
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic modification of dihydromyricetin by glucosylation and acylation, and its effect on the solubility and antioxidant activity†
David
Rodriguez-Garcia
a,
Carlos
Uceda
a,
Laura
Barahona
b,
Marta
Ruiz-Nuñez
 c,
Antonio O.
Ballesteros
a,
Tom
Desmet
d,
Julia
Sanz-Aparicio
c,
Maria
Fernandez-Lobato
b,
Jose L.
Gonzalez-Alfonso
*a and
Francisco J.
Plou
*a
c,
Antonio O.
Ballesteros
a,
Tom
Desmet
d,
Julia
Sanz-Aparicio
c,
Maria
Fernandez-Lobato
b,
Jose L.
Gonzalez-Alfonso
*a and
Francisco J.
Plou
*a
aInstituto de Catálisis y Petroleoquímica, CSIC, 28049 Madrid, Spain. E-mail: josel.g@csic.es; fplou@icp.csic.es
bCentro de Biología Molecular Severo Ochoa, Universidad Autónoma de Madrid, 28049, Madrid, Spain
cInstituto de Química Física Blas Cabrera, CSIC, 28006 Madrid, Spain
dCentre for Synthetic Biology (CSB), Department of Biotechnology, Ghent University, 9000 Ghent, Belgium
First published on 10th December 2024
Abstract
Although dihydromyricetin exhibits strong potential for pharmaceutical applications, its limited aqueous solubility, permeability and stability restrict its use. In this work, we have synthesized a series of glucosides and acyl-glucosides of dihydromyricetin that could increase the bioavailability of this molecule. First, the R134A variant of sucrose phosphorylase from Thermoanaerobacterium thermosaccharolyticum catalyzed the formation of three monoglucosides, and the major one was identified as dihydromyricetin 4′-O-α-D-glucopyranoside (>75% conversion yield). The molecular features that define this specificity for the 4′-OH phenolic group were investigated through induced-fit docking analysis of each potential derivative. Furthermore, the acylation of the 4′-monoglucoside with fatty acid vinyl esters (C8, C12, and C16) was performed with high efficiency using the lipase from Thermomyces lanuginosus. Three novel acyl derivatives of dihydromyricetin were characterized. Furthermore, the water solubility and antioxidant activity (ABTS, DPPH) of the synthesized compounds were measured, concluding that the location of the glucosyl moiety may affect their physicochemical properties and, as a result, their bioactivity.
Introduction
Vine tea (Ampelopsis grossedentata) has long been enjoyed for its appealing flavour and health benefits, especially in traditional Chinese medicine.1 Its primary bioactive compound is the flavanonol (+)-dihydromyricetin (DMY, also known as ampelopsin), which has attracted interest due to its potential uses in food, cosmetic, nutraceutical and pharmaceutical compositions.2 The tender stems and leaves of the plant are particularly rich in DMY, comprising over 35% of their dried weight (w/w). Hovenia dulcis, commonly referred to as the Japanese raisin tree, and Cedrus deodara are other natural sources of DMY.1bDMY is being explored as a natural antioxidant in the food and beverage industries, with promising potential to enhance food preservation and extend shelf life.3 Among its pharmaceutical activities, it has been reported that DMY safeguards the cardiovascular system,4 inhibits proliferation and migration of cancer cells,5 protects against neurodegenerative diseases,6 ameliorates liver diseases7 and provides anti-inflammatory,8 antimicrobial9 and skin protection effects.10 In addition, no significant cytotoxicity of DMY has been observed in normal cells.11
Although DMY shows considerable potential for development in the pharmaceutical field, it faces challenges in applications due to its low water-solubility, permeability and stability, which explain its poor bioavailability in vivo.12 DMY has five phenolic hydroxyl groups, which contribute to its strong antioxidant activity but also increase its susceptibility to instability.13 DMY is stable at an acidic pH of 1.0–5.0 but becomes easily oxidized and significantly degraded under neutral and alkaline conditions, particularly between pH 6.0 and 8.0.13 Based on the biopharmaceutical classification system (BCS) standards, DMY is categorized as a class IV compound due to its low solubility and permeability,2 with an absolute bioavailability close to 4%.14 To address its low bioavailability, different strategies have been developed, such as co-administration of DMY with other substances, along with novel formulations designed to improve its stability, solubility, permeability, and bioactivity.1b Thus, researchers have designed various new dosage forms for DMY, including gastric floating formulations,15 microemulsions,16 nanoparticles,17 emulsion gels,18 cocrystals,19 phospholipid complexes,20 and cyclodextrin inclusion complexes,21 thereby enhancing its medicinal properties.
Chemical modification is an alternative for enhancing the bioavailability of polyphenols. In particular, glucosylation involves attaching a glucose moiety to the polyphenol compound, thereby increasing its water solubility and stability.22 Enzymes offer a highly efficient, clean and selective approach for glucosylating complex compounds in comparison with traditional chemical methods.23 In this context, the R134A variant of the sucrose phosphorylase from Thermoanaerobacterium thermosaccharolyticum demonstrated a significant affinity for polyphenols compared to the native enzyme, attributed to the larger size of its catalytic pocket.24 Notably, it has shown success in the glucosylation of various polyphenols, including pyrogallol, alkyl gallates, resveratrol, quercetin, catechins and dihydrochalcones.22a,24,25 A variant (Q345F) of the sucrose phosphorylase from Bifidobacterium adolescentis was also able to glucosylate (+)-catechin and resveratrol.26
On the other hand, acylation involves the attachment of a fatty acid to the polyphenol, which can provide additional advantages. Acylation enhances the lipophilicity of polyphenols, improving their partitioning into lipid-rich environments and potentially enhancing their penetration through biological barriers, including cell membranes, the blood–brain barrier, and skin, which are mainly made up of lipids.27 We recently devised an effective strategy to acylate flavonoids based on a first α-glucosylation step using sucrose phosphorylase, followed by acylation catalyzed by a lipase.28
In this work, we have investigated the synthesis of several glucosides and acyl-glucosides of DMY. These modifications render derivatives with different solubility (in water and organic solvents), permeability and stability compared to the aglycone, which could enhance the bioavailability and functional properties of DMY.
Results and discussion
Glucosylation of dihydromyricetin
The glucosylation of dihydromyricetin was performed using the R134A variant of the sucrose 6′-phosphate phosphorylase from T. thermosaccharolyticum (TtSPP_R134A) (Fig. 1A). This enzyme has been previously shown to be effective in the glucosylation of various polyphenols.22c,25b The reaction (1 mL) was conducted at 48 °C and pH 6.5, in the absence of a cosolvent, using a 12.5-fold molar excess of sucrose relative to DMY. HPLC analysis (Fig. 1B) revealed the formation of three products: one major (1a) and two minor peaks (1b and 1c). | ||
| Fig. 1 (A) Scheme of the DMY glucosylation reaction; (B) HPLC chromatograms (at 15 min, 1 h and 4 h reaction) showing the glucosylation of dihydromyricetin catalyzed by the sucrose phosphorylase variant TtSPP_R134A. Reaction conditions: 40 mM DMY, 500 mM sucrose, 2 U mL−1 sucrose phosphorylase, 50 mM MOPS buffer (pH 6.5), 48 °C, orbital shaking. | ||
Although DMY was not fully soluble in MOPS buffer at the assayed concentration, the reaction was quite fast and the flavonoid was mostly consumed in only 4 h (Fig. 1B). Seemingly, as the reaction progresses, the polyphenol dissolves in the medium until it is completely transformed into products.
To study the effect of the molar excess of sucrose on the glucosylation yield, four concentrations of sucrose were assessed: 0.1, 0.25, 0.5 and 1 M, which corresponded to molar ratios of 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 6.25:1, 12.5:1 and 25:1 with respect to DMY. The progress of the formation of the main glucoside 1a is represented in Fig. 2.
:1, 6.25:1, 12.5:1 and 25:1 with respect to DMY. The progress of the formation of the main glucoside 1a is represented in Fig. 2.
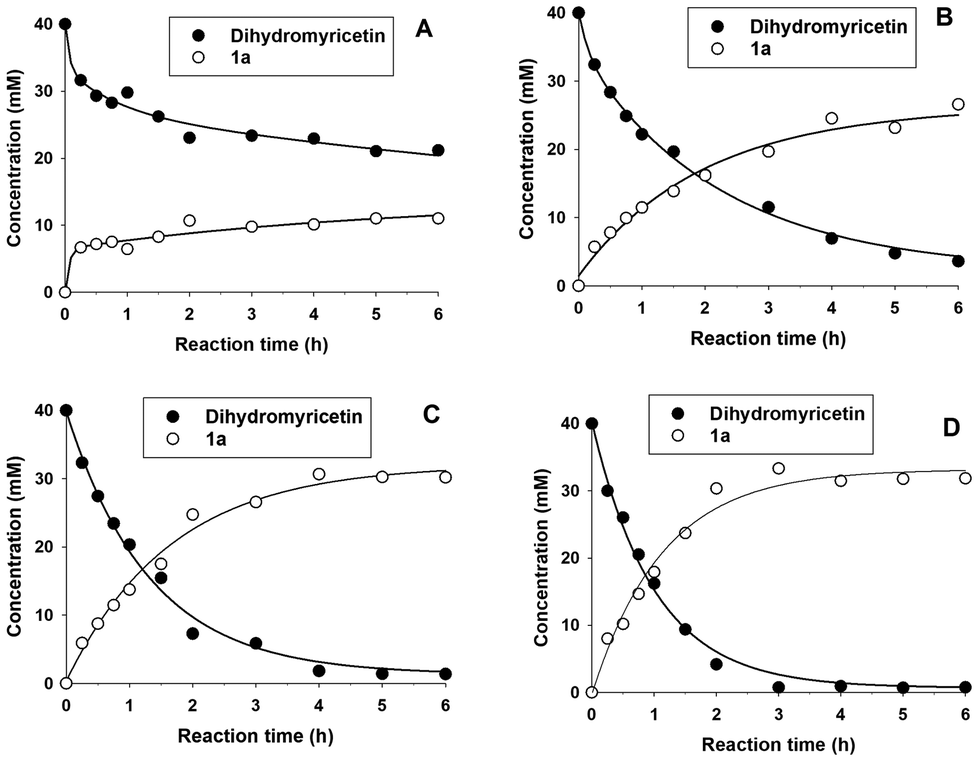 | ||
| Fig. 2 Reaction profiles of the formation of the main glucosylation product (1a) using 40 mM dihydromyricetin and different concentrations of sucrose: (A) 0.1 M; (B) 0.25 M; (C) 0.5 M; and (D) 1 M. Reaction conditions: 40 mM DMY, 2 U mL−1 sucrose phosphorylase, 50 mM MOPS buffer (pH 6.5), 48 °C, orbital shaking. | ||
As shown, it is necessary to employ at least a 6.25-fold molar excess of sucrose to achieve a notable conversion yield. Using 0.5 and 1 M sucrose, the DMY almost disappeared in 6 h. In both cases, the concentration of the main glucoside reached a plateau at around 4 hours, corresponding to a conversion yield higher than 75%. At this point, the remaining DMY was insignificant, approximately 1.0–1.5 mM. We selected 0.5 M sucrose for further studies of the glucosylation reaction.
The conversion of DMY into its glucosides was really efficient compared with other glycosylation processes described for this flavonoid. Thus, Wen et al. employed a glycosyltransferase from Helleborus thibetanus to glucosylate DMY, reporting a conversion yield lower than 10%.29
The glucosylation reaction was scaled to 72 mL, and the primary products were purified using silica gel chromatography, following the procedure outlined in the Experimental section. Two products (1a and 1b) were isolated and characterized by mass spectrometry and NMR. Although we were unable to purify compound 1c, HPLC-MS analysis revealed that it was a monoglucoside.
The molecular weights of 1a and 1b were determined using ESI-MS in negative mode, coupled with a QTOF analyzer. For 1a, the presence of a major peak with m/z 481.0989 that corresponded to the ion [M − H]− of the DMY monoglucoside was detected (Fig. S1, ESI†). In the case of 1b, the major peak was detected at an m/z value of 481.0988, which corresponded to the [M − H]− ion of another monoglucoside.
The glucosylation position of both compounds was determined by NMR. Proton (1H) and carbon (13C) NMR experiments, along with 2D-gHSQC, 2D-gCOSY and 2D-gHMBC spectroscopy, were conducted to identify and assign the signals.
For the major compound 1a (Fig. S2–S4, ESI†), the HMBC spectrum shows a correlation between the anomeric proton of the glucose and the carbon 4′, which demonstrated α-glucosylation at the 4′-O position of ring B (Fig. 3). The same α-glucoside (CAS 1391980-27-0) was obtained using a mutant dextransucrase from Lactobacillus reuteri, along with a diglucosylated (α-maltosyl) conjugate.22b In this case, the reaction was carried out in the presence of 20% (v/v) DMSO, even though no conversion yield is mentioned. The same 4′-derivative at the pyrogallol group was also synthesized using the dextransucrase from Leuconostoc mesenteroides B-1299CB4.30 In this case, although the yield was notable, at least five polyglucosylated products were obtained containing 1–5 glucose units attached to the 4′-OH.
 | ||
| Fig. 3 Molecular structure of the isolated α-glucosides: (1a) dihydromyricetin 4′-O-α-D-glucopyranoside (major product) and (1b) dihydromyricetin 3′-O-α-D-glucopyranoside (minor product). | ||
In contrast, for the minor compound 1b, the 1H-NMR spectrum showed a shift in the aromatic signals of ring B, in comparison with the aglycone, and the loss of symmetry in the signals of this ring suggested that glycosylation had occurred at the chemically equivalent positions 3′ or 5′ (Fig. 3). Moreover, this was confirmed by HMBC through the observed correlation between the anomeric proton of the glucose and carbon 3′ of ring B (Fig. S5–S7, ESI†). This anomeric proton exhibits a J value of 3.7 Hz, which indicates an α-linkage. To our knowledge, this is a novel compound.
Due to the preference of this enzyme for phenolic OHs versus aliphatic OHs,24,25,31 our hypothesis is that monoglucoside 1c, which could not be purified, must be glucosylated at ring A, corresponding to the derivatives 5-OH or 7-OH, instead of aliphatic 3-OH at ring C.
Molecular modelling of the glucosylation reaction
We investigated the specificity of TtSPP_R134A in glucosylating DMY by induced-fit docking analysis of the 3′-O-α-D-glucopyranoside and 4′-O-α-D-glucopyranoside derivatives, using the crystal structure of TtSPP25a with Arg134 replaced by Ala as the template. It is worth noting that Arg134 corresponds to Arg148 in the deposited coordinates that contain an N-terminal polyHis-tag. Autodock Vina was used to explore the accommodation of the derivatives in the active site and allowed further insight into the molecular interactions underlying the specificity of the reaction.Several residues delineating the catalytic tunnel, therefore considered key in acceptor–substrate binding, were defined as flexible, i.e., Phe146, Phe171, Tyr215, Phe225, Glu252, and His358. Around 9 solutions were calculated with the 3′-O-α-D-glucopyranoside derivative for each docking run, in a binding energy range between −12.74 and −7.164 kcal mol−1. In the case of the 4′-O-α-D-glucopyranoside, 10 solutions were calculated in a binding energy range between −8.583 and −6.782 kcal mol−1. From these conformations, we selected the pose with more conservative interactions at subsite −1, as deduced from superimposition of the model onto the reported complex of the homologous Bifidobacterium adolescentis sucrose phosphorylase with sucrose (BaSP, PDB code 2GDU).32 It should be noted that the best model of the 4′-O-α-D-glucopyranoside (Fig. 4B) shows a more conserved interacting pattern, as compared to the 3′-O-α-D-glucopyranoside derivative (Fig. 4A), which also corresponds to the first-ranked conformation in terms of energy. Thus, the glucose unit of the derivative makes the expected polar links to the TtSPP_R134A residues Arg209 (O2), His309 (O3), Asp63 (O4, O6) and His101 (O6) (Fig. 4B). Moreover, the modelled complex suggests that binding of DMY seems to involve a marked reshaping of the narrow slit, giving access to the TtSPP_R134A active site.
 | ||
| Fig. 4 Docking simulation of the dihydromyricetin glucosylated derivatives into the TtSPP_R134A variant. Polar interactions of the (A) 3′-O-α-D-glucopyranoside and (B) 4′-O-α-D-glucopyranoside in the catalytic pocket are depicted, as a result of the docking simulation from AutoDock Vina. The catalytic residues are shown in light green. (C) and (D) illustrate how several residues at the active site show large conformational changes upon binding, making room for accommodating the bulky DMY moiety. Grey sticks show the position of these residues in the unliganded enzyme. | ||
As shown in Fig. 4C and D, DMY interacts with the enzyme through hydrophobic interactions, positioned between His358 and Phe171, with both residues presenting a significant conformational change upon binding. In addition, the prominent side-chains of the residues Phe146, Tyr215 and Phe225 retract by large conformational changes, allowing space to allocate the fused ring of the bulky DMY moiety. Besides, with the exception of Tyr215, the three Phe side chains are located in loops, thus, in principle, not confined to rigid regions, which may potentially contribute to providing additional flexibility to the enzyme, allowing it to accommodate bulky acceptors such as DMY. It is plausible that, apart from the enlargement of the cavity in TtSPP_R134A, the flexibility of this region allocating subsites +1 and +2 is related to the broad acceptor promiscuity of this variant, enabling the enzyme to accept bulky donor substrates like this flavonoid.24 This flexibility is probably enhanced by the removal of the neighbouring Arg134 side-chain in the mutant (Fig. S8, ESI†).
Acylation of glucosylated dihydromyricetin
Acylating non-glycosylated polyphenols is particularly challenging, especially when long-chain fatty acids are involved.33 The previous literature reports that DMY fatty acid esters have been synthesized through chemical methods using either acid or alkaline catalysts.34 Enzymatic acylation methodologies offer several advantages over chemical methods, including milder reaction conditions, higher regioselectivity, and often fewer purification steps.35In this context, the presence of an aliphatic secondary 3-OH group in the C ring of DMY allowed Cao et al. and Li et al. to synthesize acetylated derivatives,36 but no results have been reported for longer acyl chains. Recently, we proposed a strategy for highly-efficient acylation of polyphenols based on a first α-glucosylation step—which increases the number of available reactive sites—followed by acylation of the sugar moiety catalyzed by a lipase.28,37
In light of this, we explored the acylation of dihydromyricetin 4′-O-α-D-glucoside using vinyl esters with fatty acid chains of varying lengths (C8, C12, and C16). Immobilized Thermomyces lanuginosus lipase (Lipozyme TL IM) was chosen as a biocatalyst for its exceptional regioselectivity towards the 6-OH position of glucose in acylation reactions.38 The reaction was carried out in tert-butyl alcohol, which has demonstrated great effectiveness as a solvent for enzyme-catalyzed reactions, significantly contributing to the stability and activity of the biocatalyst. Additionally, it offers remarkable solubility for dihydromyricetin monoglucoside. Vinyl esters were selected as acylating agents due to their fast transesterification rates with carbohydrates, up to 100-fold faster compared to alkyl esters.39
The acylation reactions were analyzed (Fig. 5). As illustrated, the reactions proceeded rapidly, completing within 90 min for the three acylating agents, with conversion yields exceeding 95%. The immobilized lipase exhibited exceptional efficiency.
 | ||
| Fig. 5 Reaction profiles of the acylation reactions in tert-butyl alcohol using 15 mM dihydromyricetin 4′-O-α-D-glucoside and 330 mM vinyl ester: (A) C8; (B) C12; and (C) C16. Reaction conditions: Lipozyme TL IM (7 mg mL−1), 60 °C. | ||
The acylated derivatives were purified using silica-gel chromatography and characterized by exact mass spectrometry (ESI-MS, negative mode) as shown in Fig. S9 (ESI).† The acylation position of the three acylated derivatives was determined by 2D-NMR (Fig. S10–S18, ESI†). The NMR signals of the compounds were assigned by a combination of 1D (1H and 13C) and 2D (gCOSY, gHSQC and gHMBC) experiments. In all cases, as expected, the acylation took place at the 6-OH primary hydroxyl of the glucose (Fig. 6), as inferred from the 2D-gHMBC experiment by the correlation between the two protons at position 6′′ of the glucose and the first carbon of the acyl chain.
 | ||
| Fig. 6 Molecular structure of the synthesized acylated derivatives: (2a) dihydromyricetin 4′-O-(6′′-O-octanoyl)-α-D-glucopyranoside; (2b) dihydromyricetin 4′-O-(6′′-O-lauroyl)-α-D-glucopyranoside; and (2c) dihydromyricetin 4′-O-(6′′-O-palmitoyl)-α-D-glucopyranoside. | ||
These fatty acid glucosyl derivatives of DMY are novel compounds and could also prove highly useful for applications in hydrophobic environments both in vitro and in vivo.2
Aqueous solubility
DMY has low water solubility and remains stable only at low temperatures.12a This appears to be associated with its low membrane permeability and bioavailability.40 We measured the aqueous solubility at 25 °C of dihydromyricetin, the 3′- and 4′-O-α-glucosides and the three acylated derivatives (Table 1).| Compound | Solubility (g L−1) |
|---|---|
| Dihydromyricetin | 1.1 ± 0.1 |
| 1a | 5.0 ± 0.4 |
| 1b | 666 ± 43 |
| 2a | 0.059 ± 0.001 |
| 2b | 0.030 ± 0.001 |
| 2c | 0.006 ± 0.000 |
The solubility of DMY matched well with that reported by Sun et al. (0.85 g L−1).13 However, Fan et al. reported a lower solubility for DMY of 0.2 g L−1 and 0.9 g L−1 at 25 °C and 37 °C, respectively.41
The monoglucoside at the 4′-OH position exhibited 4.5 times higher aqueous solubility compared to the aglycone. The solubility reported by Woo et al. for the same α-glucoside at room temperature was significantly higher (59.7 mM, approx. 28.7 g L−1).30 Interestingly, the minor glucoside at 3′-OH exhibited an outstanding solubility in water, nearly 600-fold compared with the aglycone. This suggests that the position of the glucosyl group may influence its physicochemical properties. As anticipated, the aqueous solubility of the acyl-glucoside was extremely low, in the ppm range. The solubility decreased with the length of the fatty acid chain.
Antioxidant activity
DMY has demonstrated strong antioxidant properties both in vitro and in vivo, even higher than those of ascorbic acid and Trolox.1b,42 This notable activity is related to the o-dihydroxy system (hydroxyl groups at the C-3′ and C-4′ positions) in the B-ring, along with the free hydroxyl group at the C-3 position in the C-ring.The synthesized compounds were tested for their ability to neutralize the radical cation ABTS˙+ or DPPH. After adding DMY or its derivatives, the reduction in colour intensity was measured using spectrophotometry (Table 2). The TEAC values were calculated from the slope of the plot showing the percentage of absorbance reduction vs. the concentration for the flavonoid divided by the slope of the equivalent plot for Trolox. A higher TEAC value indicates a greater antioxidant capacity. The TEAC value for Trolox is 1. It is worth noting that the TEAC values depend on the incubation time between the ABTS˙+ and the antioxidant.43 We observed that 15 min of incubation gave rise to consistent results.
| Compound | TEAC (ABTS˙+) | TEAC (DPPH) |
|---|---|---|
| (R)-Trolox served as the reference in both assays (TEAC value 1). #p < 0.01 vs. dihydromyricetin; *p < 0.005 vs. dihydromyricetin (n = 3). | ||
| Dihydromyricetin | 2.77 ± 0.20 | 1.96 ± 0.18 |
| 1a | 1.08 ± 0.08* | 0.020 ± 0.002* |
| 1b | 1.17 ± 0.12* | 0.98 ± 0.12* |
| 2a | 2.04 ± 0.20# | 0.0034 ± 0.0002* |
| 2b | 2.55 ± 0.23 | 0.0056 ± 0.0006* |
| 2c | 2.66 ± 0.23 | 0.0037 ± 0.0002* |
Table 2 indicates that the TEAC of DMY with ABTS˙+ is higher than that of its glucosides; however, the differences are not substantial when compared to the acylated derivatives.
In the case of DPPH, the only derivative that does not show a significant decrease in antioxidant activity is the 3′-OH monoglucoside. The low activity of acyl-glucosides could be linked to the steric accessibility of the compounds during their reaction with DPPH.44
Conclusions
In this study, we have explored the enzymatic synthesis of various glucosides and acyl-glucosides of DMY in order to vary its Hydrophilic–Lipophilic Balance (HLB). The glucosylation proceeded rapidly, producing 4′-O-α-D-glucoside as the predominant product. This α-glucosylation step enabled the subsequent acylation of the sugar moiety, which was catalyzed by a regioselective lipase. The second step exhibited remarkable speed and conversion, yielding a range of acyl-glucosides with varying chain lengths. These modifications result in derivatives that exhibit different physicochemical properties (including water solubility) compared to the aglycone, potentially altering the bioavailability of DMY. This could favor the application of DMY in the food, nutraceutical, cosmetic and pharmaceutical industries, in which more polar or nonpolar derivatives of DMY may be necessary.Experimental
Enzymes and reagents
The recombinant sucrose phosphorylase variant TtSPP_R134A from Thermoanaerobacterium thermosaccharolyticum was produced following previously established protocols,22a but in this case the Escherichia coli culture was scaled-up to a 5 L fermenter (Biostart BPlus, Braun Biotech) including 4 L of batch medium (10 g L−1 tryptone, 5 g L−1 yeast extract, 5 g L−1 NaCl, 100 mg L−1 ampicillin and 0.05 mL L−1 antifoam 204) maintained at 37 °C and 300 rpm agitation during 16 h. The lipase from Thermomyces lanuginosus, immobilized on granulated silica (Lipozyme TL IM, 250 IUN g−1), was generously supplied by Novozymes. Dihydromyricetin was sourced from Hunan MT Health Inc. (Hunan, China), while sucrose was obtained from Scharlau. Vinyl octanoate and vinyl palmitate were purchased from TCI Chemicals. Vinyl laurate, ABTS [2,2-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)], DPPH (2,2-diphenyl-1-picrylhydrazyl), and (R)-Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) were acquired from Sigma Aldrich. All other reagents and solvents used were of the highest available purity grade.Assay for sucrose phosphorylase activity
The sucrose phosphorylase activity was evaluated using the bicinchoninic acid (BCA) assay for reducing sugars described by Waffenschmidt et al.45 adapted to a 96-well microplate scale as reported previously.22a One unit of activity (1 U) corresponded to the release of one μmol of reducing sugars per minute.Glucosylation of dihydromyricetin
For the screening of best reaction conditions, the mixture contained 40 mM dihydromyricetin, 0.1–1 M sucrose, and sucrose phosphorylase (2 U mL−1 measured in the BCA assay) in 1 mL of 50 mM MOPS buffer (pH 6.5). The reaction was maintained at 48 °C during 4 h with orbital shaking (800 rpm) and followed by high-performance liquid chromatography (HPLC).For the scaling-up of the reaction, DMY (1 g, 3.13 mmol), sucrose (13.4 g, 39.2 mmol) and sucrose phosphorylase TtSP_R134A (2 U mL−1) were mixed with 72 mL of 50 mM MOPS buffer (pH 6.5) in a 100 mL Pyrex flask. The reaction was maintained at 48 °C with magnetic stirring and monitored by thin-layer chromatography (TLC) and high-performance liquid chromatography (HPLC). After 4 hours, once the reaction was complete, the reaction mixture was transferred to a 250 mL flask, and an equal volume of ethanol (72 mL) was added to stop the reaction. This ethanol was subsequently removed using a rotary evaporator (model R-210, Buchi; Flawil, Switzerland). The aqueous phase was extracted with ethyl acetate to remove the carbohydrates, until no more sugars were observed in the TLC plates. The organic phase was dried with MgSO4 and evaporated in the rotary evaporator. The glucosides were purified using silica gel chromatography. The column head was prepared by dissolving the products in methanol and adding silica gel 60 (particle size 0.06–0.2 mm, Scharlau). Subsequently, the solvent was removed using a rotary evaporator. The mobile phase consisted of a mixture of ethyl acetate, methanol and water in the proportions 60:5:4 (v/v/v). The fractions were collected and analyzed by TLC and HPLC. Finally, the solvent was evaporated using a rotary evaporator.
General procedure for enzymatic acylation of ampelopsin
Dihydromyricetin 4′-O-α-D-glucopyranoside (100 mg, 0.207 mmol), vinyl ester (4.6 mmol) and Lipozyme TL IM (98 mg) were mixed in 14 mL of tert-butyl alcohol (previously dried using 3 Å molecular sieves). The reaction mixture was incubated at 60 °C under vigorous shaking. Aliquots of 50 μL were taken at various time points and diluted with 450 μL of methanol. These samples were analyzed by TLC and HPLC. After 1 hour, when the reaction was almost complete, the mixture was kept in a refrigerator at 4 °C, where the reaction stops as the tert-butanol solvent freezes. The solvent was removed using a rotary evaporator. The acyl-glucosides were purified using silica gel chromatography. The column head was prepared by dissolving the products in methanol and adding silica gel 60 (particle size 0.06–0.2 mm, Scharlau). The mobile phase consisted of a mixture of ethyl acetate and hexane in a 1:1 (v/v) ratio, until fatty acids were no longer detected on TLC plates (revealed with bromocresol green). Subsequently, the mobile phase was switched to 100% ethyl acetate, until products were no longer detected on TLC plates (revealed under UV light). The solvents were then evaporated to obtain the acylated DMY derivatives. The purified products were collected as yellow oils and characterized by MS and 2D-NMR.
Thin-layer chromatography (TLC)
TLC analysis used 60 F254 silica gel plates (Merck). To monitor the glycosylation and acylation reactions, the mobile phase consisted of a mixture of ethyl acetate, methanol, and H2O in a 60:5:4 (v/v/v) ratio. To visualize dihydromyricetin or the synthesized products, UV light was used. For detecting sugars, the plate was dipped in a 10% H2SO4 solution and heated. Fatty acids were revealed using a solution of bromocresol green (5 mg) and NaOH (1 mg) in 25 mL of ethanol.
High-performance liquid chromatography (HPLC)
HPLC analysis was performed using a quaternary pump (model 600, Waters, Milford, MA, USA) coupled to an autosampler (model ProStar 420, Varian Inc., Palo Alto, CA, USA). The column used was a Zorbax Eclipse Plus C18 (4.6 × 100 mm, 3.5 μm, Agilent) at 40 °C, and detection was performed using a photodiode array detector (ProStar, Varian). Peaks were detected at 295 nm and analyzed using Varian Star LC workstation 6.41 software. The mobile phase consisted of water and acetonitrile, both containing 0.1% (v/v) formic acid, with a flow rate of 1 mL min−1.For the glucosylation reaction, the initial mobile phase contained 1% (v/v) acetonitrile during 1.5 min. Then, the acetonitrile increased to 10% (v/v) within the next minute and to 25% (v/v) in the following 12.5 min. For the acylation reaction, the initial mobile phase contained 1% (v/v) acetonitrile during 1.5 min. Then, the acetonitrile increased to 10% (v/v) within the next 30 seconds and to 95% in the following 13 min. In both cases, the mobile phase returned to the initial conditions, and the column was equilibrated for 5 minutes.
Mass spectrometry
The molecular weight of the main products was determined using high-resolution mass spectrometry with electrospray ionization (ESI) coupled to a hybrid QTOF analyzer (Agilent 6500 Accurate Mass) in negative reflector mode.Nuclear magnetic resonance (NMR) analysis
The structure of the products was determined using a combination of standard 1D and 2D NMR techniques (1H, 13C, gCOSY, gHSQC and gHMBC). Samples were dissolved in DMSO-d6 and their spectra were recorded using a Agilent System 500 NMR spectrometer equipped with a 5 mm HCN cold probe, at 298 K. Chemical shifts were reported in parts per million (ppm), using the residual DMSO-d6 signal as an internal reference. All pulse sequences were provided by Varian. For the 1H NMR experiment, parameters included a spectral width of 8012.8 Hz, a relaxation delay of 1.0000 s, 8 scans, and a digital resolution of 0.12. The acquisition time was 2.0447 s, and the experiment was carried out at 499.81 MHz. For the 13C NMR experiment, parameters included a spectral width of 31250.0 Hz, a relaxation delay of 1.0000 s, and a digital resolution of 0.48. The acquisition time was 1.0486 s, and the experiment was conducted at 125.69 MHz. For the 2D-gHMBC experiment, parameters included spectral widths of 6345.2 Hz for 1H and 30165.9 Hz for 13C, with a relaxation delay of 1.0000 s, 16 scans, and a digital resolution of 6.20 for 1H and 29.46 for 13C. The matrix size was 1024 × 1024 points. For the 2D-gHSQC experiment, parameters included spectral widths of 6345.2 Hz for 1H and 25133.5 Hz for 13C, with a relaxation delay of 1.0000 s, 4 scans for each of 96-time increments, and a digital resolution of 6.20 for 1H and 24.54 for 13C. The matrix size was 1024 × 1024 points. For the 2D-gCOSY experiment, parameters included spectral widths of 6345.2 Hz for both 1H dimensions, with a relaxation delay of 1.0000 s, 4 scans for each of 128-time increments, and a digital resolution of 6.20 for both dimensions. The matrix size was 1024 × 1024 points, and the experiment was conducted at 499.81 MHz for both dimensions.
Automated docking of dihydromyricetin glucosides into the TtSPP_R134A structure
Glucose coordinates were downloaded from the PDB server (https://www.rcsb.org), whilst dihydromyricetin coordinates were generated from a SMILES string using Coot.46 The program Mercury from CCDC (Cambridge Crystallographic Data Centre)47 was used to create the O-glycosidic bond between glucose and the flavonoid, generating the glucosylated derivatives, which were regularized by using Avogadro.48 The deposited coordinates of TtSPP (PDB code 6S9V) were modified to the R148A mutation using Coot.46 This Arg148 of the crystal structure corresponded to Arg134 in the wild-type TtSPP sequence due to a 14 residue-long N-terminal histidine tag. All water molecules and other heteroatoms were removed from the coordinates, and this protein was used as a receptor in the docking simulation. AutodockTools49 was used to create the pdbqt files, including polar hydrogens and Kollman charges added to the receptor coordinates and Compute Gasteiger charges added to the ligands. The glycosidic linkages between the glucose and DMY and the C–C bonds linking the three phenolic rings to the fused rings within the DMY molecule were defined as rotatable bonds.Docking studies were carried out using AutoDock Vina50 with Phe146, Phe171, Tyr215, Phe225, Glu252 and His358 defined as flexible residues. The grid dimensions were 15 × 15 × 15.75 Å, with a grid centre at 33.539 × 84.029 × 1.666 Å for 3′-O-α-D-glucopyranoside, and 12.75 × 15 × 17.25 Å, with a grid centre at 34.539 × 83.029 × −0334 Å for 4′-O-α-D-glucopyranoside. Default parameters were defined, with exhaustiveness of 8 and energy range of 3. From the solutions, the best poses of the glucosylated dihydromyricetin derivates were chosen based on minimum binding energy and conservation of the binding mode at subsite −1. The structural figures were prepared using PyMOL.
Aqueous solubility
The compounds were incubated in water at 25 °C under saturated conditions for 24 h with orbital stirring at 1000 rpm. Subsequently, the samples were centrifuged and diluted with methanol when necessary (to fit into the calibration curve). Then, 150 μL of the supernatants were analyzed in triplicate using a 96-well plate. A DMY calibration curve in methanol (ranging from 0 to 100 μM) was used for the analysis. Absorbance measurements were taken at 295 nm using a BioTek Synergy H1 microplate reader (Agilent).Antioxidant activity
The capacity of the synthesized derivatives to reduce the radical cation 2,2-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS˙+) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) was assessed using the method described by Re et al.43 adapted to 96-well plates. (R)-Trolox served as the reference in both assays. Standard solutions were prepared in methanol, with concentrations ranging from 0 to 200 μM for (R)-Trolox and from 0 to 1000 μM for DMY and its glucosides and acyl-glucosides. The compounds (20 μL) at varying concentrations were mixed with 230 μL of ABTS˙+ (diluted with methanol to achieve an absorbance of 0.7 at 655 nm) or 200 μL of DPPH (200 μM in methanol). After incubation in the dark at room temperature for 15 min, the absorbance was measured at 655 nm for ABTS˙+ and 540 nm for DPPH using a BioTek Synergy H1 microplate reader (Agilent). The degree of inhibition of ABTS˙+ or DPPH absorbance was plotted against the concentration of both Trolox and the DMY derivatives. To calculate the Trolox Equivalent Antioxidant Capacity (TEAC), the slope of the plot showing the percentage inhibition of absorbance versus concentration for the flavonoid was divided by the slope of the equivalent plot for Trolox.Author contributions
DR-G, CU, and LB performed the experimental research and the analysis. JLG-A developed the methodology. TD rationally designed the sucrose phosphorylase variant. MR-N and JS-A performed the molecular and docking simulations and the subsequent analysis. MF-L designed the enzyme production. JLG-A and FJP wrote the original draft. MF-L, JS-A and FJP obtained the funding. JLG-A, AOB and FJP designed and directed the project. All authors discussed the results and contributed to the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the individuals involved in mass spectrometry and NMR services at the Centro de Química Orgánica “Lora Tamayo” (CENQUIOR, CSIC) for their analyses and technical assistance. We thank Ramiro Martínez (Novozymes) for providing immobilized lipases. We thank Celia Andrés-Juan for interpretation of the physicochemical properties of the DMY derivatives. This work was supported by the following grants: (1) PDC2022-133134-C21/22 “ACYLGLUFLAV_APP” funded by MCIN/AEI/10.13039/501100011033 by the “European Union NextGenerationEU/PRTR” and (2) PID2022-136367OB-C31/32/33 “GLYCOENZ-GREEN” funded by MCIN/AEI/10.13039/501100011033/ and through FEDER, a Way of Making Europe. We thank the Spanish Society of Catalysis (SECAT) for awarding a Master's Scholarship to D. Rodríguez-García.References
- (a) R. C. V. Carneiro, L. Ye, N. Baek, G. H. A. Teixeira and S. F. O'Keefe, J. Funct. Foods, 2021, 76, 104317 CrossRef; (b) H. Zhang, G. Caprioli, H. Hussain, N. P. K. Le, M. A. Farag and J. Xiao, eFood, 2021, 2, 164–184 CrossRef.
- D. Liu, Y. Mao, L. Ding and X. A. Zeng, Trends Food Sci. Technol., 2019, 91, 586–597 CrossRef PubMed.
- H. Xu, T. Zhao, F. Liu, Y. Zhang, Y. Xie, X. Xiao and Y. Zhang, LWT–Food Sci. Technol., 2023, 174, 114387 CrossRef.
- S. Liu, Q. Ai, K. Feng, Y. Li and X. Liu, Apoptosis, 2016, 21, 1366–1385 CrossRef.
- (a) K.-J. Fan, B. Yang, Y. Liu, X.-D. Tian and B. Wang, Mol. Med. Rep., 2017, 16, 9758–9762 CrossRef PubMed; (b) G. B. Park, J. Y. Jeong and D. Kim, Oncol. Lett., 2017, 14, 7947–7956 Search PubMed.
- X. Kou, X. Liu, X. Chen, J. Li, X. Yang, J. Fan, Y. Yang and N. Chen, Oncotarget, 2016, 7, 74484–74495 CrossRef.
- X. Zeng, J. Yang, O. Hu, J. Huang, L. Ran, M. Chen, Y. Zhang, X. Zhou, J. Zhu, Q. Zhang, L. Yi and M. Mi, Antioxid. Redox Signal., 2019, 30, 163–183 CrossRef PubMed.
- M. Wen, X. Sun, L. Pan, S. Jing, X. Zhang, L. Liang, H. Xiao, P. Liu, Z. Xu, Q. Zhang and H. Huang, Eur. J. Pharmacol., 2024, 978, 176799 CrossRef.
- H. Liang, K. He, T. Li, S. Cui, M. Tang, S. Kang, W. Ma and L. Song, Sci. Rep., 2020, 10, 21416 CrossRef.
- C. Falckenhayn, A. Bienkowska, J. Söhle, K. Wegner, G. Raddatz, B. Kristof, D. Kuck, R. Siegner, R. Kaufmann, J. Korn, S. Baumann, D. Lange, A. Schepky, H. Völzke, L. Kaderali, M. Winnefeld, F. Lyko and E. Grönniger, Front. Aging, 2024, 4, 1258184 CrossRef PubMed.
- Y. Wang, J. Wang, H. Xiang, P. Ding, T. Wu and G. Ji, Biomed. Pharmacother., 2022, 148, 112771 CrossRef PubMed.
- (a) R. Zhang, H. Zhang, H. Shi, D. Zhang, Z. Zhang and H. Liu, Drug Delivery, 2022, 29, 3052–3070 CrossRef; (b) D. Xiang, C.-G. Wang, W.-Q. Wang, C.-Y. Shi, W. Xiong, M.-D. Wang and J.-G. Fang, Int. J. Food Sci. Nutr, 2017, 68, 704–711 CrossRef.
- C.-C. Sun, Y. Li, Z.-P. Yin and Q.-F. Zhang, J. Sci. Food Agric., 2021, 101, 3862–3869 CrossRef.
- L. Liu, X. Yin, X. Wang and X. Li, Pharm. Biol., 2017, 55, 657–662 CrossRef PubMed.
- H. Liu, C. Gan, H. Shi, K. Qu, L. Jing, M. Lu, B. Su, H. Yu, H. Yuan, J. Chen, R. Zhang and W. Zeng, J. Drug Delivery Sci. Technol., 2021, 61, 102279 CrossRef.
- T. Chen, S. Zhu, Y. Lu, H. Cao, Y. Zhao, G. Jiang, L. Zhu and T. Lu, Anti-Cancer Agents Med. Chem., 2012, 12, 919–928 CrossRef PubMed.
- C.-C. Sun, H. Su, G.-D. Zheng, W.-J. Wang, E. Yuan and Q.-F. Zhang, Food Chem., 2020, 330, 127245 CrossRef PubMed.
- S. Geng, X. Liu, H. Ma, B. Liu and G. Liang, Food Chem., 2021, 355, 129660 CrossRef PubMed.
- C. Wang, W. Xiong, S. R. Perumalla, J. Fang and C. C. Sun, Int. J. Pharm., 2016, 511, 245–252 CrossRef PubMed.
- X. Zhao, C. Shi, X. Zhou, T. Lin, Y. Gong, M. Yin, L. Fan, W. Wang and J. Fang, Eur. J. Pharm. Sci., 2019, 138, 104994 CrossRef PubMed.
- L.-P. Ruan, B.-Y. Yu, G.-M. Fu and D.-N. Zhu, J. Pharm. Biomed. Anal., 2005, 38, 457–464 CrossRef PubMed.
- (a) J. L. Gonzalez-Alfonso, Z. Ubiparip, E. Jimenez-Ortega, A. Poveda, C. Alonso, L. Coderch, J. Jimenez-Barbero, J. Sanz-Aparicio, A. Ballesteros, T. Desmet and F. J. Plou, Adv. Synth. Catal., 2021, 363, 3079–3089 CrossRef; (b) T. Klingel, M. Hadamjetz, A. Fischer and D. Wefers, Carbohydr. Res., 2019, 483, 107741 CrossRef PubMed; (c) J. L. Gonzalez-Alfonso, P. Peñalver, A. O. Ballesteros, J. C. Morales and F. J. Plou, Front. Nutr., 2019, 6, 30 CrossRef PubMed; (d) Y. S. Lee, J. B. Woo, S. I. Ryu, S. K. Moon, N. S. Han and S. B. Lee, Food Chem., 2017, 229, 75–83 CrossRef PubMed; (e) C. Febres-Molina, X. Prat-Resina and G. A. Jaña, Org. Biomol. Chem., 2023, 21, 9591–9602 RSC.
- T. Desmet, W. Soetaert, P. Bojarová, V. Kren, L. Dijkhuizen, V. Eastwick-Field and A. Schiller, Chem. – Eur. J., 2012, 18, 10786–10801 CrossRef PubMed.
- M. E. Dirks-Hofmeister, T. Verhaeghe, K. De Winter and T. Desmet, Angew. Chem., Int. Ed., 2015, 54, 9289–9292 CrossRef PubMed.
- (a) J. Franceus, N. Capra, T. Desmet and A.-M. W. H. Thunnissen, Int. J. Mol. Sci., 2019, 20, 3906 CrossRef PubMed; (b) K. De Winter, G. Dewitte, M. E. Dirks-Hofmeister, S. De Laet, H. Pelantová, V. Křen and T. Desmet, J. Agric. Food Chem., 2015, 63, 10131–10139 CrossRef PubMed.
- M. Demonceaux, M. Goux, J. Hendrickx, C. Solleux, F. Cadet, É. Lormeau, B. Offmann and C. André-Miral, Org. Biomol. Chem., 2023, 21, 2307–2311 RSC.
- (a) C. Alonso, L. Rubio, S. Touriño, M. Martí, C. Barba, F. Fernández-Campos, L. Coderch and J. L. Parra, Free Radicals Biol. Med., 2014, 75, 149–155 CrossRef; (b) L. Otaegui, J. Lehoux, L. Martin, L. Givalois, T. Durand, C. Desrumaux and C. Crauste, Org. Biomol. Chem., 2024, 22, 2877–2890 RSC.
- J. L. Gonzalez-Alfonso, C. Alonso, A. Poveda, Z. Ubiparip, A. O. Ballesteros, T. Desmet, J. Jiménez-Barbero, L. Coderch and F. J. Plou, J. Agric. Food Chem., 2024, 72, 4325–4333 CrossRef PubMed.
- C. Wen, H.-C. Wu, W.-H. Ouyang, J.-X. Nie, Y.-P. Guo, F. Wang, L.-L. Hu, J.-H. Yang, L.-J. Zheng, J.-L. Wang, W. Huang, G.-P. Liang and R.-W. Jiang, J. Agric. Food Chem., 2023, 71, 8998–9008 CrossRef PubMed.
- H.-J. Woo, H.-K. Kang, T. T. H. Nguyen, G.-E. Kim, Y.-M. Kim, J.-S. Park, D. Kim, J. Cha, Y.-H. Moon, S.-H. Nam, Y.-M. Xia, A. Kimura and D. Kim, Enzyme Microb. Technol., 2012, 51, 311–318 CrossRef PubMed.
- J. Franceus and T. Desmet, Int. J. Mol. Sci., 2020, 21, 2526 CrossRef PubMed.
- O. Mirza, L. K. Skov, D. Sprogøe, L. A. M. van den Broek, G. Beldman, J. S. Kastrup and M. Gajhede, J. Biol. Chem., 2006, 281, 35576–35584 CrossRef CAS PubMed.
- M. L. Contente, F. Annunziata, P. Cannazza, S. Donzella, C. Pinna, D. Romano, L. Tamborini, F. G. Barbosa, F. Molinari and A. Pinto, J. Agric. Food Chem., 2021, 69, 13669–13681 CrossRef CAS PubMed.
- T. Matsumoto and S. Tahara, Nippon Nogeikagaku Kaishi, 2001, 75, 659–667 CrossRef.
- P. Torres, A. Poveda, J. Jimenez-Barbero, A. Ballesteros and F. J. Plou, J. Agric. Food Chem., 2010, 58, 807–813 CrossRef PubMed.
- (a) S.-L. Cao, X. Deng, P. Xu, Z.-X. Huang, J. Zhou, X.-H. Li, M.-H. Zong and W.-Y. Lou, J. Agric. Food Chem., 2017, 65, 2084–2088 CrossRef PubMed; (b) W. Li, H. Wu, B. Liu, X. Hou, D. Wan, W. Lou and J. Zhao, J. Biotechnol., 2015, 199, 31–37 CrossRef PubMed.
- F. J. Plou, J. L. González-Alfonso, A. O. Ballesteros, C. Alonso, L. Coderch, A. Poveda, J. Jimenez-Barbero, T. Desmet and Z. Ubiparip, PatentPCT/EP2023/066107, 2022 Search PubMed.
- (a) J. Jadhav and A. P. Pratap, Tenside, Surfactants, Deterg., 2017, 54, 539–545 Search PubMed; (b) A. Chyba, V. Mastihuba and M. Mastihubová, Bioorg. Med. Chem. Lett., 2016, 26, 1567–1570 CrossRef PubMed; (c) M. Ferrer, M. A. Cruces, F. J. Plou, M. Bernabe and A. Ballesteros, Tetrahedron, 2000, 56, 4053–4061 CrossRef.
- Y. F. Wang, J. J. Lalonde, M. Momongan, D. E. Bergbreiter and C. H. Wong, J. Am. Chem. Soc., 1988, 110, 7200–7205 CrossRef.
- Q. Tong, X. Hou, J. Fang, W. Wang, W. Xiong, X. Liu, X. Xie and C. Shi, J. Pharm. Biomed. Anal., 2015, 114, 455–461 CrossRef.
- L. Fan, Q. Tong, W. Dong, G. Yang, X. Hou, W. Xiong, C. Shi, J. Fang and W. Wang, J. Agric. Food Chem., 2017, 65, 4597–4604 CrossRef.
- (a) J. Gao, B. Liu, Z. Ning, R. Zhao, A. Zhang and Q. Wu, J. Food Biochem., 2009, 33, 808–820 CrossRef; (b) C. Xie, Z. Chen, C. Zhang, X. Xu, J. Jin, W. Zhan, T. Han and J. Wang, Life Sci., 2016, 157, 131–139 CrossRef.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radicals Biol. Med., 1999, 26, 1231–1237 CrossRef PubMed.
- J. Xie and K. M. Schaich, J. Agric. Food Chem., 2014, 62, 4251–4260 CrossRef PubMed.
- S. Waffenschmidt and L. Jaenicke, Anal. Biochem., 1987, 165, 337–340 CrossRef PubMed.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrometry and NMR spectra. See DOI: https://doi.org/10.1039/d4ob01682c |
| This journal is © The Royal Society of Chemistry 2025 |