.4 | 14.2 |
| 9f | t-Bu | 250 | 7.1 | 232 | −4.1 | 11.2 |
According to the sign of the pairwise interaction between the chromophores at the 4 and 6 positions in the gg, gt and tg rotamers (Fig. 5), the general decrease in the CD Cotton effects can only be explained by a decrease in the population of the gg rotamer (positive contribution) and an increase in the population of the gt rotamer (negative contribution).
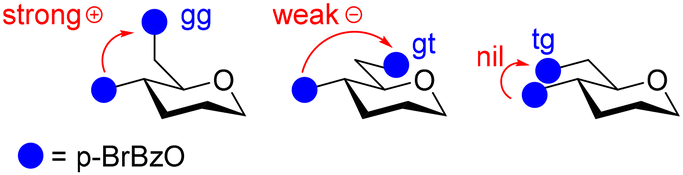 | ||
| Fig. 5 Sign of the pairwise interaction between the chromophores at the 4 and 6 positions in the gg, gt and tg rotamers. | ||
CD and 1H NMR data comparison indicated the existence of an excellent correlation between the magnitudes of the rotamer populations obtained by 1H NMR and the CD A values.
Conformational analysis of the glycosidic bond
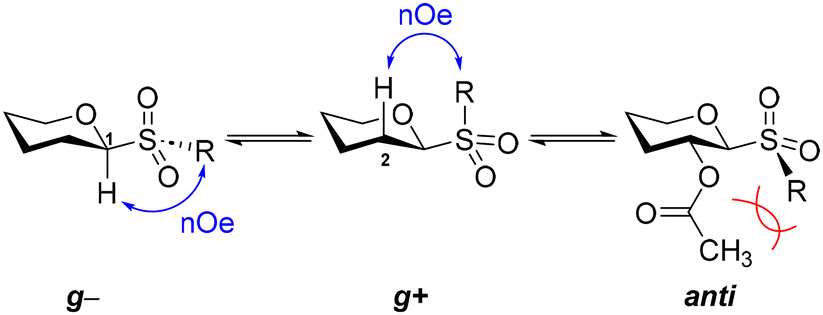
To gain a deeper insight into the flexibility of alkyl glucosyl sulfones, we examined the conformational properties about the ϕ (H1–C1–S–C) torsion angle by means of selective 1D-NOESY spectroscopy. The free rotation about C1–S, either for glucosyl sulfoxides or sulfones, generates three staggered rotamers called g−, g+ and anti (Fig. 6), based on the spatial relationship between the aglycone's carbon directly attached to the sulfur atom and the endocyclic oxygen O5. | ||
| Fig. 6 Staggered g−, g+, and anti conformers about the ϕ (C–S–C1–O5) torsion angle for glucosyl sulfoxides and sulfones and the nomenclature used herein. | ||
As can be observed in Fig. 7, for the g− conformer, we anticipate nOe couplings between H1 and the aglycone's protons closer to the sulfur, whereas for the g+ conformer, we expect similar nOe couplings between the aglycone's protons and H2. In C2-substituted β-glucosides, the anti rotamer is heavily destabilized by steric repulsions and we do not expect significant contributions from it. Therefore, we carried out selective 1D-NOESY experiments in C6D6 (apolar) and CD3CN (polar) for the per-acetylated sulfones 4a (R = Me), 4b (R = Et), 4d (R = i-Pr), and 4f (R = t-Bu), thus representing glucosides carrying methyl, primary, secondary, and tertiary alkyl aglycones.
 | ||
| Fig. 7 Expected nOe couplings between H1 and the aglycone's protons closer to the sulfur. | ||
Fig. 8 shows stacked sections of the 1D-NOESY spectra for selected sulfones. We selectively excited the alkyl's hydrogen atoms closer to H1 and H2 corresponding to the methyl group in 4a, the ethyl's methylene in 4b, the iso-propyl's methine in 4d, and the tert-butyl's methyl groups in 4f, whose signals were well isolated in the respective 1H NMR spectrum on both tested solvents, allowing for selective irradiation.
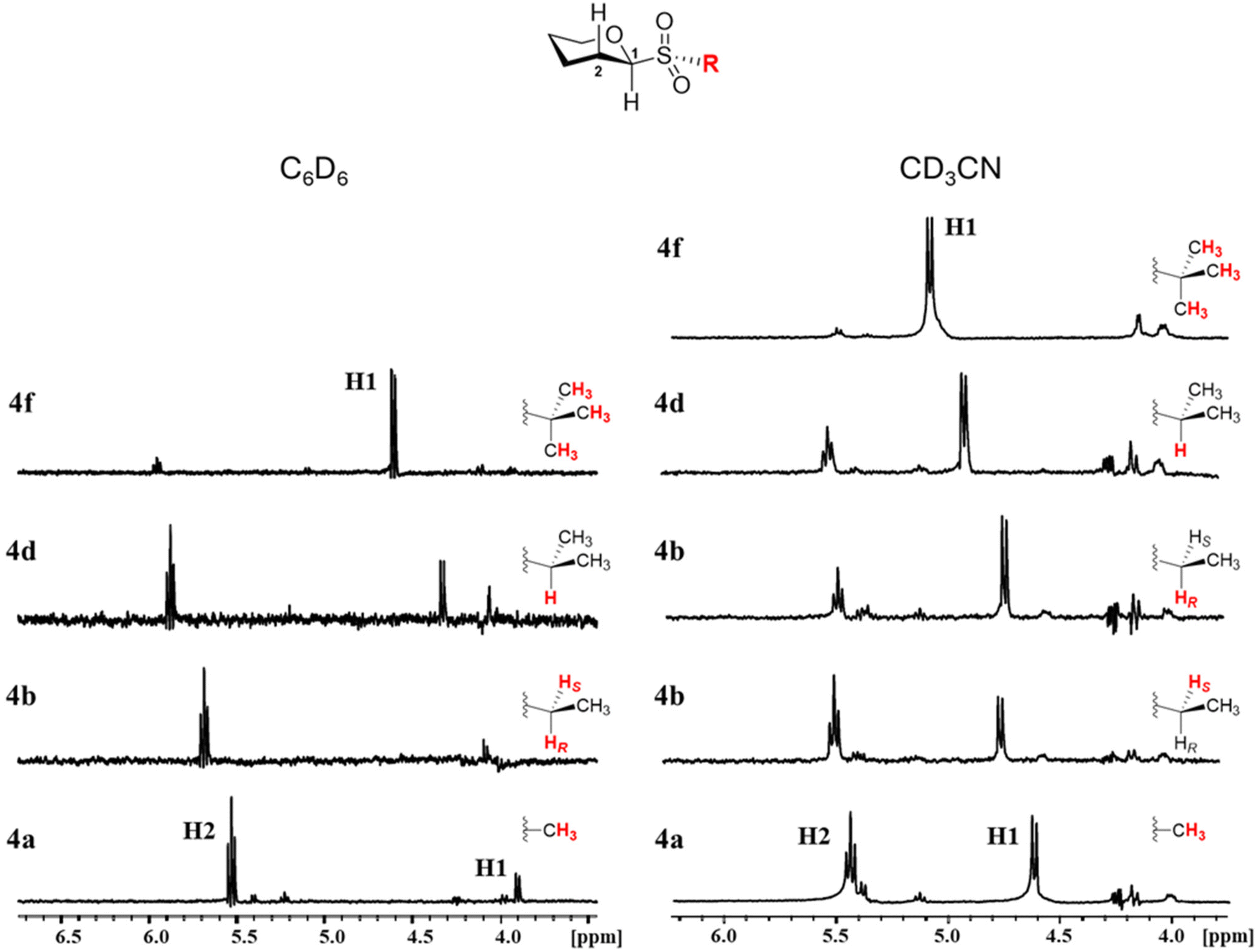 | ||
| Fig. 8 Stacked 1D-NOESY spectra for glucosyl sulfones 4a (R = Me), 4b (R = Et), 4d (R = i-Pr), and 4f (R = t-Bu) in C6D6 (A) and CD3CN (B). Irradiated hydrogens are highlighted in red. | ||
In the apolar media (C6D6), we observe that methyl and ethyl derivatives 4a and 4b show strong nOe couplings with H2 and a weak interaction with H1, suggesting that in these small-aglycone-carrying sulfones, the g+ rotamer predominates the conformation about ϕ. As the aglycone increases its degree of substitution, the intensity of the nOe peak with H2 decreases and that with H1 increases. Thus, iso-propyl sulfone 4d shows comparable nOe intensities between H1 and H2 nOe peaks, suggesting an equilibrium between the g− and g+ conformers. In tert-butyl sulfone 4f, the larger nOe peak with H1 suggests a ϕ torsion largely predominated by the g− conformer. No nOe spatial couplings with the acetyl group at C2 were observed upon irradiation of the above-mentioned protons.
In CD3CN, the conformation about ϕ shows a different pattern, especially for those glucosyl sulfones carrying smaller alkyl substituents. Thus, we observe that derivatives 4a and 4b exhibit comparable intensities on the H1 and H2 nOe peaks, which points to a glycosidic ϕ conformation distributed between the g− and g+ rotamers. The iso-propyl glucoside 4d shows nOe peaks with H1 and H2, with H1 being the most intense, suggesting a preference for the g− conformer. In tert-butyl sulfone 4f, the largely predominant H1 nOe peak evidences a glycosidic conformation anchored in g−. Thus, the effect of the media polarity over the glycosidic bond conformation seems to be more relevant in glycosides carrying small alkyl substituents, where we observe higher flexibilities. As the aglycone substituent's size increases, the glycosidic bond becomes stiffer and anchored in g− in both polar and apolar media, likely due to the steric repulsions with H2 which destabilize g+.
Conclusions
Herein we have shown by means of NMR and CD spectroscopy that the rotamer populations of the hydroxymethyl group in alkyl glucosyl sulfones depend on the structural nature of the aglycone. Thus, in both synthetic models, an increment in the alkyl substituent's bulkiness leads to progressive increments in gt contributions at the expense of gg. In addition, as the Charton value (ν) increases, the gt population linearly increases at the expense of the gg population.These results are like those obtained with alkyl O-,8S-,9 and C-10 glycosides as well as RS glucosyl sulfoxides, but opposite to those from the SS glucosyl sulfoxides.11 All these studies pointed to different values of the stereoelectronic exo-anomeric effect to be responsible for this behavior. In addition, the independent conformational properties of carbasugars12 of the aglycone reveal that the endocyclic oxygen is involved and it is necessary for this relationship.
Fig. 9 shows the general behavior of Pgtversus Taft's steric parameter (ES) of the alkyl group attached to O-, S-, RS and SS sulfinyl as well as sulfonyl groups in CDCl3. It can be observed that in all cases but the SS glucosyl sulfoxides, an increase in the absolute value of ES leads to larger gt populations. Therefore, it seems that the conformational properties of the hydroxymethyl group of ester-protected alkyl glucosyl sulfones are also directly related to the stereoelectronic exo-anomeric effect.
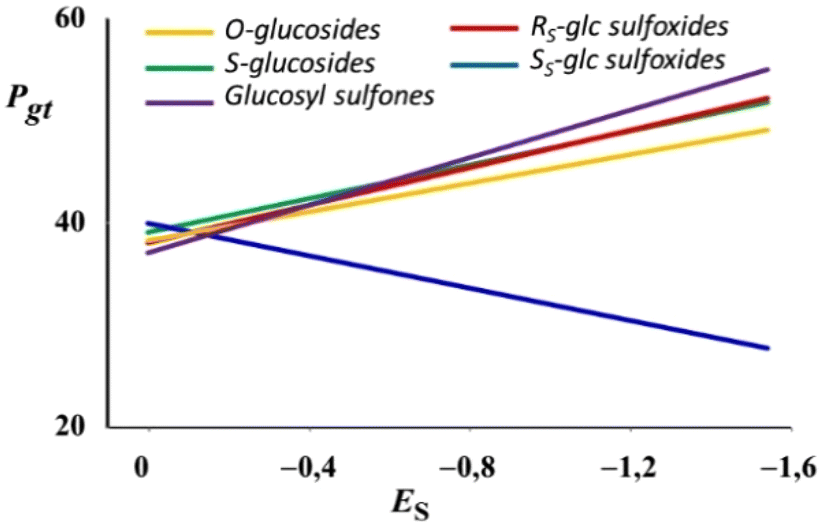 | ||
| Fig. 9 P gt versus alkyl's Taft's values (Es) for alkyl O-glucosides, thioglucosides, glucosyl sulfoxides and sulfones. | ||
Opposite to the sulfinyl group, the sulfonyl group possesses higher π character in the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.29 This property could give rise to a πSO → σ*C1–O5 interaction that, in a similar way to other glycosides, led to the existence of the exo-anomeric effect in alkyl glucosyl sulfones. Either the g− or the g+ conformation fulfills the spatial requirements for the πSO → σ*C1–O5 interaction to take place.
O bond.29 This property could give rise to a πSO → σ*C1–O5 interaction that, in a similar way to other glycosides, led to the existence of the exo-anomeric effect in alkyl glucosyl sulfones. Either the g− or the g+ conformation fulfills the spatial requirements for the πSO → σ*C1–O5 interaction to take place.
The nOe study on the conformational patterns about the glycosidic bond ϕ (H1–C1–S–C) revealed its dependency on the aglycone's nature. As the alkyl substituent increases its degree of substitution, the intensity of the nOe peak with H2 decreases and that with H1 increases, meaning that the g− conformer increases its predominance at the expense of g+.
The solvent polarity exerts an important influence over the conformational pattern of the glycosidic bond and a less pronounced one over the hydroxymethyl group's flexibility. In apolar media (C6D6), glycosides carrying small aglycones exhibit a glycosidic conformation markedly predominated by g+, whereas in a polar solvent (CD3CN), the glycosidic bond becomes more flexible with comparable g+ and g− distributions. In both polar and apolar media, we observe the same hydroxymethyl conformational trends with comparable Pgg, Pgt, and Ptg values in both glycoside series studied.
Experimental
General information
NMR spectra were recorded in Bruker AVANCE 400, 500 and 600 spectrometers. Mono-dimensional 1H NMR spectra were collected at 400, 500 and 600 MHz and 1D 13C NMR spectra were collected at 100 and 150 MHz, VTU 300.0 °K. Chemical shifts are reported in parts per million (ppm). The residual solvent peak was used as an internal reference. FIDs were processed using Bruker TopSpin software by applying a Fourier transform with phase correction combined with exponential multiplication (efp command). The 3JH,H coupling constants were measured directly from the peak's chemical shift differences of the resulting spectra or using the distance measurement tool in TopSpin. The H5,H6 coupling constants (ABX system) were obtained (i) by direct measurement of the lines of H6R, H6S, or H5 in spectrometers of different magnetic field strengths (from 400 to 600 MHz) and (ii) by simulating each spectrum by using Bruker's simulation and iteration tool (DAISY) available in TopSpin 4.0.7 (see ESI Tables S2–S4 and Fig. S26–S45†).30HRMS spectra were analyzed by TOF MS ES+. For analytical and preparative thin-layer chromatography, silica gel ready-foils and glass-backed plates (1 mm) were used, respectively, being developed with 254 nm UV light and/or spraying with AcOH/H2O/H2SO4 (80:16:4) and heating at 150 °C. Column chromatography was performed using silica gel (0.015–0.04 mm) and n-hexane/EtOAc solvent systems. All reagents were obtained from commercial sources and used without further purification. Solvents were dried and distilled before use.
For general procedures for the synthesis and characterization of the alkyl β-D-thioglucopyranosides 3a, 3b, and 3(d–f) and 8(a–f), see ref. 9, 13 and 14. For compound 3c, see the following section.
n-Propyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (3c)
To a stirred solution of 1,2,3,4,6-penta-O-acetyl-β-D-glucopyranose (855 mg, 2.2 mmol) and propanothiol (2 eq., 0.40 mL) in dry dichloromethane (5 mL mmol−1) at room temperature and under an N2 atmosphere, boron trifluoride etherate (0.1 eq.) was added dropwise. The reaction progress was followed by TLC. When the starting compound was consumed, it was quenched by the addition of Et3N and diluted with CH2Cl2. The organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The product was purified by chromatography using silica gel and n-hexane/ethyl acetate as the eluent to afford the n-propyl thioglucoside 3c (818 mg, 1.5 mmol, 92%). Rf: 0.43 (n-hex/EtOAc 3:2); [α]D: −27.4 (c 1.2, CHCl3); HRMS (FAB): calcd for C17H26O9NaS [M + Na]+: 429.1195, found: 429.1190; 1H NMR (δ, 400 MHz, CDCl3): 5.21 (dd, J = 9.4 and 9.4 Hz, H-3), 5.07 (dd, J = 9.4 and 9.4 Hz, H-4), 5.02 (dd, J = 9.7 and 9.7 Hz, H-2), 4.47 (d, J = 10.0 Hz, H-1), 4.23 (dd, J = 5.0 and 12.3 Hz, H-6R), 4.12 (dd, J = 2.2 and 12.3 Hz, H-6S), 3.69 (ddd, J = 2.2, 5.0 and 9.8 Hz, H-5), 2.71–2.57 (m, 2H), 2.07 (s, 3H), 2.05 (s, 3H), 2.01 (s, 3H), 1.99 (s, 3H), 1.67–1.57 (m, 2H), 0.97 (dd, J = 7.3 and 7.3 Hz, 3H); 13C NMR (δ, CDCl3): 170.6 (s), 170.2 (s), 169.4 (s), 169.3 (s), 83.7 (d, C-1), 75.8 (d, C-5), 73.9 (d, C-3), 69.9 (d, C-2), 68.4 (d, C-4), 62.2 (t, C-6), 32.1 (t), 23.1 (q), 20.7 (q), 20.6 (q), 20.5 (q), 13.4 (q); E.A.: calcd for C17H26O9S: C, 50.24; H, 6.45; S, 7.89, found: C, 50.34; H, 6.60; S, 7.78.
General procedure for the oxidation of alkyl S-glycosides to alkyl glycosyl sulfones
Alkyl S-glucoside (1.0 equiv.), Ac2O (3.0 equiv.), and SiO2 (200 mg mmol−1 of S-glucoside) were suspended in DCM (5.0 ml mmol−1 of S-glucoside), and H2O2 (3.0 equiv., from a 35% in H2O solution) was added at room temperature and under vigorous stirring. The reaction was maintained under stirring until achieving complete oxidation of the starting material (TLC analysis). The system was then diluted with DCM, filtered through filter paper to remove SiO2, and the filtrate was transferred to a separation funnel and subsequently washed with 10% Na2S2O3, NaHCO3 (sat.), and brine. The organic layer was collected, dried over Na2SO4, filtered, and concentrated in a rotary evaporator. The resulting crude was then purified by normal phase flash chromatography using mixtures of ethyl acetate and n-hexane as the eluent system in proportions according to the sulfone's polarity.2,3,4,6-Tetra-O-acetyl-1-(methylsulfonyl)-β-D-glucopyranoside (4a)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, methyl S-glucoside 3a (83 mg, 0.22 mmol) afforded glycosyl sulfone 4a (90 mg, 0.219 mmol, 99%) as a white solid. Rf: 0.33 (n-hex/EtOAc 2:3); [α]D: −2.7 (c 0.9, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 5.42 (dd, J = 9.6 and 9.6 Hz, H-2), 5.30 (dd, J = 9.3 and 9.3 Hz, H-3), 5.12 (dd, J = 9.7 and 9.7 Hz, H-4), 4.35 (d, J = 9.9 Hz, H-1), 4.32 (dd, J = 4.8 and 12.7 Hz, H-6R), 4.20 (dd, J = 2.2 and 12.7 Hz, H-6S), 3.85 (ddd, J = 2.2, 4.8, and 10.0 Hz, H-5), 2.95 (s, 3H), 2.08 (s, 3H), 2.06 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.4 (s), 169.9 (s), 169.5 (s), 169.2 (s), 88.4 (d, C-1), 76.8 (d, C-5), 73.0 (d, C-3), 67.4 (d, C-4), 66.7 (d, C-2), 61.3 (d, C-6), 36.4 (q), 20.6 (q), 20.6 (q), 20.5 (q), 20.4 (q). HRMS (FAB): calcd for C15H22O11S Na [M + Na]+: 433.0781, found: 433.0770.
2,3,4,6-Tetra-O-acetyl-1-(ethylsulfonyl)-β-D-glucopyranoside (4b)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, ethyl S-glucoside 3b (115 mg, 0.29 mmol) afforded glycosyl sulfone 4b (119 mg, 0.28 mmol, 97%) as a white solid. Rf: 0.37 (n-hex/EtOAc 2:3); [α]D: −9.6 (c 0.3, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 5.49 (dd, J = 9.6 and 9.6 Hz, H-2), 5.31 (dd, J = 9.4 and 9.4 Hz, H-3), 5.12 (dd, J = 9.8 and 9.8 Hz, H-4), 4.46 (d, J = 9.9 Hz, H-1), 4.26 (dd, J = 4.7 and 12.7 Hz, H-6R), 4.21 (dd, J = 2.1 and 12.7 Hz, H-6S), 3.83 (ddd, 2.1, 4.7, and 9.2 Hz, H-5), 3.14 (m, 2H), 2.08 (s), 2.07 (s), 2.04 (s), 2.03 (s), 1.40 (dd, J = 7.5 and 7.5 Hz, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.4 (s), 170.0 (s), 169.3 (s), 169.2 (s), 87.5 (d, C-1), 76.8 (d, C-5), 73.1 (d, C-3), 67.4 (d, C-4), 66.5 (d, C-2), 61.4 (t, C-6), 43.9 (t), 20.6 (q), 20.6 (q), 20.5 (q), 20.4 (q), 5.4 (q); HRMS (FAB): calcd for C16H25O11S [M + H]+: 425.1118, found: 425.1133.
2,3,4,6-Tetra-O-acetyl-1-(n-propylsulfonyl)-β-D-glucopyranoside (4c)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, n-propyl S-glucoside 3c (91 mg, 0.22 mmol) afforded sulfone 4c (92 mg, 0.21 mmol, 95%) as a white solid. Rf: 0.47 (n-hex/EtOAc 2:3); [α]D: −17.2 (c 0.9, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 5.47 (dd, J = 9.6 and 9.6 Hz, H-2), 5.30 (dd, J = 9.3 and 9.3 Hz, H-3), 5.11 (dd, J = 9.7 and 9.7 Hz, H-4), 4.42 (d, J = 9.9 Hz, H-1), 4.26 (dd, J = 4.8 and 12.6 Hz, H-6R), 4.20 (dd, J = 2.3 and 12.6 Hz, H-6S), 3.82 (ddd, J = 2.3, 4.8, and 10.0 Hz, H-5), 3.08 (m, 2H), 2.08 (s, 3H), 2.06 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 1.90 (m, 2H), 1.09 (dd, J = 7.4 and 7.4 Hz, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.4 (s), 170.0 (s), 169.3 (s), 169.2 (s), 87.8 (d, C-1), 76.7 (d, C-5), 73.1 (d, C-3), 67.4 (d, C-4), 66.4 (d, C-2), 61.4 (t, C-6), 50.9 (t), 20.6 (q), 20.6 (q), 20.5(q), 20.5(q), 14.7 (t), 13.2 (q); HRMS (FAB): calcd for C17H26O11SNa [M + Na]+: 461.1094, found: 461.1075.
2,3,4,6-Tetra-O-acetyl-1-(iso-propylsulfonyl)-β-D-glucopyranoside (4d)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, iso-propyl S-glucoside 3d (95 mg, 0.23 mmol) afforded sulfone 4d (92 mg, 0.21 mmol, 91%) as a white solid. Rf: 0.38 (n-hex/EtOAc 2:3); [α]D: −12.4 (c 0.9, CHCl3); 1H NMR (δ, 600 MHz, CDCl3): 5.56 (dd, J = 9.6 and 9.6 Hz, H-2), 5.33 (dd, J = 9.6 and 9.6 Hz, H-3), 5.11 (dd, J = 9.6 and 9.6 Hz, H-4), 4.63 (d, J = 9.6 Hz, H-1), 4.23 (dd, J = 2.4 and 12.6 Hz, H-6S), 4.20 (dd, J = 4.8 and 12.6 Hz, H-6R), 3.80 (ddd, J = 2.4, 4.8, and 9.6 Hz, H-5), 3.48 (m, 1H), 2.08 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.39 (d, J = 7.2 Hz, 3H), 1.37 (d, J = 7.2 Hz, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.3 (s), 170.1 (s), 169.2 (s), 169.0 (s), 85.9 (d, C-1), 76.7 (d, C-5), 73.2 (d, C-3), 67.5 (d, C-4), 66.5 (d, C-2), 61.6 (t, C-6), 51.2 (d), 20.6 (q), 20.6 (q), 20.5(q), 20.4(q), 16.1 (q), 13.4 (q); HRMS (FAB): calcd for C17H26O11SNa [M + Na]+: 461.1094, found: 461.1092.
2,3,4,6-Tetra-O-acetyl-1-(cyclohexylsulfonyl)-β-D-glucopyranoside (4e)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, cyclohexyl S-glucoside 3e (101 mg, 0.25 mmol) afforded sulfone 4e (110 mg, 0.23 mmol, 94%) as a white solid. Rf: 0.52 (n-hex/EtOAc 2:3); [α]D: −14.6 (c 1.2, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 5.55 (dd, J = 9.7 and 9.7 Hz, H-2), 5.32 (dd, J = 9.4 and 9.4 Hz, H-3), 5.07 (dd, J = 9.5 and 9.5 Hz, H-4), 4.60 (d, J = 9.8 Hz, H-1), 4.22 (dd, J = 2.3 and 12.5 Hz, H-6S), 4.16 (dd, J = 5.6 and 12.5 Hz, H-6R), 3.78 (ddd, J = 2.3, 5.6, and 10.1 Hz, H-5), 3.24 (m, 1H), 2.16 (m, 2H), 2.07 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H), 2.01 (s, 3H), 1.60 (m, 4H), 1.26 (m, 4H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.3 (s), 170.1 (s), 169.2 (s), 169.0 (s), 85.7 (d, C-1), 76.7 (d, C-5), 73.2 (d, C-3), 67.5 (d, C-4), 66.3 (d, C-2), 61.8 (t, C-6), 58.9 (d), 25.9 (t), 25.1 (t), 25.0 (t), 25.0 (t), 22.8 (t), 20.6 (q), 20.5 (q), 20.5(q), 20.4(q); HRMS (ESI): calcd for C20H30O11NaS [M + Na]+: 501.1422, found: 501.1407.
2,3,4,6-Tetra-O-acetyl-1-(tert-butylsulfonyl)-β-D-glucopyranoside (4f)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, tert-butyl S-glucoside 3f (110 mg, 0.26 mmol) afforded sulfone 4f (116 mg, 0.25 mmol, 96%) as a white solid. Rf: 0.40 (n-hex/EtOAc 2:3); [α]D: −3.2 (c 1.0, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 5.58 (dd, J = 9.3 and 9.3 Hz, H-2), 5.34 (dd, J = 9.3 and 9.3 Hz, H-3), 5.06 (dd, J = 9.8 and 9.8 Hz, H-4), 4.84 (d, J = 9.7 Hz, H-1), 4.24 (dd, J = 2.4 and 12.5 Hz, H-6S), 4.13 (dd, J = 6.5 and 12.5 Hz, H-6R), 3.80 (ddd, J = 2.4, 6.5, and 9.4 Hz, H-5), 2.07 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 1.47 (m, 9H); 13C{1H} (δ, 100 MHz, CDCl3): 170.2 (s), 170.1 (s), 169.2 (s), 168.9 (s), 86.3 (d, C-1), 76.5 (d, C-5), 73.2 (d, C-3), 67.7 (d, C-4), 66.5 (d, C-2), 62.6 (t, C-6), 62.1 (s), 23.9 (q, ×3C), 20.6 (q), 20.6 (q), 20.5 (q), 20.4 (q); HRMS (FAB): calcd for C18H29O11S [M + H]+: 453.1431, found: 453.1405.
2,3-Di-O-acetyl-4,6-bis-O-(4-bromobenzoyl)-1-(methylsulfonyl)-β-D-glucopyranoside (9a)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, methyl S-glucoside 8a (18 mg, 0.03 mmol) afforded sulfone 9a (19 mg, 0.027 mmol, 92%) as a white solid. Rf: 0.43 (n-hex/EtOAc 1:1); [α]D: +9.4 (c 0.2, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 7.81 (m, 4H), 7.56 (m, 4H), 5.53 (m, H-2 and H-3), 5.45 (dd, J = 9.7 and 9.7 Hz, H-4), 4.61 (dd, J = 3.0 and 12.5 Hz, H-6S), 4.47 (d, J = 9.4 Hz, H-1), 4.44 (dd, J = 4.9 and 12.5 Hz, H-6R), 4.13 (ddd, J = 3.0, 4.9, and 9.7 Hz, H-5), 2.97 (s, 3H), 2.09 (s, 3H), 1.93 (s, 3H). 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.1 (s), 169.5 (s), 165.7 (s), 164.5 (s), 132.1 (d, ×2C), 131.9 (d, ×2C), 131.3 (s, ×2C), 131.1 (s, ×2C), 129.4 (s), 128.7 (s), 128.0 (s), 127.1 (s), 88.5 (d, C-1), 76.9 (d, C-5), 72.6 (d, C-3), 68.7 (d, C-4), 66.7 (d, C-2), 62.5 (t, C-6), 36.6 (q), 20.6 (q), 20.4 (q); HRMS (FAB): calcd for C25H24 Br2O11SNa [M + Na]+: 712.9304, found: 712.9293; UV (CH3CN) λmax nm (ε): 245 (ε 38200); CD (CH3CN) λext nm (Δε): 251 (14.9), 233 (−6.6).
2,3-Di-O-acetyl-4,6-bis-O-(4-bromobenzoyl)-1-(ethylsulfonyl)-β-D-glucopyranoside (9b)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, ethyl S-glucoside 8b (19 mg, 0.03 mmol) afforded sulfone 9b (20 mg, 0.028 mmol, 95%) as a white solid: Rf: 0.47 (n-hex/EtOAc 1:1); [α]D: +8.6 (c 0.1, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 7.81 (m, 4H), 7.58 (m, 4H), 5.59 (dd, J = 9.1 and 9.1 Hz, H-4), 5.55 (dd, J = 9.0 and 9.0 Hz, H-3), 5.44 (dd, J = 9.5 and 9.5 Hz, H-4), 4.58 (d, J = 9.6 Hz, H-1), 4.60 (dd, J = 3.0 and 12.1 Hz, H-6S), 4.43 (dd, J = 5.3 and 12.1 Hz, H-6R), 4.11 (ddd, J = 3.0, 5.3, and 9.3 Hz, H-5), 3.15 (m, 2H), 2.08 (s, 3H), 1.93 (s, 3H), 1.36 (dd, J = 7.5 and 7.5 Hz, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 169.7 (s), 169.1 (s), 164.9 (s), 164.1 (s), 131.9 (d, ×2C), 131.7 (d, ×2C), 131.1 (d, ×2C), 130.9 (d, ×2C), 129.2 (s), 128.6 (s), 127.8 (s), 126 (s), 87.4 (d, C-1), 76.9 (d, C-5), 72.5 (d, C-3), 68.5 (d, C-4), 66.2 (d, C-2), 62.3 (t, C-6), 29.5 (t), 20.4 (q), 20.2 (q), 5.4 (q); HRMS (ESI) calcd for C26H26O11NaSBr2 [M + Na]+: 726.9460, found: 726.9479; UV (CH3CN) λmax nm (ε): 245 (ε 38200); CD (CH3CN) λext nm (Δε): 251 (13.5), 233 (−5.9).
2,3-Di-O-acetyl-4,6-bis-O-(4-bromobenzoyl)-1-(n-propylsulfonyl)-β-D-glucopyranoside (9c)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, n-propyl S-glucoside 8c (33 mg, 0.05 mmol) afforded sulfone 9c (35 mg, 0.049 mmol, 98%) as a white solid. Rf: 0.53 (n-hex/EtOAc 1:1); [α]D: +6.4 (c 0.3, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 7.85–7.80 (m, 4H), 7.60–7.55 (m, 4H), 5.59 (dd, J = 9.1 and 9.1 Hz, H-2), 5.55 (dd, J = 9.3 and 9.3 Hz, H-3), 5.43 (dd, J = 9.5 and 9.5 Hz, H-4), 4.60 (dd, J = 3.0 and 12.5 Hz, H-6S), 4.54 (d, J = 9.4 Hz, H-1), 4.44 (dd, J = 5.4 and 12.5 Hz, H-6R), 4.11 (ddd, J = 3.0, 5.4, and 9.5 Hz, H-5), 3.13–3.03 (m, 2H), 2.09 (s, 3H), 2.05 (s, 3H), 1.94–1.80 (m, 2H), 1.03 (dd, J = 7.5 and 7.5 Hz, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 169.9 (s), 169.3 (s), 165.1 (s), 164.3 (s), 132.1 (d), 131.9 (d), 131.3 (d), 131.1 (d), 129.4 (s), 128.7 (s), 128.0 (s), 127.1 (s), 87.8 (d, C-1), 77.2 (d, C-5), 72.7 (d, C-3), 68.7 (d, C-4), 66.4 (d, C-2), 62.6 (t, C-6), 51.1 (t), 20.6 (q), 20.4 (q), 14.8 (t), 13.2 (q); HRMS (ESI); calcd for C27H28O11NaSBr2 [M + Na]+: 742.9596, found: 742.9591; UV (CH3CN) λmax nm (ε): 245 (ε 38200); CD (CH3CN) λext nm (Δε): 251 (12.9), 235 (−5.8).
2,3-Di-O-acetyl-4,6-bis-O-(4-bromobenzoyl)-1-(iso-propylsulfonyl)-β-D-glucopyranoside (9d)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, iso-propyl S-glucoside 8d (36 mg, 0.05 mmol) afforded sulfone 9d (33 mg, 0.046 mmol, 92%) as a white solid. Rf: 0.50 (n-hex/EtOAc 1:1); [α]D: −6.4 (c 0.4, CHCl3) 1H NMR (δ, 400 MHz, CDCl3): 7.81 (m, 4H), 7.56 (m, 4H), 5.65 (dd, J = 9.4 and 9.4 Hz, H-2), 5.57 (dd, J = 9.3 and 9.3 Hz, H-3), 5.40 (dd, J = 9.8 and 9.8 Hz, H-4), 4.74 (d, J = 9.6 Hz, H-1), 4.56 (dd, J = 2.7 and 12.4 Hz, H-6S), 4.43 (dd, J = 5.8 and 12.4 Hz, H-6R), 4.09 (ddd, J = 2.7, 5.8, and 9.0 Hz, H-5), 3.47 (m, 1H), 2.07 (s, 3H), 1.92 (s, 3H), 1.36 (d, J = 7.0 Hz, 3H), 1.30 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.1 (s), 169.1 (s), 165.1 (s), 164.3 (s), 132.1 (d, ×2), 131.9 (d, ×2), 131.3 (d), 131.1 (d, ×2), 129.3 (s), 128.8 (s), 127.9 (s), 127.1 (s), 85.8 (d, C-1), 76.6 (d, C-5), 72.3 (d, C-3), 68.8 (d, C-4), 66.5 (d, C-2), 62.7 (t, C-6), 51.2 (d), 20.6 (q), 20.5 (q), 16.2 (q), 13.3 (q); HRMS (FAB): calcd for C27H28O11NaSBr2 [M + Na]+: 742.9596, found: 742.9551; UV (CH3CN) λmax nm (ε): 244 nm (ε 38200); CD (CH3CN) λext nm (Δε): 252 (10.9), 232 (−5.8).
2,3-Di-O-acetyl-4,6-bis-O-(4-bromobenzoyl)-1-(cyclohexylsulfonyl)-β-D-glucopyranoside (9e)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, cyclohexyl S-glucoside 8e (71 mg, 0.1 mmol) afforded sulfone 9e (70 mg, 0.093 mmol, 93%) as a white solid. Rf: 0.58 (n-hex/EtOAc 1:1); [α]D: −8.3 (c 0.5, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 7.82 (m, 4H), 7.57 (m, 4H), 5.66 (dd, J = 9.4 and 9.4 Hz, H-2), 5.55 (dd, J = 9.4 and 9.4 Hz, H-3), 5.39 (dd, J = 9.8 and 9.8 Hz, H-4), 4.70 (d, J = 9.6 Hz, H-1), 4.55 (dd, J = 2.4 and 12.5 Hz, H-6S), 4.42 (dd, J = 5.9 and 12.5 Hz, H-6R), 4.08 (ddd, J = 2.4, 5.9, and 9.6 Hz, H-5), 3.24 (m, 1H), 2.17 (m, 2H), 2.07 (s, 3H), 1.98 (s, 3H), 1.56 (m, 4H), 1.20 (m, 4H); 13C{1H} NMR (δ, 100 MHz, CDCl3): 170.1 (s), 169.1 (s), 165.1 (s), 164.3 (s), 132.1 (d, ×2), 131.9 (d, ×2), 131.3 (d, ×2C), 131.1 (d, ×2), 129.3 (s), 128.8 (s), 127.9 (s), 127.3 (s), 85.6 (d, C-1), 76.7 (d, C-5), 72.8 (d, C-3), 68.7 (d, C-4), 66.3 (d, C-2), 62.8 (t, C-6), 58.9 (d), 25.9 (t), 25.0 (t), 24.9 (t), 24.7 (t), 22.7 (t), 20.6 (q), 20.5 (q); HRMS (FAB): calcd for C30H32O11NaSBr2 [M + Na]+: 780.9930, found: 780.9948; UV (CH3CN) λmax nm (ε): 244 nm (ε 38200); CD (CH3CN) λext nm (Δε): 252 (9.8), 229 (−4.4).
2,3-Di-O-acetyl-4,6-bis-O-(4-bromobenzoyl)-1-(tert-butylsulfonyl)-β-D-glucopyranoside (9f)
Following the general procedure for the synthesis of alkyl glycosyl sulfones, tert-butyl S-glucoside 8f (106 mg, 0.15 mmol) afforded sulfone 9f (99 mg, 0.14 mmol, 93%) as a white solid. Rf: 0.52 (n-hex/EtOAc 1:1); [α]D: −15.2 (c 0.8, CHCl3); 1H NMR (δ, 400 MHz, CDCl3): 7.81 (m, 4H), 7.57 (m, 4H), 5.69 (dd, J = 9.2 and 9.2 Hz, H-2), 5.58 (dd, J = 9.1 and 9.1 Hz, H-3), 5.36 (dd, J = 9.6 and 9.6 Hz, H-4), 4.95 (d, J = 9.5 Hz, H-1), 4.58 (dd, J = 2.6 and 12.2 Hz, H-6S), 4.39 (dd, J = 6.8 and 12.2 Hz, H-6R), 4.12 (ddd, J = 2.6, 6.8, and 9.9 Hz, H-5), 2.07 (s, 3H), 1.94 (s, 3H), 1.44 (s, 9H); 13C{1H} (δ, 100 MHz, CDCl3): 170.1 (s), 169.0 (s), 165.1 (s), 164.4 (s), 132.1 (d, ×2C), 132.0 (d, ×2C), 131.3 (d, ×2C), 131.1 (d, ×2C), 129.3 (s), 128.8 (s), 127.9 (s), 127.1 (s), 86.4 (d, C-1), 76.5 (d, C-5), 72.8 (d, C-3), 68.8 (d, C-4), 66.5 (d, C-2), 63.2 (t, C-6), 62.6 (s), 23.9 (q, ×3C), 20.7 (q), 20.5 (q); HRMS (FAB): calcd for C28H30Br2NaO11S [M + Na]+: 756.9753; found: 756.9787; UV (CH3CN) λmax nm (ε): 244 nm (ε 38200); CD (CH3CN) λext nm (Δε): 250 (7.1), 232 (−4.1).
Conflicts of interest
There are no conflicts to declare.Data availability
All related data generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.Acknowledgements
This research was supported by the Ministerio de Ciencia e Innovación (MICINN, Spain), through grant CTQ2007-67532-C02-02/BQU, and by the Universidad de La Laguna. C. A. S. thanks the Universidad de La Laguna and Gobierno de Canarias for the doctoral fellowship, the Department of Pharmaceutical Sciences at SJU for the generous starting package, and the SJU Office of Grants and Sponsored Research for the Seed Grant FY23.References
- A. Varki, Glycobiology, 2016, 27(1), 3–49 CrossRef PubMed.
- (a) P.-C. Pang, P. C. N. Chiu, C.-L. Lee, L.-Y. Chang, M. Panico, H. R. Morris, S. M. Haslam, K.-H. Khoo, G. F. Clark, W. S. B. Yeung and A. Dell, Science, 2011, 333(6050), 1761–1764 CrossRef CAS PubMed; (b) G. F. Clark, Hum. Reprod., 2013, 28(3), 566–577 CrossRef CAS PubMed.
- (a) J. D. Marth and P. K. Grewal, Nat. Rev. Immunol., 2008, 8(11), 874–887 CrossRef CAS PubMed; (b) L. G. Baum and B. A. Cobb, Glycobiology, 2017, 27(7), 619–624 CrossRef CAS PubMed.
- (a) L. Unione, A. Gimeno, P. Valverde, I. Calloni, H. Coelho, S. Mirabella, A. Poveda, A. Arda and J. Jimenez-Barbero, Curr. Med. Chem., 2017, 24(36), 4057–4080 CrossRef CAS PubMed; (b) Y. Li, D. Liu, Y. Wang, W. Su, G. Liu and W. Dong, Front. Immunol., 2021, 12 CAS; (c) R. Matos, I. Amorim, A. Magalhães, F. Haesebrouck, F. Gärtner and C. A. Reis, Front. Mol. Biosci., 2021, 8 Search PubMed.
- (a) M. N. Christiansen, J. Chik, L. Lee, M. Anugraham, J. L. Abrahams and N. H. Packer, Proteomics, 2014, 14(4–5), 525–546 CrossRef CAS PubMed; (b) O. M. T. Pearce and H. Läubli, Glycobiology, 2015, 26(2), 111–128 CrossRef PubMed; (c) S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15(9), 540–555 CrossRef CAS PubMed; (d) D. Josic, T. Martinovic and K. Pavelic, Electrophoresis, 2019, 40(1), 140–150 CrossRef CAS PubMed.
- (a) K. Dang, S. Jiang, Y. Gao and A. Qian, Mol. Biol. Rep., 2022, 49(8), 8037–8049 CrossRef CAS PubMed; (b) M. Nauman and P. Stanley, Biochem. Soc. Trans., 2022, 50(2), 689–701 CrossRef CAS PubMed; (c) A. R. Yale, E. Kim, B. Gutierrez, J. N. Hanamoto, N. S. Lav, J. L. Nourse, M. Salvatus, R. F. Hunt, E. S. Monuki and L. A. Flanagan, Stem Cell Rep., 2023, 18(6), 1340–1354 CrossRef CAS PubMed; (d) J. C. Chatham and R. P. Patel, Nat. Rev. Cardiol., 2024, 21(8), 525–544 CrossRef PubMed.
- B. Deore, R. W. Kwok, M. Toregeldiyeva, J. T. Vázquez, M. Marianski and C. A. Sanhueza, J. Org. Chem., 2023, 88(22), 15569–15579 CrossRef CAS PubMed.
- (a) C. Mayato, R. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2004, 15(15), 2385–2397 CrossRef CAS; (b) E. Q. Morales, J. I. Padron, M. Trujillo and J. T. Vázquez, J. Org. Chem., 1995, 60(8), 2537–2548 CrossRef CAS; (c) J. I. Padrón and J. T. Vázquez, Chirality, 1997, 9(5–6), 626–637 CrossRef; (d) J. I. Padrón, E. Q. Morales and J. T. Vázquez, J. Org. Chem., 1998, 63(23), 8247–8258 CrossRef; (e) C. Nóbrega and J. T. Vázquez, Tetrahedron: Asymmetry, 2003, 14(18), 2793–2801 CrossRef.
- (a) C. A. Sanhueza, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2008, 19(2), 258–264 CrossRef CAS; (b) B. Deore, J. E. Ocando, L. D. Pham and C. A. Sanhueza, J. Org. Chem., 2022, 87, 5952–5960 CrossRef CAS PubMed.
- (a) C. Mayato, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2007, 18(8), 931–948 CrossRef CAS; (b) C. Mayato, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2007, 18(23), 2803–2811 CrossRef CAS.
- (a) C. A. Sanhueza, R. L. Dorta and J. T. Vázquez, J. Org. Chem., 2011, 76(19), 7769–7780 CrossRef CAS PubMed; (b) C. A. Sanhueza, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2013, 24(9), 582–593 CrossRef CAS.
- C. Mayato, R. L. Dorta, J. M. Palazón and J. T. Vázquez, Carbohydr. Res., 2012, 352, 101–108 CrossRef CAS PubMed.
- K. P. R. Kartha and R. A. Field, Synthesis and Activation of Carbohydrate Donors: Thioglycosides and Sulfoxides, in Carbohydrates, ed. H. Osborn, Elsevier Sciences Ltd, Oxford, 2023; pp. 121–145 Search PubMed.
- P. Tiwari, G. Agnihotri and A. K. Misra, J. Carbohydr. Chem., 2005, 24(7), 723–732 CrossRef CAS.
- R. Kakarla, R. G. Dulina, N. T. Hatzenbuhler, Y. W. Hui and M. J. Sofia, J. Org. Chem., 1996, 61(23), 8347–8349 CrossRef CAS PubMed.
- (a) H. Ohrui, H. Horiki, H. Kishi and H. Meguro, H., Agric. Biol. Chem., 1983, 47(5), 1101–1106 CAS; (b) Y. Nishida, H. Ohrui and H. Meguro, Tetrahedron Lett., 1984, 25(15), 1575–1578 CrossRef CAS; (c) Y. Nishida, H. Hori, H. Ohrui and H. Meguro, J. Carbohydr. Chem., 1988, 7(1), 239–250 CrossRef CAS.
- H. Amarasekara, S. Dharuman, T. Kato and D. Crich, J. Org. Chem., 2018, 83(2), 881–897 CrossRef CAS PubMed.
- Linear regression equations: Pgg=−23.8v+76.9 (R2=0.92); Pgt=23.5v+22.1 (R2=0.62).
- Linear regression equations: Pgg=−25.6v+77.7 (R2=0.90); Pgt=25.6v+22.1 (R2=0.87).
- Linear regression equations: Pgg=−18.1v+70.9 (R2=0.97); Pgt=18.1v+29.1 (R2=0.97).
- Linear regression equations: Pgg=−17.5v+66.5 (R2=0.91); Pgt=6.7v+39.3 (R2=0.21).
- Linear regression equations: Pgg=−13.8v+61.2 (R2=0.85); Pgt=13.4v+32.6 (R2=0.53).
- Linear regression equations: Pgg=−13.1v+62.7 (R2=0.81); Pgt=24.4v+22.4 (R2=0.97).
- M. Charton, J. Am. Chem. Soc., 1975, 97(6), 1552–1556 CrossRef CAS.
- (a) R. W. Jr. Taft, J. Am. Chem. Soc., 1952, 74(11), 2729–2732 CrossRef; (b) R. W. Jr. Taft, J. Am. Chem. Soc., 1952, 74(12), 3120–3128 CrossRef; (c) R. W. Jr Taft, J. Am. Chem. Soc., 1953, 75(18), 4538–4539 CrossRef; (d) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91(2), 165–195 CrossRef CAS.
- The Charton values used in this work are: methyl v=0.52, ethyl v=0.56, n-propyl v=0.68, iso-propyl v=0.79, cyclohexyl v=0.87, and tert-butyl v=1.24.
- (a) N. Harada and K. Nakanishi, Circular Dichroic Spectroscopy. Exciton Coupling in Organic Stereochemistry, University Science Books, California, 1983 Search PubMed; (b) Circular Dichroism: Principles and Applications, ed. N. Berova, K. Nakanishi and R. W. Woody, Wiley-VCH, New York, N. Y., 2nd edn, 2000 Search PubMed; (c) N. Harada, K. Nakanishi and N. Berova, Electronic CD Exciton Chirality Method: Principles and Applications in Comprehensive Chiroptical Spectroscopy: Applications in Stereochemical Analysis of Synthetic Compounds, Natural Products, and Biomolecules, ed. N. Berova, P. L. Polavarapu, K. Nakanishi and R. W. Woody, John Wiley & Sons, New York, NY, USA, 2012 Search PubMed.
- Circular Dichroism and the Conformational Analysis of Biomolecules, ed. G. D. Fasman, Plenum Press, New York, 1996, p. 25–157 Search PubMed.
- (a) A. E. Reed and P. von Ragué, J. Am. Chem. Soc., 1990, 112, 1434 CrossRef CAS; (b) C. H. Patterson and R. P. Messmer, J. Am. Chem. Soc., 1990, 112, 4138 CrossRef CAS; (c) R. P. Messmer, J. Am. Chem. Soc., 1991, 113, 433–440 CrossRef CAS; (d) D. B. Chesnut and L. D. Quin, J. Comput. Chem., 2004, 25, 734–738 CrossRef CAS PubMed; (e) F. A. M. Rudolph, A. L. Fuller, A. M. Z. Slawin, M. B. Bühl, R. A. Aitken and J. D. Woollins, J. Chem. Crystallogr., 2010, 40, 253–265 CrossRef CAS.
- (a) T. D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Elsevier Science, 3rd edn, 2016. DOI:10.1016/C2015-0-04654-8; (b) T. R. Hoye, P. R. Hanson and J. R. Vyvyan, J. Org. Chem., 1994, 59, 4096–4103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all alkyl glucosyl sulfones, including expansions of their H5 and H6 signals; some selected 1D NOESY experiments; tables with experimental and calculated NMR data; overlaid regions of experimental and simulated 1H NMR spectra, as well as plots of calculated rotamer populations from simulated spectra vs. Charton parameters. See DOI: https://doi.org/10.1039/d4ob02056a |
| This journal is © The Royal Society of Chemistry 2025 |