
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransformative reactions in nitroarene chemistry: C–N bond cleavage, skeletal editing, and N–O bond utilization
Keiichiro
Iizumi
and
Junichiro
Yamaguchi
 *
*
Department of Applied Chemistry, Waseda University, 513 Wasedatsurumakicho, Shinjuku, Tokyo 162-0041, Japan. E-mail: junyamaguchi@waseda.jp
First published on 10th January 2025
Abstract
Nitroarenes are highly versatile building blocks in organic synthesis, playing a pivotal role in various reactions. Common transformations involving nitroarenes include nucleophilic aromatic substitution (SNAr) reactions, where the nitro group functions both as a potent electron-withdrawing group that activates the aromatic ring and as a leaving group facilitating the substitution. Additionally, the direct transformation of nitro groups, such as reduction-driven syntheses of amines and carboxylic acids, as well as ipso-substitution SNAr reactions, have been extensively explored. Interactions between ortho-nitro groups and neighboring substituents also provide unique opportunities for selective transformations. However, beyond these well-established processes, direct transformations of nitro groups have been relatively limited. In recent years, significant advancements have been made in alternative methodologies for nitro group transformations. This review focuses on the latest progress in novel transformations of nitroarenes, with emphasis on three major categories: (i) functional group transformations involving C–N bond cleavage in nitroarenes, (ii) skeletal editing via nitrene intermediates generated by N–O bond cleavage, and (iii) the utilization of nitroarenes as an oxygen source through N–O bond cleavage. These developments under-score the expanding utility of nitroarenes in modern organic synthesis.
 Keiichiro Iizumi | Keiichiro Iizumi was born in Tokyo in 1997. He enrolled at Waseda University, where he earned his bachelor's degree in 2020. He then continued to graduate studies at Waseda University, receiving his master's degree in 2022. Currently, in his final year of the doctoral program under Prof. Junichiro Yamaguchi |
 Junichiro Yamaguchi | Junichiro Yamaguchi earned a B.S. in 2002 from Tokyo University of Science and then a Ph.D. in 2007 under the direction of Prof. Yujiro Hayashi. Following postdoctoral studies with Prof. Phil S. Baran at The Scripps Research Institute, he began his career at Nagoya University in 2008 as an assistant professor working with Prof. Kenichiro Itami, and was promoted to associate professor in 2012. He moved to Waseda University in 2016 as a principal investigator and was promoted to full professor in 2018. |
1. Introduction
Nitroarenes a crucial class of compounds in organic chemistry, valued for their high reactivity and versatility in diverse chemical transformations. The presence of the nitro group (–NO2) imparts unique electronic properties to the aromatic ring, making nitroarenes indispensable intermediates across a broad range of synthetic applications.1 Traditionally, nitroarenes have been utilized in nucleophilic aromatic substitution (SNAr) reactions, where the electron-withdrawing nature of the nitro group facilitates the substitution of leaving groups, particularly at the ortho- and para-positions (Fig. 1A).2 These reactions often proceed via the formation of a Meisenheimer complex, a delocalized anionic intermediate that stabilizes the transition state and promotes reaction progression. Additionally, the nitro group itself can act as a leaving group in ipso-substitution SNAr reactions, highlighting its dual functionality as both an activating and a leaving group.3 Another unique transformation is vicarious nucleophilic substitution (VNS), in which the leaving group is present on the nucleophile rather than the aromatic ring, resulting in the formal substitution of a hydride on the aromatic ring.4 These transformations further demonstrate the functional versatility of nitroarenes in synthetic methodologies, underpinned by their ability to enable selective functionalization, particularly in adjacent positions. Interactions between ortho-nitro groups and neighboring substituents, such as hydrogen bonding or chelation effects, can also facilitate regioselective transformations, broadening the scope of nitroarene reactivity.1a In addition to their role in SNAr reactions, nitroarenes are commonly employed in reductions, where the nitro group is converted into valuable functional groups such as amines and carboxylic acids.5 The reductive transformation of nitroarenes has been extensively studied and remains one of the most widely used methods for generating amine-containing compounds.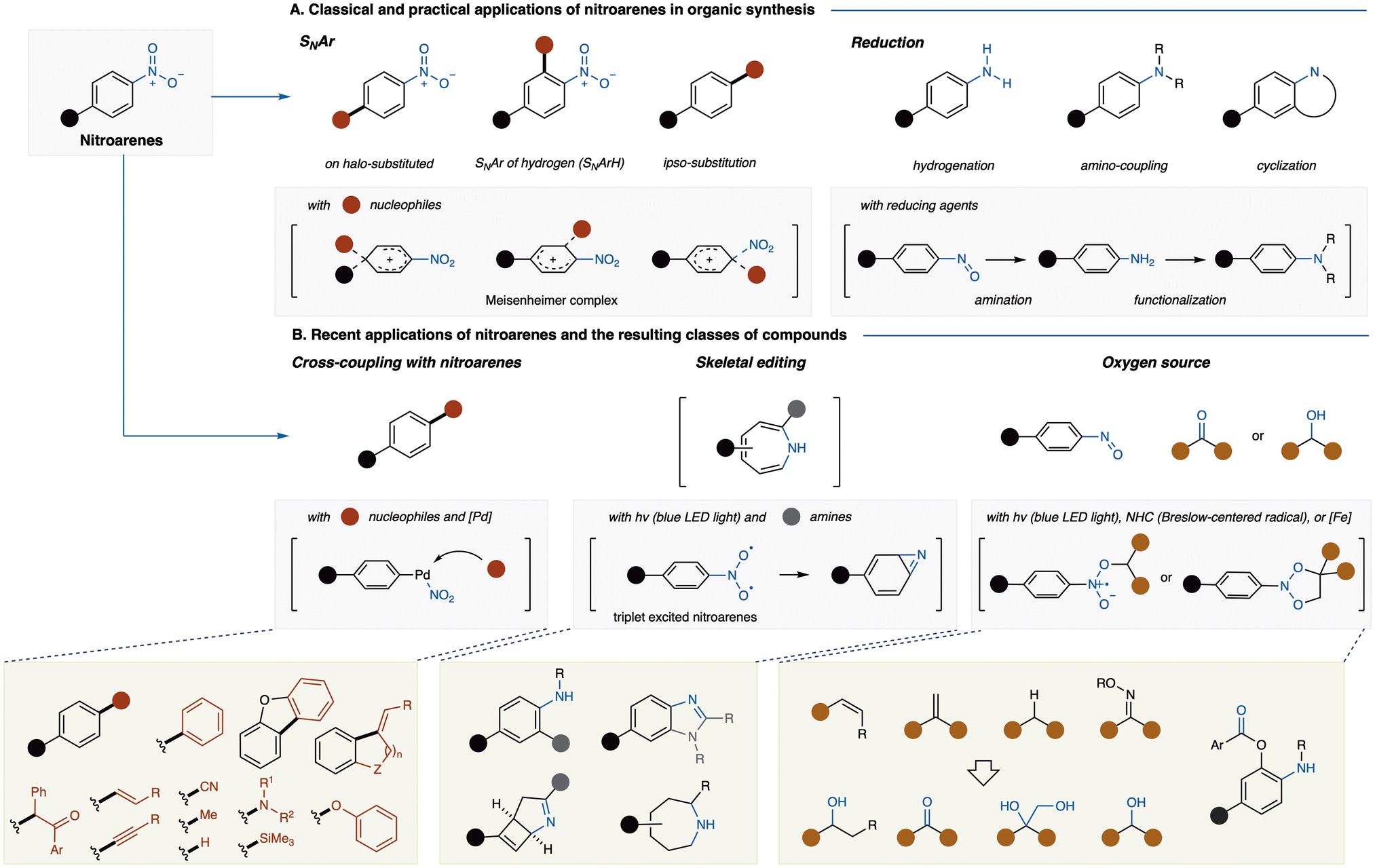 | ||
| Fig. 1 Synthetic applications of nitroarenes in organic chemistry. (A) Traditional and widely used applications of nitroarenes in nucleophilic aromatic substitution (SNAr) and reduction reactions for the synthesis of amines, carboxylic acids, and functionalized aromatic compounds. (B) Emerging applications of nitroarenes in modern synthetic methodologies, including denitrative cross-coupling, skeletal editing, and oxygen transfer reactions, along with examples of the resulting compound classes. | ||
While traditional transformations of nitroarenes—primarily substitution and reduction—are well-explored, the past decade has witnessed remarkable advancements in alternative transformation methods. Notably, novel approaches in C–N and N–O bond cleavage, often catalyzed or mediated by nitrene intermediates, have introduced efficient and sustainable routes to complex molecular architectures. These innovations not only expand the synthetic utility of nitroarenes but also align with the growing demand for greener and more selective methodologies in organic synthesis.
In this review, we focus on three emerging areas of nitroarene chemistry that have garnered considerable attention (Fig. 1B): (i) functional group transformations involving C–N bond cleavage, particularly through palladium-catalyzed cross-coupling with nucleophiles; (ii) skeletal editing using triplet excited-state nitroarenes,6 which proceed via diazirine intermediates; and (iii) the utilization of nitroarenes as an oxygen source through N–O bond cleavage. These three cutting-edge reactions share the common feature of utilizing inexpensive nitroarenes, but their reaction sites differ entirely. Denitrative coupling targets the carbon adjacent to the nitro group, skeletal editing leverages the nitrogen atom of the nitro group, and oxygen transfer reactions utilize the oxygen atoms of the nitro group. However, they all share the clever exploitation of the electronic polarization inherent in aromatic nitro compounds. These transformations represent a significant shift in how nitroarenes can be applied in modern organic synthesis, offering new strategies for molecular editing and functionalization. By exploring these recent developments, this review aims to provide a comprehensive overview of the expanding utility of nitroarenes and their potential for future applications in synthetic organic chemistry.
2. Functional group transformations involving C–N bond cleavage in nitroarenes
Cross-coupling reactions utilizing haloarenes are well-established and widely employed in organic synthesis, serving as a foundational tool for constructing diverse organic compounds.7 Over the years, numerous cross-coupling methodologies have been developed, enabling access to a broad range of molecular architectures. However, recent attention has increasingly shifted towards identifying alternative electrophiles that minimize halogen-containing waste, a major byproduct of traditional haloarene couplings.8 Aromatic nitro compounds have emerged as promising alternatives, and transition metal-catalyzed denitrative couplings have been increasingly reported (Fig. 2A). | ||
| Fig. 2 Advances in catalytic denitrative transformations of nitroarenes. (A) Timeline highlighting key developments in catalytic denitrative reactions of nitroarenes. (B) Pd-Catalyzed Suzuki–Miyaura coupling enabled by C–NO2 bond activation. (C) Structures of effective ligands and representative nucleophiles utilized in denitrative coupling reactions. | ||
In 2011, Wu and Chen reported a rhodium-catalyzed denitrative cross-coupling reaction between electron-deficient nitroarenes and aryl boronic acids.9 This work was followed by several studies documenting etherification and sulfidation reactions of nitroarenes using various transition metal catalysts. However, these metal-catalyzed denitrative couplings have primarily been limited to nitroarenes bearing electron-withdrawing substituents, with transformations largely restricted to the formation of C–O or C–S bonds, thereby limiting the broader utility of nitroarenes as aryl electrophiles.10
A breakthrough came with the work of Nakao and co-workers, who demonstrated that Pd/BrettPhos catalysts could effectively activate C–NO2 bonds in nitroarenes, enabling Suzuki–Miyaura coupling (Fig. 2B).10a This catalytic system has since proven effective in facilitating C–NO2 bond cleavage, expanding the synthetic potential of nitroarenes in cross-coupling reactions. Subsequent studies have highlighted the utility of N-heterocyclic carbene (NHC) ligands, including imidazopyridine-based and pyridine-fused triazolylidene carbenes, which have shown significant efficacy in C–NO2 bond activation.11 Diverse denitrative transformations of nitroarenes, including amination,10b hydrogenation,10c intramolecular C–H arylation,10d methylation,10e Mizoroki–Heck reaction,10f,g and Sonogashira coupling,10h have been reported (Fig. 2C). A comprehensive summary of these transformations up to 2021 was provided by Yamaguchi group.12a In this review, we focus on denitrative coupling reactions involving a range of nucleophiles reported since 2021, with particular attention to recent advances in reactions that proceed without the use of transition metal catalysts.12b
2.1. Etherification
Diaryl ethers are essential structural motifs commonly found in natural products and bioactive molecules.13 Methods for their synthesis typically involve Ullmann reactions catalyzed by copper or iron, as well as Pd-catalyzed Buchwald–Hartwig etherifications.14 However, these conventional methods rely on haloarenes as substrates, which can present environmental concerns due to halogenated byproducts. As a complementary, halogen-free approach, etherification from aromatic nitro compounds has been developed, notably by a Nakao group (Scheme 1A).15 Using Pd(acac)2/L1 catalyst system with K3PO4 as a base, nitroarenes 1 were reacted with arenols 2 to afford diaryl ethers 3 in good to moderate yields. | ||
| Scheme 1 Pd-Catalyzed denitrative etherification. (A) Reaction conditions and ligand screening for the etherification of nitroarenes with arenols (Nakao group). (B) Substrate scope of nitroarenes and alcohols applicable to Pd-catalyzed etherification. | ||
An extensive ligand screening revealed that ligands L1 and L2, characterized by bulky substituents on the oxygen atom, demonstrated catalytic activity in this transformation, whereas BrettPhos, a commonly used ligand in denitrative coupling reactions, showed no activity (Scheme 1A). This method proved effective for various nitroarenes, including those with electron-donating and electron-withdrawing substituents, converting them into the corresponding diaryl ethers 3 (Scheme 1B). Additionally, substrates such as 1-nitronaphthalene and 2-methoxy-3-nitropyridine were successfully applied in this reaction. In terms of alcohol substrates, not only substituted arenols but also benzyl alcohol and even water were compatible, demonstrating the method's versatility.
The Nakao group further investigated the nitrite–phenoxide exchange step by adding premixed p-cresol and K3PO4 to a solution of A in 1,4-dioxane-d8 (Scheme 2). After stirring the mixture at room temperature for 24 h, a new signal was observed in the 31P NMR spectrum, shifted 3 ppm downfield from that of A. Although the product could not be fully characterized, this signal is presumed to correspond to the formation of (L1)PdII(Ph)(O-p-tol) (B) as diaryl ether 3a was detected upon heating the complex to 80 °C. The thermal stability of this complex below 80 °C suggests that reductive elimination is the rate-determining step. Additionally, the %Vbur values were calculated for each ligand to evaluate the steric hindrance around the metal center.16 These findings support the observation that reactions are less favorable with bulkier ligands and suggest a correlation between the progression of reductive elimination and the steric bulk of the ligands.
 | ||
| Scheme 2 Mechanistic studies of the nitrite–phenoxide exchange and reductive elimination steps. | ||
2.2. α-Arylation of ketones
Catalytic α-arylation of carbonyl compounds with aryl halides is an efficient strategy for the direct synthesis of α-aryl carbonyl compounds, which serve as pivotal intermediates in pharmaceuticals and π-conjugated materials.17 Recently, Wu reported the α-arylation of ketone 4 using nitroarenes 1 as arylating agents under a Pd/BrettPhos catalytic system in the presence of K3PO4, affording a variety of α-arylated carbonyl compounds 5 (Scheme 3).18 his methodology demonstrated good compatibility with various functional groups. The reaction tolerated a broad range of ketones 4, from aryl phenyl ketones to alkyl phenyl ketones, illustrating the versatility of the α-arylation process. | ||
| Scheme 3 Pd-Catalyzed α-arylation of ketones with nitroarenes (Wu group). | ||
Utilizing the inherent reactivity of 4-halonitrobenzene in SNAr reactions, a one-pot, three-component denitrative α-arylation was performed (Scheme 4). Specifically, 4-fluoronitroarene (6), 1,2-diphenylethan-1-one (7), and phenol (8) were subjected to Pd/BrettPhos catalytic conditions, enabling a sequential one-pot SNAr and denitrative α-arylation to afford 9 in a good yield.
 | ||
| Scheme 4 Three-component coupling by SNAr/denitrative α-arylation. | ||
2.3. Migita–Kosugi–Stille coupling
The Migita–Kosugi–Stille reaction has become a cornerstone in organic synthesis for constructing carbon–carbon single bonds.19 This base-free, transition metal-catalyzed reaction between organostannanes and organic electrophiles is one of the most efficient and widely used methods for synthesizing highly functionalized molecules. Its outstanding selectivity, robust functional group tolerance, and broad applicability make it indispensable for various applications, such as the synthesis of natural products and the development of conjugated polymers for electronic devices.In 2022, the Hudhomme group reported a Pd-catalyzed biaryl synthesis of nitroarenes 10 with arylstannanes 11 in the presence of LiCl (Scheme 5).20 This study focuses on the functionalization of perylene–diimide (PDI) derivatives 10, which are anticipated to exhibit unique electronic properties in organic electronics.21 Accordingly, the nitroarene substrates are limited to a select few examples, primarily compound 10 and its analogues. Conversely, the reaction tolerates a diverse array of organostannanes, enabling the synthesis of a wide range of PDI derivatives 12, including those containing heteroaromatic rings and alkynes.
 | ||
| Scheme 5 Denitrative Migita–Kosugi–Stille coupling of nitoroarenes (Hudhomme group). | ||
2.4. Silylation
In organic synthesis chemistry, silicon-containing compounds are valuable intermediates due to their ability to introduce a variety of functional groups.22 Despite numerous advancements in C–Si bond formation, denitrative silylation of aromatic nitro compounds has remained a significant challenge. In 2023, Yu, Duan and Li reported a method for denitrative silylation of aromatic compounds using a palladium catalyst in combination with 1 and disilane 13 (Scheme 6).23 They identified hexamethyldisilane (13) as an effective silylation reagent, which, under Pd/BrettPhos catalysis with K3PO4 as a base, produced silylated arenes 14 in good yields. This protocol demonstrated broad applicability across a range of nitroarenes, including heteroarenes, under the optimized conditions. | ||
| Scheme 6 Pd-Catalyzed denitrative silylation of nitroarenes (Duan and Li group). | ||
To showcase the synthetic versatility of the aryl silane products 14, Scheme 7 illustrates several representative transformations of aryl silanes into valuable building blocks. Borylation of 14 with BBr3 afforded dibromo(p-tolyl)borane (15) in 73% yield (I). Oxidative homocoupling of 14 using a palladium catalyst and o-chloranil yielded biaryl 16 in 86% yield (II). Additionally, 14 was successfully converted into fluoroarene 17 and diiodinated product 18, demonstrating the transformation of the C(sp2)–Si bond into the C(sp2)–halogen bond (III and IV).
 | ||
| Scheme 7 Derivatizations of aryl trimethylsilane compounds. | ||
2.5. Buchwald–Hartwig amination
The Buchwald–Hartwig amination, a Pd-catalyzed coupling of aryl halides with amines, is well-established method for synthesizing substituted arylamines.24 Traditionally, the synthesis of arylamines from nitroarenes has required multiple steps, including nitro group reduction followed by N-substitution. To simplify this process, the Nakao and Wu groups independently developed Buchwald–Hartwig amination methods for nitroarenes.11b However, these methods did not address the N-arylation of N–H heteroarenes.In response, Yu and Duan developed a Buchwald–Hartwig amination oprotocol for nitroarenes with N–H heteroarenes (Scheme 8).25 Using Pd(acac)2/BrettPhos catalyst system and K3PO4 as a base, nitroarenes 1 were coupled with N–H heteroarene 19 to give N-arylated heterocycles 20 in moderate to good yields. The reaction tolerated various nitroarenes 1 with electron-donating and electron-withdrawing substituents, successfully converting them into the corresponding arylamines 20. Other nitroarenes, including 2-methoxy-3-nitropyridine and 5-nitroindole, also proved compatible with the reaction conditions. For the amine component, a range of N–H heteroarenes, including pyrrole, indole, and carbazole, were effective substrates.
 | ||
| Scheme 8 Pd-Catalyzed denitrative amination of nitroarenes with N–H heteroarenes (Yu and Duan group). | ||
More recently, the Li group achieved the formation of C(sp2)–N bonds via a Buchwald–Hartwig-type amination using synthetically upstream nitroarenes 1 as the sole starting materials, by reducing a portion of the nitroarenes in situ to amines (Scheme 9A).26 Using Pd/BrettPhos as the optimal catalyst and B2pin2 as the most effective reductant, they successfully synthesized the desired diarylamine 21. Nitroarenes 1 bearing both electron-donating and electron-withdrawing substituents afforded symmetrical diarylamines 21 in good yields. Additionally, nitroarenes containing heteroaromatic rings, such as pyridine and benzofuran, were effectively converted to the corresponding hetero diarylamines 21.
 | ||
| Scheme 9 Pd-Catalyzed denitrative amination of nitroarenes as sole starting materials (Li group). (A) In situ reduction of nitroarenes 1 to amines for C(sp2)–N bond formation, yielding symmetrical diarylamines 21. (B) Investigation of potential intermediates, with aniline identified as a key intermediate for diarylamine formation 24. | ||
To investigate potential intermediates, they tested compounds 23 including aniline, nitrosobenzene, N-phenylhydroxylamine, azobenzene, and azoxybenzene in reactions with nitroarene 22 in the absence of the reductant B2pin2 (Scheme 9B). Only aniline led to the formation of the desired diarylamine product 24, achieving a yield of 62%. These results strongly support the hypothesis that arylamine is likely a key intermediate in this transformation.
In addition, by adjusting the equivalents of the reductant B2pin2, they successfully synthesized symmetrical triarylamines 25 directly from nitroarenes 1 (Scheme 10). This reaction was compatible with nitroarenes bearing both electron-donating and electron-withdrawing substituents.
 | ||
| Scheme 10 Substrate scope of nitroarenes to form symmetrical triarylamines. | ||
Almost concurrently, Chen and co-workers developed a method for synthesizing diarylamines via the reduction and coupling of nitroarenes 1 using palladium catalysts with NHC ligands (Scheme 11).11g They identified Pd/NHC A complex as the optimal catalyst, with Et3SiH serving as the most effective reductant for nitroarenes 1 in this transformation, affording the desired diarylamine 21. This reaction consistently produced symmetrical diarylamines 21 in good yields from nitroarenes 1 containing both electron-donating and electron-withdrawing substituents.
 | ||
| Scheme 11 Pd-Catalyzed denitrative amination using nitroarenes as the sole starting material (Chen group). | ||
2.6. Mizoroki–Heck reaction
The Mizoroki–Heck reaction between aryl halides and alkenes is a well-established method for constructing aryl alkenes.27 Recently, Yamaguchi and You introduced a Pd-catalyzed denitrative Mizoroki–Heck reaction.10f,g Although denitrative Mizoroki–Heck reactions have been explored, their scope has generally been limited to activated alkenes, such as acrylates and styrenes. Expanding this methodology, the Wu group developed a palladium-catalyzed regioselective Mizoroki–Heck reaction using unactivated alkenes 26 and nitroarenes 1. (Scheme 12).28 The authors reoptimized previously reported conditions for use with unactivated alkenes, employing anisole as the solvent to facilitate the reaction at higher temperatures. This practical protocol enabled the synthesis of a wide range of denitrative alkenylated products 27 with high E/Z selectivity. | ||
| Scheme 12 Denitrative Mizoroki–Heck reaction of nitroarenes with unactivated alkenes (Wu group). | ||
More recently, the Yamaguchi group reported a Pd-catalyzed denitrative intramolecular Mizoroki–Heck reaction of nitroarenes 1 (Fig. 3A).29 Since a tethered-alkene group can be introduced onto the nitroarene core through nucleophilic aromatic substitution (SNAr), this denitrative approach provides a convergent synthetic route to benzo-fused cyclic compounds 28. They found that a combination of Pd(acac)2, BrettPhos and Cs2CO3 as the base provided the desired cyclized products 28 in moderate yields.
 | ||
| Fig. 3 Denitrative intramolecular Mizoroki–Heck reaction (Yamaguchi group). (A) Substrate scope. (B) Plausible mechanism. | ||
Based on previous reports, they proposed the following mechanism for intramolecular Mizoroki–Heck reaction (Fig. 3B). The reaction begins with oxidative addition of Pd(0) species I to nitroarene 1, generating intermediate II. Intramolecular carbopalladation then forms the alkyl–Pd(II) species III, which undergoes β-hydrogen elimination to produce intermediate IV. Subsequent base-assisted reductive elimination regenerates the active Pd(0) species I, while product 28 dissociates from IV. Under high-temperature conditions, exo–endo isomerization of the cyclic olefin can occur via a reversible Pd–H migratory insertion between IV and IV′, facilitated by β-hydrogen elimination of V.30
2.7. Cyanation
Aryl nitriles are essential intermediates in the development of pharmaceuticals and organic electronic materials, playing a key role in the synthesis of therapeutic compounds such as citalopram, used for treating major depression, and etravirine, used for HIV treatment.31 The cyanation of nitroarenes offers a pathway to aryl nitriles; however, traditional methods typically require multiple steps, including reduction, diazotization, and the Sandmeyer reaction. In 2024, the Yamaguchi group reported a single-step, Pd-catalyzed denitrative cyanation using nitroarenes 1 as electrophilic partners and aminoacetonitriles 29 as non-metal cyanating agents.32aTraditionally, transition-metal-catalyzed cyanation reactions rely on metal cyanides as the cyanide source, which generate hazardous HCN and metal waste. Organic cyanide sources, such as morpholin-4-ylacetonitrile (29), offer a safer alternative. Using Pd(acac)2/BrettPhos as the catalytic system and a base, nitroarenes 1 react with 29 to afford aryl nitriles 30 in moderate yields (Scheme 13A). In addition to its safety profile, this organic cyanide reagent serves as a “cyanide shuttle”, gradually releasing cyanide ions into the reaction mixture (Scheme 13B). Notably, reactions using metal cyanides can be hindered due to catalyst deactivation by excess cyanide.33 In contrast, the gradual release from the organic cyanide source may allow for more effective cyanation of less reactive aromatic electrophiles, such as aryl esters, which typically undergo oxidative addition more slowly than haloarenes.34
 | ||
| Scheme 13 Pd-Catalyzed denitrative cyanation of nitroarenes (Yamaguchi group) (A) one-step cyanation of nitroarenes with aminoacetonitrile 29 (B) Role of aminoacetonitriles as cyanide shuttles, gradually releasing cyanide ions for effective cyanation. | ||
Since nitro groups are generally stable under conventional cross-coupling conditions, they conducted a sequential cross-coupling of 31 (Scheme 14A). Initially, Suzuki–Miyaura coupling of 31 with 32 produced biaryl 33 smoothly. The resulting nitroarene 33 was then subjected to the current C–C bond-forming cyanation with 29, affording 34 in 61% yield over two steps. In another example, a sequential aromatic C–H cyanation was performed without the need for a chelation directing group (Scheme 14B).35 A previously reported aromatic C–H nitration of 35 followed by the present denitrative cyanation of the resulting 36, successfully generated 37 in a moderate yield. More recently, Chen, Cao, and Kong reported a one-step synthesis of aryl nitriles via electrochemically driven denitrative cyanation of nitroarenes.32b See Scheme 20 for further details.
 | ||
| Scheme 14 Sequential coupling reactions Involving nitroarenes (A) sequential Suzuki–Miyaura coupling and denitrative cyanation of nitroarene (B) sequential C–H nitration followed by denitrative cyanation. | ||
2.8. α-C–H Arylation of heteroarenes
α-Arylated heteroarenes are essential structural motifs in natural products, pharmaceuticals, and functional materials.36 Heteroarenes such as indoles and pyrroles with α-aryl groups substituents are particularly valuable frameworks in drug discovery, where the α-aryl group is often critical for pharmacological activity.37 In 2019, Yamaguchi and Nakao reported a Pd-catalyzed denitrative intramolecular C–H arylation of nitroarenes.10d Recently, Yu achieved an intermolecular C–H arylation of nitroarenes 1 and heteroarenes 38 using a Pd/EPhos catalyst in the presence of Cs2CO3 and CuCl (Scheme 15A).38 This method proved applicable to various nitroarenes 1 and heteroarenes 38, including benzofuran, indole, furan, and pyrrole, resulting in the formation of the corresponding diaryl products 39. | ||
| Scheme 15 Intermolecular α-C–H arylation of heteroarenes with nitroarenes (Yu group) (A) Pd-catalyzed intermolecular C–H arylation (B) synthesis of SKF-64346. | ||
With this Pd-catalyzed intermolecular C–H arylation method established, the Yu group applied it to the synthesis of SKF-64346, a compound with therapeutic potential for Alzheimer's disease (Scheme 15B).39 The synthesis began with a two-step SN2 reaction of nitroarene 40 to produce alkoxynitroarene 41. This intermediate was then subjected to Pd-catalyzed denitrative intermolecular C–H arylation with benzofuran, followed by Friedel–Crafts acylation under acyl chloride and SnCl4 conditions, ultimately yielding SKF-64346.
2.9. Denitrative coupling without metal catalyst
Recent studies have reported metal-free denitrative coupling reactions. Unlike metal-catalyzed processes, which rely on the oxidative addition of nitroarenes to metals as a driving force, these reactions proceed via radical functionalization through the formation of aryl radicals. It should be noted that dearomative/denitrative cyclization reactions are also known without metal catalyst;40 however, these reactions are not covered in this review. | ||
| Scheme 16 Metal-free denitrative hydroxylation of nitroarenes (James group). | ||
The James group investigated the formation of arenol 46 by reacting oxime 43 with 4-nitrobiphenyl 45 in the presence of the radical and redox scavenger Galvinoxyl (Scheme 17A). The significant inhibition of arenol 46 formation suggested a radical chain mechanism. Furthermore, repeating the reaction with sodium metal, a sacrificial single-electron donor, increased the yield of arenol 46, indicating that the reaction likely proceeds via one-electron reduction.
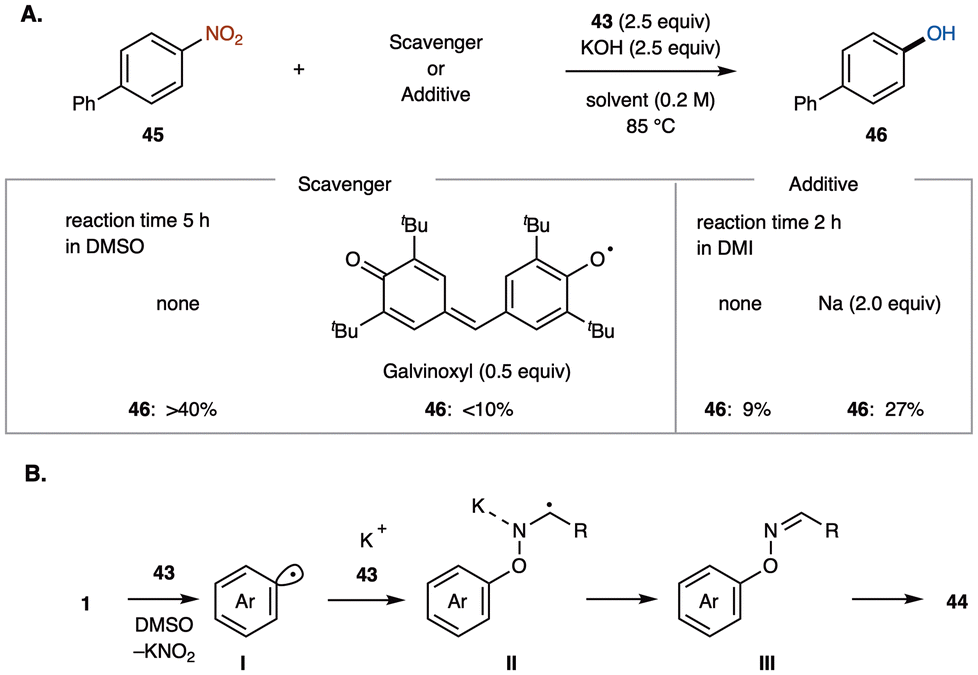 | ||
| Scheme 17 Mechanistic studies of metal-free denitrative hydroxylation. (A) Radical trapping experiments. (B) Plausible mechanism. | ||
Based on these observations, they proposed the following denitrative hydroxylation mechanism (Scheme 17B). The radical-nucleophilic substitution (SRN1) chain reaction is proposed to begin with the interaction between oxime 43 and nitroarene 1, leading to the formation of aryl radical I.44 This aryl radical then reacts with the oxime anion 43 to form radical anion II. Subsequently, radical anion II transfers an electron to nitroarene 1via intermolecular electron transfer, regenerating aryl radical I and releasing coupling product III, which fragments in situ to produce the observed arenol 44.
 | ||
| Scheme 18 Electrochemical borylation of nitroarenes (Cai and Zhang group). | ||
Based on mechanistic studies, they proposed a borylation mechanism (Scheme 19). Initially, nitrobenzene 1 undergoes electron transfer at the cathode, combining with protons to form nitrosobenzene I, which then reacts with B2pin2 and H2O to give aniline II and (Bpin)2 O. Subsequent diazotization of II with t-BuONO generates phenyldiazonium salt III, which undergoes single electron transfer (SET) to release N2, tert-butoxide anion, and phenyl radical IV. Finally, radical IV reacts with B2pin2 and the tert-butoxide anion to form the borylated product 48.
 | ||
| Scheme 19 Plausible mechanism. | ||
Subsequently, Chen, Cao, and Kong reported the electrochemical cyanation of nitroarene 1 (Scheme 20).47 By reacting nitroarene 1 with t-BuNC (49), t-BuONO, and [Min]TolSO3 as the ionic liquid under various electrochemical conditions, they successfully synthesized the desired aryl nitriles 30 in moderate to high yields, with excellent tolerance for a wide range of functional groups. They proposed that in this reaction, nitroarenes 1 are electrochemically reduced to anilines, which subsequently form aryl radical intermediates. These radicals then react with t-BuNC, the radical trapping agent, to yield the aryl nitriles 30.
 | ||
| Scheme 20 Electrochemical cyanation of nitroarenes (Chen, Cao, and Kong groups). | ||
While denitrative coupling has significantly broadened the synthetic utility of nitroarenes, challenges remain in expanding substrate generality, particularly for electron-rich nitroarenes, and in minimizing catalyst loadings to improve sustainability. Additionally, the high reaction temperatures often required for these transformations present a significant obstacle. Developing catalysts with higher activity and efficiency is crucial to address this issue. Future advancements could focus on designing more robust catalytic systems or exploring photocatalytic approaches to enhance reaction efficiency and selectivity under milder conditions.
3. Skeletal editing of nitroarenes using nitrene precursors
Recent advancements in organic synthesis have increasingly focused on molecular rearrangement and skeletal editing, enabling selective and systematic modification of molecular frameworks by adding or removing atoms. The development of new redox-active organophosphorus catalysts has broadened the scope of nitroarene transformations, enhancing both functional group tolerance and substrate generality, and enabling more efficient and selective skeletal rearrangement reactions. Furthermore, it has been discovered that nitroarenes can be photochemically reduced to nitrenes using blue LED irradiation, facilitating skeletal editing reactions that not only leverage classic principles but also significantly expand functional group compatibility and applicability across diverse substrates. This section discusses these recent advancements and the unique mechanisms underlying these transformations.3.1. o-Phenylenediamine synthesis
The incorporation of amine groups onto aromatic rings is essential in medicinal chemistry and synthetic applications, especially given that anilines are key components of numerous bioactive compounds. Developing methods for their efficient synthesis holds considerable societal impact.48 Among nitrogen-containing aromatic compounds, ortho-phenylenediamines represent a particularly valuable yet challenging derivatives due to their presence in many high-impact pharmaceuticals.49 Traditional synthetic methods for these derivatives often require multi-step sequences, beginning with ortho-halogenation of nitroarenes or ortho-nitration of haloarenes, typically involving a minimum of four steps.50 Recent strategies using C–H activation and functionalization via metal or radical species have provided partial solutions to these synthetic challenges.51 However, these methods still necessitate the installation and removal of directing groups, adding complexity and limiting practical applications due to the increased number of synthetic steps.In 2023, Radosevich introduced a novel strategy for the reductive C–H amination of nitroarenes 1, enabling tandem C- and N-functionalization to synthesize 2-aminoanilides 51 and benzimidazoles 52 from readily available starting materials (Fig. 4A).52 Using a redox-active organophosphorus catalyst (P·[O]) and PhSiH3 as the terminal reductant, nitroarenes 1 were reacted with secondary amines 50 at 120 °C, followed by the addition of acyl chloride, affording aniline derivatives 51 in moderate to good yields. When primary amines 50 were used, benzimidazoles 52 were obtained in similar yields. The reaction demonstrated compatibility with a variety of nitroarenes containing electron-donating and electron-withdrawing substituents, as well as a range of primary and secondary amines, including morpholine. Based on previous studies, it was proposed that the redox-active organophosphorus catalyst could facilitate complete deoxygenation of the nitroarene via a P(III)/P(V) cycle, leading to the formation of a high-energy arylnitrene intermediate I (Fig. 4B).53 Phenylnitrene I, typically generated via photolytic decomposition of phenylazide, is known to undergo isomerization to benzazirine II and dihydroazepine III valence tautomers.54 These intermediates can react with nucleophiles such as amines to form 2-amino-3H-azepines IV and V through ring expansion, a transformation confirmed by isolating V in the absence of acyl chloride. The proposed mechanism involves an azepine-to-azanorcaradiene 6π electrocyclization (VI–VII), which is promoted by N-acylation, followed by decomposition of the fused bicyclic aminal VII with rearomatization of the arene core. Consequently, 2-aminoanilide 51 can undergo thermal cyclization under established conditions to yield the target benzimidazole 52.
 | ||
| Fig. 4 (A) Reductive C/N-difunctionalization of nitroarenes as a modular approach to synthesize 2-aminoanilides and benzimidazoles. (B) Proposed mechanism illustrating key intermediates in the skeletal editing process (Radosevich group). | ||
More recently, the Leonori group also reported a strategy for ortho-phenylenediamine synthesis via dearomative-rearomative coupling of nitrobenzenes with amines using bule LED (Fig. 5A).55 This one-pot procedure involved the initial reaction of nitroarenes 1 and amine 50 with P(OiPr)3 under blue LED irradiation, followed by a dark reaction with trifluoroacetic anhydride (TFAA), affording aniline derivatives 53 in moderate to good yields. The reaction proceeds through the generation of an aryl nitrene intermediate from nitroarenes under mild blue light irradiation at room temperature. This method showed broad substrate compatibility, successfully transforming various nitroarenes bearing electron-donating and electron-withdrawing substituents into the desired phenylenediamines 53. Additionally, fluoro, boryl, and bromo substituents were well-tolerated. Both primary and secondary amines with diverse structures were also effective as reaction partners.
 | ||
| Fig. 5 (A) Synthesis of ortho-phenylenediamines via dearomative–rearomative coupling of nitroarenes with amines. (B) Proposed mechanistic outline involving triplet nitroarene and key intermediates (Leonori group). | ||
Based on prior studies, they proposed a mechanism in which blue light irradiation excites nitroarene 1 to its triplet excited state I (Fig. 5B).52,56 The triplet nitroarene, acting as a strong electrophile, undergoes a stepwise radical [3 + 1] cycloaddition with electron-rich phosphite, forming the deoxygenated singlet nitrene IIvia intermediate II-int. Nitrene II then undergoes cyclization to form azirine III, which undergoes a 6π electrocyclic ring opening, yielding the seven-membered ketimine IV. In its strained, reactive state, ketimine IV is readily intercepted by an amine, forming a second C–N bond to give 1H-azepine V, which subsequently isomerizes to the more thermodynamically stable 3H-isomer VI. In the final steps, VI reacts with TFAA as an electrophilic trapping agent, producing the heteroaromatic compound VII through enolization. This thermodynamic driving force initiates a second 6π electrocyclization in a ring-closing direction, forming N–TFA aziridine VIII. The push–pull characteristics of this intermediate facilitate aromatization, affording the ortho-aniline 53. Thus, although the reactivity and mechanism of nitroarenes in these transformations have been known for some time,56 the use of blue LED light as an energy source has significantly expanded functional group tolerance and substrate generality, making this a prime example of a classic reactivity reintroduced as a novel reaction.
3.2. Bicyclic pyrrolines synthesis
Saturated nitrogen heterocycles are frequently used scaffolds in the discovery of bioactive compounds,57 with sp3-rich fragments particularly valued for enhancing the biological activity of lead molecules. Among these, saturated bicyclic amines stand out due to their structural rigidity, which allows for precise three-dimensional (3D) orientation of substituents, making them especially relevant in this context.58 The Leonori group recently developed an efficient method for synthesizing 2-azabicyclo[3.2.0]heptanes (54) from nitroarenes 1via a triplet excited state (Scheme 21A).59 Although this transformation is based on a known reaction, the use of blue LED irradiation, as in the previous section, enabled a significant expansion of substrate scope, enhancing functional group compatibility.60 Under blue LED irradiation, nitroarenes 1 reacted with P(OiPr)3 and amines to afford the corresponding 2-azabicyclo[3.2.0]heptanes 54. In a mechanism similar to their previously reported ortho-diamine synthesis (Fig. 5B), the triplet-excited state of nitroarene I undergoes reaction with an amine to produce the 1H-azepine IIvia dearomatization. Subsequently, II isomerizes to the 3H-azepine II′, which then undergoes a 4π electrocyclization to give the desired 2-azabicyclo[3.2.0]heptane 54. | ||
| Scheme 21 (A) Proposed mechanistic pathway for the synthesis of 2-azabicyclo[3.2.0]heptanes. (B) Comparative reactivity of 3-Ph and 4-Ph substituted nitroarenes under photochemical conditions. | ||
This reaction explores a photochemical strategy that leverages the excited-state reactivity of both nitroarene 1 and azepine intermediate II′, along with two electrocyclic processes: one thermal and one photochemical (Scheme 21B). The process requires two photoexcitations, which may proceed either in a single step or as two sequential steps, depending on the absorption properties of the intermediates. Using meta- and para-phenyl-substituted nitroarenes (1a[3-Ph] and 1b[4-Ph]), they found that irradiation of 1a with 350 nm light afforded the desired product 54a in moderate yield, whereas similar conditions for 1b failed to produce trace amounts of the pyrroline isomer 54b.
To further investigate the reactivity, they isolated the N-insertion and 4π electrocyclization steps. Irradiating 1a under 427 nm light afforded intermediate II′a in 43% yield, while 1b led to a complex mixture. However, using iPrOH as the solvent enabled the isolation of II′b in 42% yield. Subsequent irradiation of II′a and II′b with 350 nm light produced the desired cyclized products, with II′a smoothly converting to 54a in yields comparable to the direct method. Although the 4π electrocyclization of II′b was more challenging, it still afforded 54b in 58% under diluted conditions. These findings indicate that a stepwise approach, separating azepine formation from electrocyclization, can effectively give the target C6 pyrroline isomer 54b. Further analysis of the calculated singlet excited-state HOMO–LUMO energy gap of 3H-azepine II′ revealed that II′a (3-Ph) is more reactive than II′b (4-Ph), providing insights into the reactivity differences observed.
Two synthetic protocols (A or B) for converting nitroarenes 1 into bicyclic pyrrolines 54 have successfully provided C6-functionalized derivatives 54 in good to moderate yields (Scheme 22). This approach is compatible with nitroarenes 1 bearing various aromatic substituents, including halogens and electron-withdrawing groups. The use of meta-substituted nitroarenes proceeds smoothly under the direct protocol (A), affording the desired products in moderate yields and often resulting in two isomeric forms. Additionally, this method is applicable to polysubstituted nitroarenes, efficiently converting the corresponding C6-functionalized derivatives 54.
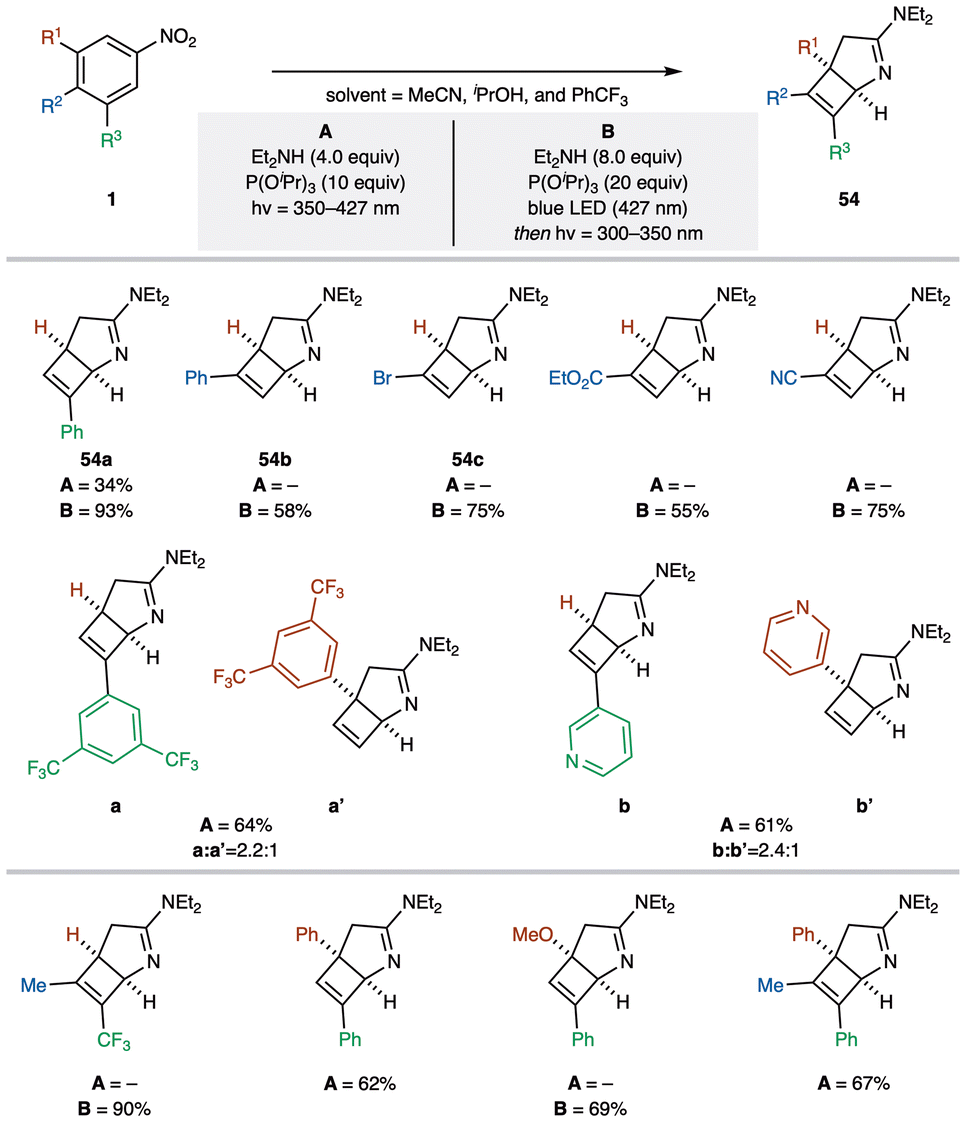 | ||
| Scheme 22 Substrate scope for the conversion of nitroarenes 1 to C6-functionalized bicyclic pyrrolines 54 (Leonori group). | ||
They further explored derivatization of the C6–Br derivative 54c, demonstrating its conversion to the desired pyrroline products 55 and 56 in high yields using standard Ullmann and Buchwald–Hartwig cross-coupling methods (Scheme 23). Additionally, coupling product 56 under standard hydrogenolysis conditions afforded amino-substituted heterocycles 57. Notably, 57 serves as a potential precursor for the synthesis of conformationally constrained analogues of the antihistaminic drug methdilazine 58. The 3D spatial arrangement of anti-5-substituted-2-azabicyclo[3.2.0]heptanes allows for “cyclobutanation” to access highly strained conformations, with distortion energies exceeding 34 kcal mol−1.
 | ||
| Scheme 23 Derivatizations of pyrroline 54c by cross-coupling reactions. | ||
3.3. Synthesis of polysubstituted azepanes
Nitrogen-containing heterocycles are essential building blocks in the discovery and development of bioactive materials.61 Saturation and ring substitution are critical features in medicinal chemistry, as they produce molecules with globular shapes and multiple vectors for exploring three-dimensional (3D) chemical space. However, the prevalence of certain nitrogen heterocycles in high-value products is often limited by their commercial availability, which depends on ease of synthesis and functionalization. For example, piperidine, a commonly used saturated nitrogen heterocycle, can be converted to pyrrolidine by one-carbon deletion and is readily accessible. In contrast, adding one carbon forms azepane, a far less common structure (<1% of N-heterocycles)49c due to the synthetic challenges associated with seven-membered rings compared to more accessible six- and five-membered rings. Existing methods for synthesizing azepanes typically involve multi-step transformations from linear precursors or Beckmann rearrangement of functionalized piperidones, limiting versatility and hindering the preparation of highly substituted derivatives.62Recently, the Leonori group developed a method for converting commercially available nitroarenes 1 into saturated azepane heterocycles 59 (Scheme 24).63 This process involves generating nitrene intermediates via blue light irradiation, followed by a one-atom ring expansion to form an intermediate, which is then subjected to hydrogenolysis. While this method effectively provides azepanes, it primarily involves simple hydrogenation of intermediates like those seen in Fig. 5. Although similar reductions of azepine intermediates to related compounds have been reported in the past,56 few reports have emerged since then.64 This strategy enabled the synthesis of several para-substituted derivatives, yielding C4-substituted azepanes in good yields. While para-bromonitrobenzene underwent efficient ring expansion, subsequent hydrogenolysis presented challenges, as it led to nearly complete dehalogenation under standard conditions. Nevertheless, they achieved successful C4 arylation through Buchwald–Hartwig amination following ring expansion, thus enabling access to previously unattainable aminated derivatives.
 | ||
| Scheme 24 Substrate scope for the synthesis of polysubstituted azepanes (Leonori group). | ||
Furthermore, a three-step synthetic sequence using para-substituted nitroarenes with phenyl lithium nucleophiles enabled the synthesis of disubstituted azepanes in good yields. The reaction also proceeded smoothly with meta-substituted nitrobenzenes, producing the desired products along with C4 isomers. Despite difficulties in achieving N-insertion with ortho-substituted nitrenes, they successfully conducted the reaction with ester-containing derivatives, obtaining C3-functionalized azepanes in useful yields. Additionally, the method proved effective with pentadeuterated nitrobenzene, resulting in a tetradeuterated azepane with near-quantitative yield. Multi-substituted nitroarenes also afforded the corresponding azepanes in good yields.
Skeletal editing via nitrene intermediates offers a powerful tool for molecular rearrangement and functionalization. However, controlling the reactivity of high-energy intermediates such as nitrenes remains a challenge, often requiring specific reaction conditions. Future efforts should explore strategies to achieve greater regioselectivity and to extend these transformations to more complex and biologically relevant substrates.
4. The utilization of nitroarenes with N–O bond cleavage as an oxygen source
This chapter discusses recent radical reactions in which nitroarenes serve as the oxygen source, reacting with substrates and subsequently undergoing N–O bond cleavage. It is important to note that this review does not cover reactions where the addition step proceeds through an ionic mechanism (e.g., the reduction of nitro compounds to nitroso compounds followed by addition)65 or where nitro compounds react via a radical mechanism but do not primarily yield products resulting from N–O bond cleavage.664.1. NHC-catalyzed β-hydroxylation of enals
β-Hydroxyl (α-unsubstituted) esters and their derivatives are valuable building blocks, commonly found as subunits in biologically active compounds and natural products.67 facilitates acyl anion reactivity, and numerous transformations employing α,β-unsaturated aldehydes as substrates have been developed.68 However, most of these reactions rely on two-electron mechanisms. In contrast, recent research has investigated single-electron pathways as an alternative. Rovis demonstrated that by using nitropyridine N-oxide (1c) as a single-electron oxidant, β-hydroxy ester 61 can be synthesized from cinnamaldehyde 60 and NHC catalyst B in methanol (Scheme 25A).69 This approach highlights how selecting single-electron oxidants can expand reaction pathways. | ||
| Scheme 25 (A) Substrate scope for the racemic β-hydroxy ester formation. (B) Substrate scope for the asymmetric β-hydroxy ester formation using chiral NHC catalyst (Rovis group). | ||
The scope of this reaction is remarkably broad, proceeding efficiently with both aryl and alkyl substitutions at the β-position of the enal 60. Aliphatic substrates, in particular, yield high quantities of the desired product under these conditions. Additionally, β,β-disubstituted substrates also participate in the reaction, affording tertiary alcohols in moderate yields. Furthermore, using a chiral NHC C catalyst enables asymmetric transformations, rendering the reaction applicable to both aryl and aliphatic enals (Scheme 25B).
The Rovis group proposed a reaction mechanism involving Breslow-centered radical cation species IIIa (Scheme 26A). The reaction begins with the formation of the Breslow intermediate II, which undergoes single-electron oxidation by nitrobenzene 1, generating a Breslow-centered radical cation IIIa and a nitrobenzene-derived radical anion IIIb. These radicals then combine, where the homoenolate-centered radical and oxygen-centered radical react to form intermediate IV. This intermediate decomposes to release an NHC-bound alkoxide and nitrosobenzene V. The NHC-bound alkoxide subsequently reacts with methanol to regenerate the catalyst and produce the β-hydroxy ester 61. Additionally, α,β-unsaturated methyl ester VII may form through deprotonation of intermediate III, followed by a second single-electron abstraction, resulting in the formation of α,β-unsaturated acyl azolium VI, which is then intercepted by methanol.
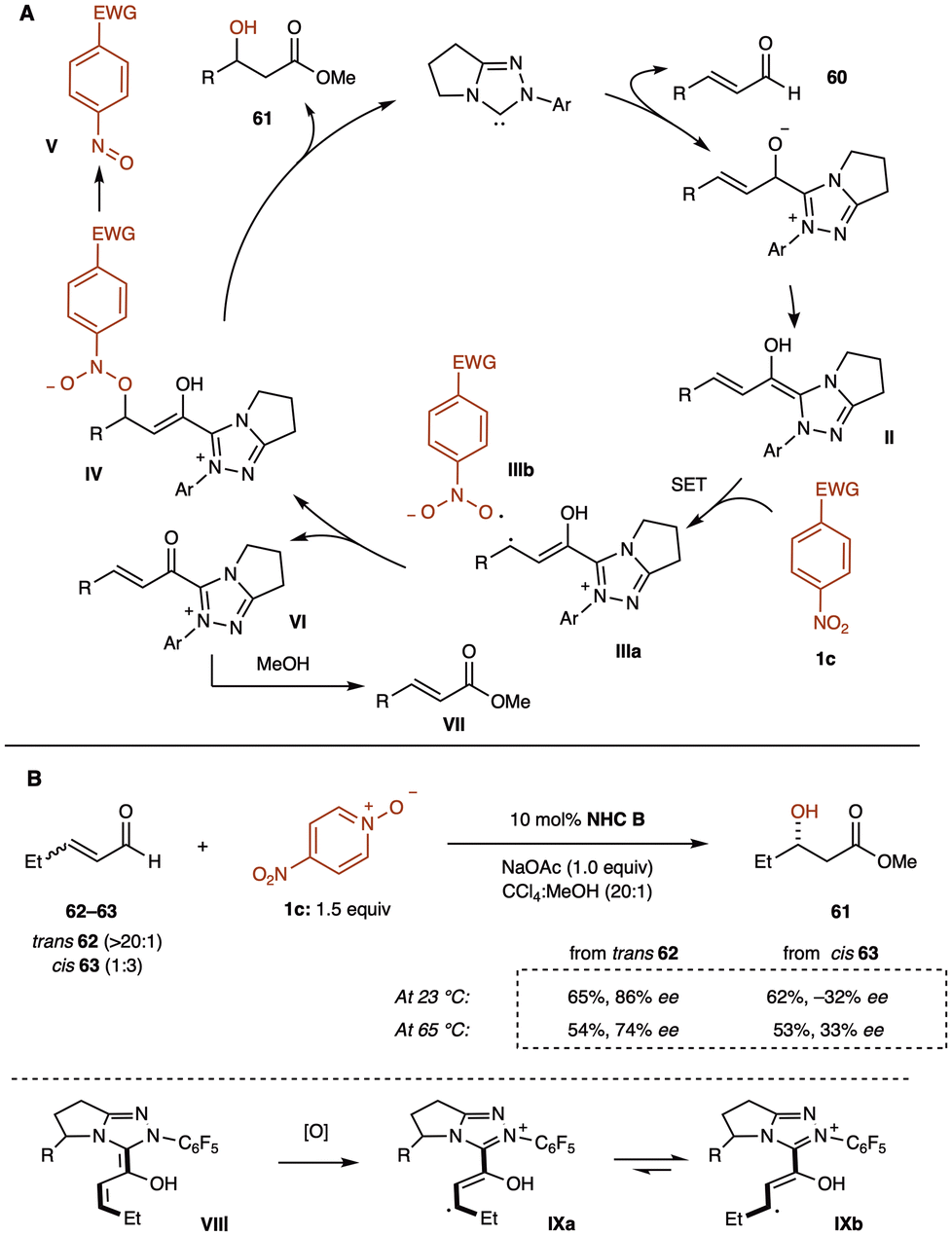 | ||
| Scheme 26 (A) Proposed mechanism for the β-hydroxylation reaction. (B) Stereochemical investigation using cis- and trans-enals as probes. | ||
To investigate the stereochemical outcome of the reaction, the group tested both cis- and trans-enals (62 and 63). If the reaction proceeds via a radical mechanism, with radical recombination slower than bond rotation, the cis- and trans-enals should produce the same major enantiomer 61′ through interconversion of intermediates IXa and IXb (Scheme 26B). At room temperature, however, the cis- and trans-olefin isomers produced opposite major enantiomers. At 65 °C, enantiomeric convergence was observed, supporting the hypothesis of a discrete radical intermediate in the reaction mechanism.
More recently, the Chi group reported an NHC-catalyzed oxidative single-electron transfer (SET) of enals, introducing a hydroxyl group at the β-carbon of enals 60 (Scheme 27A).70 They found that nitrobenzenesulfonic carbamate (1d) served as an effective oxidant when combined with NHC catalyst D and K2CO3, successfully affording β-hydroxy esters 61 from enals 60. This catalytic system exhibits broad applicability, accommodating a wide range of enals, including β-(hetero)aryl and β-alkyl enals, and enabling the highly enantioselective synthesis of β-hydroxy esters with valuable applications. Mechanistic studies suggest that the reaction proceeds via a radical pathway involving multiple radical intermediates.
 | ||
| Scheme 27 (A) Substrate scope for the chiral NHC-catalyzed β-hydroxylation of enals (Chi group). (B) Substrate scope for the NHC-catalyzed β-hydroxylation of enals using a chiral nitoroarene as an oxidant (Chi group). | ||
Subsequently, Chi and coworkers reported an asymmetric oxidation using a chiral nitroarene as the oxidant (Scheme 27B).71 When the chiral oxidant 1e was reacted with enal 60 in the presence of NHC catalyst E, the corresponding ester 61 was obtained in high yield and with excellent enantioselectivity. The key to achieving high enantioselectivity was the introduction of the nitro group at the ortho position, which brings the reaction site into close proximity to the chiral environment. Additionally, introducing a trifluoromethyl group at the para position further improved both the yield and enantioselectivity. The reaction proceeded efficiently with various cinnamaldehyde derivatives bearing various functional groups. High enantioselectivity was maintained even when the benzene ring was replaced with furan or naphthalene. For alkyl-substituted enals, although enantioselectivity slightly decreased, moderate to high yields of the desired ester 61 were still achieved. Since 1e acts as the oxidant, it must be used in stoichiometric amounts. However, a significant advantage is that it can be easily prepared by simply mixing the corresponding sulfonyl chloride and amine.
4.2. Fe-Catalyzed anaerobic Mukaiyama-type hydration of alkenes
The transformation of alkenes into alcohols is a key process in synthesis, as alcohol functionalities are prevalent in pharmaceuticals and natural products. While acid-catalyzed hydration is the simplest approach, it often presents practical challenges, including ether formation and cationic rearrangements. Consequently, Mukaiyama-type radical Markovnikov hydration and anti-Markovnikov hydration via hydroboration–oxidation are commonly employed at the laboratory scale.72Recently, the Studer group developed an anaerobic Mukaiyama-type alkene hydration reaction using nitroarenes 1f as non-gaseous oxygen donors under catalytic conditions (Scheme 28).73 Using an iron catalyst and PhSiH3 as a stoichiometric reductant, they achieved high selectivity and stereoselectivity in converting alkenes 64 into secondary and tertiary alcohols 65. This method accommodates internal alkenes, including natural products like β-citronellol, and provides benzylic alcohols in moderate to good yields from styrene derivatives. Additionally, the reaction demonstrates excellent diastereoselectivity, producing diastereomerically enriched alcohols.
 | ||
| Scheme 28 Fe-Catalyzed Mukaiyama-type hydration of alkenes using nitroarenes as oxygen donors (Studer group). | ||
Mechanistically, the Fe(III)–H species first undergoes hydrogen atom transfer (HAT) with alkene 64, forming a radical adduct (Scheme 29). This radical is then trapped by nitroarene 1f, resulting in the formation of a heteroatom-centered radical and an Fe(II) species. The radical intermediate is subsequently reduced by Fe(II), leading to ionic fragmentation and protonation to give 65 while regenerating the Fe(III) species, which is then converted back to Fe(III)–H. Unlike previous studies by the Baran group and by Cheung and Hu, where nitroarenes are reduced early in the reaction and serve as radical amination reagents, here, the nitroarene acts as a trapping agent for alkyl radicals.74
 | ||
| Scheme 29 Proposed mechanism for Fe-catalyzed Mukaiyama-type hydration of alkenes. (A) Mechanistic pathway involving Fe(III)–H and nitroarene as radical trapping agents. (B) Radical probe experiments supporting the proposed mechanism. | ||
To confirm that nitroarenes 1f can function as alkyl radical acceptors, they thermally decomposed dodecanoyl peroxide in the presence of 1f, confirming the formation of alkoxyamine 66 (80%) and undecanol (45%) (Scheme 29A). The formation of undecanol suggests that the nitro group in electron-deficient nitroarenes reacts with the C-radical at the oxygen atom, indicating that the alcohol is unlikely to originate from a hydroxylamine or alkoxyamine intermediate under oxidative conditions. To further verify the involvement of radical pathways, they conducted radical probe experiments using 1,6-diene 66 (Scheme 29B). The product 68, formed via radical 5-exo cyclization, was obtained in good yield and moderate cis/trans selectivity, supporting the proposed mechanism.
More recently, Studer group reported an intramolecular variant of the Mukaiyama hydration reaction (Scheme 30).75 Using substrate 1g, in which a linker and an olefin are attached ortho to the nitro group of a nitroarene, the reaction proceeds in the presence of Fe catalyst and PhSiH3 to afford the hydrated product 69. Unlike the intermolecular reaction, this transformation does not require an electron-deficient nitroarene. Functional groups such as methoxy or unprotected amino groups are well tolerated, and substrates bearing cyano or bromo substituents also react smoothly, undergoing a 1,6-oxygen shift to give the desired product 69. Additionally, the reaction proceeds efficiently even when the linker contains nitrogen or sulfonylamide moieties, producing not only secondary alcohols but also tertiary alcohols. When chiral alkenes with stereogenic centers are employed, the reaction proceeds diastereoselectively. For example, phenyl-substituted alkene exhibit a diastereomeric ratio (dr) of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, while smaller substituents like a hydroxyl group reduce the selectivity (dr = 3:1). However, using a bulky silyl-protected hydroxyl group restores high diastereoselectivity, enabling efficient oxygen-transfer reactions. The position of the nitro group on the aromatic ring is also critical. While C2 substitution facilitates the reaction, substrates with the nitro group at the C4 position fail to undergo the transformation. This observation suggests that the reaction involves an intramolecular radical oxygen-transfer mechanism rather than an intermolecular process.
:1, while smaller substituents like a hydroxyl group reduce the selectivity (dr = 3:1). However, using a bulky silyl-protected hydroxyl group restores high diastereoselectivity, enabling efficient oxygen-transfer reactions. The position of the nitro group on the aromatic ring is also critical. While C2 substitution facilitates the reaction, substrates with the nitro group at the C4 position fail to undergo the transformation. This observation suggests that the reaction involves an intramolecular radical oxygen-transfer mechanism rather than an intermolecular process.
 | ||
| Scheme 30 Intramolecular radical oxygen-transfer reactions using nitroarenes (Studer grpup). | ||
4.3. Photoinduced oxygen transfer using nitroarenes for the anaerobic cleavage of alkenes
The oxidative cleavage of alkenes to produce key carbonyl groups is a fundamental transformation in synthetic organic chemistry, essential for natural products and drug candidates.76 Although ozonolysis is a common method, it poses challenges due to the explosive nature of ozone, specialized equipment requirements, and difficulty in controlling stoichiometry.77 While other oxidative methods exist, they are limited in scope and produce significant toxic waste.78 Recently, Parasram and co-workers developed a visible light-induced anaerobic cleavage of alkenes using nitroarenes as oxygen transfer reagents, achieving the synthesis of ketones and overcoming traditional limitations.79 In a similar reaction reported by Buchi et al. in the 1950s using UV light, the ketone yields were notably low.80 In contrast, the present method employing 390 nm (purple light) irradiation successfully converts various olefins to ketones with significantly improved yields. This reaction enables the efficient formation of acetophenones and benzophenones 71 by reacting alkenes 70 with cyanonitrobenzene in MeCN under 390 nm (purple light) irradiation (Fig. 6Aa). The method also works with less reactive aliphatic alkenes 71, affording carbonyl compounds effectively.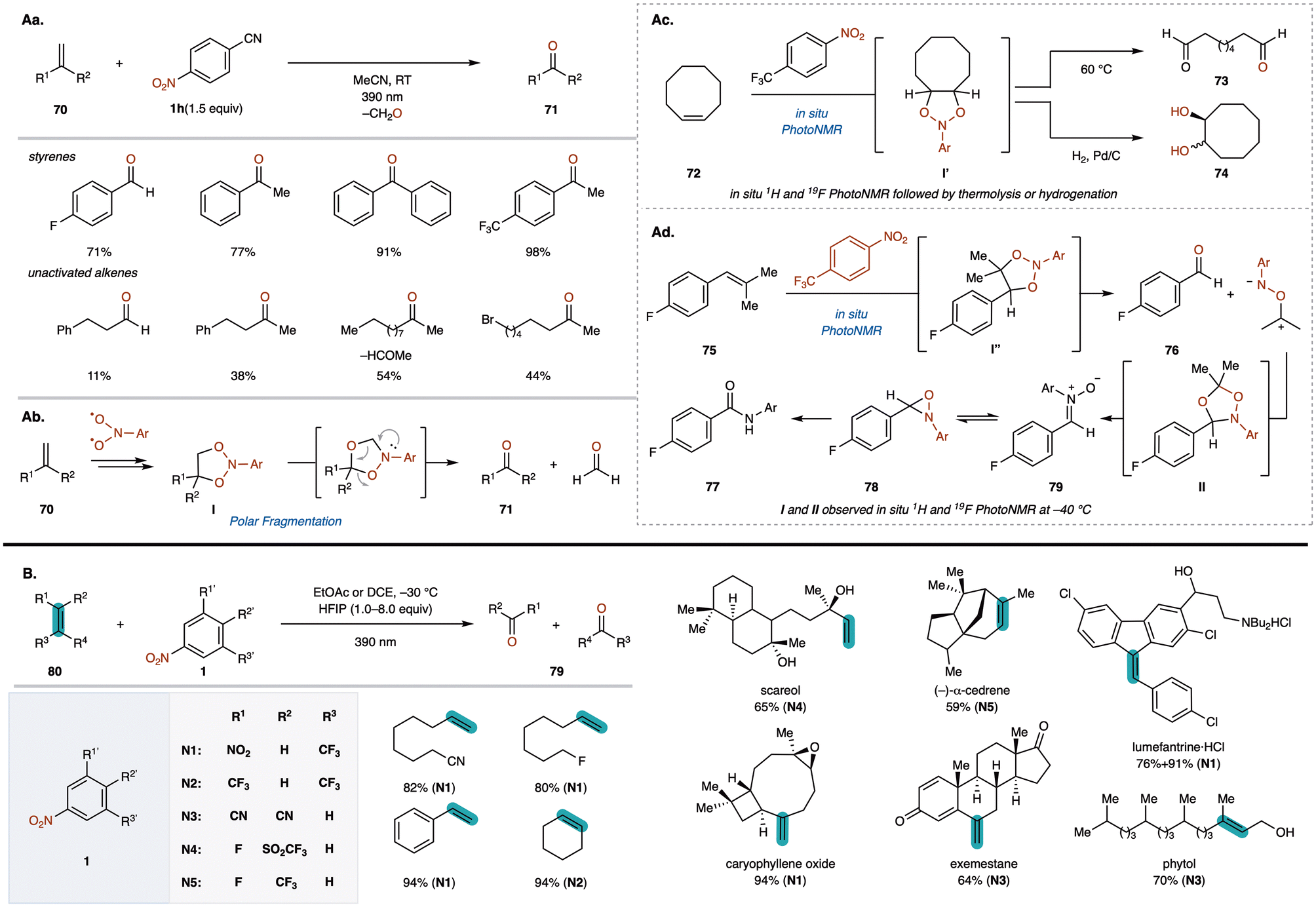 | ||
| Fig. 6 (A) Visible light-induced anaerobic cleavage of alkenes using nitroarenes as oxygen sources (Parasram group). (B) Anaerobic oxidative cleavage of alkenes with nitroarenes under purple light irradiation (Leonori group). | ||
Mechanistically, the reaction begins with the excitation of nitroarene 1h upon absorbing 390 nm light, leading to a radical addition to alkenes 70 and the formation of a transient dioxazolidine intermediate I (Fig. 6Ab). Subsequent intramolecular radical ring closure affords another intermediate, which decomposes via a radical or polar pathway to produce carbonyl compounds 71. Using in situ1H and 19F photoNMR, they confirmed that the reaction of cis-cyclooctene (72) with 4-trifluoromethylnitrobenzene gave dioxazolidine I′ at 25 °C (Fig. 6Ac). Thermolysis of I′ at 60 °C produced dicarbonyl compound 73, indicating that its decomposition is not photochemically promoted. Furthermore, hydrogenation of I′ resulted in a 1:1 mixture of cis- and trans-diols 74, supporting a non-stereospecific radical ring closure.
To determine if additional intermediates form during the reaction, they monitored the reaction progress of cis-styrene (75) with nitroarene 1f in CD2Cl2 at −40 °C using photoNMR (Fig. 6Ad). Upon irradiation at 395 nm, two proton resonances (I′′ and II) and corresponding 19F signals increased in the initial 2 h, with a modest increase in aldehyde 76. Intermediates I′′ and II are likely due to 1,3-dipolar cycloaddition between 76 and a carbonyl imine, resembling ozonolysis. Increasing the temperature from −40 °C to −20 °C caused rapid decomposition of I′′ and II, affording 76 and additional products 77–79.
Almost simultaneously, Leonori reported a similar ketone synthesis from alkenes 80 using nitroarenes 1 under 390 nm purple light irradiation (Fig. 6B).81 They found that using CH2Cl2 with HFIP as an additive was crucial for good reactivity. Since excited-state nitroarenes can abstract hydridic α-C(sp3)–H bonds, HFIP is thought to suppress this side reaction through hydrogen bonding with the heteroatom, enhancing reactivity with unactivated alkenes. By tuning nitroarene substituents, this method exhibited high functional group compatibility and site selectivity, making it applicable to a broad range of pharmaceutical compounds and natural products.
4.4. Olefin dihydroxylation using nitroarenes
Vicinal diols are frequently found in various bioactive compounds and are essential in high-value products across the pharmaceutical, agrochemical, and fragrance industries.77b Numerous methods for olefin dihydroxylation have been developed to synthesize vicinal diols, yet it remains challenging to convert olefins into vicinal diols under mild conditions, especially when working with a broad range of unactivated olefins.As previously mentioned, Leonori and colleagues reported the synthesis of ketones through oxidative cleavage of alkenes using nitroarenes (Fig. 6B). Building on this work, they have more recently developed a method for synthesizing vicinal diols by applying a similar approach. Specifically, their method involves the formation of a dioxazolidine intermediate from alkenes 81, followed by reductive cleavage of the N–O bond under light irradiation, using nitroarene 1i as the oxygen source to give vicinal diols 82 (Scheme 31A).82 This method successfully converts various alkenes, including terminal, internal, and cyclic alkenes such as cyclooctene and cyclohexene, to their corresponding diols, achieving high yields even with geminally disubstituted alkenes.
 | ||
| Scheme 31 Olefin dihydroxylation via nitroarenes as an oxygen source (Leonori group). | ||
In examining stereoselectivity, they found that syn-diol 84 was preferentially obtained over anti-diol 84′ when using (E)-5-decene or (Z)-5-decene 83 as substrates (Scheme 31B). This stereoselectivity is attributed to the rapid bond rotation of triplet biradicals I and I′, which equilibrate to the less sterically hindered I. This intermediate then undergoes cyclization, producing the syn-cycloadduct II as the major component.
4.5. Anaerobic hydroxylation of C(sp3)–H bonds
The direct transformation of aliphatic C–H bonds into alcohol groups is a significant challenge in modern organic chemistry.83 This process requires selective activation of robust C(sp3)–H bonds, efficient C–O bond formation, and compatibility with oxidation-sensitive functional groups. Various strategies have emerged for incorporating oxygen into aliphatic frameworks, including direct C–H oxidation.84 However, this approach often necessitates harsh reaction conditions and faces challenges with site-selectivity and preferential alcohol formation. Although transition-metal catalysts and enzyme-based methods have been explored, these methods frequently require directing groups, incur high costs associated with precious metals, and suffer from low efficiency.85In 2023, the Parasram group introduced an anaerobic C–H hydroxylation method for aliphatic systems using photoexcited nitroarenes 1 (Scheme 32A).86 Although this type of reaction has been reported previously, it traditionally required harsh UV irradiation conditions or was limited to intramolecular reactions under visible light.87 Under visible light irradiation in CH2Cl2/HFIP, nitroarenes 1 facilitate benzylic C–H hydroxylation, producing cyclic benzylic alcohols 86 in various ring sizes, as well as ethylbenzene and toluene derivatives. This method also achieves efficient hydroxylation of tertiary C–H bonds and C–H bonds in cyclic alkanes 85, yielding alcohols 86 in good to excellent yields.
 | ||
| Scheme 32 (A) Anaerobic hydroxylation of C(sp3)–H bonds using nitroarenes (Parasram group). (B) Proposed mechanism for the hydroxylation process. | ||
The proposed mechanism involves the direct photoexcitation of the nitroarene to generate a triplet biradical intermediate (Scheme 32B). This intermediate undergoes hydrogen atom transfer (HAT) with the C(sp3)–H bond of the hydrocarbon, forming an alkyl radical and an oxygen-centered dihydroxyaniline radical. Radical recombination then affords intermediate I, which decomposes to produce the oxygen-transfer product 86. The possibility of alcohol formation through hydrolysis of intermediate I is considered low, as demonstrated by the lack of 18O incorporation when H218O was added to the reaction.
More recently, Miyake and colleagues reported the synthesis of keto-functionalized low-density polyethylene (Keto-LDPE) through the oxidation of C(sp3)–H bonds in LDPE using nitroarenes as oxidants under LED irradiation.88 While methods such as carbon monoxide copolymerization and transition metal-catalyzed C–H oxidation are well-established, this approach stands out for its simplicity and its potential to oxidize waste polyolefins, making it a more practical and versatile alternative.
4.6. Access 2-aminophenols via excited-state Cu catalysis and nitroarenes
2-Aminophenol derivatives are essential scaffolds in many top-selling pharmaceuticals, and synthesizing them from readily available and inexpensive nitroarenes significantly enhances their practicality. Ngai and colleagues successfully demonstrated the first direct synthesis of 2-aminophenols 88 from nitroarenes 1 using excited-state copper catalysis (Scheme 33).89 They found that 2-aminophenol derivatives 88 could be obtained by reacting nitroarenes 1 and acyl chlorides 87 with the copper catalyst Cu(IMes)Cl and rac-BINAP under visible light irradiation in THF at 60 °C. This mild reaction is compatible with a wide range of substrates, including aromatic nitroarenes with diverse substituents as well as both aromatic and aliphatic acyl chlorides. It also accommodates complex structures, such as derivatives of marketed drugs, natural products, and bio-relevant compounds. | ||
| Scheme 33 Excited-state copper-catalyzed synthesis of 2-aminophenols from nitroarenes (Nugai group). | ||
The proposed reaction mechanism involves several key steps (Scheme 34). Photoexcitation of the Cu(I)–BINAP complex forms an excited-state [Cu*(I)–BINAP] complex, which reduces the acyl chloride to generate a Cu(II)Cl–BINAP complex and releases an acyl radical. Simultaneously, the photoexcited nitroarene 1 undergoes intersystem crossing to form a triplet-state radical intermediate I, which reacts with the acyl radical to produce a short-lived radical intermediate II. Intermediate II then decomposes to give nitrosobenzene III and a carboxyl radical. Nitrosobenzene III acts as an effective radical trap, capturing a second acyl radical to form a persistent radical IV. Radical IV then combines with another acyl radical to generate the bis-acylated intermediate V, which undergoes in-cage 1,3-acyl migration and tautomerization to afford the desired 2-aminophenol 88. Concurrently, the Cu(II)Cl–BINAP complex undergoes ligand-to-metal charge transfer (LMCT) under visible light, releasing a chlorine radical and regenerating the Cu(I)–BINAP catalyst, thereby completing the copper catalytic cycle.
 | ||
| Scheme 34 Proposed reaction mechanism for excited-state copper-catalyzed synthesis of 2-aminophenols. | ||
Studies using 18O-labeled benzoyl chloride as the acyl radical source produced an acylated aminophenol 88′, which, upon debenzoylation, cyclization, and dehydration, yielded a benzoxazole 90 with 23.3% incorporation of 18O from nitroanisole 89. These findings suggest that acyloxy migration proceeds via an in-cage contact ion pair VI mechanism.
4.7. Oxidative cleavage of ketoximes to ketones using photoexcited nitroarenes
4.7. and 4.8. sections: Although the ultimate oxygen source is water rather than nitroarene, nitroarenes are utilized in these reactions for the conversion of ketoximes to ketones and in the hydration of alkynes.As part of ongoing research into the total synthesis of natural products, including cephalotane family members, developing methods to convert methoximes to ketones is critically important.90 Over the past decade, methoximes have gained attention as effective directing groups for C–H functionalization, enabling the formation of value-added products with diverse substituents.91 However, unlike O–acetyl oximes, methoximes are notoriously difficult to convert back to ketones, often requiring harsh conditions that result in low yields.
In 2023, Sarpong reported a photoirradiation reaction using nitrobenzene 1j to efficiently cleave benzyl ketoximes 91 and generate the corresponding ketones 92 (Scheme 35A).92 This reaction demonstrated high efficiency across a range of alkyl ketoximes, including substrates with halogens, boron groups, and cyclic ketones. Notably, the oxime cleavage was applicable to complex substrates, with a cephanolide D derivative affording 43% after deprotection.
 | ||
| Scheme 35 (A) Oxidative cleavage of ketoximes to ketones using nitroarenes (Sarpong group). (B) Proposed mechanism for the nitroarene-mediated cleavage reaction. | ||
The proposed mechanism involves a cycloaddition reaction between photoexcited nitroarene 1j and oxime 91, followed by cleavage and subsequent reaction with water to give the ketone 92 (Scheme 35B). To investigate this mechanism, they performed experiments using 18O-labeled nitroarene (1j-18O) and H218O. Treating adamantanone methoxime 93 with 18O-labeled nitroarene in a mixture of acetonitrile and 18O-labeled water resulted in 6% 18O incorporation in ketone 94-18O. Using unlabeled nitroarene with 18O-labeled water led to 69% 18O incorporation, indicating rapid oxygen exchange in the ketone group. When both 1j-18O and 18O-labeled water were used, the 18O content in the product increased to 77%, suggesting that both the nitroarene and water contribute to the oxygen atom in the ketone.
4.8. Triplet excited nitroarene converts linear alkynes to bent ketones by deleting a carbon atom
The ozonolysis of alkynes typically produces a mixture of products, with the composition influenced by reaction conditions and substrate electronic characteristics.93 Traditional methods for alkyne cleavage include using stoichiometric amounts of transition metal oxides, hypervalent iodine, peracids, or pressurized molecular oxygen. Previous studies involving photoexcited nitrobenzene under mercury arc lamp irradiation for three days resulted in complex mixtures, including benzophenone anil, carbon dioxide, nitrosobenzene, dibenzanilide, 2-hydroxyazobenzene, and β-lactam.94 However, these harsh conditions and product complexity have limited the practicality of such approaches, particularly in fine chemical industries and fields requiring high-throughput techniques.To address these limitations, Maji and co-workers proposed using triplet-excited nitroarenes 1k as an alternative to ozone, enabling oxygen atom introduction under mild, visible-light irradiation (Scheme 36).95 They irradiated alkynes 95 with nitrobenzene 1 as an oxygen atom donor in 1,2-dichloroethane, using purple LEDs (λmax = 390 nm) at 25 °C, producing ketone 96. This method is effective with both symmetrical diaryl and unsymmetrical aryl–alkyl alkynes. Notably, complex molecules, including derivatives of ibuprofen and flurbiprofen, reacted smoothly, yielding products in 67–86% while preserving a variety of sensitive functional groups.
 | ||
| Scheme 36 Conversion of linear alkynes to bent ketones via triplet-excited nitroarenes (Maji group). | ||
They propose a reaction mechanism (Scheme 37). The excited state of nitroarene, generated through irradiation with purple light, is expected to participate in a stepwise [3 + 2] cycloaddition with alkyne 95. This initially forms transient species I, which then undergoes cleavage of a weak O–N bond to give zwitterion II. Zwitterion II rearranges to form strained intermediate III, which decomposes to produce ketene IV and nitrosoarene. Control experiments have validated the presence of these intermediates. At this stage, ketene IV and nitrosoarene are proposed to undergo a [2 + 2] cycloaddition, generating the four-membered intermediate V. A retro [2 + 2] cycloaddition of V then releases imine VI and CO2, while subsequent hydrolysis of VI produce the final product 96, which has one less carbon atom than the original alkyne.
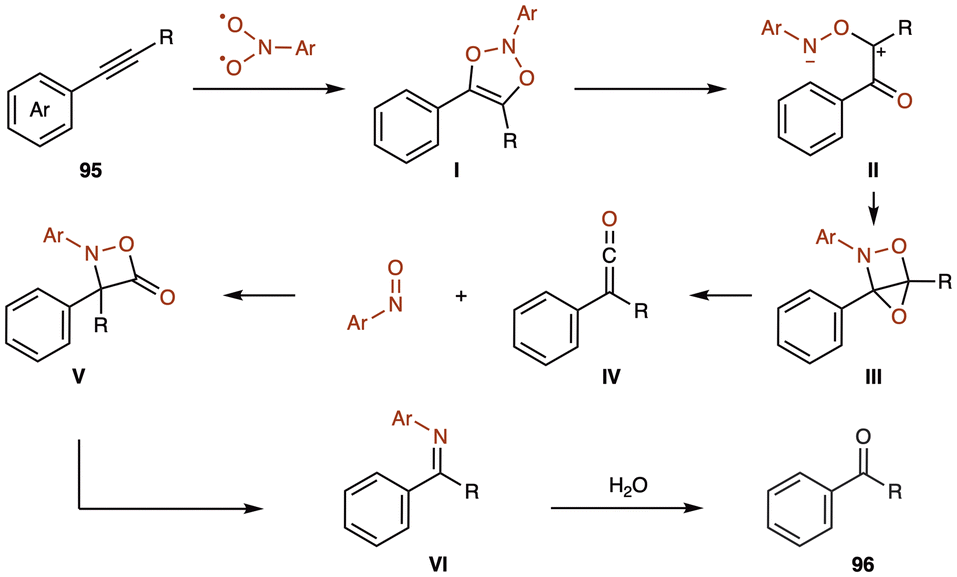 | ||
| Scheme 37 Proposed reaction mechanism for the conversion of alkynes to ketones using triplet-excited nitroarenes. | ||
The use of nitroarenes as an oxygen source in oxidation reactions has introduced a more sustainable alternative to traditional oxidants. Nonetheless, achieving higher atom economy and expanding the applicability to unactivated alkenes or C–H bonds in a broader range of substrates remain areas requiring further exploration. Innovations in catalyst design or the development of dual catalytic systems could address these limitations and enable more efficient transformations.
5. Conclusions
Over the past decade, remarkable progress has been made in the chemistry of nitroarenes, particularly in transition metal-catalyzed denitrative transformations that enable the formation of diverse bonds, including C–C, C–N, C–H, C–O, C–B, and C–Si. The development of advanced catalytic systems, such as Pd/BrettPhos, has not only expanded the scope of these reactions but also deepened our mechanistic understanding. Moreover, the advent of metal-free denitrative coupling reactions has introduced new possibilities for nitroarene transformations. In parallel, the photoexcitation of nitroarenes to their triplet excited state has enabled unprecedented aromatic ring expansions, facilitating the synthesis of a wide range of aniline derivatives and aliphatic heterocycles. Additionally, the innovative use of nitro groups as an oxygen source has paved the way for anaerobic oxidation protocols, providing sustainable alternatives to traditional methodologies, such as alkene hydration and oxidative cleavage, which often rely on oxygen or ozone. Despite these substantial advancements, the full potential of nitroarenes in large-scale applications remains underexplored. Challenges such as cost-effectiveness, scalability, and compatibility with sensitive functional groups must be addressed to unlock their broader adoption. Leveraging computational tools for reaction design and exploring tandem or cascade processes involving nitroarenes hold significant promise for future breakthroughs in this field.As discussed throughout this review, the transformative power of nitroarene chemistry lies in its ability to target entirely different elements—carbon, nitrogen, and oxygen—within the aromatic nitro group. This precise control has been made possible by the development of novel catalysts (e.g., Pd-based systems) and innovative reaction conditions (e.g., blue LED irradiation). While many of these transformations trace their origins to classical reactions, modern technologies have reimagined nitroarenes, transforming them from “classic materials” into a new generation of versatile building blocks. These advancements have profoundly enhanced the utility of nitroarenes, offering sustainable and environmentally friendly methodologies for organic synthesis. The growing body of research underscores the evolving role of nitroarenes, highlighting their potential for continued innovation, broad applications, and their unique ability to bridge historical foundations with cutting-edge technologies.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.Conflicts of interest
There are no conficts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP21H05213 (Digi-TOS) (to J. Y.). This work was partly supported by JST ERATO Grant Number JPMJER1901 (to J. Y.).References
- (a) S. Sengupta and P. Das, Org. Biomol. Chem., 2021, 19, 8409–8424, 10.1039/d1ob01455b; (b) G. Calderari and D. Seebach, Helv. Chim. Acta, 1985, 68, 1592–1604, DOI:10.1002/hlca.19850680611; (c) K. Schofiled, Aromatic Nitrations, Cambridge University Press, Cambridge, 1980 Search PubMed; (d) G. A. Olah, R. Malhotra and S. C. Narang, in World Scientific Series in 20th Century Chemistry, World Scientific Publishing Company, 2003, vol. 11, pp. 975–979. DOI:10.1142/9789812791405_0192.
- (a) F. Terrier, Modern Nucleophilic Aromatic Substitution, Wiley, 1st edn, 2013. DOI:10.1002/9783527656141; (b) J. Miller, Aromatic Nucleophilic Substitution, Elsevier, Amsterdam, 1968 Search PubMed; (c) J. F. Bunnett and R. E. Zahler, Chem. Rev., 1951, 49, 273–412, DOI:10.1021/cr60153a002.
-
(a) J. R. Beck, Tetrahedron, 1978, 34, 2057–2068, DOI:10.1016/0040-4020(78)89004-8;
(b) T. Takekoshi, Polym. J., 1987, 19, 191–202, DOI:10.1295/polymj.19.191;
(c) Y. Shen, H. Liu and Y. Chen, J. Org. Chem., 1990, 55, 3961–3962, DOI:10.1021/jo00299a051;
(d) M. M
![[a with combining cedilla]](https://www.rsc.org/images/entities/char_0061_0327.gif) kosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631–2666, DOI:10.1021/cr020086+;
(e) M. Mąkosza, Chem. – Eur. J., 2014, 20, 5536–5545, DOI:10.1002/chem.201400097;
(f) R. Loska and M. Mąkosza, Synthesis, 2020, 3095–3110, DOI:10.1055/s-0040-1707149.
kosza and K. Wojciechowski, Chem. Rev., 2004, 104, 2631–2666, DOI:10.1021/cr020086+;
(e) M. Mąkosza, Chem. – Eur. J., 2014, 20, 5536–5545, DOI:10.1002/chem.201400097;
(f) R. Loska and M. Mąkosza, Synthesis, 2020, 3095–3110, DOI:10.1055/s-0040-1707149. - (a) S. S. Gawali, Rev. Adv. Chem., 2023, 13, 416–430, DOI:10.1134/S263482762360024X; (b) M. Mąkosza, Chem. – Eur. J., 2020, 26, 15346–15353, DOI:10.1002/chem.202003770; (c) M. Mąkosza, Chem. Soc. Rev., 2010, 39, 2855, 10.1039/b822559c; (d) M. Makosza and J. Winiarski, Acc. Chem. Res., 1987, 20, 282–289, DOI:10.1021/ar00140a003.
- (a) G. Booth, Nitro Compounds, Aromatic, John Wiley & Sons, Inc., New York, 2000 Search PubMed; (b) N. Ono, The Nitro Group In Organic Synthesis, WILEY-VCH, 2001 CrossRef; (c) R. Ballini, G. Bosica, D. Fiorini and M. Petrini, Tetrahedron Lett., 2002, 43, 5233–5235, DOI:10.1016/S0040-4039(02)01039-0; (d) L. Larina and V. Lopyrev, Nitroazoles: Synthesis, Structure and Applications, Springer Science, 2009. DOI:10.1007/978-0-387-98070-6; (e) S. G. Zlotin, I. L. Dalinger, N. N. Makhova and V. A. Tartakovsky, Russ. Chem. Rev., 2020, 89, 1 Search PubMed; (f) F. Hao and N. Nishiwaki, Molecules, 2020, 25, 673, DOI:10.3390/molecules25030673; (g) D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680, DOI:10.1021/acs.chemrev.8b00547; (h) S. G. Zlotin, I. L. Dalinger, N. N. Makhova and V. A. Tartakovsky, Russ. Chem. Rev., 2020, 89, 1–54, DOI:10.1070/RCR4908; (i) J. Cadogan, Synthesis, 2002, 11–17, DOI:10.1055/s-1969-34189; (j) G. Bartoli, R. Dalpozzo and M. Nardi, Chem. Soc. Rev., 2014, 43, 4728–4750, 10.1039/c4cs00045e; (k) D. Zou, W. Wang, Y. Hu and T. Jia, Org. Biomol. Chem., 2023, 21, 2254–2271, 10.1039/D3OB00064H.
- (a) P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Chem, 2023, 9, 3401–3414, DOI:10.1016/j.chempr.2023.09.008; (b) R. Jana and K. Pradhan, Chem. Commun., 2024, 60, 8806–8823, 10.1039/D4cc02582b; (c) K. Teng, Q. Liu, J. Lv and T. Li, ChemPhotoChem, 2024, 8, e202300236, DOI:10.1002/cptc.202300236.
- N. Miyaura, in Cross-Coupling Reactions: A Practical Guide, Springer Berlin Heidelberg, Berlin, Heidelberg, 2002, vol. 219 Search PubMed.
- (a) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741–5829, DOI:10.1021/acs.chemrev.0c01030; (b) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19–30, DOI:10.1002/ejoc.201200914; (c) Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515, DOI:10.1021/acs.chemrev.0c00088; (d) J. Lou, Q. Wang, P. Wu, H. Wang, Y.-G. Zhou and Z. Yu, Chem. Soc. Rev., 2020, 49, 4307–4359, 10.1039/C9CS00837C; (e) R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888, 10.1039/C7CS00182G.
- X. Zheng, J. Ding, J. Chen, W. Gao, M. Liu and H. Wu, Org. Lett., 2011, 13, 1726–1729, DOI:10.1021/ol200251x.
- (a) M. R. Yadav, M. Nagaoka, M. Kashihara, R.-L. Zhong, T. Miyazaki, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2017, 139, 9423–9426, DOI:10.1021/jacs.7b03159; (b) F. Inoue, M. Kashihara, M. R. Yadav and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 13307–13309, DOI:10.1002/anie.201706982; (c) M. Kashihara, M. R. Yadav and Y. Nakao, Org. Lett., 2018, 20, 1655–1658, DOI:10.1021/acs.orglett.8b00430; (d) K. K. Asahara, T. Okita, A. N. Saito, K. Muto, Y. Nakao and J. Yamaguchi, Org. Lett., 2019, 21, 4721–4724, DOI:10.1021/acs.orglett.9b01593; (e) B. Feng, Y. Yang and J. You, Chem. Sci., 2020, 11, 6031–6035, 10.1039/D0SC01641A; (f) T. Okita, K. K. Asahara, K. Muto and J. Yamaguchi, Org. Lett., 2020, 22, 3205–3208, DOI:10.1021/acs.orglett.0c00983; (g) F. Zhou, F. Zhou, R. Su, Y. Yang and J. You, Chem. Sci., 2020, 11, 7424–7428, 10.1039/D0SC02058C; (h) B. Feng, Y. Yang and J. You, Chem. Commun., 2020, 56, 790–793, 10.1039/C9CC08663C.
- (a) M. Kashihara, R.-L. Zhong, K. Semba, S. Sakaki and Y. Nakao, Chem. Commun., 2019, 55, 9291–9294, 10.1039/C9CC05055H; (b) W. Chen, K. Chen, W. Chen, M. Liu and H. Wu, ACS Catal., 2019, 9, 8110–8115, DOI:10.1021/acscatal.9b02760; (c) K. Iizumi, K. P. Nakayama, K. Kato, K. Muto and J. Yamaguchi, J. Org. Chem., 2022, 87, 11909–11918, DOI:10.1021/acs.joc.2c01562; (d) T. Zhou, P. Gao, E. Bisz, B. Dziuk, R. Lalancette, R. Szostak and M. Szostak, Catal. Sci. Technol., 2022, 12, 6581–6589, 10.1039/D2CY01136K; (e) W. Chen, W. Chen, M. Liu and H. Wu, Org. Lett., 2022, 24, 6983–6987, DOI:10.1021/acs.orglett.2c02796; (f) Y. Ma, A. A. Hussein and Z. Wang, J. Org. Chem., 2022, 87, 531–539, DOI:10.1021/acs.joc.1c02536; (g) W. Chen, K. Chen, X. Wang, L. Yang and W. Chen, RSC Adv., 2024, 14, 16624–16628, 10.1039/D4RA00921E.
- For reviews, see: (a) K. Muto, T. Okita and J. Yamaguchi, ACS Catal., 2020, 10, 9856–9871, DOI:10.1021/acscatal.0c02990; (b) H. Li, Z. Lei, X. Yang, Y. Xiao, Y. Yin, J. Du, W. Duan and L. Yu, Eur. J. Org. Chem., 2024, e202400787, DOI:10.1002/ejoc.202400787; (c) M. Kashihara and Y. Nakao, Acc. Chem. Res., 2021, 54, 2928–2935, DOI:10.1021/acs.accounts.1c00220; (d) S. Maddah-Roodan, R. Soltani and A. Ghaderi, J. Iran. Chem. Soc., 2021, 18, 519–542, DOI:10.1007/s13738-020-02054-2; (e) L. Peng, Z. Hu, Z. Tang, Y. Jiao and X. Xu, Chin. Chem. Lett., 2019, 30, 1481–1487, DOI:10.1016/j.cclet.2019.04.008.
- (a) E. N. Pitsinos, V. P. Vidali and E. A. Couladouros, Eur. J. Org. Chem., 2011, 1207–1222, DOI:10.1002/ejoc.201001520; (b) F. Bedos-Belval, A. Rouch, C. Vanucci-Bacqué and M. Baltas, MedChemComm, 2012, 3, 1356, 10.1039/c2md20199b; (c) T. Chen, H. Xiong, J.-F. Yang, X.-L. Zhu, R.-Y. Qu and G.-F. Yang, J. Agric. Food Chem., 2020, 68, 9839–9877, DOI:10.1021/acs.jafc.0c03369.
- (a) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400–5449, DOI:10.1002/anie.200300594; (b) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971, DOI:10.1002/anie.200804497; (c) G. Mann, C. Incarvito, A. L. Rheingold and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 3224–3225, DOI:10.1021/ja984321a.
- N. Matsushita, M. Kashihara, M. Formica and Y. Nakao, Organometallics, 2021, 40, 2209–2214, DOI:10.1021/acs.organomet.1c00183.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841, 10.1039/b922984a.
- (a) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082–1146, DOI:10.1021/cr9000836; (b) Y.-J. Hao, X.-S. Hu, Y. Zhou, J. Zhou and J.-S. Yu, ACS Catal., 2020, 10, 955–993, DOI:10.1021/acscatal.9b04480.
- Z. Li, Y. Peng and T. Wu, Org. Lett., 2021, 23, 881–885, DOI:10.1021/acs.orglett.0c04104.
- (a) M. Kosugi, K. Sasazawa, Y. Shimizu and T. Migita, Chem. Lett., 1977, 6, 301–302, DOI:10.1246/cl.1977.301; (b) D. Milstein and J. K. Stille, J. Am. Chem. Soc., 1978, 100, 3636–3638, DOI:10.1021/ja00479a077; (c) C. Cordovilla, C. Bartolomé, J. M. Martínez-Ilarduya and P. Espinet, ACS Catal., 2015, 5, 3040–3053, DOI:10.1021/acscatal.5b00448.
- A. Makhloutah, D. Hatych, T. Chartier, L. Rocard, A. Goujon, F.-X. Felpin and P. Hudhomme, Org. Biomol. Chem., 2022, 20, 362–365, 10.1039/D1OB02291A.
- F. Fernández-Lázaro, N. Zink-Lorre and Á. Sastre-Santos, J. Mater. Chem. A, 2016, 4, 9336–9346, 10.1039/C6TA02045C.
- (a) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651, DOI:10.1021/acscatal.6b02374; (b) Z. Rappoport and Y. Apeloig, The Chemistry of Organic Silicon Compounds, Wiley, New York, 2000 Search PubMed.
- J. Yao, L. Yu, W. Duan and C.-J. Li, Org. Chem. Front., 2023, 10, 524–530, 10.1039/D2QO01764D.
- (a) J. P. Wolfe, S. Wagaw, J.-F. Marcoux and S. L. Buchwald, Acc. Chem. Res., 1998, 31, 805–818, DOI:10.1021/ar9600650; (b) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067, DOI:10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L.
- L. Feng, J. Yao, L. Yu and W. Duan, Org. Chem. Front., 2022, 9, 2351–2356, 10.1039/D2QO00010E.
- Z. Lei, J. Yao, Y. Xiao, W. H. Liu, L. Yu, W. Duan and C.-J. Li, Chem. Sci., 2024, 15, 3552–3561, 10.1039/D3SC06618E.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066, DOI:10.1021/cr9903048.
- F. Zhang, F. Wang, Y. Zhao, R. Chen and X. Wu, Org. Chem. Front., 2023, 10, 2193–2197, 10.1039/D3QO00132F.
- K. K. Asahara, K. Muto and J. Yamaguchi, Chem. Lett., 2023, 52, 299–302, DOI:10.1246/cl.230056.
- The olefin-isomerization of Mizoroki–Heck products was previously observed, see: X. Wu and J. (Steve) Zhou, Chem. Commun., 2013, 49, 4794, 10.1039/c3cc41722k.
- (a) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917, DOI:10.1021/jm100762r; (b) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2078, DOI:10.1021/cr020045d.
- (a) K. Iizumi, H. Tanaka, K. Muto and J. Yamaguchi, Org. Lett., 2024, 26, 3977–3981, DOI:10.1021/acs.orglett.4c01118; (b) Y. Chen, Q. Chen, S. Zhang, K. Feng, Y.-Q. Xu, X. Chen, Z.-Y. Cao and X. Kong, Org. Lett., 2024, 26, 7555–7559, DOI:10.1021/acs.orglett.4c02552.
- S. Erhardt, V. V. Grushin, A. H. Kilpatrick, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2008, 130, 4828–4845, DOI:10.1021/ja078298h.
- K. Iizumi, M. B. Kurosawa, R. Isshiki, K. Muto and J. Yamaguchi, Synlett, 2021, 1555–1559, DOI:10.1055/s-0040-1706555.
- (a) S. Pimparkar, A. Koodan, S. Maiti, N. S. Ahmed, M. M. M. Mostafa and D. Maiti, Chem. Commun., 2021, 57, 2210–2232, 10.1039/D0CC07783F; (b) G. Xu, L. Zhou, C. Lv, M. Xian and Q. Wang, J. Saudi Chem. Soc., 2019, 23, 896–902, DOI:10.1016/j.jscs.2019.01.002.
- (a) X. Hu, J.-W. Wu, M. Wang, M.-H. Yu, Q.-S. Zhao, H.-Y. Wang and A.-J. Hou, J. Nat. Prod., 2012, 75, 82–87, DOI:10.1021/np2007318; (b) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859, DOI:10.1021/jm4017625; (c) Z. Peng, K. Huang, Y. Tao, X. Li, L. Zhang, P. Lu and Y. Wang, Mater. Chem. Front., 2017, 1, 1858–1865, 10.1039/C7QM00145B.
- J.-Y. Mérour, F. Buron, K. Plé, P. Bonnet and S. Routier, Molecules, 2014, 19, 19935–19979, DOI:10.3390/molecules191219935.
- J. Yao, Y. Xiao, H. Li, X. Yang, J. Du, Y. Yin, L. Feng, W. Duan and L. Yu, Org. Lett., 2024, 26, 7307–7312, DOI:10.1021/acs.orglett.4c02340.
- H.-J. Ha, D. W. Kang, H.-M. Kim, J.-M. Kang, J. Ann, H. J. Hyun, J. H. Lee, S. H. Kim, H. Kim, K. Choi, H.-S. Hong, Y. Kim, D.-G. Jo, J. Lee and J. Lee, J. Med. Chem., 2018, 61, 396–402, DOI:10.1021/acs.jmedchem.7b00844.
- K. Mo, X. Zhou, J. Wu and Y. Zhao, Chem. Commun., 2021, 58, 1306–1309, 10.1039/D1CC05724C.
- B. Floris, P. Galloni, V. Conte and F. Sabuzi, Biomolecules, 2021, 11, 1325, DOI:10.3390/biom11091325.
- R. D. Knudsen and H. R. Snyder, J. Org. Chem., 1974, 39, 3343–3346, DOI:10.1021/jo00937a007.
- L. Duff, H. Meakin, A. Richardson, A. J. Greener, G. W. A. Smith, I. Ocaña, V. Chechik and M. J. James, Chem. – Eur. J., 2023, 29, e202203807, DOI:10.1002/chem.202203807.
- R. D. Guthrie, C. Hartmann, R. Neill and D. E. Nutter, J. Org. Chem., 1987, 52, 736–740, DOI:10.1021/jo00381a005.
- D. G. Hall, in Boronic Acids, ed. D. G. Hall, Wiley-VCH, Weinheim, Germany, 2005, pp. 1–99 Search PubMed.
- L. Du, B. Zhang, S. Ji, H. Cai and H. Zhang, Sci. China: Chem., 2023, 66, 534–539, DOI:10.1007/s11426-022-1470-8.
- Y. Chen, Q. Chen, S. Zhang, K. Feng, Y.-Q. Xu, X. Chen, Z.-Y. Cao and X. Kong, Org. Lett., 2024, 26, 7555–7559, DOI:10.1021/acs.orglett.4c02552.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge UK, 2004 Search PubMed.
- (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394, DOI:10.1038/s41557-018-0021-z; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479, DOI:10.1021/jm200187y; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274, DOI:10.1021/jm501100b; (d) D. G. Brown, M. M. Gagnon and J. Boström, J. Med. Chem., 2015, 58, 2390–2405, DOI:10.1021/jm501894t.
- A. Nilova, L.-C. Campeau, E. C. Sherer and D. R. Stuart, J. Med. Chem., 2020, 63, 13389–13396, DOI:10.1021/acs.jmedchem.0c00915.
- (a) H. M. Begam, R. Choudhury, A. Behera and R. Jana, Org. Lett., 2019, 21, 4651–4656, DOI:10.1021/acs.orglett.9b01546; (b) T.-J. Cheng, J.-J. Chen, P. Wu, H. Xu and H.-X. Dai, Org. Lett., 2021, 23, 8505–8509, DOI:10.1021/acs.orglett.1c03223; (c) J. E. Gillespie, C. Morrill and R. J. Phipps, J. Am. Chem. Soc., 2021, 143, 9355–9360, DOI:10.1021/jacs.1c05531; (d) Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764–1767, DOI:10.1021/ol500464x; (e) Á. M. Martínez, N. Rodríguez, R. G. Arrayás and J. C. Carretero, Chem. Commun., 2014, 50, 2801, 10.1039/c3cc49633c; (f) P. Wu, W. Huang, T.-J. Cheng, H.-X. Lin, H. Xu and H.-X. Dai, Org. Lett., 2020, 22, 5051–5056, DOI:10.1021/acs.orglett.0c01632; (g) Q.-L. Yang, X.-Y. Wang, J.-Y. Lu, L.-P. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2018, 140, 11487–11494, DOI:10.1021/jacs.8b07380.
- G. Li, M. N. Lavagnino, S. Z. Ali, S. Hu and A. T. Radosevich, J. Am. Chem. Soc., 2023, 145, 41–46, DOI:10.1021/jacs.2c12450.
- T. V. Nykaza, A. Ramirez, T. S. Harrison, M. R. Luzung and A. T. Radosevich, J. Am. Chem. Soc., 2018, 140, 3103–3113, DOI:10.1021/jacs.7b13803.
- (a) H. Inui, K. Sawada, S. Oishi, K. Ushida and R. J. McMahon, J. Am. Chem. Soc., 2013, 135, 10246–10249, DOI:10.1021/ja404172s; (b) O. L. Chapman and J. P. Le Roux, J. Am. Chem. Soc., 1978, 100, 282–285, DOI:10.1021/ja00469a049.
- R. Sánchez-Bento, B. Roure, J. Llaveria, A. Ruffoni and D. Leonori, Chem, 2023, 9, 3685–3695, DOI:10.1016/j.chempr.2023.10.008.
- R. J. Sundberg, B. P. Das and R. H. Smith, J. Am. Chem. Soc., 1969, 91, 658–668, DOI:10.1021/ja01031a024.
- (a) D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394, DOI:10.1038/s41557-018-0021-z; (b) H. Pellissier, in Synthetic Approaches to Nonaromatic Nitrogen Heterocycles, ed. A. M. M. M. Faisca Phillips, Wiley, 1st edn, 2020, pp. 399–435 Search PubMed; (c) L. D. Pennington and D. T. Moustakas, J. Med. Chem., 2017, 60, 3552–3579, DOI:10.1021/acs.jmedchem.6b01807.
- (a) A. D. G. Lawson, M. MacCoss and J. P. Heer, J. Med. Chem., 2018, 61, 4283–4289, DOI:10.1021/acs.jmedchem.7b01120; (b) M. R. Bauer, P. Di Fruscia, S. C. C. Lucas, I. N. Michaelides, J. E. Nelson, R. I. Storer and B. C. Whitehurst, RSC Med. Chem., 2021, 12, 448–471, 10.1039/D0MD00370K.
- E. Matador, M. J. Tilby, I. Saridakis, M. Pedrón, D. Tomczak, J. Llaveria, I. Atodiresei, P. Merino, A. Ruffoni and D. Leonori, J. Am. Chem. Soc., 2023, 145, 27810–27820, DOI:10.1021/jacs.3c10863.
- F. R. Atherton and R. W. Lambert, J. Chem. Soc., Perkin Trans. 1, 1973, 1079, 10.1039/p19730001079.
- A. Mann, in The Practice of Medicinal Chemistry, ed. C. G. Wermuth, Academic Press, New York, 3rd edn, 2008, pp. 363–379 Search PubMed.
- M. A. Chiacchio, L. Legnani, U. Chiacchio and D. Iannazzo, in More Synthetic Approaches to Nonaromatic Nitrogen Heter-ocycles, ed. A. M. F. Phillips, 2022, pp. 529–558 Search PubMed.
- R. Mykura, R. Sánchez-Bento, E. Matador, V. K. Duong, A. Varela, L. Angelini, R. J. Carbajo, J. Llaveria, A. Ruffoni and D. Leonori, Nat. Chem., 2024, 16, 771–779, DOI:10.1038/s41557-023-01429-1.
- (a) L. A. Paquette, J. Am. Chem. Soc., 1962, 84, 4987–4988, DOI:10.1021/ja00883a079; (b) Y. Liu, F. Chen, Y.-M. He, C. Li and Q.-H. Fan, Org. Biomol. Chem., 2019, 17, 5099–5105, 10.1039/C9OB00770A; (c) S. J. Taylor, E. H. Demont, J. Gray, N. Deeks, A. Patel, D. Nguyen, M. Taylor, S. Hood, R. J. Watson, R. A. Bit, F. McClure, H. Ashall and J. Witherington, J. Med. Chem., 2015, 58, 8236–8256, DOI:10.1021/acs.jmedchem.5b01102.
- For examples: (a) Y. Gao, S. Yang, W. Xiao, J. Nie and X.-Q. Hu, Chem. Commun., 2020, 56, 13719–13730, 10.1039/d0cc06023b; (b) J. Xiao and X. Li, Angew. Chem., 2011, 123, 7364–7375, DOI:10.1002/ange.201100148; (c) S. Zhou, Q. Liu, M. Bao, J. Huang, J. Wang, W. Hu and X. Xu, Org. Chem. Front., 2021, 8, 1808–1816, 10.1039/D1QO00134E; (d) F. Ragaini, S. Cenini, E. Borsani, M. Dompé, E. Gallo and M. Moret, Organometallics, 2001, 20, 3390–3398, DOI:10.1021/om010076j; (e) K. Teng, Q. Liu, J. Lv and T. Li, ChemPhotoChem, 2024, 8, e202300236, DOI:10.1002/cptc.202300236.
- (a) D. Zou, L. Gan, F. Yang, H. Wang, Y. Pu, J. Li and P. J. Walsh, Nat. Commun., 2021, 12, 7060, DOI:10.1038/s41467-021-26767-x; (b) M. Li, M. Zhao, X. Zhong, X. Wen, X. Liu, H. Jiang, R. Tan and J. Li, Org. Chem. Front., 2025, 12, 636–640, 10.1039/D4QO01418A; (c) H. Zhu, J. N. Powell, V. A. Geldchen, A. S. Drumheller and T. G. Driver, Angew. Chem., Int. Ed., 2024, e202416126, DOI:10.1002/anie.202416126; (d) X.-Z. Shao, G.-Y. Xu, W. Fan, S. Zhang and M.-B. Li, Org. Chem. Front., 2023, 10, 624–631, 10.1039/D2QO01743A; (e) B. Wang, M.-J. Zhou and Q.-L. Zhou, Org. Lett., 2022, 24, 2894–2898, DOI:10.1021/acs.orglett.2c00831.
- H.-T. Kuo, C.-F. Peng, H.-Y. Huang, C.-H. Lin, I.-S. Chen and I.-L. Tsai, Planta Med., 2011, 77, 736–741, DOI:10.1055/s-0030-1250534.
- (a) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402–10405, DOI:10.1021/ja203810b; (b) A. Chan and K. A. Scheidt, Org. Lett., 2005, 7, 905–908, DOI:10.1021/ol050100f; (c) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208, DOI:10.1002/anie.200461572; (d) M. He and J. W. Bode, Org. Lett., 2005, 7, 3131–3134, DOI:10.1021/ol051234w; (e) V. Nair, C. R. Sinu, B. P. Babu, V. Varghese, A. Jose and E. Suresh, Org. Lett., 2009, 11, 5570–5573, DOI:10.1021/ol901918x; (f) B. E. Maki and K. A. Scheidt, Org. Lett., 2008, 10, 4331–4334, DOI:10.1021/ol8018488.
- N. A. White and T. Rovis, J. Am. Chem. Soc., 2014, 136, 14674–14677, DOI:10.1021/ja5080739.
- Y. Zhang, Y. Du, Z. Huang, J. Xu, X. Wu, Y. Wang, M. Wang, S. Yang, R. D. Webster and Y. R. Chi, J. Am. Chem. Soc., 2015, 137, 2416–2419, DOI:10.1021/ja511371a.
- H. Wang, Y. Wang, X. Chen, C. Mou, S. Yu, H. Chai, Z. Jin and Y. R. Chi, Org. Lett., 2019, 21, 7440–7444, DOI:10.1021/acs.orglett.9b02736.
- H. J. Arpe, in Industrial Organic Chemistry, Wiley-VCH, Weinheim, Germany, 5th edn, 2010 Search PubMed.
- A. Bhunia, K. Bergander, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 8313–8320, DOI:10.1002/anie.202015740.
- (a) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891, DOI:10.1126/science.aab0245; (b) C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 12494, DOI:10.1038/ncomms12494.
- J. Elfert, A. Bhunia, C. G. Daniliuc and A. Studer, ACS Catal., 2023, 13, 6704–6709, DOI:10.1021/acscatal.3c00958.
- A. Rajagopalan, M. Lara and W. Kroutil, Adv. Synth. Catal., 2013, 355, 3321–3335, DOI:10.1002/adsc.201300882.
- (a) Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Angew. Chem., Int. Ed., 2021, 60, 15138–15152, DOI:10.1002/anie.202014974; (b) S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989, DOI:10.1021/cr040679f; (c) T. J. Fisher and P. H. Dussault, Tetrahedron, 2017, 73, 4233–4258, DOI:10.1016/j.tet.2017.03.039.
- (a) B. R. Travis, R. S. Narayan and B. Borhan, J. Am. Chem. Soc., 2002, 124, 3824–3825, DOI:10.1021/ja017295g; (b) K. C. Nicolaou, V. A. Adsool and C. R. H. Hale, Org. Lett., 2010, 12, 1552–1555, DOI:10.1021/ol100290a.
- D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437–15442, DOI:10.1021/jacs.2c05648.
- G. Buchi and D. E. Ayer, J. Am. Chem. Soc., 1956, 78, 689–690, DOI:10.1021/ja01584a048.
- A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81–86, DOI:10.1038/s41586-022-05211-0.
- C. Hampton, M. Simonetti and D. Leonori, Angew. Chem., Int. Ed., 2023, 62, e202214508, DOI:10.1002/anie.202214508.
- (a) M. K. Goetz, J. E. Schneider, A. S. Filatov, K. A. Jesse and J. S. Anderson, J. Am. Chem. Soc., 2021, 143, 20849–20862, DOI:10.1021/jacs.1c09280; (b) K. Yoshizawa, Y. Shiota and Y. Kagawa, Bull. Chem. Soc. Jpn., 2000, 73, 2669–2673, DOI:10.1246/bcsj.73.2669.
- (a) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362–3374, DOI:10.1002/anie.201006368; (b) Y.-F. Liang and N. Jiao, Acc. Chem. Res., 2017, 50, 1640–1653, DOI:10.1021/acs.accounts.7b00108; (c) D. Miller, Free Radical Biol. Med., 1990, 8, 95–108, DOI:10.1016/0891-5849(90)90148-C; (d) P. Stavropoulos, R. Çelenligil-Çetin and A. E. Tapper, Acc. Chem. Res., 2001, 34, 745–752, DOI:10.1021/ar000100+; (e) W. G. Shuler, S. L. Johnson and M. K. Hilinski, Org. Lett., 2017, 19, 4790–4793, DOI:10.1021/acs.orglett.7b02178.
- (a) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912, 10.1039/c1cs15085e; (b) Z. Iqbal, A. Joshi and S. Ranjan De, Adv. Synth. Catal., 2020, 362, 5301–5351, DOI:10.1002/adsc.202000762; (c) Z. Li, Z. Wang, N. Chekshin, S. Qian, J. X. Qiao, P. T. Cheng, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, Science, 2021, 372, 1452–1457, DOI:10.1126/science.abg2362.
- J. M. Paolillo, A. D. Duke, E. S. Gogarnoiu, D. E. Wise and M. Parasram, J. Am. Chem. Soc., 2023, 145, 2794–2799, DOI:10.1021/jacs.2c13502.
- UV light conditions: (a) J. W. Weller and G. A. Hamilton, J. Chem. Soc. D, 1970, 1390–1391, 10.1039/c29700001390; (b) S. Negele, K. Wieser and T. Severin, J. Org. Chem., 1998, 63, 1138–1143, DOI:10.1021/jo971617a; (c) J. Libman and E. Berman, Tetrahedron Lett., 1977, 18, 2191–2192, DOI:10.1016/S0040-4039(01)83717-5 Intramolecular: (d) B. Wang, H. Ren, H.-J. Cao, C. Lu and H. Yan, Chem. Sci., 2022, 13, 11074–11082, 10.1039/D2SC03590A.
- X. Liu, Z. Hu, B. S. Portela, E. M. Rettner, A. Pineda, J. Miscall, N. A. Rorrer, A. T. Krummel, R. S. Paton and G. M. Miyake, Angew. Chem., Int. Ed., 2024, e202418411, DOI:10.1002/anie.202418411.
- J. A. Shah, A. Banerjee, U. Mukherjee and M.-Y. Ngai, Chem, 2024, 10, 686–697, DOI:10.1016/j.chempr.2023.11.005.
- (a) M. Haider, G. Sennari, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2021, 143, 2710–2715, DOI:10.1021/jacs.1c00293; (b) G. Sennari, K. E. Gardner, S. Wiesler, M. Haider, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 19173–19185, DOI:10.1021/jacs.2c08803.
- (a) Y.-F. Liang, X. Wang, Y. Yuan, Y. Liang, X. Li and N. Jiao, ACS Catal., 2015, 5, 6148–6152, DOI:10.1021/acscatal.5b01700; (b) C. Ma, C.-Q. Zhao, Y.-Q. Li, L.-P. Zhang, X.-T. Xu, K. Zhang and T.-S. Mei, Chem. Commun., 2017, 53, 12189–12192, 10.1039/C7CC07429H; (c) Y. Park, S. Jee, J. G. Kim and S. Chang, Org. Process Res. Dev., 2015, 19, 1024–1029, DOI:10.1021/acs.oprd.5b00164; (d) Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956–1963, DOI:10.1021/cs502126n; (e) B. S. Pilgrim, A. E. Gatland, C. H. A. Esteves, C. T. McTernan, G. R. Jones, M. R. Tatton, P. A. Procopiou and T. J. Donohoe, Org. Biomol. Chem., 2016, 14, 1065–1090, 10.1039/C5OB02320C; (f) S. Yu, B. Wan and X. Li, Org. Lett., 2013, 15, 3706–3709, DOI:10.1021/ol401569u.
- L. T. Göttemann, S. Wiesler and R. Sarpong, Chem. Sci., 2024, 15, 213–219, 10.1039/D3SC05414D.
- D. Habrant, V. Rauhala and A. M. P. Koskinen, Chem. Soc. Rev., 2010, 39, 2007, 10.1039/b915418c.
- (a) S. Lai and D. G. Lee, Tetrahedron, 2002, 58, 9879–9887, DOI:10.1016/S0040-4020(02)01227-9; (b) D. Yang, F. Chen, Z.-M. Dong and D.-W. Zhang, J. Org. Chem., 2004, 69, 2221–2223, DOI:10.1021/jo0357925; (c) A. P. Krapcho, J. R. Larson and J. M. Eldridge, J. Org. Chem., 1977, 42, 3749–3753, DOI:10.1021/jo00443a026; (d) K. Miyamoto, Y. Sei, K. Yamaguchi and M. Ochiai, J. Am. Chem. Soc., 2009, 131, 1382–1383, DOI:10.1021/ja808829t; (e) K. Aravinda Kumar, V. Venkateswarlu, R. Vishwakarma and S. Sawant, Synthesis, 2015, 3161–3168, DOI:10.1055/s-0034-1381026; (f) T. Yamaguchi, T. Nobuta, Y. Kudo, S. Hirashima, N. Tada, T. Miura and A. Itoh, Synlett, 2013, 607–610, DOI:10.1055/s-0032-1318308; (g) H. Muramatsu, K. Inukai and T. Ueda, J. Org. Chem., 1964, 29, 2220–2223, DOI:10.1021/jo01031a027.
- A. Dixit, S. K. Jana, A. Jati, A. Das, P. Kumar, P. Dey, N. Aditya, K. Sarkar and B. Maji, 2023. DOI:10.26434/chemrxiv-2023-t1cvq-v2.
| This journal is © The Royal Society of Chemistry 2025 |