
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric isochalcogenourea-catalysed (4 + 2)-cycloadditions of ortho-quinone methides and allenoates†
Anna
Scheucher
a,
Christoph
Gross
b,
Magdalena
Piringer
a,
Johanna
Novacek
c,
Armin R.
Ofial
 b and
Mario
Waser
*a
b and
Mario
Waser
*a
aInstitute of Organic Chemistry, Johannes Kepler University Linz, Altenbergerstrasse 69, 4040 Linz, Austria. E-mail: mario.waser@jku.at
bDepartment Chemie, Ludwig-Maximilians-Universität München, Butenandtstr. 5-13, 81377 München, Germany
cInstitute of Analytical and General Chemistry, Johannes Kepler University Linz, Altenbergerstrasse 69, 4040 Linz, Austria
First published on 3rd December 2024
Abstract
Chiral isochalcogenoureas (i.e. isothioureas and isoselenoureas) catalyse the asymmetric (4 + 2)-cycloaddition of various allenoates with ortho-quinone methides. This approach provides straightforward access to different chromane derivatives with high enantioselectivities, good yields, and control of the configuration of the exocyclic double bond. Furthermore, some of the novel ortho-quinone methides used herein were successfully integrated into the Mayr reactivity scale by determining their electrophilicity parameter.
Introduction
Allenoates 1 have been established as versatile reagents for various (formal) cycloaddition reactions over the course of the last three decades.1,2 Upon using chiral Lewis base (LB) organocatalysts,3 which form an activated betaine intermediate via addition to the β-carbon of the allenoate (Scheme 1A), reactions between allenoates and dipolar (vinylogous) acceptors (E-Nu) can lead to structurally diverse carbo- and heterocycles in a stereoselective manner.4 Remarkably, the nature of the catalyst usually has a strong influence on the reaction pathway, thus allowing for orthogonal outcomes depending on the used class of catalysts.5–10 Most commonly, (chiral) tertiary phosphines are the Lewis bases of choice for allenoate activations.5,6 Besides, also tertiary amines7 or N-heterocyclic carbenes (NHCs)8 have proven their potential for allenoate-based cycloadditions. Very recently, we have shown that chiral isochalcogenoureas (IChUs, Scheme 1B), i.e. isothioureas (ITUs)11 and isoselenoureas (ISeU),12 hold much potential for the activation of allenoates, too.10 Interestingly, in our studies we found that these easily accessible bench-stable Lewis bases allow for complementary reaction pathways as compared to the established phosphine- and amine-catalysed protocols.9,10 More specifically, so far we have investigated cycloaddition reactions between allenoates and four different classes of Michael acceptors and in all cases we exclusively observed (4 + 2)-heterocycloadditions leading to the formation of highly functionalised dihydropyrans with a (Z)-configured exocyclic double bond.10 In sharp contrast, phosphine catalysis often leads to (3 + 2)-carbocyclisations2,13 while tertiary amines can give analogous (4 + 2)-heterocycloadditions but with (E)-configured double bonds instead.14 Thus, IChUs represent a powerful alternative catalyst platform for asymmetric allenoate cycloadditions which provides an entry to chiral targets that are not easily accessible with the classically used Lewis base organocatalysts.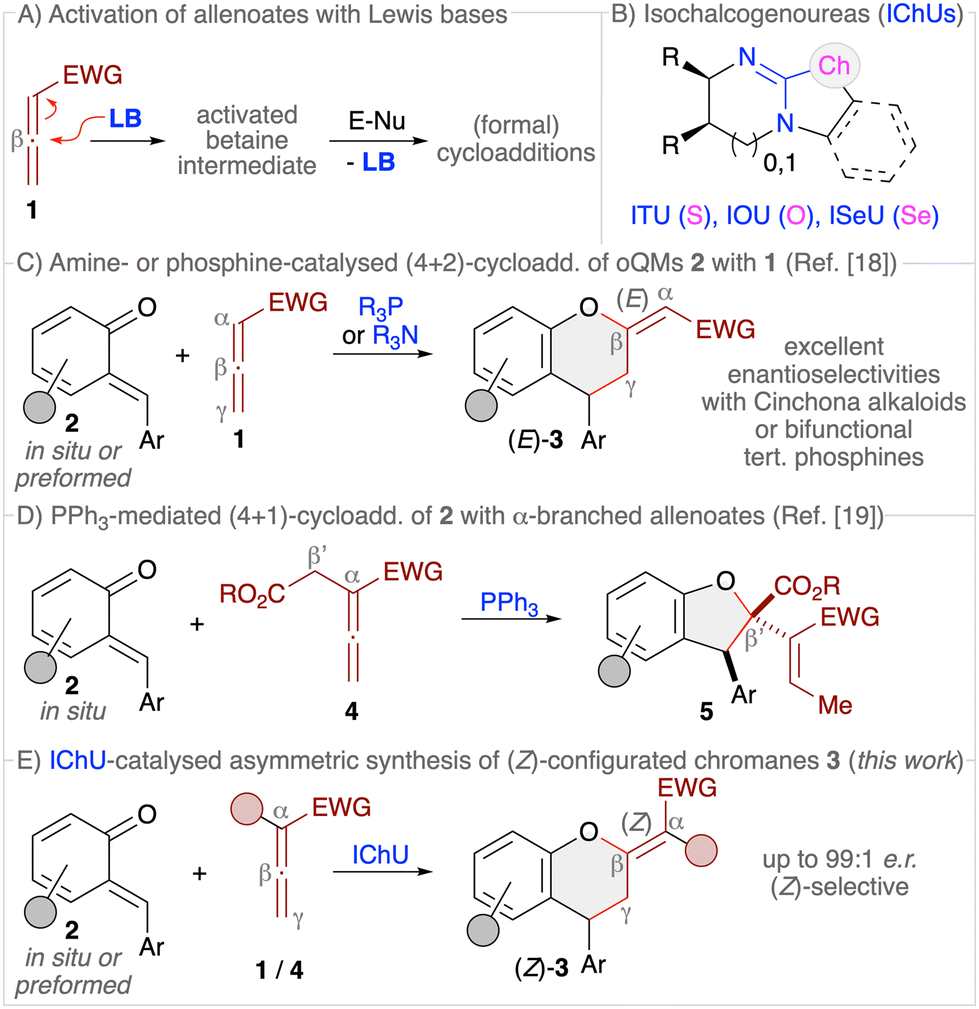 | ||
| Scheme 1 (A) General concept of LB-activation of allenoates; (B) isochalcogenoureas; (C) established amine- and phosphine-catalysed (4 + 2)-cycloadditions of o-quinone methides 2 with allenoates 1; (D) our recently developed PPh3-mediated (4 + 1)-cycloaddition of α-branched allenoates 4; (E) the herein investigated IChU-catalysed synthesis of chromanes 3. | ||
ortho-Quinone methides (oQMs, 2) are intensively investigated building blocks that can undergo various heterocycloaddition reactions.15 These reactive and often rather unstable compounds can either be formed in situ and then immediately be trapped by nucleophiles or dienophiles. Or, less frequently, oQMs have been preformed and used directly. (4 + 2)-Cycloadditions of oQMs16 can lead to highly functionalised chiral chromane17 derivatives 3, and it was recently shown that reactions between allenoates and oQMs in the presence of either chiral tertiary amines (i.e. Cinchona alkaloids) or chiral bifunctional tertiary phosphines can lead to the formation of chromanes 3 with an (E)-configured exocyclic double bond (Scheme 1C).18 Interestingly, we recently found that the reaction of α-alkyloxycarbonylmethyl-substituted allenoates 4 with oQMs 2 in the presence of triphenylphosphine resulted in the formation of dihydrobenzofurans 5via a (4 + 1)-cycloaddition instead (Scheme 1D),19 thus underscoring the diversity of possible reaction pathways that allenoates can enter. Considering these recent results from other groups and ourselves, which demonstrate that cycloadditions of allenoates and oQMs can lead to various highly decorated aryl-fused oxygenated heterocycles straightforwardly, we were now wondering whether it is possible to carry out such reactions under IChU catalysis as well. Based on our recent observations,10 we concluded that this approach should give us predominately access to the (Z)-configured chromanes 3 instead of the already established (E)-configured ones.18 In addition, we were wondering if the presence of α- or γ-substituents will be tolerated without affecting the overall cycloaddition pathway too, in contrast to the mentioned differences when using phosphine catalysis (compare Scheme 1C and D).18c,19 Overall such an approach should thus provide an entry to the densely functionalised chiral products 3 in a highly selective manner (Scheme 1E).
Results and discussion
Electrophilic reactivities of oQMs
According to the general mechanism depicted in Scheme 1A, Lewis base addition to the electrophilic allenoates 16 as well as the subsequent trapping of the nucleophilic betaine intermediate by sufficiently reactive electrophiles are the key bimolecular reactions that need to be understood to rationally optimise the cycloaddition reactions. In the context of this work, insight into the electrophilic reactivity of ortho-quinone methides 2 is crucial to define the scope of the IChU-catalysed synthesis of chromanes.The Munich team recently synthesised a series of prototypical oQMs 2 formally derived from sesamol (= 3,4-methylendioxyphenol) and various acceptor- and donor-substituted benzaldehydes, studied their reactivities toward reference nucleophiles, and finally characterised the electrophilicities E of oQMs 2 on the Mayr reactivity scale.20 To cover further oQMs used in this work, we set out to include heteroaryl-substituted oQMs with furanyl (oQM1), pyrrolyl (oQM2), and indolyl (oQM3) moieties as well as with extended π-system (oQM4) (Scheme 2). The second-order rate constants k2 of addition reactions of carbanions (reference nucleophiles) to these oQMs in DMSO at 20 °C were determined by using (stopped-flow) photometric methods. Then, the electrophilicities E of oQM1–oQM4 were calculated from the experimentally determined k2 and the reported nucleophilicity parameters (N and sN)21 of the reference nucleophiles according to the Mayr–Patz equation (see ESI, Section 1† for details).
 | ||
| Scheme 2 Comparing the Mayr electrophilicities E of oQM1–oQM4 with those of aryl-substituted oQMs (such as 2a), para-quinone methides, and further Michael acceptors (with data from ref. 20 and 21c). Compounds are ordered according to their E parameters with increasing reactivity from bottom to the top. Top line: blue dots mark the electrophilic positions of oQM1–oQM4. | ||
The p-anisyl-substituted oQM 2a (E = −15.20)20 was used in the optimisation and screening studies of this work. Supplementing the Mayr electrophilicity scale21c by the ortho-quinone methides oQM1–oQM4 shows that all oQMs are located in a narrow reactivity range (Scheme 2). For oQM1 (E = −15.73), oQM3 (E = −15.55), and oQM4 (E = −16.00) it can be anticipated that they may perform comparably well as 2a or the only slightly less electrophilic p-(dimethylamino)-substituted oQM (E = −16.07).20 Allene ketones were successfully shown to undergo phosphine-catalysed (4 + 2) annulations with oQM4.18c However, due to the low regioselectivity of the ambident oQM4 in reactions with nucleophiles (see ESI, Section 12†), we excluded oQM4 from further studies in this work. Similarly, low regioselectivities for the attack of C-nucleophiles at vinyl para-quinone methides were reported previously.22 The N-methylpyrrol-2-yl-substituted oQM2 is by almost two orders of magnitude less electrophilic than the standard oQM 2a. Successful (4 + 2)-heterocycloadditions with oQM2 would, therefore, significantly enhance the reactivity range of oQMs that could be used for the IChU-catalysed reactions with allenoates.
Cycloadditions with preformed stabilised oQMs
We started our investigations on the allenoate cycloadditions by using the stabilised electron-rich benzodioxole-based oQMs 2. A first screening and optimisation of reaction conditions was carried out using oQM 2a and the unbranched allenoate 1a (Table 1).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | IChU (mol%) | Solv. | T (°C) | (Z)-3ab (%) |
(Z)-3a erc | (E)-3ab (%) |
6ab (%) |
| a Unless otherwise stated, reactions were run for 24 h using 0.15 mmol 1a and 0.1 mmol 2a in the presence of the given catalyst in the indicated solvent (c = 0.02 M with respect to 2a) under N2 at the indicated temperature (tol. = toluene; DCM = dichloromethane; THF = tetrahydrofuran). b Calculated from the 1H NMR spectrum of the crude product using mesitylene as an internal standard (IST). c Enantiomeric ratio determined by HPLC using a chiral stationary phase. d Isolated yield. e With added Cs2CO3 (1 equiv.). | |||||||
| 1 | ITU1 (20%) | Tol. | 80 | — | — | — | — |
| 2 | ITU2 (20%) | Tol. | 80 | 48 | 99:1 |
8 | 32 |
| 3 | ITU3 (20%) | Tol. | 80 | 61 (59) |
99:1 |
7 (83:17)c |
28 (74:26)c |
| 4 | ISeU (20%) | Tol. | 80 | 59 | 99:1 |
13 | 26 |
| 5 | ITU3 (10%) | Tol. | 80 | 54 | 99:1 |
9 | 28 |
| 6 | ITU3 (20%) | Tol. | 40 | 52 | 99:1 |
7 | 20 |
| 7 | ITU3 (20%) | Tol. | 120 | 51 | 99:1 |
8 | 25 |
| 8 | ITU3 (20%) | DCM | 80 | 33 | 99:1 |
8 | 29 |
| 9 | ITU3 (20%) | THF | 80 | 58 | 99:1 |
10 | 29 |
| 10 | ITU3 (20%) | Tol.e | 80 | 29 | 99:1 |
4 | 26 |

Testing the four different catalysts depicted in Fig. 1 under conditions similar to those established for IChU-catalysed allenoate (4 + 2)-cycloadditions with various Michael acceptors,10 we observed a likewise reactivity trend herein as well (entries 1–4). While BTM (ITU1, entry 1) did not allow for any product formation, the 6-ring-based HBTM (ITU2, entry 2), HyperBTM (ITU3, entry 3), and its selenium-containing analogue (ISeU, entry 4) promoted the (4 + 2)-cycloaddition well. This difference in reactivity between the BTM motif and the HBTM/HyperBTM scaffold can most likely be rationalised by the lower nucleophilicity of 5-ring-based isothioureas,23 thus slowing down the initial addition to the allenoate (we recently showed that this step has a rather high activation barrier, which most likely also explains the need for higher reaction temperatures10). Interestingly, we not only observed the formation of the anticipated chromane 3a [with the (Z)-isomer being the major one; the configuration of the double bond was assigned by NOESY NMR experiments], but also notable amounts of the chromene 6a (originating from an initial α-attack of the allenoate to the benzylic position of the oQM). This observation is in sharp contrast to our previous studies, where analogous α-addition-based products were obtained in minute amounts only (if formed at all). Noteworthy, the enantioselectivity for the targeted (Z)-3a was very high, independent of the used catalyst (entries 2–4). On the other hand, the catalyst scaffold, as well as the reaction conditions (entries 2–10) had an influence on the product distribution. Overall, it turned out that HyperBTM (ITU3) is the Lewis base of choice. Using 20 mol% of this catalyst in toluene at 80 °C allows for around 60% (Z)-3a selectivity, besides approx. 10% of the (E)-diastereomer and slightly less than 30% of 6a (entry 3). Interestingly, the two side-products (E)-3a (83:17 er) and 6a (74:26 er) were obtained with significantly lower enantioselectivities as compared to the major product (Z)-3a (99:1 er). Changing the solvent (entries 8 and 9), adding base (as exemplified for Cs2CO3; entry 10), and varying the temperature (entries 6 and 7) did not allow for any better results and lower catalyst loading (entry 5) resulted in a reduced yield too. Gratifyingly, the (Z)-isomer could easily be separated from the other two cycloaddition side-products by means of a simple silica gel column chromatography, thus giving (Z)-3a in a moderate isolated yield of 59% with excellent enantioselectivity (entry 3).
 | ||
| Fig. 1 IChUs used herein. | ||
With suited asymmetric conditions for the synthesis of (Z)-3a at hand, we next investigated the application scope by using various sesamol-derived oQMs 2 as well as different allenoates 1 and 4 (Scheme 3). Varying the ester group of allenoates 1 first (see products 3a–f) showed that t-butyl esters allow for the highest yields with reduced amounts of the α-addition products 6. This can be explained by the higher steric shielding of the α-position, thus preventing formation of compounds 6 while making the γ-position more accessible. Accordingly, testing of different oQMs was then carried out with the t-butyl allenoate as the cycloaddition partner. As shown for products 3g–3p various (hetero)aryl groups were well tolerated and in each case the level of enantioselectivity was very high. However, the method came to its limits when using γ-substituted allenoates. In this case the reaction was found to be rather messy and the only distinct product that could be obtained in trace amounts and with very low er was the chromene 6s (formed via α-attack of the activated allenoate to QM). Remarkably when using α-branched allenoates 4 instead again the formation of the chromane skeleton 3 was the dominant transformation (see products 3q and 3r). This is in sharp contrast to our previous observations when reacting such allenoates with in situ generated oQMs (Scheme 1D)19 and OH-containing para-QMs24 in the presence of phosphine catalysts, thus underscoring the generality and functional group tolerance of the IChU-catalysed (4 + 2)-cycloaddition. Interestingly, herein the isoselenourea derivative ISeU allowed for higher yields and again the enantioselectivity was nearly perfect.
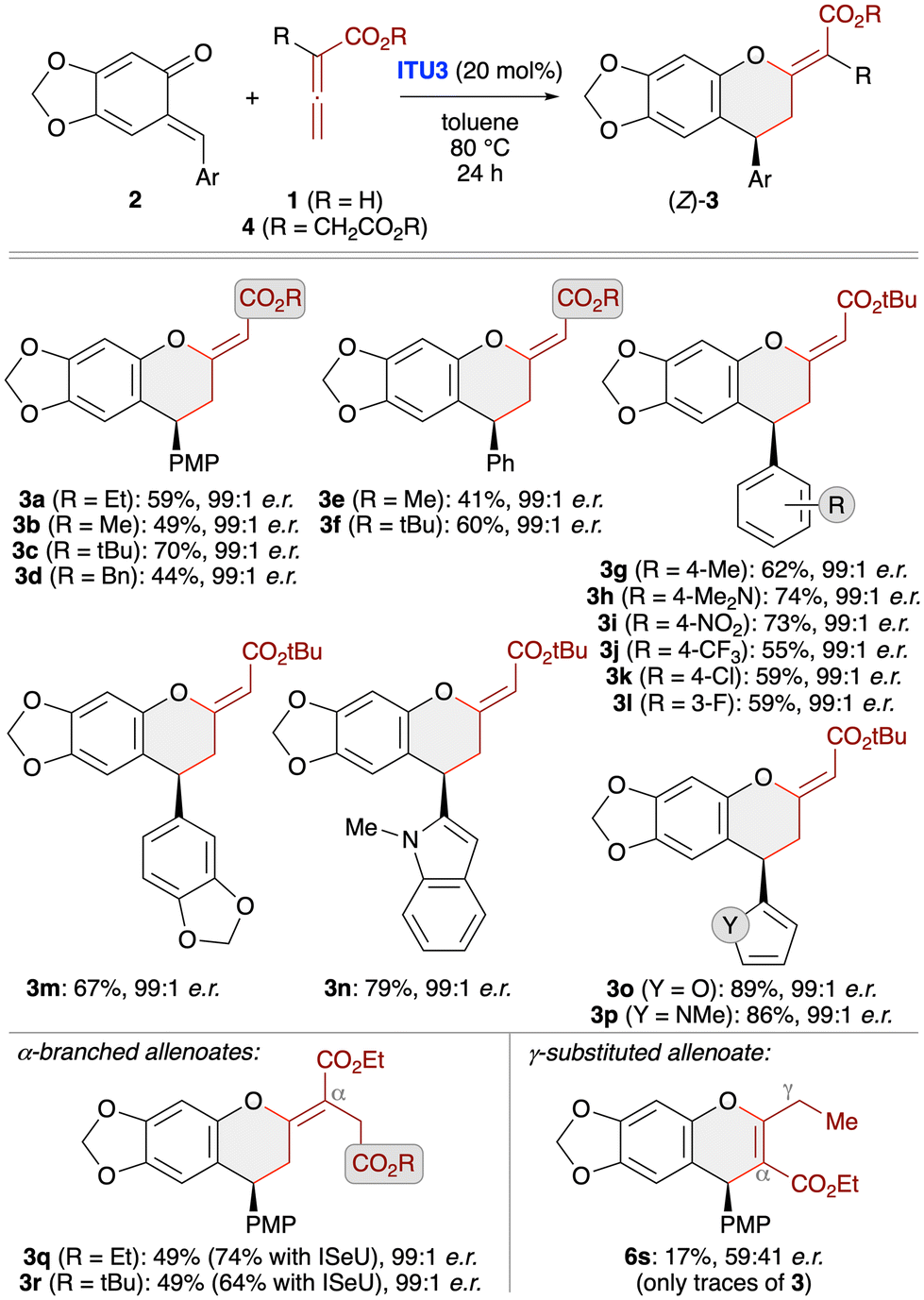 | ||
| Scheme 3 Asymmetric application scope testing various stabilised oQMs and different allenoates using (2S,3R)-HyperBTM (ITU3; conditions as specified in entry 3, Table 1). | ||
Cycloadditions with in situ generated oQMs
When using less stabilised oQMs these transient species are usually generated in situ from different easily accessible precursors.13 One straightforward approach that should be compatible with our Lewis base-catalysis strategy relies on the use of α-(arylsulfonyl)methyl-substituted β-naphthols 7 under basic conditions, which leads to the formation of the corresponding oQMs via elimination of an arylsulfinic acid. This strategy was recently also utilised in our (4 + 1)-cycloaddition of allenoates 4 (Scheme 1D)19 and we thus set out to explore the possibility of applying this to IChU-catalysed allenoate cycloadditions. By starting from the precursor 7a and the parent allenoate 1a we first optimised the synthesis of product 3aa (Table 2). We initially set the reaction temperature to 40 °C as we know from previous investigations with compounds 7 that higher temperatures usually lead to a relatively fast decomposition of the corresponding oQMs. First test reactions with the three isothiourea catalysts ITU1–3 under basic conditions showed a similar reactivity trend as compared to the stabilised oQM 2a (compare entries 1–3 of both tables). Again HyperBTM (ITU3) was best suited giving the targeted (Z)-3aa with moderate NMR yield and excellent enantioselectivity (entry 3). Interestingly, formation of the (E)-diastereoisomer and the α-addition product 6aa was less pronounced as compared to the use of 2a. Unfortunately, lower catalyst loading was again not well tolerated (entry 4). Interestingly however, in this case ISeU was found to be higher yielding (entry 5). Some further optimisation of reaction conditions was first carried out with ITU3 (entries 6–8) showing that an excess of 3 equiv. of allenoate is beneficial, while increased or decreased temperature did not allow for any improvement (a few other solvents or bases were tested too but did not allow for any improvement). Finally, using ISeU in combination with 3 equiv. of 1a and 1 equiv. of Cs2CO3 in toluene at 40 °C allowed for the synthesis of (Z)-3aa in 77% isolated yield with 99:1 er and only minor quantities of the other cycloaddition side products (entry 9).
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | IChU (mol%) | T (°C) | (Z)-3aab (%) |
(Z)-3aa erc | (E)-3aab (%) |
6aab (%) |
| a Unless otherwise stated, reactions were run for 24 h using 0.15 mmol 1a and 0.1 mmol 7a in the presence of the given catalyst in toluene under N2 at the indicated temperature. b Calculated from the 1H NMR spectrum of the crude product using mesitylene as an internal standard (IST). c Enantiomeric ratio determined by HPLC using a chiral stationary phase. d Using 3 equiv. of allenoate 1a. e Isolated yield. | ||||||
| 1 | ITU1 (20%) | 40 | — | — | — | — |
| 2 | ITU2 (20%) | 40 | 29 | 97:3 |
8 | 5 |
| 3 | ITU3 (20%) | 40 | 54 | 99:1 |
8 | 4 |
| 4 | ITU3 (10%) | 40 | 19 | 98:2 |
5 | 3 |
| 5 | ISeU (20%) | 40 | 77 | 99:1 |
12 | 4 |
| 6 | ITU3 (20%)d | 40 | 72 | 99:1 |
12 | 4 |
| 7 | ITU3 (20%)d | 60 | 60 | 99:1 |
10 | 5 |
| 8 | ITU3 (20%)d | 25 | 56 | 99:1 |
11 | 4 |
| 9 | ISeU (20%) | 40 | 79 (77) |
99:1 |
12 (63:37)c |
3 (56:44)c |

Investigating the asymmetric application scope for the cycloaddition starting from oQM precursors 7 (Scheme 4) showed that various naphthol-based derivatives are well tolerated, as exemplified for products 3aa–aj. Unfortunately, however, the method comes to its limits when utilising simple phenol-based QM precursors that yield less stable and thus more easily decomposing oQMs. While products 3ak and 3al could not be accessed at all and 3an was only detected in trace amounts, the 8-Me-containing 3am could at least be obtained in low yield (but with very high enantioselectivity). In all these cases we observed a very pronounced formation of unidentified side-products originating from the decomposition of the in situ formed quinone methides. Using α-branched allenoates 4 allowed for the selective (4 + 2)-cycloaddition as well, as demonstrated for the synthesis of chromanes 3ao and 3ap. Again, this result is in sharp contrast to our recent phosphine-catalysed (4 + 1)-cycloaddition protocol (Scheme 1D),19 underscoring the orthogonal catalytic potential of different Lewis bases for allenoate activation. Finally, analogous to the use of preformed oQMs (see product 6r, Scheme 3), the use of γ-branched allenoates resulted in the formation of the chromene skeleton herein as well, but with low yield and unsatisfying enantioselectivity only (product 6aq).
 | ||
| Scheme 4 Asymmetric application scope using in situ generated oQMs (accessed from compounds 7) and different allenoates in the presence of (2S,3R)-SeHyperBTM (ISeU; conditions as detailed in entry 9, Table 2). | ||
We have not been able to obtain crystals of the enantioenriched products 3 that would have allowed for an unambiguous assignment of their absolute configuration by means of single crystal X-ray diffraction. Thus we recorded vibrational circular dichroism (VCD)25 spectra of both enantiomers of compound 3e and compared the experimental spectra with those calculated from DFT optimised structures which strongly supports the absolute configuration depicted in Scheme 3.26 This sense of configuration of the major enantiomer is in full accordance with our recent observations of IChU-catalysed allenoate-based (4 + 2)-cycloadditions where we always observed this orientation of the substituent on the stereogenic center in position 4 of tetrahydropyran ring when using the (2S,3R)-configured ITU3 or ISeU.10 Furthermore, comparison of the optical rotation of our (Z)-configured products 3 with reported structurally similar (E)-configured ones18 support this sense of configuration as well and we therefore assigned all other products in analogy.
Mechanistically, we proposed that the herein reported stereoselective syntheses of chromanes 3 follow the established pathway for our recently introduced IChU-catalysed (4 + 2)-cycloadditions of allenoates with different Michael acceptors (Scheme 5).10 More specifically, the IChU first activates the allenoates 1 giving the resonance-stabilised Int-A. This chiral intermediate then undergoes 1,4-addition to the oQM 2 with its γ-carbon, giving Int-B. This is also the step that controls the configuration of products 3. As stated above, so far we have always observed the same sense of induction when using the same enantiomers of our catalysts, thus substantiating a very high level of catalyst control and a very well defined Int-A. Int-B then undergoes ring-closure and final IChU-elimination which also sets the configuration of the exocyclic double bond. Interestingly, while other Lewis base catalysts usually favour (E)-configurated double bonds in such transformations,18 IChUs show pronounced (Z)-selectivity, a kinetic phenomenon which we could also recently support by DFT calculations.10a
 | ||
| Scheme 5 Mechanistic proposal.10 | ||
Follow-up transformations
Finally, we also carried out some test reactions to demonstrate the suitability of products 3 to serve as starting materials for further transformations. As outlined in Scheme 6, the exocyclic double bond can be selectively hydrogenated under homogeneous conditions by using Wilkinson's catalyst. In this way product 8 was obtained with high diastereoselectivity; the cis configuration of the 2-acetyl group and the 4-indolyl substituent was determined by NOESY experiments. Furthermore, the ester functionality can be reduced to the primary alcohol 9 by using an excess of LiAlH4. It should be stated that neither of these two transformations have been optimised further, but in our opinion they represent a proof-of-concept to demonstrate the suitability of compounds 3 for further manipulations. | ||
| Scheme 6 Reductive follow-up transformations of compounds 3. | ||
Conclusions
Isothioureas (ITUs) and isoselenoureas (ISeUs) were successfully employed as chiral Lewis base catalysts for asymmetric (4 + 2)-cycloadditions of various allenoates with different ortho-quinone methides. This approach allows for the synthesis of chromanes 3 with (Z)-configured exocyclic double bonds in high enantioselectivities and good isolated yields. Accompanying VCD studies supported the assignment of the absolute configuration of the products. Furthermore, the electrophilicity parameters of some heteroaryl-substituted and π-extended ortho-quinone methides were successfully determined. These quantitative reactivity data hint to the reactivity range which the electrophilic component in the (4 + 2)-cycloadditions needs to cover. This knowledge should allow us to identify additional suitable reaction partners for such allenoate-based cycloadditions in the future, which can be selected based on their reported electrophilicity parameters E.Experimental details26
General procedure using preformed oQMs 2
A flame dried N2-flushed flask was charged with ITU3 (20 mol%), the respective oQM 2 (0.1 mmol, 1 equiv.) and toluene (5 mL, 0.02 mol L−1), directly followed by the addition of the allenoate 1 or 4 (0.15 mmol, 1.5 equiv.). The reaction mixture was then stirred at 80 °C for 24 h. After cooling, the mixture was filtered through a Na2SO4 plug. The solvent was removed under reduced pressure to yield the crude products 3a–q. Purification via preparative TLC (heptane:EtOAc = 2:1) gave the products in the reported yields and enantiopurities.
General procedure using oQM precursors 7
A N2-flushed and flame-dried flask was charged with ISeU (20 mol%), Cs2CO3 (0.1 mmol, 1 equiv.), the respective oQM precursor 7 (0.1 mmol, 1 equiv.) and toluene (5 mL, 0.02 mol L−1). The allenoate (0.3 mmol, 3 equiv.) was added and the mixture was heated to 40 °C and stirred for 24 h. After cooling to r.t., the mixture was filtered through a Na2SO4 plug and the solvent was removed under reduced pressure to give the crude products 3. Purification via preparative TLC (heptane:EtOAc = 2:1) gave the products in the reported yields and enantiopurities.
Author contributions
A. S., C. G., and M. P. carried out all the syntheses, method development and analysis of the compounds. C. G. characterised the electrophilicites of the oQMs. J. N. carried out the VCD measurements and accompanying calculations. A. R. O. and M. W. initiated and supervised the project and wrote the manuscript with contributions from all authors.Data availability
The data supporting this article have been included as part of the ESI.† Raw data of the individual kinetic measurements that support the findings of this study are openly available in Open Data LMU at https://doi.org/10.5282/ubm/data.545.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The LMU team gratefully acknowledges generous support by the Dept. Chemie, LMU München. Research at JKU Linz was generously supported by the Austrian Science Funds (FWF): Project No. P36004 (financial support through the matching funds program by the Austrian National Foundation for Research, Technology and Development and the Research Department of the State of Upper Austria). This used VCD spectrometer was acquired through funding of the Linz Institute of Technology (LIT-INVEST-2020-002).References
- (a) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (b) S. Ma, Chem. Rev., 2005, 105, 2829 Search PubMed; (c) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC; (d) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074 CrossRef CAS PubMed; (e) Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC; (f) Y. Xiao, Z. Sung, H. Guo and O. Kwon, Beilstein J. Org. Chem., 2014, 10, 2089 CrossRef; (g) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC; (h) F. López and J. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904 RSC; (i) Y. Wei and M. Shi, Org. Chem. Front., 2017, 4, 1876 CAS; (j) E.-Q. Li and Y. Huang, Chem. Commun., 2020, 56, 680 RSC; (k) R. Hajinasiri, Tetrahedron, 2022, 126, 133053 CAS; (l) M. Alonso and P. Almendros, Adv. Synth. Catal., 2023, 365, 1332 Search PubMed.
- Pioneering report introducing Lewis base-catalysed cycloadditions of allenoates: C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CAS.
- (a) M. J. Gaunt and C. C. C. Johansson, Chem. Rev., 2007, 107, 5596 CrossRef CAS PubMed; (b) G. C. Fu, Acc. Chem. Res., 2000, 33, 412 CrossRef CAS; (c) S. France, D. J. Guerin, S. J. Miller and T. Lectka, Chem. Rev., 2003, 103, 2985 CrossRef CAS; (d) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed; (e) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109 RSC.
- M. Shi, Y. Wei, M.-X. Zhao and J. Zhang, Organocatalytic Cycloadditions for Synthesis of Carbo- and Heterocycles, Wiley-VCH, Weinheim, 2018 Search PubMed.
- For illustrative reviews on chiral phosphine organocatalysis including allenoate applications, too: (a) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (b) A. Voituriez, A. Marinetti and M. Gicquel, Synlett, 2015, 142 CAS; (c) H. Li and Y. Lu, Asian J. Org. Chem., 2017, 6, 1130 CrossRef CAS; (d) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344 CrossRef CAS PubMed; (e) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CrossRef CAS PubMed; (f) Y. Huang, J. Liao, W. Wang, H. Liu and H. Guo, Chem. Commun., 2020, 56, 15235 RSC; (g) C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536 CrossRef CAS PubMed.
- For reactivities of tertiary phosphines towards allenoates: (a) F. An, H. Jangra, Y. Wei, M. Shi, H. Zipse and A. R. Ofial, Chem. Commun., 2022, 58, 3358 RSC; (b) F. An, J. Brossette, H. Jangra, Y. Wei, M. Shi, H. Zipse and A. R. Ofial, Chem. Sci., 2024, 15, 18111 RSC.
- Early reports describing the use of chiral tert. amines for allenoate reactions: (a) J.-B. Denis, G. Masson, P. Retailleau and J. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5356 CrossRef CAS PubMed; (b) X. Wang, T. Fang and X. Tong, Angew. Chem., Int. Ed., 2011, 50, 5361 CrossRef CAS; (c) K. D. Ashtekar, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 5732 CrossRef CAS PubMed; (d) C.-K. Pei, Y. Jiang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2012, 51, 11328 CAS; (e) C.-K. Pei and M. Shi, Chem. – Eur. J., 2012, 18, 6712 CAS.
- Early examples on the use of NHCs for allenoate-based cycloadditions: (a) L. Sun, T. Wang and S. Ye, Chin. J. Chem., 2012, 30, 190 CAS; (b) Y. Hu, S. Li, Z. Wang, Y. Yao, T. Li, C. Yu and C. Yao, J. Org. Chem., 2018, 83, 3361 CAS; (c) S. S. Lopez, A. A. Jaworski and K. A. Scheidt, J. Org. Chem., 2018, 83, 14637 CAS.
- Illustrative studies reporting complementary outcomes when using different Lewis bases for allenoate cycloadditions: (a) G.-T. Huang, T. Lankau and C.-H. Yu, J. Org. Chem., 2014, 79, 1700 CAS; (b) L.-J. Yang, S. Li, S. Wang, J. Nie and J.-A. Ma, J. Org. Chem., 2014, 79, 3547 CAS; (c) G.-T. Huang, Z. Lankau and C.-H. Yu, Org. Biomol. Chem., 2014, 12, 7297 CAS; (d) Y. Li, S. Du, Z. Du and C. Chen, RSC Adv., 2016, 6, 82260 RSC.
- (a) L. S. Vogl, P. Mayer, R. Robiette and M. Waser, Angew. Chem., Int. Ed., 2024, 63, e202315345 CAS; (b) M. Piringer, M. Hofer, L. S. Vogl, P. Mayer and M. Waser, Adv. Synth. Catal., 2024, 366, 2115 CAS; (c) M. Waser, Chem. Lett., 2024, 53, upae168 CrossRef.
- For reviews on isothiourea catalysis: (a) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 5589 CrossRef CAS; (b) V. B. Birman, Aldrichimica Acta, 2016, 49, 23 CAS; (c) W. C. Hartley, T. J. C. O'Riordan and A. D. Smith, Synthesis, 2017, 49, 3303 CrossRef CAS; (d) A. Biswas, H. Mondal and M. S. Maji, J. Heterocycl. Chem., 2020, 57, 3818 CrossRef CAS; (e) C. McLaughlin and A. D. Smith, Chem. – Eur. J., 2021, 27, 1533 CrossRef CAS; (f) J. Bitai, M. T. Westwood and A. D. Smith, Org. Biomol. Chem., 2021, 19, 2366 RSC; (g) A. J. Nimmo, C. M. Young and A. D. Smith, Isothiourea Catalysis – New Opportunities for Asymmetric Synthesis, in Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities, ed. Ł. Albrecht, A. Albrecht and L. Dell'Amico, Wiley-VCH, Weinheim, 2023, ch. 5, p. 151 Search PubMed.
- Seminal study on the use of isoselenoureas: C. M. Young, A. Elmi, D. J. Pascoe, R. K. Morris, C. McLaughlin, A. M. Woods, A. B. Frost, A. Houpliere, K. B. Ling, T. K. Smith, A. M. Z. Slawin, P. H. Willoughby, S. Cockroft and A. D. Smith, Angew. Chem., Int. Ed., 2020, 59, 3705 Search PubMed.
- (a) R. Ma, G. Song, Q. Xi, L. Yang, E.-Q. Li and Z. Duan, Chin. J. Org. Chem., 2019, 39, 2196 Search PubMed; (b) S. Anwar, L.-T. Lin, V. Srinivasadesikan, V. B. Gudise and K. Chen, RSC Adv., 2021, 11, 38648 CAS.
- (a) K. D. Ashtekar, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 5732 CAS; (b) X. Wang, T. Fang and X. Tong, Angew. Chem., Int. Ed., 2011, 50, 5361 Search PubMed; (c) C. K. Pei, Y. Jiang and M. Shi, Org. Biomol. Chem., 2012, 10, 4355 CAS; (d) C.-K. Pei, Y. Jiang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2012, 51, 11328 CAS; (e) C. Wang, H. Jia, C. Zhang, Z. Gao, L. Zhou, C. Yuan, Y. Xiao and H. Guo, J. Org. Chem., 2017, 82, 633 CAS.
- (a) W.-J. Bai, J.-G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655 CAS; (b) M. S. Singh, A. Nagaraju, N. Anand and S. Chowdhury, RSC Adv., 2014, 4, 55924 CAS; (c) L. Caruana, M. Fochi and L. Bernardi, Molecules, 2015, 20, 11733 CrossRef CAS PubMed; (d) D. V. Osipov, V. A. Osyanin and Y. N. Klimochkin, Russ. Chem. Rev., 2017, 86, 625 CrossRef CAS; (e) X. Li, Z. Li and J. Sun, Nat. Synth., 2022, 1, 426 CrossRef CAS.
- Early asymmetric (4 + 2)-annulations of oQMs using different starting materials and catalysis modes: (a) C.-C. Hsiao, H.-H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258 CrossRef CAS PubMed; (b) O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923 CrossRef CAS PubMed; (c) S. Saha and C. Schneider, Org. Lett., 2015, 17, 648 CrossRef CAS PubMed; (d) L. Caruana, M. Mondatori, V. Corti, S. Morales, A. Mazzanti, M. Fochi and L. Bernardi, Chem. – Eur. J., 2015, 21, 603 Search PubMed; (e) D. Zhou, K. Mao, J. Zhang, B. Yan, W. Wang and H. Xie, Tetrahedron Lett., 2016, 57, 5649 CAS; (f) Y. Zhu, W.-Z. Zhang, L. Zhang and S. Luo, Chem. – Eur. J., 2017, 23, 1253 CAS; (g) H. Hu, Y. Liu, J. Guo, L. Lin, Y. Xu, X. Liu and X. Feng, Chem. Commun., 2015, 51, 3835 CAS; (h) S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392 CAS.
- (a) Chemistry of Heterocyclic Compounds: Chromans and Tocopherols, ed. G. P. Ellis and I. M. Lockhart, Wiley Interscience, New York, 1981, vol. 36 Search PubMed; (b) H. C. Shen, Tetrahedron, 2009, 65, 3931 CrossRef CAS; (c) T. Rosenau and S. Böhmdorfer, ortho-Quinone Methides in Tocopherol Chemistry, in Quinone Methides, ed. S. Rokita, Wiley, Hoboken (NJ), 2009, ch. 6, p. 163 Search PubMed.
- (4 + 2)-Cycloadditions of allenoates with oQMs: (a) Y.-H. Deng, W.-D. Chu, X.-Z. Zhang, X. Yan, K.-Y. Yu, L.-L. Yang, H. Huang and C.-A. Fan, J. Org. Chem., 2017, 82, 5433 CrossRef CAS PubMed; (b) P. Chen, K. Wang, W. Guo, X. Liu, Y. Liu and C. Li, Angew. Chem., Int. Ed., 2017, 56, 3689 CrossRef CAS; (c) Z. Wang, T. Wang, W. Yao and Y. Lu, Org. Lett., 2017, 19, 4126 CrossRef CAS PubMed.
- K. Zielke and M. Waser, Org. Lett., 2018, 20, 768 CAS.
- C. Gross, A. Eitzinger, N. Hampel, P. Mayer and A. R. Ofial, Chem. – Eur. J., 2024, e202403785, DOI:10.1002/chem.202403785.
- (a) R. Lucius, R. Loos and H. Mayr, Angew. Chem., Int. Ed., 2002, 41, 91 CrossRef CAS; (b) H. Mayr, Tetrahedron, 2015, 71, 5095 CAS; (c) A collection of published reactivity parameters N, sN, and E is freely accessible at: https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank2/ (accessed Nov 5, 2024).
- A. Eitzinger, R. J. Mayer, N. Hampel, P. Mayer, M. Waser and A. R. Ofial, Org. Lett., 2020, 22, 2182 CrossRef CAS.
- B. Maji, C. Joanesse, T. A. Nigst, A. D. Smith and H. Mayr, J. Org. Chem., 2011, 76, 5104 CrossRef CAS PubMed.
- K. Zielke, O. Kováć, M. Winter, J. Pospíšil and M. Waser, Chem. – Eur. J., 2019, 25, 8163 CrossRef CAS PubMed.
- For illustrative reviews: (a) C. Merten, T. P. Golub and N. M. Kreienborg, J. Org. Chem., 2019, 84, 8797 CrossRef CAS PubMed; (b) C. Merten, Eur. J. Org. Chem., 2020, 5892 CrossRef CAS.
- See ESI for further details.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical details, VCD investigations, kinetic measurements. See DOI: https://doi.org/10.1039/d4ob01855a |
| This journal is © The Royal Society of Chemistry 2025 |