
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Axially chiral dimethylaminobenzimidazoles and their preliminary evaluation as acyl-transfer reagents†
Daniel
Toman
 a,
Ivan
Nemec
b,
Ondřej
Kurka
c,
Adam
Přibylka
a and
Petr
Cankař
*a
a,
Ivan
Nemec
b,
Ondřej
Kurka
c,
Adam
Přibylka
a and
Petr
Cankař
*a
aDepartment of Organic Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 77900 Olomouc, Czech Republic. E-mail: petr.cankar@upol.cz
bDepartment of Inorganic Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 77900 Olomouc, Czech Republic
cDepartment of Analytical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 77900 Olomouc, Czech Republic
First published on 6th February 2025
Abstract
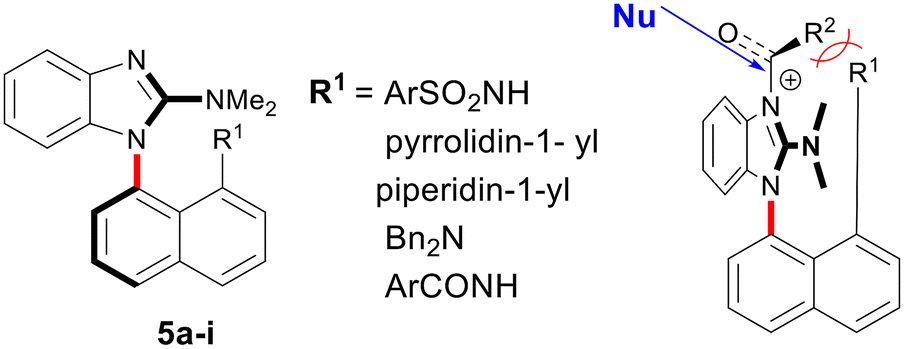
The synthesis of axially chiral benzimidazoles with a peri-substituted naphthalene and a dimethylamino group at positions 1 and 2, respectively, was developed. We evaluated these compounds in the desymmetrization reaction of cis-tetrahydrophthalic anhydride with benzyl alcohol, as the nucleophilic sp2 imidazole nitrogen atom is able to catalyze the acyl-transfer reaction. The prepared benzimidazoles demonstrated catalytic activity and showed that their axial chirality impacts stereoselectivity.
Introduction
The acyl group transfer reaction represents one of the most common chemical transformations, occurring widely in both natural enzymatic processes and organic syntheses.1,2 One family of chiral acyl transfer catalysts comprises derivatives based on the DMAP structure,3 introduced by Vedejs in the late 1990s for the kinetic resolution of alcohols (Fig. 1).4,5 Later, this family of catalysts was expanded to include other derivatives, differing in the variety of functional groups, the position of substitution at the pyridine ring and in the type of chirality. Examples of 3- or 4-substituted DMAP derivatives are shown in Fig. 1.6–17 | ||
| Fig. 1 Examples of chiral acyl transfer catalysts. | ||
Another important group of acyl transfer catalysts is N-alkyl imidazole. These catalysts benefit from the sterically more accessible, nucleophilic sp2 nitrogen atom of the imidazole ring, which kinetic studies have shown to be more efficient in acyl transfer reactions than pyridine-based catalysts.18,19 The imidazole moiety is naturally present in enzymes through histidine, which inspired Miller and colleagues to develop small peptide molecules incorporating histidine as acyl transfer catalysts.20–24 Zhang and colleagues developed a series of bicyclic imidazole catalysts (Fig. 1), featuring a rigid structure in which a chiral center is directly adjacent to the nucleophilic sp2 nitrogen atom. These imidazole catalysts were shown to catalyze asymmetric Steglich rearrangement with high enantioselectivities.25
Further, the importance of the nucleophilic sp2 nitrogen atom of the imidazole ring is not only associated with natural enzymatic acyl transfer reaction but also with hydrogen bonding as can be seen in acetyltransferases26 and, for example, cyclin-dependent kinases interacting with the imidazole ring containing inhibitors.27
While various chiral acyl transfer catalysts have been reported, the importance and broad application of the acylation reaction continues to drive research efforts. These efforts aim to extend the substrate scope, enable milder reaction conditions, improve stereochemical outcomes, and, more generally, a search for novel attractive chiral scaffolds with multiple chiral elements is also emerging.28–32 Herein, we present a preliminary study of axially chiral N-naphthalenebenzimidazoles as a novel class of imidazole-based acyl transfer catalysts (Fig. 2). The chiral environment of the catalyst 5 is a result of restricted rotation around the C–N bond that connects the naphthalene and benzimidazole rings. We hypothesize that the chiral environment of the N-acylbenzimidazolium intermediates can be significantly modulated by the structure of the substituent R1 at the peri position of the naphthalene ring. Additionally, the benzene ring of the benzimidazole and dimethylamino group are also expected to influence the stereochemical outcome. The presence of the dimethylamino group at position 2 of the benzimidazole ring not only enhances the stability of atropisomers but is primarily designed to increase the nucleophilicity of the catalyst. Moreover, the supposed distortion in the N-acyl bond of intermediate 5, induced by steric hindrance, may accelerate the kinetics of acyl transfer.6 In this report, we present the synthesis of benzimidazole-based catalysts, an evaluation of their catalytic activity, the stability of selected atropisomers, and preliminary results for the enantioselective desymmetrization reaction of a meso-anhydride.
 | ||
| Fig. 2 Design of the catalyst and rationale for the enantioselective N-acyl transfer reaction. | ||
Results and discussion
Synthesis of dimethylaminobenzimidazoles
The synthesis was initiated with naphthalene-1,8-diamine 1 and 1-fluoro-2-nitrobenzene, yielding diamine 2 as depicted in Scheme 1. Subsequently, the primary amine was sulfonylated using a slight molar excess of the corresponding sulfonyl chloride to provide derivatives 3a–e with good overall yields. Reduction of the nitro group to the corresponding amine was accomplished using powdered zinc in a mixture of methanol and acetic acid. The reaction proceeded smoothly without any side products. Typically, only a simple work up was needed to furnish intermediates 4a–e. | ||
Scheme 1 Synthesis of benzimidazoles 5a–e and 6a–b functionalized with sulfonamide moieties. Reagents and conditions: (a) 1-fluoro-2-nitrobenzene, KOH, DMSO, rt, 4 h; (b) RSO2Cl, DCM/pyr 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, rt, 2 h; (c) Zn, MeOH/AcOH 25:2, rt, 2 h; (d) phosgene dimethyliminium chloride, CHCl3, reflux, 2 h; (e) Zn, DMA–DMA, MeOH/AcOH 25:1, 60 °C, 4 h. :2, rt, 2 h; (c) Zn, MeOH/AcOH 25:2, rt, 2 h; (d) phosgene dimethyliminium chloride, CHCl3, reflux, 2 h; (e) Zn, DMA–DMA, MeOH/AcOH 25:1, 60 °C, 4 h. | ||
In the final cyclization step, phosgene dimethyliminium chloride was utilized to form benzimidazoles 5a–e with dimethylamino group at position 2. The yields were moderate to good after purification by column chromatography.
According to NMR experiments, compound 5c exists as a mixture of its free and zwitterionic forms. This behaviour can be attributed to the strongly electron-withdrawing nature of the 3,5-bis(trifluoromethyl)benzenesulfonyl group. As a result, obtaining a clear 1H NMR spectrum of 5c proved challenging under standard conditions. The spectrum became well-resolved only after the addition of a few drops of trifluoroacetic acid to the sample.
Two additional derivatives, 6a and 6b, bearing a methyl group at position 2 of the benzimidazole ring, were synthesized from nitroamines 3a and 3b. The transformation was achieved using a one-pot reduction/cyclization protocol with N,N-dimethylacetamide dimethylacetal as the cyclization reagent to expand the library of prepared compounds.33
The synthesis of benzimidazoles 5f and 5g, functionalized with pyrrolidine and piperidine moieties at the peri position relative to the benzimidazole ring, commenced with the double alkylation of the primary amine using 1,4-dibromobutane or 1,5-dibromopentane (Scheme 2). The resulting compounds, 3f and 3g, were isolated in moderate yields. In the next step, an attempt to reduce the nitro group using powdered zinc, as described in Scheme 1, led to the formation of significant amounts of impurities, complicating the isolation and results in reduced overall yields. Consequently, catalytic hydrogenation was employed to yield diamines 4f and 4g in quantitative yield. Finally, the target benzimidazoles 5f and 5g, were obtained under the same conditions used for sulfonamides 5a–e.
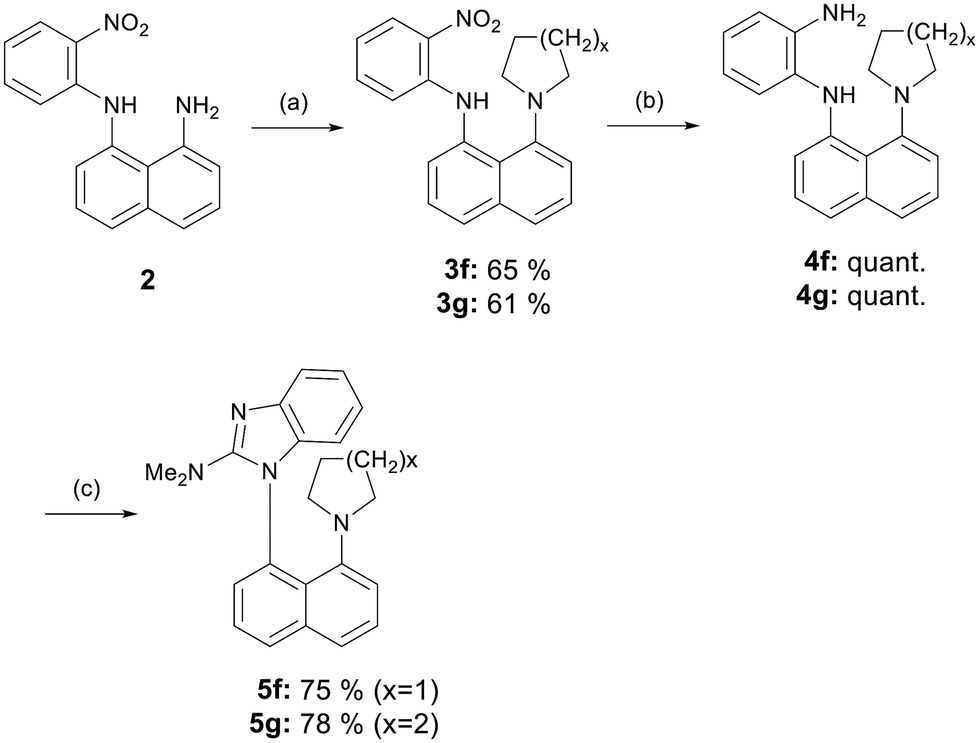 | ||
| Scheme 2 Synthesis of benzimidazoles 5f and 5g functionalized with pyrrolidine and piperidine moieties. Reagents and conditions: (a) 1,4-dibromobutane or 1,5-dibromopentane, K2CO3, DMSO, rt, 24 h; (b) 10% Pd/C, hydrogen atmosphere, EtOAc, rt, 4 h; (c) phosgene dimethyliminium chloride, CHCl3, reflux, 2 h. | ||
The synthesis of dibenzylated derivative 5h closely followed the procedure for the synthesis benzimidazoles 5f and 5g; however, the reaction conditions required modification. An excess of benzyl bromide was employed, and the temperature was increased. Despite the presence of some unreacted starting material 2 in the reaction mixture, the reaction time could not be prolonged, as benzylation also occurred at the secondary amine, and the resulting impurity was challenging to separate from the desired product 3h. The subsequent reduction of the nitro group was carried out with powdered zinc, and the resulting diamine 4h was converted to the final benzimidazole 5h using the same methodology as for derivatives 5a–g (Scheme 3).
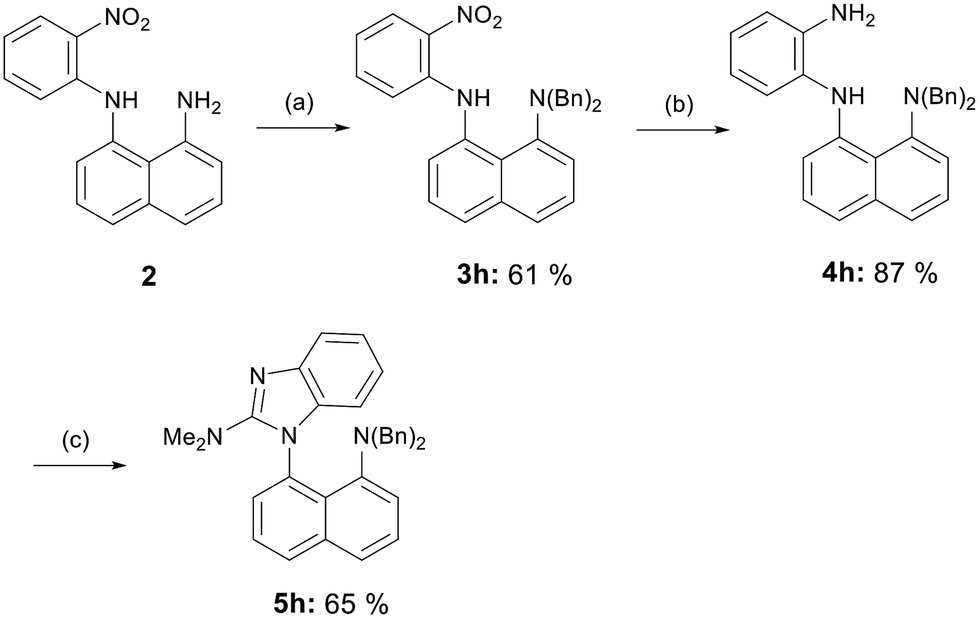 | ||
| Scheme 3 Synthesis of dibenzylated benzimidazole 5h. Reagents and conditions: (a) BnBr, K2CO3, MeCN, 70 °C, 16 h; (b) Zn, MeOH/AcOH 25:2, rt, 2 h; (c) phosgene dimethyliminium chloride, CHCl3, reflux, 2 h. | ||
We also synthesized a derivative with the amide functionality, 5i (Scheme 4). The 3,5-bis(trifluoromethyl)benzoyl group was introduced in the first step. The reduction of nitro group with zinc initially resulted in the formation of a pyrimidine ring derivative rather than compound 4i. Additionally, iron-catalyzed reduction with hydrazine was unsuccessful.34 Consequently, we opted for catalytic hydrogenation to achieve the desired intermediate. The final step was performed using phosgene diminium chloride in the presence of pyridine to prevent the formation of impurities.
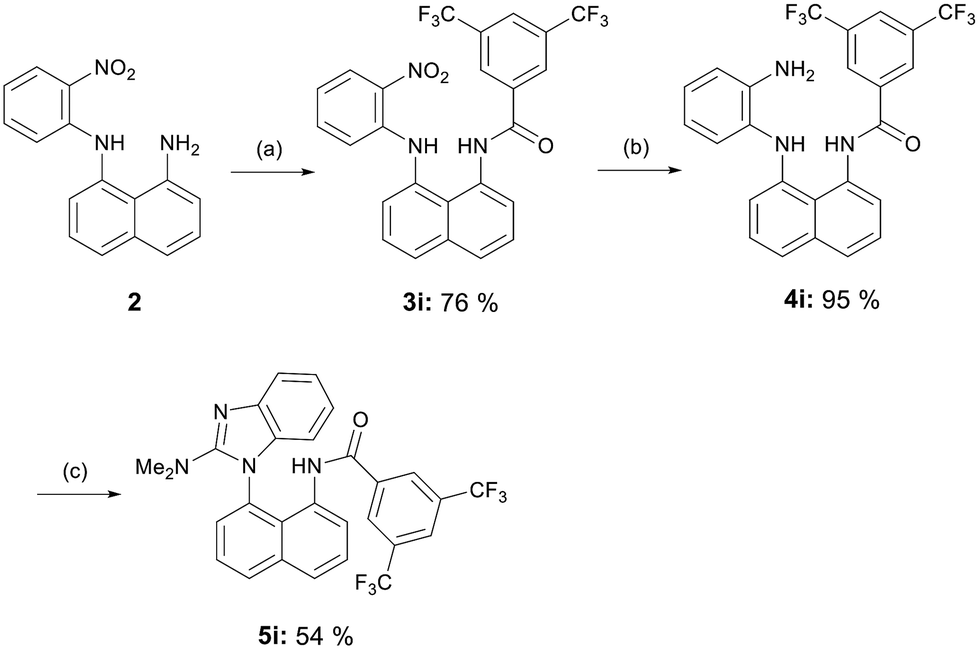 | ||
| Scheme 4 Synthesis of benzimidazole 5i with amide functionality. Reagents and conditions: (a) 3,5-(bis)trifluoromethyl benzoyl chloride, DCM/pyr 3:2, rt, 2 h; (b) 10% Pd/C, hydrogen atmosphere, EtOAc, rt, 4 h; (c) phosgene dimethyliminium chloride, pyr, CHCl3, rt, 2 h. | ||
Evaluation of catalytic activity
We then decided to evaluate the catalytic activity of benzimidazoles 5a–i in the reaction of cis-tetrahydrophthalic anhydride 6 with benzyl alcohol to obtain monoester 7. The experiments were performed using racemic catalysts (Table 1).|
|
||
|---|---|---|
| Entry | Catalyst | Reaction timea (h) |
| a Monoesterification was monitored by 1H NMR spectroscopy and was terminated once the anhydride 6 was completely consumed. The reaction yielded only monoester 7, which was not isolated. | ||
| 1 | — | 6 days (21% of 6 left) |
| 2 | 5a | 20 |
| 3 | 5b | 21 |
| 4 | 5c | 36 |
| 5 | 5d | 26 |
| 6 | 5e | 36 (partly soluble in ether) |
| 7 | 5f | 22 |
| 8 | 5g | 22 |
| 9 | 5h | 50 |
| 10 | 5i | 48 |
| 11 | 6a | >6 days (insoluble in ether) |
| 12 | 6b | >6 days (insoluble in ether) |

The first experiment was conducted without a catalyst to demonstrate that the reaction was incomplete after 6 days (entry 1). Benzimidazoles with the sulfonamide moiety significantly accelerated the reaction rate (entries 1–6), with 5a and 5b proving to be the most efficient. The catalyst 5f, containing the pyrrolidine moiety, achieved full conversion after 22 hours (entry 7). A similar result was observed with 5g (entry 8). The steric hindrance of the dibenzylamino groups slowed the reaction rate, with full conversion of starting material occurring only after 50 hours (entry 9). The benzamide functionality did not improve the reaction rate (entry 10).
Additionally, we attempted to catalyze the monoesterification reaction with benzimidazoles 6a and 6b, expecting lower catalytic activity due to the substitution of the dimethylamino group with a methyl group. The reaction rate was found to be comparable to the conditions in entry 1 (without the catalyst), likely due to their insolubility in diethyl ether.
Furthermore, both atropisomers of compound 5f were successfully separated by semi-preparative chiral HPLC. The thermal stability of atropisomer (−)-(Ra)-5f was evaluated by heating in ethylene glycol at three different temperatures: 80, 120, and 160 °C. Our investigation revealed that racemization occurred only at 160 °C, accompanied by partial thermal decomposition. The absolute configuration of both atropisomers of 5f was determined through single-crystal X-ray crystallography (please see ESI†).
Desymmetrization experiments
Preliminary stereoselective desymmetrization experiments were conducted under the catalysis of benzimidazole (+)-(Sa)-5f at three concentrations, using the reaction conditions outlined in Table 1. Monobenzyl ester 7 was isolated as a mixture of enantiomers with a ratio of 11:89 when anhydride 6 was used at a concentration of 0.05 M. A lower concentration at 0.025 M resulted in worse enantioselectivity, with an enantiomeric ratio of 27:73, although the reaction time was extended to 48 hours. Further dilution of anhydride 6 to 0.01 M led to a significant decrease in reactivity, as residual anhydride 6 remained in the reaction mixture after 8 days, and racemic monoester 7 was formed. The dextrorotatory optical rotation of 7 (c = 1.0, MeOH), isolated as a mixture of enantiomers with a 27:73 enantiomeric ratio, indicated that (1R,6S)-6-((benzyloxy)carbonyl)cyclohex-3-ene-1-carboxylic acid was the predominant enantiomer.35,36
Conclusions
In summary, we successfully developed the synthesis of axially chiral benzimidazoles with a dimethylamino group at position 2, designed to mimic guanidine moiety. In total, 11 atropisomeric benzimidazoles were synthesized from readily available and inexpensive starting materials. The potential use of these benzimidazoles as acyl-transfer reagents was tested in a desymmetrization reaction of cis-tetrahydrophthalic anhydride with benzyl alcohol. The best catalytic activity was observed for benzimidazoles 5a, 5b, 5f, and 5g. Benzimidazole 5f was separated by semi-preparative HPLC into two atropisomers, which allowed us to confirm the high racemization barrier and evaluate the efficiency of chiral transfer from the axially chiral benzimidazole to the benzyl monoester 7, obtained by desymmetrization of cis-tetrahydrophthalic anhydride 6. In conclusion, it was demonstrated that benzimidazoles with a dimethylamino group at position 2 are capable of catalyzing acyl transfer reactions. Furthermore, their axial chirality presents potential for chiral transfer in asymmetric variants of these reactions. However, further optimization and development are necessary to improve their stereoselectivity.Experimental
All starting materials, solvents, and reagents were bought from commercial suppliers (Merck, Fluorochem, Acros Organic, VWR, Lach-Ner, Apollo Scientific) and were used without further purification. NMR spectra were recorded on the spectrometer Jeol ECH400II (400 MHz for 1H spectra, 100 MHz for 13C spectra), Jeol ECX-500SS (500 MHz for 1H spectra, 126 MHz for 13C spectra) or Bruker Avance III HD 500 (500 MHz for 1H spectra, 126 MHz for 13C spectra) at ambient temperature. Chemical shifts are quoted as particles per million (ppm) and are referenced as follows: CDCl3: 7.26 ppm (1H), 77.0 ppm (13C) and DMSO-d6: 2.50 ppm (1H), 39.52 ppm (13C). Coupling constants (J) are quoted in hertz. HRMS spectra were recorded by Thermo Exactive Plus high-resolution mass spectrometer with electrospray ionization (ESI) and Orbitrap analyser operating at a positive or negative full scan mode in the range of 60–800 m/z. Thin-layer chromatography was performed on alumina sheets coated with Merck Silica gel 60 F254. The plates were developed and analyzed under UV light. Separations by column chromatography were carried out using SiliaFlash Irregular Silica Gel P60 (40–63 μm) as the stationary phase. Melting points were measured on a Boetius stage and are uncorrected. Chiral HPLC analyses were performed using HPLC Agilent 1260 II equipped with DA (diode array) detector. IR spectra were measured by DRIFT (Diffuse Reflectance Infrared Fourier Transform) on Thermo Nicolet AVATAR 370 FTIR spectrometer.Synthetic procedures
:1). Mp 112–113 °C; 1H NMR (500 MHz, CDCl3) δ 9.60 (s, 1H), 8.24 (dd, J = 8.6, 1.6 Hz, 1H), 7.75 (dd, J = 8.2, 1.3 Hz, 1H), 7.39 (dd, J = 8.3, 7.3 Hz, 1H), 7.35–7.27 (m, 3H), 7.21 (dd, J = 7.3, 1.3 Hz, 1H), 6.88–6.75 (m, 2H), 6.68 (dd, J = 6.0, 2.7 Hz, 1H), 4.60 (br. s, 2H); 13C NMR (126 MHz, CDCl3) δ 144.87, 143.79, 137.18, 136.07, 135.29, 133.63, 128.79, 127.22, 126.41, 125.84, 125.02, 121.04, 118.85, 118.12, 117.80, 111.83; HRMS (ESI): m/z [M + H]+ calcd for C16H14N3O2: 280.1081, found: 280.1083.
General procedure for preparation of sulfonamides (3a–e)
Nitroamine 2 (279 mg, 1 mmol) was dissolved in a mixture of dichloromethane (3 mL) and pyridine (2 mL). The corresponding sulfonyl chloride (1.2 mmol) was added in one portion and the mixture was allowed to stir at room temperature for 2 hours. After the reaction was completed according to TLC, it was diluted with DCM (5 mL) and washed with 1 M aqueous hydrochloric acid (2 × 5 mL) and with water (1 × 5 mL). The organic layer was dried over magnesium sulfate and the crude compound was purified using column chromatography (dichloromethane) to yield the corresponding sulfonamide.General procedure for preparation of nitro compounds (3f–g)
Nitroamin 2 (1 g, 3.58 mmol) and potassium carbonate (2.18 g, 15.77 mmol) were suspended in dimethyl sulfoxide (10 mL). The corresponding dibromoalkane was subsequently added. The reaction mixture was allowed to stir at room temperature for 24 hours. After the reaction was completed, the resulting mixture was poured into water (50 mL) and extracted with dichloromethane (3 × 30 mL). The combined extracts were dried over magnesium sulfate and the solvent was evaporated on a rotavap. A crude oily residue was purified by column chromatography (dichloromethane/n-hexane, 1:1) to yield corresponding nitro compound 3f or 3g.
:1) to yield 402 mg of 9 as a red solid (61%). Mp = 127–128 °C; 1H NMR (400 MHz, CDCl3) δ 12.60 (s, 1H), 8.20 (dd, J = 8.4, 1.5 Hz, 1H), 7.63 (dd, J = 8.6, 0.6 Hz, 1H), 7.58–7.49 (m, 1H), 7.51–7.39 (m, 1H), 7.44 (dd, J = 7.7, 1.1 Hz, 1H), 7.38–7.25 (m, 2H), 7.18 (t, J = 7.8 Hz, 1H), 7.11–6.96 (m, 6H), 7.02–6.92 (m, 4H), 6.89–6.76 (m, 2H), 4.45 (d, J = 13.4 Hz, 2H), 4.29 (d, J = 13.4 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 144.7, 141.9, 137.6, 137.1, 137.0, 135.7, 134.9, 130.3, 127.9, 127.2, 126.8, 126.0, 125.2, 124.9, 123.9, 123.6, 123.0, 118.4, 118.0, 116.6, 59.8; HRMS (ESI): m/z [M + H]+ calcd for C30H26N3O2: 460.2020, found: 460.2025.
:1) to yield 564 mg of nitroamine 3i as an orange solid (76%). Mp = 206–207 °C; 1H NMR (400 MHz, CDCl3) δ 10.81 (s, 1H), 9.34 (s, 1H), 8.36 (dd, J = 7.7, 1.2 Hz, 1H), 7.98–7.91 (m, 4H), 7.88 (dd, J = 8.6, 1.5 Hz, 1H), 7.83 (dd, J = 8.3, 1.0 Hz, 1H), 7.63 (t, J = 8.0 Hz, 1H), 7.52 (dd, J = 8.2, 7.3 Hz, 1H), 7.33–7.27 (m, 2H), 6.78 (ddd, J = 8.5, 7.1, 1.3 Hz, 1H), 6.58 (dd, J = 8.5, 1.2 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 163.0, 144.2, 137.4, 136.4, 136.4, 134.3, 134.2, 132.9, 132.3 (q, J = 34.0 Hz), 129.8, 127.8, 126.8, 126.6, 126.4, 126.1, 125.3, 123.8, 122.9 (q, J = 273.0 Hz), 121.9, 119.7, 118.0; HRMS (ESI): m/z [M − H]− calcd for C25H14F6N3O2: 518.0934, found: 518.0945.
General procedure for preparation of diamines (4a–e)
Sulfonamide 3a–e (1 mmol) and powdered zinc (262 mg; 4 mmol) were suspended in methanol (4.5 mL). Acetic acid (0.36 mL; 6.3 mmol) was added in one portion and the resulting mixture was allowed to stir at room temperature for 2 hours. After the reaction was completed, residual zinc was filtered off, and the filtrate was diluted with 5% aqueous solution of sodium carbonate (10 mL). Product was extracted with dichloromethane (3 × 10 mL vol.) and combined extracts were dried over magnesium sulfate. A solvent was evaporated to yield corresponding diamine 4a–e. The isolated compound was purified using column chromatography, where noted.:1). Mp 149–150 °C; 1H NMR (400 MHz, CDCl3) δ 11.06 (br. s, 1H), 7.75 (t, J = 8.3, 1.1 Hz, 1H), 7.65 (dd, J = 7.6, 1.1 Hz, 1H), 7.62 (dd, J = 8.3, 0.9 Hz, 1H), 7.49–7.38 (m, 2H), 7.13 (dd, J = 7.3, 1.2 Hz, 1H), 6.91–6.82 (m, 2H), 6.57 (ddd, J = 8.0, 5.9, 2.9 Hz, 1H), 6.04 (dd, J = 7.6, 0.9 Hz, 1H), 5.79 (br. s, 1H), 2.38 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 142.9, 142.2, 136.9, 134.1, 129.1, 127.3, 127.3, 126.0, 125.2, 123.6, 121.5, 120.9, 120.8, 117.1, 115.6, 115.1, 38.6; HRMS (ESI): m/z [M + H]+ calcd for C17H18N3O2S: 328.1114, found: 328.1112.
General procedure for preparation of diamines (4f–g)
10% palladium on activated charcoal (53 mg, 0.01 mmol) was placed at the bottom of the flask. The solution of nitro compound 3f or 3g (1 mmol) in ethyl acetate (25 mL) was carefully added. The resulted suspension was stirred at RT for 4 hours under hydrogen atmosphere. After the reaction was completed, palladium on activated charcoal was removed using filtration and the filtrate was evaporated on a rotavap to yield the corresponding diamine 4f or 4g.General procedure for preparation of benzimidazoles (5a–e)
Diamine 4a–e (1 mmol) was dissolved in chloroform (5 mL). Phosgene dimethyliminium chloride (244 mg; 1.5 mmol) was added in one portion and the reaction mixture was refluxed for 2 hours. After the reaction was completed, the resulting mixture was diluted with dichloromethane (10 mL) and washed with 5% aqueous solution of sodium carbonate (2 × 5 mL) and water (1 × 5 mL). The organic layer was dried over magnesium sulfate and the solvent was evaporated on a rotavap. A crude solid was purified by column chromatography to yield corresponding benzimidazole 5a–e.:1). Mp 185–187 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.93 (dd, J = 8.3, 1.1 Hz, 1H), 7.70 (d, J = 8.1 Hz, 2H), 7.64 (d, J = 7.9 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.46 (t, J = 8.0 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.29–7.16 (m, 2H), 7.12–7.03 (m, 2H), 6.90 (t, J = 7.6 Hz, 1H), 6.40 (d, J = 7.8 Hz, 1H), 2.89 (s, 6H), 2.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 158.4, 143.8, 142.3, 138.2, 136.6, 132.7, 130.9, 129.6, 128.5, 127.6, 126.9, 126.2, 125.3, 123.5, 122.2, 121.0, 117.9, 117.2, 109.7, 40.8, 21.6; IR (FTIR): 2941, 2831, 2362, 2343, 1570, 1462, 1297, 1158, 1035, 918, 841, 740, 658 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C26H25N4O2S: 357.1693, found: 357.1688.
:1). Mp = 187–188 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 8.17 (d, J = 1.7 Hz, 1H), 7.91 (dd, J = 8.2, 1.1 Hz, 1H), 7.88–7.79 (m, 3H), 7.75–7.64 (m, 2H), 7.65–7.51 (m, 3H), 7.53–7.42 (m, 2H), 7.32 (dd, J = 8.7, 1.9 Hz, 1H), 7.18 (ddd, J = 15.9, 7.9, 1.2 Hz, 2H), 6.77 (td, J = 7.9, 1.1 Hz, 1H), 6.31 (d, J = 7.7 Hz, 1H), 2.85 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 158.5, 142.6, 138.2, 136.5, 135.0, 132.8, 132.6, 132.0, 130.8, 129.5, 129.3, 128.9, 128.9, 128.4, 127.8, 127.4, 126.9, 126.2, 125.4, 123.5, 122.6, 122.0, 120.9, 117.7, 117.3, 109.5, 40.7; IR (FTIR): 2941, 2832, 2362, 2343, 1685, 1569 1462, 1297, 1158, 1034, 919, 842, 764, 740, 669 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C29H25N4O2S: 493.1693, found: 493.1686.
:1). Mp 210–212 °C; 1H NMR (400 MHz, CDCl3 with one drop of TFA) δ 8.23 (dd, J = 8.0, 1.5 Hz, 1H), 8.11–8.05 (m, 2H), 7.95 (s, 2H), 7.87–7.77 (m, 2H), 7.68 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.45–7.36 (m, 2H), 7.27–7.16 (m, 1H), 6.79 (d, J = 8.1 Hz, 1H), 6.60 (dd, J = 7.3, 0.8 Hz, 1H), 2.99 (s, 6H); 13C NMR (126 MHz, CDCl3 with one drop of TFA) δ 150.4, 141.7, 136.7, 133.6, 132.8 (q, J = 34.5 Hz), 132.7, 131.1, 130.3, 129.8, 129.6, 129.0, 128.7, 128.4, 128.3 (q, J = 2.7 Hz), 126.9, 126.6, 126.5, 125.5, 124.1, 122.4 (q, J = 272.6 Hz), 112.7, 111.5, 40.9; IR (FTIR): 2923, 2938, 2362, 2342, 1570, 1458, 1358, 1277, 1123, 1054, 1033, 1017, 904, 843, 817, 764, 737 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C27H21F6N4O2S: 579.1284, found: 579.1295.
:1). Mp 196–197 °C; 1H NMR (500 MHz, CDCl3) δ 8.55 (s, 1H), 8.25 (dd, J = 7.3, 1.4 Hz, 1H), 8.24–8.14 (m, 2H), 8.01 (dd, J = 8.2, 1.2 Hz, 1H), 7.96 (dd, J = 8.2, 1.1 Hz, 1H), 7.86 (dd, J = 8.2, 1.0 Hz, 1H), 7.76 (dd, J = 7.5, 0.8 Hz, 1H), 7.58 (dd, J = 7.9, 7.4 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.49 (dd, J = 8.0, 7.4 Hz, 1H), 7.38 (dd, J = 8.2, 4.3 Hz, 1H), 7.19 (dd, J = 7.3, 1.2 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.96 (ddd, J = 8.3, 7.7, 1.0 Hz, 1H), 6.78 (ddd, J = 8.1, 7.7, 0.9 Hz, 1H), 6.40 (d, J = 7.8 Hz, 1H), 2.75 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 157.8, 151.2, 142.3, 141.8, 138.0, 137.2, 136.6, 136.0, 133.9, 132.3, 131.4, 130.3, 129.0, 128.5, 128.4, 126.7, 126.5, 126.3, 125.8, 124.1, 122.6, 121.8, 121.4, 119.7, 116.1, 108.6; IR (FTIR): 2942, 2832, 2361, 2343, 1686, 1566, 1458, 1429, 1166, 1147, 1034, 921, 766, 736, 669 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C28H23N5O2S: 494.1844, found: 494.1645.
General procedure for preparation of benzimidazoles (5f and 5g)
Diamine 4f or 4g (0.91 mmol) was dissolved in chloroform (6 mL). Phosgene dimethyliminium chloride (221 mg, 1.36 mmol) was added in one portion and the reaction mixture was refluxed for 2 hours. After the reaction was completed, the resulting mixture was diluted with dichloromethane (12 mL) and washed with 5% aqueous solution of sodium carbonate (2 × 10 mL) and water (1 × 10 mL). The organic layer was dried over magnesium sulfate and the solvent was evaporated on a rotavap. A crude yellow solid was purified by column chromatography (dichloromethane/ethyl acetate, 3:2) to yield benzimidazole 5f or 5g.
:MeOH:EtOH 40:1:1 (v/v/v), flow rate: 5 mL min−1), T = 35 °C, loading: 1000 μl, UV detection: λ = 274 nm. Amount of racemic 5f was 110 mg.
:1) to yield 36 mg of 5i as a white solid (54%). Mp = 74–75 °C; 1H NMR (400 MHz, CDCl3) δ 8.09 (dd, J = 8.3, 1.1 Hz, 1H), 7.98 (dd, J = 8.2, 0.9 Hz, 1H), 7.94 (s, 1H), 7.85 (d, J = 7.4 Hz, 1H), 7.73 (s, 1H), 7.67 (t, J = 7.7 Hz, 1H), 7.65–7.58 (m, 3H), 7.47 (dd, J = 7.3, 1.2 Hz, 1H), 7.13 (d, J = 7.8 Hz, 1H), 7.08 (td, J = 7.3, 0.9 Hz, 1H), 6.97 (td, J = 7.9, 1.3 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 2.71 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 163.3, 137.2, 136.5, 136.0, 132.0 (q, J = 33.7 Hz), 131.6, 131.1, 130.7, 129.1, 128.3, 127.2, 127.0, 126.9, 126.1, 125.3, 125.3, 123.4, 121.5 (q, J = 273.0 Hz), 121.1, 118.1, 117.1, 108.6, 40.7; IR (FTIR): 2942, 2831, 2362, 2344, 1686, 1496 1462, 1277, 1129, 1031, 904, 844, 764, 739, 681 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C28H21F6N3O: 544.1614, found: 544.1624.
General procedure for preparation of benzimidazoles (6a–b)
The corresponding nitro compound 3a or 3b (0.29 mmol) and powdered zinc (75 mg, 1.16 mmol) were suspended in methanol (1.8 mL). N,N-Dimethylacetamide dimethyl acetal (0.424 mL, 2.9 mmol) and AcOH (0.148 mL, 2.59 mmol) were subsequently added. The reaction mixture was allowed to stir at 60 °C for 4 hours. After the reaction was completed, residual zinc was filtered off, and the filtrate was diluted with 5% aqueous solution of sodium bicarbonate (5 mL). The product was extracted with dichloromethane (3 × 6 mL). The combined extracts were dried over magnesium sulfate and the solvent was evaporated to yield an oily residue which was purified by column chromatography.:1). Mp 229–230 °C; 1H NMR (400 MHz, CDCl3) δ 8.02 (dd, J = 8.4, 1.2 Hz, 1H), 7.87 (dt, J = 8.0, 0.8 Hz, 1H), 7.78 (dd, J = 8.3, 1.1 Hz, 1H), 7.63–7.53 (m, 2H), 7.46 (t, J = 7.9 Hz, 1H), 7.42–7.31 (m, 3H), 7.29 (dd, J = 7.3, 1.3 Hz, 1H), 7.20 (ddd, J = 8.1, 7.3, 1.0 Hz, 1H), 7.17–7.13 (m, 2H), 6.81 (dt, J = 8.0, 0.9 Hz, 1H), 6.15 (s, 1H), 2.35 (s, 3H), 2.27 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 152.2, 142.7, 142.5, 137.6, 136.2, 136.0, 131.4, 131.1, 129.9, 129.2, 128.9, 128.8, 127.6, 127.4, 127.2, 126.1, 125.8, 121.6, 121.1, 118.1, 110.4, 20.9, 14.5; IR (FTIR): 3261, 2945, 2833, 2361, 2343, 1653, 1404, 1332, 1157, 1093, 765, 737, 663 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C25H21N3O2S: 428.1425, found: 428.1427.
:1). Mp 300–302 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 1.8 Hz, 1H), 8.01 (dd, J = 8.3, 1.3 Hz, 1H), 7.90–7.86 (m, 2H), 7.85–7.81 (m, 1H), 7.80–7.76 (m, 2H), 7.64–7.54 (m, 4H), 7.44 (t, J = 7.9 Hz, 1H), 7.39–7.33 (m, 2H), 7.29 (dd, J = 7.3, 1.3 Hz, 1H), 7.16 (ddd, J = 8.2, 7.3, 1.1 Hz, 1H), 6.77 (dt, J = 8.1, 0.9 Hz, 1H), 6.25 (s, 1H), 2.26 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ 152.3, 142.4, 137.6, 136.1, 134.1, 131.4, 131.4, 130.9, 130.0, 129.1, 128.9, 128.8, 128.7, 128.2, 127.9, 127.8, 127.5, 127.4, 126.2, 125.8, 122.8, 121.6, 121.2, 118.1, 110.5, 14.6; IR (FTIR): 2972, 2940, 2865, 2844, 2362, 1597, 1399, 1330, 1054, 1016, 1031, 761, 740 cm−1; HRMS (ESI): m/z [M + H]+ calcd for C28H21N3O2S: 484.1427, found: 484.1427.
General procedure for monoester (7) by desymmetrization of cis-tetrahydrophthalic anhydride (6)
cis-Tetrahydrophthalic anhydride (30 mg, 0.197 mmol) and the corresponding benzimidazole catalyst (0.0197 mmol) were dissolved in dry diethyl ether (3.9 mL). Benzyl alcohol (0.204 mL, 1.97 mmol) was added drop-wise and the resulting solution was stirred at room temperature until meso-anhydride 6 was not detected (monitored by TLC). Then, diethyl ether was evaporated and the residue was dissolved in dichloromethane (5 mL) and extracted with saturated aqueous solution of sodium bicarbonate (2 × 5 mL). The aqueous layer was washed with dichloromethane (1 × 5 mL; to remove traces of benzyl alcohol and catalyst), acidified with conc. hydrochloric acid to pH = 1, and extracted with ethyl acetate (2 × 5 mL). Organic layer was dried over magnesium sulfate and concentrated on a rotovap to give benzyl ester 7.
1H NMR (500 MHz, CDCl3) δ 7.42–7.27 (m, 5H), 5.69 (t, J = 1.8 Hz, 2H), 5.21–5.08 (m, 2H), 3.14–3.06 (m, 2H), 2.67–2.53 (m, 2H), 2.45–2.32 (m, 2H);35 [α]22D +4.51° (c = 1.0, MeOH), ratio: 27:73.35,36
Author contributions
D. T. performed all synthesis and wrote the original draft. I. N. did the X-ray crystallography. O. K. developed chiral analytical and semi-preparative methods and separated both individual atropisomers (−)-5f and (+)-5f. A. P. developed separation method at analytical level and analysed products after desymmetrization reaction by chiral HPLC. P. C. supervised the project and participated in writing – review and editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI† or upon request from the corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Czech Science Foundation (25-16634S) and Internal Grant Agency of Palacký University Olomouc (IGA_PrF_2024_028) is gratefully acknowledged. I.N. would like to acknowledge the financial support from institutional sources of the Department of Inorganic Chemistry, Palacký University Olomouc, Czech Republic.References
- C. R. Shugrue and S. J. Miller, Chem. Rev., 2017, 117, 11894–11951 CrossRef PubMed.
- C. E. Müller and P. R. Schreiner, Angew. Chem., Int. Ed., 2011, 50, 6012–6042 CrossRef PubMed.
- R. P. Wurz, Chem. Rev., 2007, 107, 5570–5595 CrossRef PubMed.
- E. Vedejs and X. Chen, J. Am. Chem. Soc., 1996, 118, 1809–1810 CrossRef.
- E. Vedejs and X. Chen, J. Am. Chem. Soc., 1997, 119, 2584–2585 CrossRef.
- E. A. Stone, B. Q. Mercado and S. J. Miller, Org. Lett., 2019, 21, 2346–2351 CrossRef PubMed.
- A. C. Spivey, T. Fekner and H. Adams, Tetrahedron Lett., 1998, 39, 8919–8922 CrossRef.
- C. Ó. Dálaigh, S. J. Hynes, D. J. Maher and S. J. Connon, Org. Biomol. Chem., 2005, 3, 981–984 RSC.
- D. Díez, M. J. Gil, R. F. Moro, N. M. Garrido, I. S. Marcos, P. Basabe, F. Sanz, H. B. Broughton and J. G. Urones, Tetrahedron: Asymmetry, 2005, 16, 2980–2985 CrossRef.
- T. A. Duffey, S. A. Shaw and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14–15 CrossRef.
- M. R. Crittall, H. S. Rzepa and D. R. Carbery, Org. Lett., 2011, 13, 1250–1253 CrossRef CAS PubMed.
- M. R. Crittall, N. W. G. Fairhurst and D. R. Carbery, Chem. Commun., 2012, 48, 11181–11183 RSC.
- J. C. Ruble, H. A. Latham and G. C. Fu, J. Am. Chem. Soc., 1997, 119, 1492–1493 CrossRef CAS.
- Q. Luo, Z. Tian, J. Tang, J. Wang, Y. Tian, C. Peng, G. Zhan and B. Han, ACS Catal., 2022, 12, 7221–7232 CrossRef CAS.
- S. Placidi, T. Majnoni D'Intignano and R. Salvio, J. Heterocycl. Chem., 2022, 59, 1626–1634 CrossRef.
- R. Nishino, S. Hamada, E. E. Elboray, Y. Ueda, T. Kawabata and T. Furuta, Chirality, 2020, 32, 588–593 CrossRef PubMed.
- R. Nishino, T. Furuta, K. Kan, M. Sato, M. Yamanaka, T. Sasamori, N. Tokitoh and T. Kawabata, Angew. Chem., Int. Ed., 2013, 52, 6445–6449 CrossRef.
- K. A. Connors and N. K. Pandit, Anal. Chem., 1978, 50, 1542–1545 CrossRef.
- N. K. Pandit and K. A. Connors, J. Pharm. Sci., 1982, 71, 485–491 CrossRef.
- G. T. Copeland, E. R. Jarvo and S. J. Miller, J. Org. Chem., 1998, 63, 6784–6785 CrossRef PubMed.
- E. R. Jarvo, G. T. Copeland, N. Papaioannou, P. J. Bonitatebus and S. J. Miller, J. Am. Chem. Soc., 1999, 121, 11638–11643 CrossRef.
- E. R. Jarvo, M. M. Vasbinder and S. J. Miller, Tetrahedron, 2000, 56, 9773–9779 CrossRef.
- E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759–5812 CrossRef PubMed.
- C. E. Müller, L. Wanka, K. Jewell and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 6180–6183 CrossRef.
- Z. Zhang, F. Xie, J. Jia and W. Zhang, J. Am. Chem. Soc., 2010, 132, 15939–15941 CrossRef.
- I. A. Murray, A. Lewendon and W. V. Shaw, J. Biol. Chem., 1991, 266, 11695–11698 CrossRef CAS.
- R. Jorda, K. Paruch and V. Krystof, Curr. Pharm. Des., 2012, 18, 2974–2980 CrossRef CAS.
- J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS PubMed.
- H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef PubMed.
- W. Tan, X.-Y. Wu and F. Shi, ChemCatChem, 2024, 16, e202401022 CrossRef.
- G. Centonze, C. Portolani, P. Righi and G. Bencivenni, Angew. Chem., Int. Ed., 2023, 62, e202303966 CrossRef PubMed.
- A. Gaucherand, E. Yen-Pon, A. Domain, A. Bourhis, J. Rodriguez and D. Bonne, Chem. Soc. Rev., 2024, 53, 11165–11206 RSC.
- D. Toman and P. Cankař, ChemistrySelect, 2022, 7, e202203144 Search PubMed.
- M. Tomanová, L. Jedinák, J. Košař, L. Kvapil, P. Hradil and P. Cankař, Org. Biomol. Chem., 2017, 15, 10200–10211 RSC.
- C. Bolm, I. Schiffers, I. Atodiresei and C. P. R. Hackenberger, Tetrahedron: Asymmetry, 2003, 14, 3455–3467 CrossRef.
- H. Wang, L. Yan, Y. Wu, Y. Lu and F. Chen, Org. Lett., 2015, 17, 5452–5455 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2407032–2407035. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob02052a |
| This journal is © The Royal Society of Chemistry 2025 |