
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monoterpenoid selenophenes derived from (−)-carvone with GPx-like activity†
João P.
Telo
 *a,
Luis F.
Veiros
a,
Vânia
André
ab,
João Ferreira
da Silva
a,
Gonçalo C.
Justino‡
a and
Alexandra M. M.
Antunes
a
*a,
Luis F.
Veiros
a,
Vânia
André
ab,
João Ferreira
da Silva
a,
Gonçalo C.
Justino‡
a and
Alexandra M. M.
Antunes
a
aCentro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: jptelo@tecnico.ulisboa.pt
bAssociação do Instituto Superior Técnico para a Investigação e Desenvolvimento (IST-ID), Avenida António José de Almeida, 12, 1000-043 Lisboa, Portugal
First published on 17th January 2025
Abstract
Organoselenium compounds have been recognized as potential therapeutic agents against several diseases. Specifically, the incorporation of selenium into natural products has been reported to produce positive synergistic biological effects. We report herein the one-pot reaction of the natural monoterpenoid (−)-carvone with selenium bromide, which yields mentoselenophenone 1, together with minor amounts of phenols 2 and 3. A number of derivatives of 1 have also been prepared: the α,α dimer 6, oxime 7 and its Beckmann rearrangement product lactam 8. All except lactam 8 showed antioxidant GPx-like activity, with dimer 6 being the most active compound, followed by phenol 2 and oxime 7.
Introduction
Selenium was discovered in 1817 by the Swedish chemists Berzelius and Gahn, but the organoselenium chemistry only gained momentum after the discovery of selenoxide elimination by Karl B. Sharpless, a syn elimination similar to sulfoxide and nitroxide pyrolysis.1,2 Although initially considered a toxin for humans, selenium is now recognized as an essential micronutrient due to its incorporation, mostly in the form of selenocysteine, in a number of enzymes that play an important role in the antioxidant defense of cells. The two main groups of selenoenzymes are glutathione peroxidases, which catalyse the decomposition of peroxides by glutathione, and thioredoxin reductases.3 Selenoproteins are believed to prevent oncogene activation and cancer cell differentiation, and may also interfere with the tumour microenvironment, influencing cancer progression through the activation of inflammatory and immune responses.The interest in the organoselenium chemistry has significantly increased due to the discovery that these compounds can mimic the effect of glutathione peroxidase (GPx) and reproduce its catalytic activity, thus operating as strong antioxidants.4–6 Moreover, a significant number of selenium compounds show antitumor, antimicrobial, and antiviral properties and have been proposed as promising agents for cancer chemoprevention and treatment.7
Considerable efforts have been made in the synthesis of selenoderivatives of natural products such as terpenoids, coumarins, steroids or vitamins, since this has been shown to generate enhanced or synergistic pharmacological activities.8 We have synthesised selenoderivatives of chrysin and tetramethylquercetin with improved antioxidant activity and cytotoxicity, exhibiting a high differential behavior toward malignant and nonmalignant cells.9 This difference in toxicity led us to further investigate organoselenium compounds derived from other natural compounds. This work reports the formation of selenophene derivatives by the reaction of the naturally occurring terpenoid (−)-carvone with SeBr2. Most syntheses of selenophenes involve the reaction of a selenium electrophile or nucleophile with an acyclic precursor containing a π-system, typically a diene or a diyne, in either one or, most frequently, two steps.10,11 In the one-pot reaction described here, the π-system reacting with the selenium electrophile comprises an alkene and an enol.
Results and discussion
Synthesis of mentoselenophenone 1
The reaction of (−)-carvone with SeBr2 in CH2Cl2 produced mainly mentoselenophenone 1 as a racemic mixture, together with minor amounts of the fully aromatic phenols 2 and 3 (Scheme 1 and Table 1, entries 1 and 2). The structure of 1 was confirmed by single crystal X-ray diffraction data. The reaction is slow and continues after the total consumption of carvone, as shown by the constant release of gaseous HBr. By stopping the reaction after a few hours, an inseparable complex mixture is obtained. Carvone isomerizes into carvacrol in the presence of strong acids like HBr,12 and since two equivalents of HBr are produced in the reaction, the absence of carvacrol among the products suggests that the slow step is not the initial reaction of carvone with SeBr2. | ||
| Scheme 1 | ||
| Entry | Solvent | Equivalents Se | Equivalents Br2 | Time (days) | 1 | 2 | 3 |
|---|---|---|---|---|---|---|---|
| a Yields reflect the amount of isolated product, but the yields of 2 and 3 were computed from the isolated mass of the phenol mixture and the ratio of 2/3 determined by 1H NMR analysis. | |||||||
| 1 | CH2Cl2 | 1.0 | 1.0 | 2 | 32% | 9% | 4% |
| 2 | CH2Cl2 | 1.0 | 1.0 | 3 | 40% | 14% | 8% |
| 3 | CH2Cl2 | 2.0 | 1.0 | 2 | 44% | 8% | — |
| 4 | CH2Cl2 | 2.0 | 1.0 | 3 | 49% | 9% | 1% |
| 5 | CH2Cl2 | 2.0 | 1.0 | 4 | 43% | 12% | 4% |
| 6 | CH2Cl2 | 3.0 | 1.0 | 3 | 23% | 10% | — |
| 7 | CHCl3 | 2.0 | 1.0 | 1 | 41% | 10% | 4% |
| 8 | CHCl3 | 2.0 | 1.0 | 2 | 50% | 10% | 4% |
| 9 | CHCl3 | 2.0 | 1.0 | 3 | 40% | 13% | 6% |
| 10 | CHCl3 | 2.0 | 2.0 | 3 | 16% | 38% | 4% |
| 11 | C6H6 | 2.0 | 1.0 | 2 | 35% | 27% | 3% |
| 12 | C6H6 | 2.0 | 2.0 | 3 | — | 56% | 5% |
By adding equimolar amounts of molecular bromine and elemental selenium, the dark red-brown color of selenium dibromide appears immediately (eqn (1)). However, it has been shown both spectrophotometrically and by 77Se NMR that, in solution, selenium dibromide disproportionates into Se2Br2 and bromine (eqn (2)), with K2 = 0.0252 in CCl4 and K2 = 0.14 in acetonitrile.13,14 The latter value might explain why the reaction does not proceed in acetonitrile and other polar solvents. Se2Br2 is also involved in further disproportionation equilibria, as shown in eqn (3) (K3 = 2.2 × 10−4 in CH2Cl2).
 | (1) |
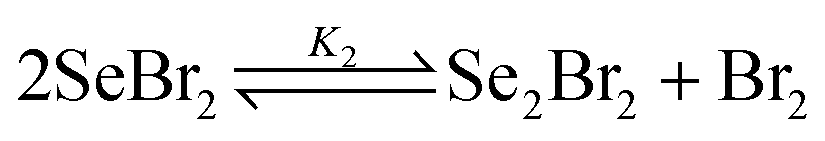 | (2) |
 | (3) |
This shows that free bromine is always present in the reaction mixture, which accounts for the presence of 2 and 3. Phenol 2 is produced by the α-bromination of ketone 1, followed by HBr elimination (see below). Aromatic bromination of 2 in the activated selenophene ring produces 3. Consistently, using two equivalents of selenium improves the yield of 1 (entries 3–5) by reducing the concentration of free Br2 (eqn (1)), but above this ratio, the yield decreases (entry 6). In this case, the main species in solution should be Se2Br2, but the reactive (electrophilic) selenium compound is probably still SeBr2. The reaction is somewhat faster in chloroform, with similar yields (entries 7–9). It is also faster in benzene (entry 11), but with a lower yield of 1 and a higher yield of phenol 2.
The reaction of (−)-carvone with SeBr2 occurs through the addition of the exocyclic double bond and the ketone enol to the selenium electrophile, but the unusual migration of the endocyclic double bond to the selenophene moiety made us study the mechanism by DFT calculations15 (see the ESI† for computational details). A simplified version of the mechanism is presented in Scheme 2, while the complete free energy profile can be found in the ESI (Fig. S1–S4†).
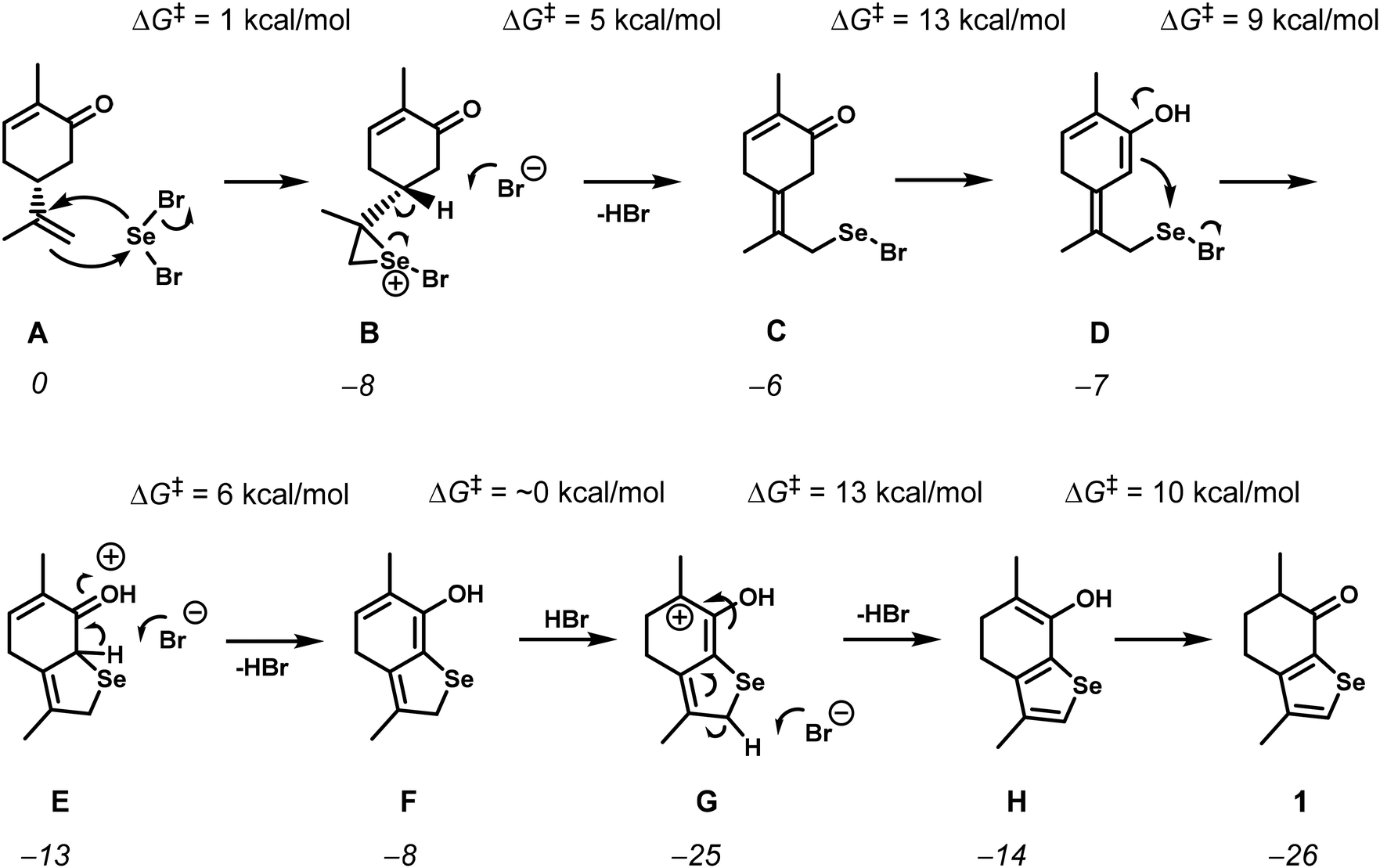 | ||
| Scheme 2 Simplified mechanism for the formation of 1 calculated using DFT. Free energy values of the intermediates (italics) and barriers (ΔG‡) are given in kcal mol−1. | ||
The reaction starts with the addition of selenium dibromide to the isopropenyl group double bond, forming the usual seleniranium ion B,16 with a negligible barrier of only 1 kcal mol−1. The alternative would be the addition to the enol double bond, but this would have a barrier of 14 kcal mol−1, primarily due to the costly energy conversion from the keto to the enol form of carvone (Fig. S5†). In fact, the enol intermediate (I in Fig. S5†) is 13 kcal mol−1 less stable than the corresponding keto form, present in A. The seleniranium ion B eliminates HBr to yield the exocyclic double bond in C, in a simple process with a barrier of 5 kcal mol−1. The next two steps are the enolization of the ketone and the attack of the enol on the selenium atom to yield the cyclic selenide F. The enolization of C has a barrier of 13 kcal mol−1 and yields intermediate D, followed by the attack of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the enol to the Se atom and the consequent formation of the 5-member ring in intermediate E, which is subsequently deprotonated to give F. The formation of the Se–C bond is the rate determining step of the mechanism. The overall barrier of the reaction is 18 kcal mol−1, measured from C′′, the protonated ketone with the exocyclic double bond (Fig. S1†), to the transition state for the formation of the Se–C bond, D′′* (Fig. S2†). The migration of the C5–C6 double bond to the selenophene ring is achieved by protonation of the enol F at C5, followed by deprotonation at the C-atom adjacent to Se in the cycle, resulting in the enol intermediate H. The series of steps, from E to H, present low free energy barriers, with the highest one (13 kcal mol−1) corresponding to C–H deprotonation, from G to H. The reaction is completed through tautomerization from the enol in intermediate H to the keto form in the final product, 1. This is, again, an easy step with a barrier of 10 kcal mol−1. Importantly, all proton transfer steps, namely the enolization processes, are facilitated by the high concentration of HBr released in the previous steps. The overall reaction from A to 1 is highly exergonic with ΔGR = −26 kcal mol−1.
C double bond of the enol to the Se atom and the consequent formation of the 5-member ring in intermediate E, which is subsequently deprotonated to give F. The formation of the Se–C bond is the rate determining step of the mechanism. The overall barrier of the reaction is 18 kcal mol−1, measured from C′′, the protonated ketone with the exocyclic double bond (Fig. S1†), to the transition state for the formation of the Se–C bond, D′′* (Fig. S2†). The migration of the C5–C6 double bond to the selenophene ring is achieved by protonation of the enol F at C5, followed by deprotonation at the C-atom adjacent to Se in the cycle, resulting in the enol intermediate H. The series of steps, from E to H, present low free energy barriers, with the highest one (13 kcal mol−1) corresponding to C–H deprotonation, from G to H. The reaction is completed through tautomerization from the enol in intermediate H to the keto form in the final product, 1. This is, again, an easy step with a barrier of 10 kcal mol−1. Importantly, all proton transfer steps, namely the enolization processes, are facilitated by the high concentration of HBr released in the previous steps. The overall reaction from A to 1 is highly exergonic with ΔGR = −26 kcal mol−1.
Synthesis of mentoselenophenone derivatives
As stated before, the presence of free bromine in the reaction mixture produces phenol 2 by α-bromination of ketone 1 and elimination of HBr (Scheme 1). Further aromatic bromination of 2 on the activated selenophene ring yields phenol 3. With two equivalents of bromine, the yield of the two aromatic phenols increases, as expected (Table 1, entries 10 and 12). This allows the isolation of phenol 2 as the main product in benzene (entry 12).Improved yields of 2 and 3 were obtained upon preparation of their α-bromo ketone precursors by acid-catalysed bromination of mentoselenophenone 1 at 0 °C (Scheme 3). Both products 4 and 5 are stable enough for characterization, including crystal structure determination from single crystal X-ray diffraction data, in the case of 4. Nevertheless, some decomposition was noticed for long storage times, even under cold conditions and a nitrogen atmosphere, as both compounds develop a darker yellow hue. Refluxing the product mixture in toluene/pyridine yields 2 and 3 quantitatively.
 | ||
| Scheme 3 | ||
The same mixture of 4 and 5 was also obtained in similar yields by bromination of 1 under base catalysis with LDA in THF, but when the same reaction was carried out with potassium t-butoxide, the products contained a significant portion of dimer 6 (Scheme 4). Dimer 6 could be isolated in 78% yield by using one-half equivalents of Br2. X-ray crystallography shows 6 to be a racemic mixture. There is no evidence for the formation of the meso stereoisomer. The potassium enolate of 1 probably reacts with the tertiary α-bromo ketone, which might be one of the few examples of an SN2-type reaction on a tertiary carbon atom.17–19 Having the structure of the dimer, the reaction path for an SN2-type reaction was studied by DFT calculations (Fig. S6 in the ESI†). Surprisingly, the transition state was found to be only 5.6 kcal mol−1 above the energy of the separated reactants, an activation barrier that is compatible with the reaction conditions. However, a radical SNR1-type mechanism cannot be excluded, and further studies would be needed to unequivocally determine the mechanism of this reaction.
 | ||
| Scheme 4 | ||
To maximize the number of available selenium derivatives with potential biological effects, the oxime derivative of 1 was prepared in 76% yield. Following treatment of oxime 7 with thionyl chloride, its Beckmann rearrangement product lactam 8 was obtained in 60% yield. The structure of the lactam shows that the oxime has a Z configuration, since the group migrating to the nitrogen atom in the Beckmann rearrangement is the one anti-periplanar to the hydroxyl group.

Crystal structures
Single crystal X-ray diffraction analysis confirmed the structure of compounds 1, 4 and 6 without ambiguity (Fig. 1). Relevant details of the X-ray data analysis are displayed in Table S1.†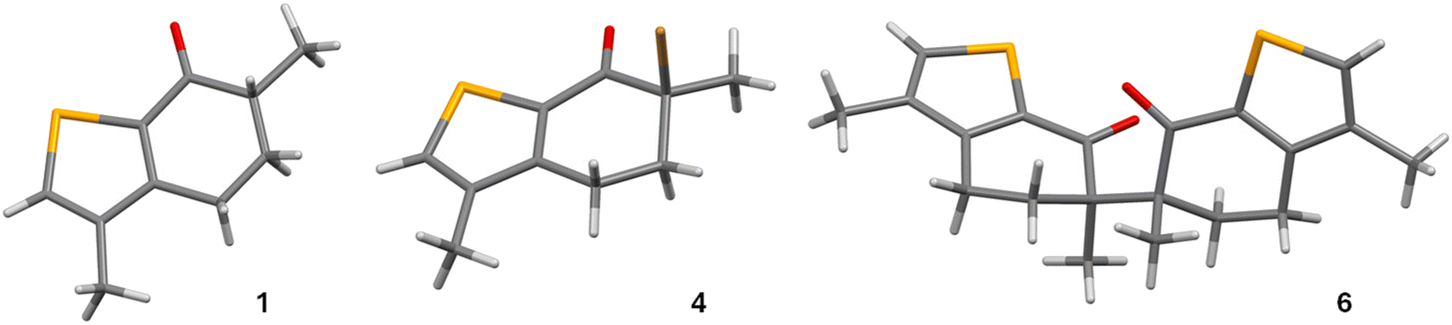 | ||
| Fig. 1 Diagrams of the molecular structures of compounds 1, 4 and 6 (atom colour code: Se, yellow; Br, brown; O, red; C, grey; H, light grey). | ||
Bond lengths and angles are comparable in the three molecules and are within the expected range, as judged from extensive analysis of the values included in the CSD.20 In compound 1, the selenophene ring can be considered planar, according to the very small values of deviation from the plane of all the five atoms. In compounds 4, these values are slightly larger, but the root-mean-square deviation is still small enough for the ring to be considered quasi-planar. In 6, one of the rings has a slightly higher deviation from planarity, but it is still very close to planar (see Fig. 1 and Table S2†). Furthermore, in compound 6, the angle between two Se ring planes is 70.76(6)°.
The bond lengths of all bonds forming the five-membered rings in the three compounds are in good agreement with the planar geometry of these rings. The values of Se(1)–C(2) and Se(1)–C(7a) in the three compounds are similar to the mean value reported in the literature for Se–Csp2 bonds (1.893 Å). The C–C bonds show values typical of conjugated bonds connecting Csp2 atoms (see Table S3†).21
The three compounds crystallize in centrosymmetric space groups (P21/n for 1 and 4 and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) for 6), which means they have inversion symmetry. This symmetry precludes the crystallization of a single enantiomer, as chiral molecules do not have an inversion centre. Therefore, these compounds are racemic mixtures, with equal amounts of left- and right-handed enantiomers coexisting in the crystal lattice. These molecules pack in such a way that each enantiomer is paired with its mirror image, effectively cancelling out chirality at the macroscopic level.
for 6), which means they have inversion symmetry. This symmetry precludes the crystallization of a single enantiomer, as chiral molecules do not have an inversion centre. Therefore, these compounds are racemic mixtures, with equal amounts of left- and right-handed enantiomers coexisting in the crystal lattice. These molecules pack in such a way that each enantiomer is paired with its mirror image, effectively cancelling out chirality at the macroscopic level.
Compounds 1 and 4 crystallize in the P21/n space group, indicating that the molecules are organized with a glide plane and a two-fold screw axis along with an inversion centre. The molecules are arranged such that each chiral molecule has a corresponding molecule of the opposite chirality, leading to a centrosymmetric arrangement in the unit cell, in which each enantiomer occupies specific sites related by symmetry operations (Fig. 2). In 6, which crystallizes in the P space group, molecules are organized in pairs of opposite enantiomers related by the inversion centre within the crystal, which also leads to a centrosymmetric arrangement (Fig. 2).
 | ||
| Fig. 2 Unit cell of compounds 1 (A), 4 (B) and 6 (C) depicting the presence of both left- and right-handed enantiomers (axis: red – a; green – b; blue – c). | ||
GPx-like activity
Selenocompounds can act as antioxidants by reacting with peroxides and be regenerated by glutathione, thus mimicking the catalytic activity of the selenoenzyme GPx. The GPx-like activity of selenocompounds 1–3 and 6 was assessed using the method of Iwaoka et al.,22 where dithiothreitol (DTTred) is used as a reducing cofactor instead of glutathione. The rate of the reaction was monitored by measuring the increase of the disulfide form (DTTox) using 1H NMR spectroscopy in CD3OD. Compounds 4 and 5 are too labile in solution and were not considered in this study. The results are shown in Fig. 3. | ||
| Fig. 3 Percentage of DTTred as a function of time in the oxidation of DTTred (0.1 M) with H2O2 (0.1 M) in the absence (blank) or in the presence of several selenophene catalysts (0.01 M). | ||
To compare the catalytic activity of the different compounds, we calculated the time necessary to oxidize 50% of DTTred, t1/2. Both phenol 2 (t1/2 = 42 min) and oxime 7 (t1/2 = 52 min) exhibit higher catalytic activity than mentoselenophenone 1 (t1/2 = 71 min). The presence of extra OH redox units in both 2 and 7 probably facilitates the catalytic cycle of the selenium atom. In the case of 7, the H atom of the oxime OH group is directly accessible by the selenium atom due to spatial proximity. Phenol 3 has a lower activity (t1/2 = 110 min) due to the electron withdrawing effect of the bromo atom, making the oxidation of the molecule more difficult. The most active selenocompound is dimer 6, with t1/2 = 20 min. The presence of two Se atoms and, possibly, the interaction between the two selenophenone moieties, where the two carbonyls are closely positioned in the crystal structure, might explain this result. Surprisingly, lactam 8, although structurally similar to the other selenocompounds, had no catalytic effect on the oxidation of DTTred. Although the experiments have been repeated twice, we think that the fluctuation of the curve for 8 compared to the blank results from experimental error.
Conclusions
Mentoselenophenone 1 is the main product of the one-pot reaction of (−)-carvone with SeBr2, yielding phenols 2 and 3 as minor products. The synthesis of 1 proceeds through the reaction of the exocyclic carbon–carbon double bond and the endocyclic enol of (−)carvone with selenium bromide. Although in selenoxide elimination, the β-ketoselenide precursor is usually prepared by the reaction of an enol or enolate with selenium bromide, the reactivity of enols towards selenium electrophiles has not been much used in the synthesis of selenophenes or other selenium compounds, and we think this might be an area worth exploring.Several derivatives of compound 1 have also been prepared, and their GPx-like catalytic activity is measured. All but lactam 8 have GPx-like activity, with dimer 6 being the most active.
Experimental
Starting materials
(−)-Carvone was prepared from (+)-limonene according to known procedures.23 Commercial molecular sieves were dried at 200 °C under vacuum before use and kept under nitrogen. Dichloromethane was dried over molecular sieves. Chloroform was freed from ethanol by shaking with concentrated sulphuric acid, washed with water, dried with anhydrous magnesium sulphate, distilled, and kept for short periods over molecular sieves. Benzene was dried by refluxing with sodium wire using the blue benzophenone radical anion as an indicator, distilled, and kept under nitrogen. All other reactants were commercially available and used without purification.
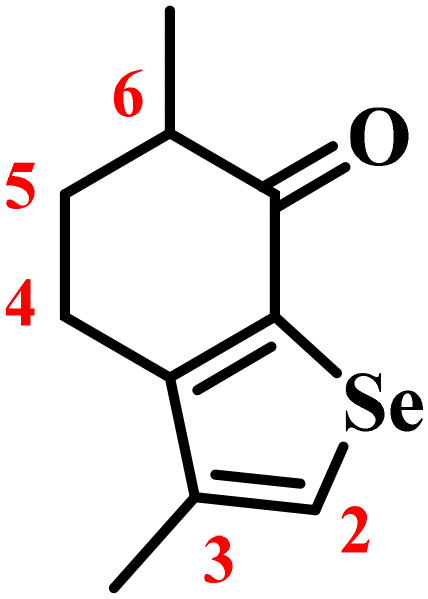
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 2.78–2.71 (dt, H, J = 17.2 Hz, J = 4.7 Hz; 4-C2), 2.65–2.58 (m, 2H; 4-C2, 6-C), 2.28–2.23 (m, H; 5-C2), 2.14 (s, 3H; 3-CC3) 1.99–1.89 (m, H; 5-C2), 1.26 ppm (d, 3H; J = 6.9 Hz; 6-CC3); 13C NMR (400 MHz, CDCl3): δ = 195.9, 154.2, 141.5, 139.9, 134.0, 41.6, 32.2, 25.9, 16.3, 15.4 ppm; HR-EI/MS: m/z (int.) calcd for C10H12OSe [M + H]+ 229.0126 (100); found 229.0133 (100).
), 2.78–2.71 (dt, H, J = 17.2 Hz, J = 4.7 Hz; 4-C2), 2.65–2.58 (m, 2H; 4-C2, 6-C), 2.28–2.23 (m, H; 5-C2), 2.14 (s, 3H; 3-CC3) 1.99–1.89 (m, H; 5-C2), 1.26 ppm (d, 3H; J = 6.9 Hz; 6-CC3); 13C NMR (400 MHz, CDCl3): δ = 195.9, 154.2, 141.5, 139.9, 134.0, 41.6, 32.2, 25.9, 16.3, 15.4 ppm; HR-EI/MS: m/z (int.) calcd for C10H12OSe [M + H]+ 229.0126 (100); found 229.0133 (100).
Alternatively, both products can be obtained in low yield from the aqueous extracts obtained in the synthesis of 1. These were made acidic with concentrated HCl and extracted three times with 25 ml of CH2Cl2. The combined organic fraction was dried and evaporated, and the mixture was separated as described above.
2: mp = 98–99 °C (needles from n-hexane); TLC: Rf = 0.34 (toluene); 1H NMR (400 MHz, CDCl3): δ = 7.46 (s, H; 2-C), 7.24 (d, H, J = 7.9 Hz; 4-C), 7.18 (d, H, J = 8.0 Hz; 5-C), 4.93 (s, H; O), 2.37 (s, 3H; 6-CC3), 2.36 ppm (s, 3H; 3-CC3); 13C NMR (400 MHz, CDCl3): δ = 150.4, 142.8, 135.9, 128.7, 128.3, 122.4, 117.4, 116.4, 16.2, 15.5 ppm; HR-EI/MS: m/z (int.) calcd for C10H10OSe [M + H]+ 226.9970 (100); found 226.9965 (100).
3: mp = 105–106 °C; TLC: Rf = 0.45 (toluene); 1H NMR (400 MHz, CDCl3): δ = 7.16 (AB q, 2H; 4-C, 5-C), 4.94 (s, H; O), 2.33 (s, 3H; 6-CC3), 2.29 ppm (s, 3H; 3-CC3); 13C NMR (400 MHz, CDCl3): δ = 149.5, 141.6, 135.6, 128.5, 128.3, 117.9, 116.5, 111.2, 15.4, 15.0 ppm; HR-EI/MS: m/z (int.) calcd for C10H9BrOSe [M + H]+ 304.9072 (100); found 304.9069 (100).
4: mp = 86–87 °C (from n-hexane); Rf = 0.53 (toluene); 1H NMR (400 MHz, CDCl3): δ = 7.99 (s, H; 2-C), 2.88–2.67 (m, 2H; 4-C2), 2.62–2.55 (m, H; 5-C2), 2.29–2.19 (m, H; 5-C2), 2.17 (s, 3H; 6-CC3), 2.01 ppm (s, 3H; 3-CC3); 13C NMR (400 MHz, CDCl3): δ = 186.8, 153.5, 139.9, 138.4, 136.2, 62.6, 41.0, 27.9, 25.7, 16.6 ppm HR-EI/MS: m/z (int.) calcd for C10H11BrOSe [M + H]+ 306.9228 (100); found 306.9225 (100).
5: mp = 90–91 °C (from n-hexane); Rf = 0.66 (toluene); 1H NMR (400 MHz, CDCl3): 2.88–2.67 (m, 2H; 4-C2), 2.62–2.55 (m, H; 5-C2), 2.29–2.19 (m, H; 5-C2), 2.17 (s, 3H; 6-CC3), 2.01 ppm (s, 3H; 3-CC3); 13C NMR (400 MHz, CDCl3): δ = 186.8, 153.5, 139.9, 138.4, 136.2, 62.6, 41.0, 27.9, 25.7, 16.6 ppm; HR-EI/MS: m/z (int.) calcd for C10H10Br2OSe [M + H]+ 386.8316 (100); found 386.8307 (100).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10).
:10).
Mp = 207–209 °C; Rf = 0.11 (toluene); 1H NMR (400 MHz, CDCl3): δ = 7.59 (s, H; 2-C), 2.99–3.06 (m, H; 5-C2), 2.61–2.77 (m, 2H; 4-C2), 2.13 (s, 3H; 3-CC3), ∼2.12 (m, H; 5-C2), 1.35 ppm (s, 3H; 6-CC3); 13C NMR (400 MHz, CDCl3): δ = 198.4, 151.9, 142.3, 139.5, 134.3, 50.2, 32.9, 23.9, 17.7, 16.3 ppm; HR-EI/MS: m/z (int.) calcd for C10H12OSe [M + H]+ 455.00286 (100); found 455.0013 (100).
Mp = 160–162 °C; 1H NMR (400 MHz, CDCl3): δ = 7.85 (s, H; 2-C), 2.86–2.90 (m, H; 6-C), 2.62–2.70 (m, 2H; 4-C2), 2.15 (s, 3H; 3-CC3), 2.07–2.10 (m, H; 5-C2), 1.84–1.89 (m, H; 5-C2), 1.30 ppm (d, 3H; J = 6.8 Hz; 6-CC3); 13C NMR (400 MHz, CDCl3): δ = 154.3, 145.4, 137.7, 132.0, 126.0, 34.4, 30.1, 25.0, 18.0, 16.3 ppm; HR-EI/MS: m/z (int.) calcd for C10H13NOSe [M + H]+ 244.0235 (100); found 244.0190 (100).
Mp = 144–145 °C; 1H NMR (400 MHz, CDCl3): δ = 7.73 (s, H; 2-C), 5.89 (s, H; N), 3.59–3.60 (m, H; 6-C), 2.70–2.77 (m, 2H; 4-C2), 2.08–2.13 (m, H; 5-C2), 2.11 (s, 3H; 3-CC3), 1.98–2.02 (m, H; 5-C2), 1.31 ppm (d, 3H; J = 6.7 Hz; 6-CC3); 13C NMR (400 MHz, CDCl3): δ = 166.0, 144.4, 141.4, 140.5, 131.0, 48.6, 34.5, 30.4, 22.2, 18.0 ppm; HR-EI/MS: m/z (int.) calcd for C10H13NOSe [M + H]+ 244.0235 (100); found 244.0190 (100).
Single crystal X-ray diffraction
X-ray crystallographic data for compounds 1, 4 and 6 (CCDC 2394794–2394796†) were collected from single crystals using an area detector diffractometer (Bruker AXS-KAPPA APEX II) at room temperature with graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation. Further details on data collection and structure determination and refinement are available in the ESI.†Table 2 lists the main crystallographic details, and complete crystallographic details are provided in Table S1 (see the ESI†).| 1 | 4 | 6 | |
|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. | |||
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21/n |
P |
| a (Å) | 7.300(7) | 7.767(3) | 10.0831(14) |
| b (Å) | 10.3528(9) | 10.906(4) | 10.4303(15) |
| c (Å) | 12.964(12) | 12.122(4) | 10.7237(16) |
| α (°) | 90 | 90 | 71.641(6) |
| β (°) | 91.02 | 90.88(2) | 70.303(6) |
| γ (°) | 90 | 90 | 63.501(6) |
| V (Å3) | 980.1(15) | 1026.7(6) | 932.0(2) |
| Z | 4 | 4 | 2 |
| GoF | 1.019 | 1.022 | 1.012 |
| Final R indicesa,b [I > 2σ(I)] | R 1 = 0.0650 | ||
| wR2 = 0.1539 | R 1 = 0.0376 | ||
| wR2 = 0.0827 | R 1 = 0.0561 | ||
| wR2 = 0.0915 | |||
Data availability
Crystallographic data for compounds 1, 4 and 6 have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2394794–2394796†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Fundação para a Ciência e a Tecnologia (FCT), I.P/MCTES, through national funds PIDDAC – CQE UIDB/00100/2020 (https://doi.org/10.5449/UIDP/00100/2020) and UIDP/00100/2020 (https://doi.org/10.54499/UIDB/00100/2020), and IMS, the Associated Laboratory, funded by Project LA/P/0056/2020 (https://doi.org/10.54499/LA/P/0056/2020). VA acknowledges FCT for contract CEECIND/00283/2018/CP1572/CT0004 (https://doi.org/10.54499/CEECIND/00283/2018/CP1572/CT0004).References
- D. N. Jones, D. Mundy and R. D. Whitehouse, Steroidal selenoxides diastereoisomeric at selenium; syn-elimination, absolute configuration, and optical rotatory dispersion characteristics, J. Chem. Soc., Chem. Commun., 1970, 86–87, 10.1039/C29700000086.
- K. B. Sharpless, M. W. Young and R. F. Lauer, Reactons of Selenoxides – Thermal syn-elimination and H2O-18 Exchange, Tetrahedron Lett., 1973, 1979–1982, DOI:10.1016/S0040-4039(01)96098-8.
- L. B. Maia, B. K. Maiti, I. Moura and J. J. G. Moura, Selenium-More than Just a Fortuitous Sulfur Substitute in Redox Biology, Molecules, 2024, 29, 120, DOI:10.3390/molecules29010120.
- V. Nascimento, E. E. Alberto, D. W. Tondo, D. Dambrowski, M. R. Detty, F. Nome and A. L. Braga, GPx-Like Activity of Selenides and Selenoxides: Experimental Evidence for the Involvement of Hydroxy Perhydroxy Selenane as the Active Species, J. Am. Chem. Soc., 2012, 134, 138–141, DOI:10.1021/ja209570y.
- N. V. Barbosa, C. W. Nogueira, P. A. Nogara, A. F. de Bem, M. Aschner and J. B. T. Rocha, Organoselenium compounds as mimics of selenoproteins and thiol modifier agents, Metallomics, 2017, 9, 1703, 10.1039/c7mt00083a.
- J. M. Anghinoni, P. T. Birmann, M. J. da Rocha, C. S. Gomes, M. J. Davies, C. A. Brüning, L. Savegnago and E. J. Lenardão, Recent Advances in the Synthesis and Antioxidant Activity of Low Molecular Mass Organoselenium Molecules, Molecules, 2023, 28, 7349, DOI:10.3390/molecules28217349.
- D. Bartolini, L. Sancineto, A. Fabro de Bem, K. D. Tew, C. Santi, R. Radi, P. Toquato and F. Galli, Selenocompounds in Cancer Therapy: An Overview, Adv. Cancer Res., 2017, 136, 259–302, DOI:10.1016/bs.acr.2017.07.007.
- W. Hou and H. Xu, Incorporating Selenium into Heterocycles and Natural Products—From Chemical Properties to Pharmacological Activities, J. Med. Chem., 2022, 65, 4436–4456, DOI:10.1021/acs.jmedchem.1c01859.
- I. L. Martins, C. Charneira, V. Gandin, J. L. Ferreira da Silva, G. C. Justino, J. P. Telo, A. J. S. C. Vieira, C. Marzano and A. M. M. Antunes, Selenium-Containing Chrysin and Quercetin Derivatives: Attractive Scaffolds for Cancer Therapy, J. Med. Chem., 2015, 58, 4250–4265, DOI:10.1021/acs.jmedchem.5b00230.
- P. S. Hellwig, T. J. Peglow, F. Penteado, L. Bagnoli, G. Perin and E. J. Lenardão, Recent Advances in the Synthesis of Selenophenes and Their Derivatives, Molecules, 2020, 25, 5907, DOI:10.3390/molecules25245907.
- W. Wu, W. Liu, D. Song and L. Yan, Synthetic routes to selenophenes (biologically valuable molecules), Synth. Commun., 2021, 51, 2924–2943, DOI:10.1080/00397911.2021.1958229.
- R. A. Kjonaas and S. P. Mattingly, Acid-Catalyzed Isomerization of Carvone to Carvacrol, J. Chem. Educ., 2005, 82, 1813, DOI:10.1021/ed082p1813.
- N. W. Tideswell and J. D. McCullough, Selenium Bromides. I. A Spectrophotometric Study of the Dissociation of Selenium Tetrabromide and Selenium Dibromide in Carbon Tetrachloride Solution, J. Am. Chem. Soc., 1956, 78, 3026–3029, DOI:10.1021/ja01594a025.
- M. Lamoureux and J. Milne, Selenium chloride and bromide equilibria in aprotic solvents; a 77Se NMR study, Polyhedron, 1990, 9, 589–595, DOI:10.1016/S0277-5387(00)86238-5.
- (a) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed; (b) Free energy values were obtained at the PBE0-D3/6-311++G(d,p)//PBE0/6-31+G(d,p) level using the Gaussian 09 package. All calculations included solvent effects using the PCM/SMD model. A full account of the computational details and a complete list of references are provided in the ESI.†.
- T. Wirth, G. Fragale and M. Spichty, Mechanistic Course of the Asymmetric Methoxyselenenylation Reaction, J. Am. Chem. Soc., 1998, 120, 3376–3381, DOI:10.1021/ja974177d.
- O. E. Edwards and C. Grieco, Displacement at Tertiary Carbon, Can. J. Chem., 1974, 52, 3561, DOI:10.1139/v74-531.
- M. Mascal, N. Hafezi and M. D. Toney, 1,4,7-Trimethyloxatriquinane: SN2 Reaction at Tertiary Carbon, J. Am. Chem. Soc., 2010, 132, 10662–10664, DOI:10.1021/ja103880c.
- Y.-Q. Zhang, C. Poppel, A. Panfilova, F. Bohle, S. Grimme and A. Gansäuer, SN2 Reactions at Tertiary Carbon Centers in Epoxides, Angew. Chem., Int. Ed., 2017, 56, 9719–9722, DOI:10.1002/anie.201702882.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179, DOI:10.1107/S2052520616003954.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19, 10.1039/P298700000S1.
- F. Kumakura, B. Mishra, K. I. Priyadarsini and M. Iwaoka, A Water-Soluble Cyclic Selenide with Enhanced Glutathione Peroxidase-Like Catalytic Activities, Eur. J. Org. Chem., 2010, 440–445, DOI:10.1002/ejoc.200901114.
- J. P. Telo, Synthesis of (-)-carvone, in Comprehensive Organic Chemistry Experiments for the Laboratory Classroom, ed. C. A. M. Afonso, N. R. Candeias, D. P. Simão, A. F. Trindade, J. A. S. Coelho, B. Tan and R. Franzén, The Royal Society of Chemistry, 2016, pp. 244–246 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2394794–2394796. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01942c |
| ‡ Current address: Escola Superior de Tecnologia do Barreiro, Instituto Politécnico de Setúbal, Rua Américo da Silva Marinho, 2839–001 Barreiro, Portugal. |
| This journal is © The Royal Society of Chemistry 2025 |