
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses of differentially fluorinated triazole-based 1-deoxysphingosine analogues en route to SphK inhibitors†
Adrià
Cardona‡
 a,
Varbina
Ivanova‡
b,
Raúl
Beltrán-Debón
c,
Xavier
Barril
b,
Sergio
Castillón
a,
Yolanda
Díaz
*a and
M. Isabel
Matheu
*a
a,
Varbina
Ivanova‡
b,
Raúl
Beltrán-Debón
c,
Xavier
Barril
b,
Sergio
Castillón
a,
Yolanda
Díaz
*a and
M. Isabel
Matheu
*a
aUniversitat Rovira i Virgili, Departament de Química Analítica i Química Orgànica, Faculty of Chemistry, C/Marcel·lí Domingo 1, 43007 Tarragona, Spain. E-mail: maribel.matheu@urv.cat
bUniversitat de Barcelona, Department of Physical Chemistry, Faculty of Pharmacy, Av. Joan XXIII s/n, Barcelona 08028, Spain
cUniversitat Rovira i Virgili, Departament de Bioquímica i Biotecnologia, Faculty of Chemistry, C/Marcel·lí Domingo 1, 43007 Tarragona, Spain
First published on 11th November 2024
Abstract
This study focuses on the stereoselective syntheses of 1-deoxysphingosine analogues as potential inhibitors of sphingosine kinase (SphK), particularly targeting its isoforms SphK1 and SphK2, which are implicated in cancer progression and therapy resistance. The research builds on previous work by designing a series of analogues featuring systematic structural modifications like the incorporation of a triazole ring, varying degrees of fluorination, and different head groups (e.g., guanidino, N-methylamino, and N,N-dimethylamino). These modifications aimed to enhance polar and hydrophobic interactions especially with SphK2. The synthesized compounds were evaluated for their inhibitory activity, revealing that certain derivatives, particularly those with guanidino groups and heptafluoropropyl fragments at the lipidic tail, exhibited significant potency and selectivity towards SphK2. Docking studies supported these findings by showing favorable binding interactions within the SphK2 active site, which were less pronounced in SphK1, correlating with the observed selectivity. This work contributes to the development of novel 1-deoxysphingosine analogues targeting SphK inhibition, as well as to the knowledge of the differential topology of the active sites in SphK1 and SphK2.
Introduction
Sphingosine kinase (SphK) plays a pivotal role in regulating the sphingolipid rheostat1 (Scheme 1), which governs the dynamic balance between ceramide (Cer) and sphingosine 1-phosphate (S1P). Depending on the position of this rheostat, programmed cell death (Scheme 1, top) or cell survival (Scheme 1, bottom) is promoted,2 since Cer and sphingosine (Sph) induce cell apoptosis and growth arrest, whereas S1P promotes cell survival and proliferation. | ||
| Scheme 1 Sphingolipid rheostat and SphK inhibition effects. | ||
In this equilibrium, SphK catalyzes the ATP-dependent phosphorylation of sphingosine (Sph) to produce sphingosine 1-phosphate (S1P). This enzyme exists in two isoforms, SphK1 and SphK2,3 which differ in their substrate preference, subcellular localizations and tissue distributions.4
The focus on SphK inhibitors as potential drugs arises from the observation that SphK1 is often overexpressed in a wide range of tumors, including solid tumours5 and leukaemia.6 Hence, SphK inhibition is associated with tumor cell apoptosis. Moreover, SphK1 is involved in induction of chemotherapeutic resistance5b and radiotherapy-resistant tumour cells or those with acquired chemoresistance exhibit elevated expression of SphK1.7 Besides, the potential of SphK2 as a target for tumor cells has emerged more recently.8 Thus, significant efforts have been made looking for effective sphingosine kinase inhibitors.9
Interest in 1-deoxysphingolipids has grown due to their remarkable biological properties, including antiproliferative and cytotoxic activities, as well as their influence on sphingolipid biosynthesis and metabolism.10 Thus, the isolation of the first antiproliferative 1-deoxysphingolipid, spisulosine,11 and its close structural relationship with other related 1-deoxysphingoid bases with remarkable cytotoxic properties, such as clavaminols,12 crucigasterins,13 xestoaminols14 or enigmol15 (Fig. 1), make this class of compounds an attractive lead for anticancer-oriented drug discovery.
 | ||
| Fig. 1 Representative compounds, previous work on sphingosine analogues, and structural modifications proposed in this work. | ||
The rationale for designing 1-deoxy analogues is to mimic the antiproliferative cytotoxic effects of sphingosine, while preventing the phosphorylation of the primary alcohol group that leads to unwanted mitogenic and anti-apoptotic activities. In this sense, one representative compound is enigmol (Fig. 1), distinguished from other 1-deoxysphingolipids by an additional hydroxyl group at the C-5 position, which imparts to this compound a polarity similar to that of sphingosine. Biological studies have shown that enigmol inhibits sphingosine kinase,15 ceramide synthase16 and protein kinase,17 exhibiting a broad spectrum of cytotoxicity (0.4 μM ≤ IC50 ≤ 14 μM) against 57 human cancer cell lines including colon, breast, brain and prostate.15 Additionally, enigmol's modest in vitro potency is compensated by its favourable pharmacokinetic properties in vivo.15
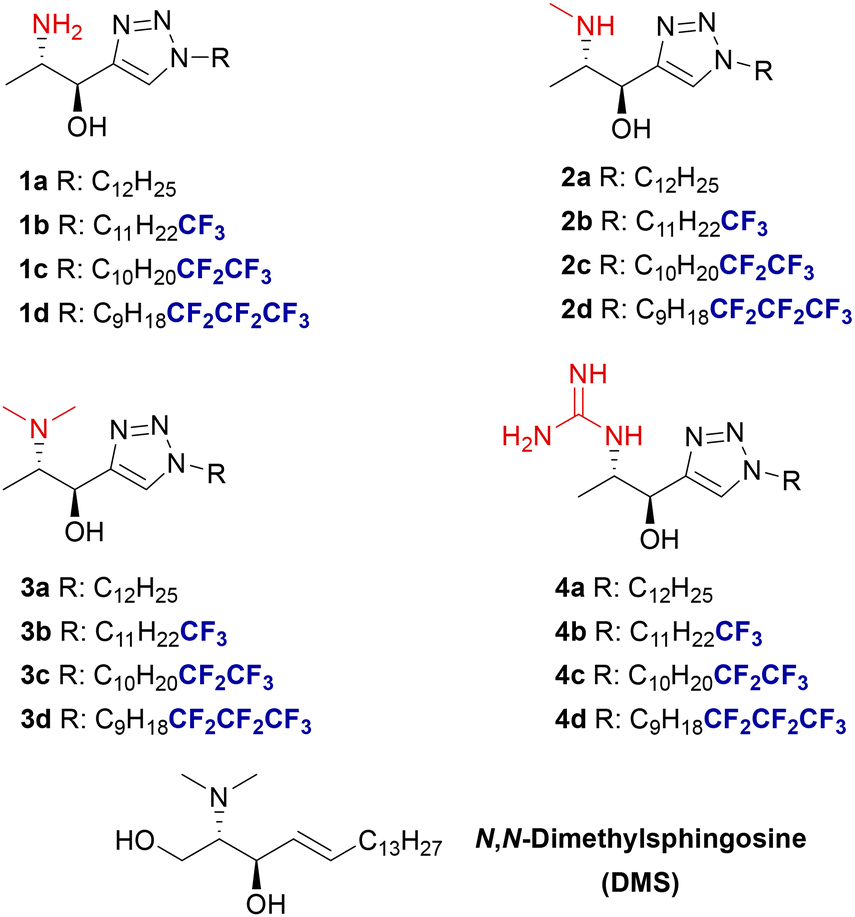
During the past years, we have been involved in developing methods for the synthesis of sphingoid bases,18 intended to be efficient sphingolipid-derived SphK inhibitors.19 In this context, we had explored the synthesis of sphingosine analogues incorporating a rigid triazole moiety in the aliphatic chain mimicking the conformational restriction provided by the 4,5-double bond in sphingosine.§ These analogues featured systematic modifications in the polar head and varying degrees of fluorination at the terminus of the aliphatic chain (Fig. 1, X = CH2OH).19a Compounds with a heptafluoro tail (Fig. 1) displayed the highest inhibitory activity against SphK2 in the low micromolar range while presenting the highest SphK2/SphK1 selectivity.
Reasons for introducing these structural modifications (Fig. 1) were: (a) the heterocyclic scaffold should be capable of establishing additional interactions with residues in the throat of the binding site of the enzyme and display a strong dipole moment;20 (b) derivatization of the free amino group into N,N-dimethylamino, guanidino, and N-methylamino moieties is expected to enhance polar interactions at the enzyme's binding site while likely preventing the acylation of the N-moiety, thereby slowing down enzymatic diversion to other sphingolipids,15 and (c) modification of the lipophilic end via a gradual increase in the degree of fluorination with perfluorinated terminal fragments to exploit interactions21 of the fatty tail of the sphingolipid with the hydrophobic bottom of the SphK binding site.22 Additionally, fluorine's stereoelectronic effects can influence the conformation of the flexible alkyl chain. Thus, strategically incorporating perfluorinated lipid fragments and finding the appropriate degree of fluorination may enhance affinity for the SphK binding site through entropy gain.23
In this context, we present here the syntheses and SphK1/SphK2 inhibitory effect of 1-deoxysphingosine analogues (Fig. 1, X = CH3) in which the above-mentioned modifications—1,2,3-triazole unit, derivatization of the amino group, and fluorination of the lipophilic tail—introduced in our previous work are explored.
Results and discussion
The synthesis of target compounds 1–4a–d (Scheme 2) was envisaged through a diastereoselective synthesis starting from commercially available L-alaninol. The construction of the anti-2,3-aminoalcohol segment present in 1-deoxysphingosine was envisioned via diastereoselective nucleophilic addition of an organoalkynylide species to L-alaninal, by the use of appropriate protecting groups on the amino group. Addition of Grignard reagents to chiral N-disubstituted amino aldehydes with bulky substituents such as benzyl moieties usually proceeds under Felkin conditions to afford 1,2-amino alcohols in excellent anti diastereomeric ratios.24 Thus, the syntheses of the corresponding N,N-dibenzyl anti-2-amino-3-alcohols 8 started with the protection of the amino group at L-alaninol, subsequent Swern oxidation and in situ addition of the acetylide Grignard reagent to the protected α-amino aldehyde intermediate (based on a one-pot methodology by Silveira-Dorta et al.)25 (Scheme 3). For the sake of comparison, the analogous processes using phthalimido and NHBoc functionalities in alcohols 6 and 7 were also explored. As expected, one-pot oxidation/addition of ethynyl magnesium bromide starting from dibenzylamino derivative 5 proceeded with high selectivity to form anti-configured alcohol 8. In contrast, the reaction of phthalimido derivative 6 led to the formation of the amino alcohol 9 with null stereoselectivity, whereas the monoprotected Boc derivative 7 led to the preferential formation of the syn-diastereoisomer 10 (Scheme 3). | ||
| Scheme 2 Retrosynthetic pathway of triazole-based 1-deoxysphingolipid analogues. | ||
 | ||
Scheme 3 Syntheses of amino-alcohol intermediates 8–10. For 5: (a) BnBr, K2CO3, H2O/acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); for 6: (b) phthalic anhydride, PhMe; for 7: (c) Boc2O, Et3N, THF. aDetermined by NMR. :1); for 6: (b) phthalic anhydride, PhMe; for 7: (c) Boc2O, Et3N, THF. aDetermined by NMR. | ||
Syntheses of the alkyltriazole derivatives
Propargyl alcohol 8 was then selected for following the synthetic scheme. The reaction of dodecyl azide (11a)19a with 8 under typical conditions for copper-catalysed azide–alkyne cycloaddition (CuAAC) (CuSO4·5H2O and sodium ascorbate) in DMSO:H2O as a solvent at 65 °C furnished triazole 12a in a 43% yield as a single diastereomer after purification of the diastereomeric mixture obtained. A 55% yield of diastereomerically pure 12a was obtained using a CH2Cl2/H2O mixture at room temperature (Scheme 4). Debenzylation with H2 and Pd/C in methanol afforded amine 1a in 88% yield.
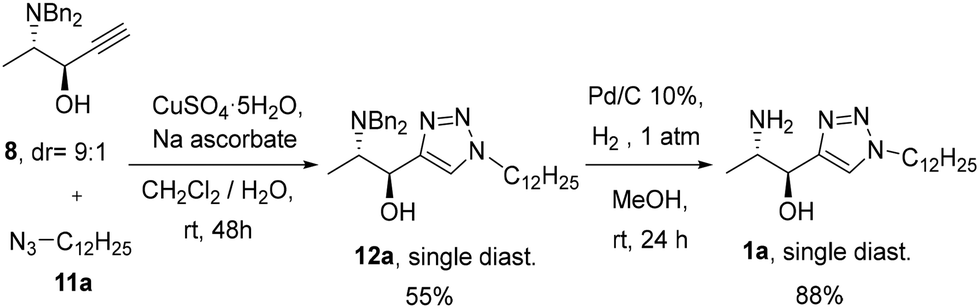 | ||
| Scheme 4 Preparation of 1avia CuAAC reaction and deprotection. | ||
The preparation of the N-methyl derivative 2a was assayed from 1avia reductive amination with aqueous formaldehyde under various reactions conditions such as NaBH3CN in MeOH, or zinc under neutral conditions (NaH2PO4);26 however, a mixture of the starting amine and dimethyl- and monomethyl amino derivatives was obtained. The best conditions were found by reduction of the N-Boc protected compound 13a with LiAlH4 to render compound 2a in 61% yield over the two steps (Scheme 5). Additionally, compound 1a was also reacted with p-formaldehyde under reductive conditions to afford N,N-dimethyl amino compound 3a in 76% yield.
 | ||
| Scheme 5 Derivatisation of the amino moiety in 1a to give 1-deoxysphingolipid analogues 2a, 3a and 4a. | ||
Parallelly, treatment of 1a with N,N′-di-Boc-1H-pyrazole-1-carboxiamidine (14) in the presence of triethylamine followed by reaction with TFA in CH2Cl2 rendered a guanidine derivative as a zwitterionic trifluoroacetate species, which by washings with aqueous NaOH gave free guanidine derivative 4a (Scheme 5).
Syntheses of partially fluorinated triazole sphingolipid analogues
Propargyl alcohol 8 was subjected to CuAAC with partially fluorinated azides 11b, 11c and 11d,19a followed by concomitant amino deprotection and hydrogenation of the alkene moiety in 12b–d to render sphingolipid analogues 1b–d, respectively, in good yields (Scheme 6).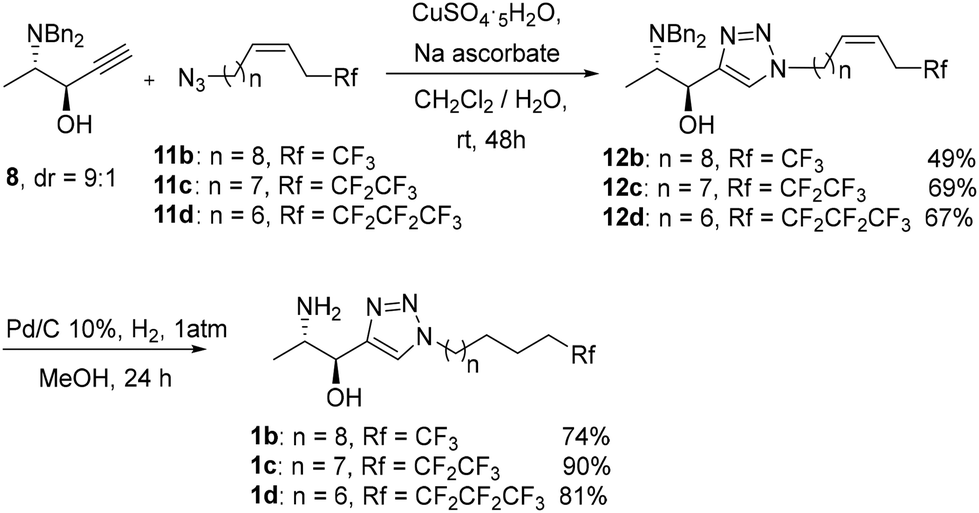 | ||
| Scheme 6 Syntheses of fluorinated 1-deoxysphingolipids 1b–d. | ||
In line with the derivatization of the parent non-fluorinated sphingolipid 1a, fluorinated compounds 1b–d were converted into the N-methyl, N,N-dimethyl and guanidinium trifluoroacetates derivatives 2b–d, 3b–d and 4b–d, respectively (Scheme 7).
 | ||
| Scheme 7 Derivatisation of the amino moiety in 1b–d to give fluorinated 1-deoxysphingolipid analogues 2b–d, 3b–d and 4b–d. | ||
In vitro sphingosine kinase assays
In vitro inhibition potency in front of SphK of compounds 1a–d, 2a–d, 3a–d, 4a–d was measured via time-resolved fluorescence resonance energy transfer (TR-FRET) analysis that relies on the immunodetection of adenosine diphosphate (ADP).27,28 The half maximal inhibitory concentrations of the synthesized 1-deoxysphingosine analogues were determined independently for SphK1 and SphK2 using dimethyl sphingosine (DMS) as a reference (Table 1).|
|
|||||||
|---|---|---|---|---|---|---|---|
| Inh. | IC50a SphK1 | IC50a SphK2 | Sel.b | Inh. | IC50a SphK1 | IC50a SphK2 | Sel.b |
| a Values are expressed as concentrations (μM). b Sel. refers to the SphK2/SphK1 selectivity ratio (IC50Sphk1/IC50Sphk2). | |||||||
| 1a | 34.8 | 18.9 | 1.8 | 3a | 92.3 | 10.0 | 9.2 |
| 1b | 127.0 | 57.4 | 2.2 | 3b | 112.0 | 57.3 | 2.0 |
| 1c | 118.0 | >200 | <0.6 | 3c | >200 | 39.4 | >5.0 |
| 1d | 71.3 | 18.8 | 3.8 | 3d | >200 | >200 | — |
| 2a | 86.9 | 20.8 | 4.2 | 4a | 50.1 | 5.0 | 10.0 |
| 2b | >200 | 47.2 | 2.5 | 4b | 77.6 | 29.2 | 2.7 |
| 2c | 95.3 | 23.6 | 4.0 | 4c | 51.4 | 9.0 | 5.7 |
| 2d | 62.1 | 25.4 | 2.4 | 4d | 19.9 | 6.4 | 3.1 |
| DMS | 27.7 | 1.4 | 19.8 | ||||
The 1-deoxysphingolipid derivatives synthesized acted as dual inhibitors against SphK1 and Sphk2, with activities in the micromolar range (Table 1), comparable to those of the sphingosine derivatives previously synthesized in our group.19a As a general trend, compounds 1a–d, 2a–d, 3a–d, and 4a–d demonstrated greater activity against SphK2 compared to SphK1, with selectivity ratios reaching up to 10 for the non-fluorinated dimethyl and guanidinium derivatives 3a and 4a respectively. However, this selectivity did not surpass that of the reference dual inhibitor DMS. The selectivity for sphingosine kinase 2 (IC50SphK1/IC50Sphk2) does not show a consistent correlation with the degree of fluorination in the lipophilic tail across the series of compounds, highlighting the complex nature of interactions within the SphK binding sites.
The least active derivatives for each series towards both SphK isoforms were invariably those with the terminal trifluoromethyl residues (1b, 2b, 3b and 4b). In some cases, the pentafluoroethyl or heptafluoropropyl fragment also had a negative impact on the inhibitory activity (1c, 3c, 3d).
For each headgroup, the most active derivatives towards SphK2 were the native non-fluorinated sphingolipids (a, IC50 = 5.0–20.8 μM) and in most of the cases the heptafluorinated derivatives (1d, IC50 = 18.8 μM; 2d, IC50 = 25.4 μM; 4d, IC50 = 6.4 μM). Within the same degree of fluorination, the inhibitory activity was not very sensitive to the nature of the polar headgroup, and especially against SphK2.
The most active sphingolipid inhibitor against SphK2 was the guanidino derivative 4a (IC50 = 5.0 μM, SphK2/SphK1 selectivity ratio of 10.0), followed by heptafluoro and pentafluoro guanidino derivatives 4d (IC50 = 6.4 μM) and 4c (IC50 = 9.0 μM), and by the non-fluorinated dimethylamino derivative 3a (IC50 = 10.0 μM, SphK2/SphK1 selectivity ratio of 9.2).
The inhibitory activity against SphK1 was, as mentioned above, comparatively lower, and in parallel to the results against SphK2, the best inhibitor is among the guanidino series (4d, IC50 = 19.9 μM), followed by the non-fluorinated free-amino derivative 1a (IC50 = 34.8 μM).
In summary, the compounds that showed the best inhibitory activity against SphK2 (IC50 ≤ 10 μM) were 3a, 4a, 4c, and 4d. Regarding SphK2/SphK1 selectivity, the highest selectivity was observed for 4a (10.0) and 3a (9.2). For SphK1 inhibitory activity, the most active compounds (IC50 < 35 μM) were 4d and 1a.
Docking studies
To better understand the data obtained from the inhibitory activity assays, the potential binding modes of a selected group of synthesized compounds were evaluated by employing a combination of homology modelling and molecular docking. Specifically, we focused on the best SphK2 inhibitors among the products synthesized, the N,N-dimethyl analogue 3a and guanidino derivatives 4a, 4c, and 4d (3a and 4a are also the most selective), and 3b and 4b, for comparative purposes.Ligand flexibility is a key factor in molecular docking, as increased degrees of freedom may compromise exhaustive sampling. For very flexible ligands (more than 20 rotatable bonds), it is necessary to increase sampling parameters.29 In this study, the compounds have moderate flexibility, with up to 15 rotatable bonds and we could observe convergence. However, protein flexibility is not considered, and therefore we interpret binding poses and docking scores with caution. Additionally, as polar interactions drive selective molecular recognition, we focused our analysis on the interactions formed by the polar heads, while the flexible aliphatic tails of the compounds were included in the docking studies to better match the binding pocket's size and shape.
Most amino acids in the binding sites of SphK1 and SphK2 are conserved,19b,30 except for key differences at the throat and toe of the binding channel. In SphK1, Ile174, Met272, and Phe288 are replaced by Val340, Leu553, and Cys569 in SphK2. These substitutions cause structural changes in the binding channel, with bulkier residues in SphK1 narrowing the space, while smaller ones in SphK2 create a wider channel. This also affects the orientation of key residues, such as Arg357 in SphK2, which is more exposed for polar interactions compared to Arg191 in SphK1 at the mouth of the binding site.
 | ||
| Fig. 2 Example of one of the best scored poses in Sphk2 for compound 4a. | ||
First, as a result of the recontouring of SphK2's throat in relation to that of SphK1,30 the triazole ring is able to stabilize the binding through π-stacking interactions with Phe358.
The hydroxyl group at position 3 of the ligand is another common feature that consistently forms a hydrogen bond with Asp344 in all the most active compounds. This interaction pattern stabilizes the compounds’ binding poses and contributes to their lower IC50 values.
Regarding the polar headgroups of these compounds, the N,N-dimethyl head in 3a adopts a linear conformation, facilitating the formation of polar OH-3 interactions. For 4a, 4c and 4d, the guanidino group adopts a bent orientation with respect to the main chain and is thus exposed to polar residues in the mouth of the binding site, forming additional hydrogen bonds with Asp247 (via a primary amine group) and the backbone carbonyl group of Leu549 (via primary and secondary amine groups), further strengthening the interactions and enhancing activity when compared to N,N-dimethyl amino derivative 3a.
Finally, the interaction of the hydrophobic tail with the toe of the binding site varies among the compounds. In 3a and 4a, the tail adopts a J-shaped orientation that fits well into the hydrophobic pocket. For 4c and 4d, the presence of polyfluorinated fragments in the tail provides additional rigidity, enhancing the fit within the channels.
The best-scoring docking poses of the less active compounds 3b and 4b (Fig. 6 in the ESI†) reveal a flipped orientation of the heteroaromatic ring, potentially disrupting π-stacking interactions with Phe358 and resulting in a suboptimal alignment of the hydroxyl hydrogen, which hinders hydrogen bond formation with Asp344. No docking poses were observed with the optimal alignment of this hydrogen atom, which likely contributes to their reduced activity.
It is noteworthy that for compound 3b, some of the best scored poses have the fluorinated tail in the polar part of the pocket.¶ This could explain the unvaryingly higher IC50 value of the trifluoromethyl derivatives in the different series of compounds.
 | ||
| Fig. 3 Example of one of the best scored poses in Sphk1 for compound 4a. | ||
With respect to the heterocyclic moiety, in compounds 3a and 4a, the triazole ring occupies a higher position, closer to the polar head, within the binding cavity of SphK1 compared to SphK2, moving closer to Asp178. This is due to structural differences among the two isoforms (available surface area and shape) in the binding pocket as a result of the presence of Ile174 and Met272 in SphK1, compared to Val340 and Leu553 in SphK2. This altered positioning of the ligands in the throat of the protein channel diminishes interactions with Phe192, which withdraws from the cavity with respect to Phe358 in SphK2, reducing the binding strength compared to that seen in the SphK2 isoform. As the triazole moiety is not anchored by Phe192 in Sphk1, the compounds are more flexible in the kinase binding site and can adopt different binding modes. For 3a, an additional hydrogen bond interaction involving one triazole nitrogen and a structural water molecule is observed (Fig. 7, ESI†). In the trifluoromethylated compound 3b, however, the orientation of the triazole group shifts, leading to a suboptimal alignment of the hydroxyl hydrogen compared to that observed in 3a. For compounds 4c and 4d, the triazole ring shifts higher within the pocket, although it still plays a role in stabilizing the compound's position in the binding site.
With regard to the hydroxyl group at the C3 position, this functionality establishes hydrogen bonds with Ser168 (3a), or with Asp178 (4a, 4c, 4d) helping to stabilize the ligand within SphK1's binding site.
Regarding the participation of the polar headgroups of these compounds in binding interactions, the dimethylamine group in 3a adopts a more folded conformation relative to the main chain compared to that observed in SphK2, thus facilitating the hydrogen bonding involving OH-3 and the triazole ring mentioned above. Meanwhile, the guanidino group in 4a–d forms a two-hydrogen-bond motif with the backbone carbonyl group of Leu268, reinforcing their binding to SphK1 in a similar way to that observed for SphK2. Compared to the Sphk2 binding motif, in Sphk1, the third hydrogen bond with the aspartate amino acid is lost. Thus, the guanidino group interaction could be a key factor in the potency of these compounds.
Lastly, the hydrophobic tail in the non-fluorinated derivative 3a fits well within the hydrophobic toe of the binding site, adopting a different orientation to that of the trifluoromethylated lipophilic tail in 3b. As the degree of fluorination increases in compounds 4c and 4d (see comparative figures in the ESI†), the tail becomes more rigid and fits well into the pocket.
Among the N,N-dimethyl amino series 3, the fact that 3c and 3d are bad inhibitors may be related to the extended conformation of the N,N-dimethylamino moiety (in contrast to the folded conformation of the guanidino group), which extends the fatty tail deeper in the hydrophobic cavity at the toe of the binding site, leading to loss of surface contact for those analogues with exceedingly high bulk, going from 3a to 3d.
Conclusions
In this study a series of 1-deoxysphingosine analogues incorporating a 1,2,3-triazole ring, modifications in the amino group (N-methyl, N,N-dimethyl and guanidino), and differently fluorinated tails have been successfully synthesized as potential sphingosine kinase inhibitors. The anti-2,3-aminoalcohol segment in the target molecules was constructed through diastereoselective nucleophilic addition of an organoalkynylide to L-alaninal derivatives with excellent anti diastereoisomeric ratios from the N,N-dibenzylated compound.The synthesized compounds exhibited dual inhibition of SphK1 and SphK2, with a general trend of higher selectivity towards SphK2. Among the different analogues, the guanidino compounds, particularly heptafluoro derivative 4a, emerged as the most potent inhibitor (IC50 = 5.0 μM), displaying significant selectivity for SphK2 (SphK2/SphK1 selectivity ratio = 10.0). The study also revealed that increasing the degree of fluorination in the lipophilic tail did not always correlate with increased selectivity or potency, underscoring the complexity of the interactions within the SphK binding sites.
The homology model built for Sphk2 shows that differences in amino acid residues between SphK1 and SphK2 at key positions, such as Val340, Leu553, and Cys569 in SphK2 replacing Ile174, Met272, and Phe288 in SphK1, lead to structural changes that affect the contact surface and orientation of key residues. These include Phe192 in SphK1, which protrudes, and Phe358 in SphK2, which buries into the J-channel. Additionally, Arg357 in SphK2 is more exposed than its homolog in SphK1, Arg191.
Docking studies provided valuable insights into the binding modes of the most potent inhibitors, mainly dependent on the polar head group. Thus, the guanidino derivatives showed distinct binding interactions, including hydrogen bonding with key residues such as Asp247 and Leu549 in SphK2 and Leu268 in SphK1. The orientation of the triazole ring, favoring π-stacking interactions with Phe358 in Sphk2, is also a critical factor influencing the optimal binding conformation of the molecules.
As a general trend, the presence of a terminal trifluoromethyl fragment at the hydrophobic tail resulted in detrimental activity within the same series of compounds, whereas compounds with a terminal heptafluoropropyl fragment showed an enhancement of SphK binding probably by rigidifying the end of the lipophilic tail, positioning it to perfectly fit the toe of the binding site.
Overall, this study advances the understanding of sphingosine kinase inhibition, as well as the knowledge of the differential topology of the active pockets of SphK1 and SphK2 and lays additional foundations for the development of novel 1-deoxysphingosine analogues targeting this pathway.
The results presented here suggest opportunities for further studies, particularly exploring modifications to chain length and functionalities, and in this sense, prospective studies are underway. For the time being, however, these results point to the potential use of terminal heptafluoropropyl groups and polar guanidinium heads as structural motifs in designing 1-deoxysphingosine analogues towards improving SphK2 inhibition.
Author contributions
Adrià Cardona: formal analysis, investigation, and methodology. Varbina Ivanova: formal analysis, investigation, and methodology. Raúl Beltrán-Debón: investigation. Xavier Barril: methodology. Sergio Castillón: supervision and funding acquisition. Yolanda Díaz: investigation, methodology, supervision, writing – original draft, and writing – review & editing. M. Isabel Matheu: investigation, methodology, supervision, writing – original draft, writing – review & editing, visualization, project administration, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Grant PID2021-125923OB-I00 funded by Ministerio de Ciencia e Innovación, Spain (MCIN) and Agencia Estatal de Investigación, Spain (AEI), MCIN/AEI/10.13039/501100011033, and by “European Regional Development Fund (ERDF), A way of making Europe”. A.C. acknowledges Universitat Rovira i Virgili, Spain, for a predoctoral fellowship (E-43-2015-0002625). V.I. acknowledges the EU Horizon 2020 MSCA Program for grant agreement 956314 (ALLODD).References
- T. A. Taha, Y. A. Hannun and L. M. Obeid, J. Biochem. Mol. Biol., 2006, 39, 113 CAS.
- (a) M. M. Young, M. Kester and H.-G. Wang, J. Lipid Res., 2013, 54, 5 CrossRef CAS; (b) M. Maceyka, K. B. Harikumar, S. Milstien and S. Spiegel, Trends Cell Biol., 2012, 22, 50 CrossRef CAS PubMed; (c) S. Ponnusamy, M. Meyers-Needham, C. E. Senkal, S. A. Saddoughi, D. Sentelle, S. P. Selvam, A. Salas and B. Ogretmen, Future Oncol., 2010, 6, 1603 CrossRef CAS PubMed; (d) W. Jiang and B. Ogretmen, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2014, 1841, 783 CrossRef CAS PubMed.
- (a) N. C. Hait, C. A. Oskeritzian, S. W. Paugh, S. Milstien and S. Spiegel, Biochim. Biophys. Acta, 2006, 1758, 2016 CrossRef CAS; (b) S. M. Pitson, Trends Biochem. Sci., 2011, 36, 97 CrossRef CAS PubMed.
- D. Hatoum, N. Haddadi, Y. Lin, N. T. Nassif and E. M. McGowan, Oncotarget, 2017, 8, 36898 CrossRef PubMed.
- (a) K. Shirai, T. Kaneshiro, M. Wada, H. Furuya, J. Bielawski, Y. A. Hannun, L. M. Obeid, B. Ogretmen and T. Kawamori, Cancer Prev. Res., 2011, 4, 454 CrossRef CAS; (b) W. Li, C. P. Yu, J. T. Xia, L. Zhang, G. X. Weng, H.-Q. Zheng, Q. L. Kong, L.-J. Hu, M.-S. Zeng, Y.-X. Zeng, M. Li, J. Li and L. B. Song, Clin. Cancer Res., 2009, 15, 1393 CrossRef CAS PubMed; (c) K. R. Johnson, K. Y. Johnson, H. G. Crellin, B. Ogretmen, A. M. Boylan, R. A. Harley and L. M. Obeid, Histochem. Cytochem., 2005, 53, 1159 CrossRef CAS; (d) R. Erez-Roman, R. Pienik and A. H. Futerman, Biochem. Biophys. Res. Commun., 2010, 391, 219 CrossRef CAS PubMed; (e) E. Ruckhäberle, A. Rody, K. Engels, R. Gaetje, G. von Minckwitz, S. Schiffmann, S. Grösch, G. Geisslinger, U. Holtrich, T. Karn and M. Kaufmann, Breast Cancer Res. Treat., 2008, 112, 41 CrossRef; (f) T. Kawamori, T. Kaneshiro, M. Okumura, S. Maalouf, A. Uflacker, J. Bielawski, Y. A. Hannun and L. M. Obeid, FASEB J., 2009, 23, 405 CrossRef CAS.
- (a) C. T. Wallington-Beddoe, J. A. Powell, D. Tong, S. M. Pitson, K. F. Bradstock and L. J. Bendall, Cancer Res., 2014, 74, 2803 CrossRef CAS; (b) S. W. Paugh, B. S. Paugh, M. Rahmani, D. Kapitonov, J. A. Almenara, T. Kordula, S. Milstien, J. K. Adams, R. E. Zipkin, S. Grant and S. A. Spiegel, Blood, 2008, 112, 1382 CrossRef CAS; (c) E. Bonhoure, A. Lauret, D. J. Barnes, C. Martin, B. Malavaud, T. Kohama, J. V. Melo and O. Cuvillier, Leukemia, 2008, 22, 971 CrossRef CAS PubMed.
- (a) W. Li, Z. Tian, H. Qin, N. Li, X. Zhou, J. Li, B. Ni and Z. Ruan, Biochem. Biophys. Res. Commun., 2015, 460, 341 CrossRef CAS; (b) H. Xiong, J. Wang, H. Guan, J. Wu, R. Xu, M. Wang, X. Rong, K. Huang, J. Huang, Q. Liao, Y. Fu and J. Yuan, Oncol. Rep., 2014, 32, 1369 CrossRef CAS PubMed.
- L. Hasanifard, R. Sheervalilou, M. Majidinia and B. Yousefi, J. Cell. Physiol., 2019, 234, 8162 CrossRef CAS PubMed.
- (a) E. Magli, A. Corvino, F. Fiorino, F. Frecentese, E. Perissutti, I. Saccone, V. Santagada, G. Caliendo and B. Severino, Curr. Pharm. Des., 2019, 25, 956 CrossRef CAS PubMed; (b) V. K. R. Tangadanchu, H. Jiang, Y. Yu, T. J. A. Graham, H. Liu, B. E. Rogers, R. Gropler, J. Perlmutter and Z. Tu, Eur. J. Med. Chem., 2020, 206, 112713 CrossRef CAS PubMed; (c) A. Corvino, R. Rosa, G. M. Incisivo, F. Fiorino, F. Frecentese, E. Magli, E. Perissutti, I. Saccone, V. Santagada, G. Cirino, M. A. Riemma, P. A. Temussi, P. Ciciola, R. Bianco, G. Caliendo, F. Roviezzo and B. Severino, Int. J. Mol. Sci., 2017, 18, 2332 CrossRef PubMed; (d) J. Cho, Y. M. Lee, D. Kim and S. Kim, J. Org. Chem., 2009, 74, 3900 CrossRef CAS; (e) J.-W. Kim, Y.-W. Kim, Y. Inagaki, Y.-A. Hwang, S. Mitsutake, Y.-W. Ryu, W. K. Lee, H.-J. Ha, C.-S. Parka and Y. Igarashi, Bioorg. Med. Chem., 2005, 13, 3475 CrossRef CAS; (f) K. Kim, Y.-L. Kim, S. J. Sacket, H.-L. Kim, M. Han, D. S. Park, B. K. Lee, W. K. Lee, H.-J. Ha and D.-S. Im, J. Pharm. Pharmacol., 2007, 59, 1035 CrossRef CAS PubMed.
- M. A. Lone, T. Santos, I. Alecu, L. C. Silva and T. Hornemann, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2019, 1864, 512 CrossRef CAS PubMed.
- R. Cuadros, E. Montejo de Garcini, F. Wandosell, G. Faircloth, J. M. Fernández-Sousa and J. Avila, Cancer Lett., 2000, 152, 23 CrossRef CAS.
- (a) A. Aiello, E. Fattorusso, A. Giordano, M. Menna, C. Navarrete and E. Muñoz, Bioorg. Med. Chem., 2007, 15, 2920 CrossRef CAS PubMed; (b) A. Aiello, E. Fattorusso, A. Giordano, M. Menna, C. Navarrete and E. Muñoz, Tetrahedron, 2009, 65, 4384 CrossRef CAS.
- E. A. Jares-Erijman, C. P. Bapat, A. Lithgow-Bertelloni, K. L. Rinehart and R. Sakai, J. Org. Chem., 1993, 58, 5732 CrossRef CAS.
- C. Jimenez and P. Crews, J. Nat. Prod., 1990, 53, 978 CrossRef CAS.
- H. Symolon, A. Bushnev, Q. Peng, H. Ramaraju, S. G. Mays, J. C. Allegood, S. T. Pruett, M. C. Sullards, D. L. Dillehay, D. C. Liotta and A. H. Merrill Jr., Mol. Cancer Ther., 2011, 10, 648 CrossRef CAS PubMed.
- H. U. Humpf, E.-M. Schmelz, F. I. Meredith, H. Vesper, T. R. Vales, E. Wang, D. S. Menaldino, D. C. Liotta and A. H. Merrill Jr., J. Biol. Chem., 1998, 273, 19060 CrossRef CAS.
- D. S. Menaldino, A. Bushnev, A. Sun, D. C. Liotta, H. Symolon, K. Desai, D. L. Dillehay, Q. Peng, E. Wang, J. Allegood, S. Trotman-Pruett, M. C. Sullards and A. H. Merrill Jr., Pharmacol. Res., 2003, 47, 373 CrossRef CAS PubMed.
- (a) J. Guasch, I. Giménez-Nueno, I. Funes-Ardoiz, M. Bernús, M. I. Matheu, F. Maseras, S. Castillón and Y. Díaz, Chem. – Eur. J., 2018, 24, 4635 CrossRef CAS PubMed; (b) J. Llaveria, A. Beltrán, W. M. C. Sameera, A. Locati, M. M. Díaz-Requejo, M. I. Matheu, S. Castillón, F. Maseras and P. J. Pérez, J. Am. Chem. Soc., 2014, 136, 5342 CrossRef CAS; (c) J. Guasch, Y. Díaz, M. I. Matheu and S. Castillón, Chem. Commun., 2014, 50, 7344 RSC; (d) J. A. Morales-Serna, J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillón, Curr. Org. Chem., 2010, 14, 2483 CrossRef CAS; (e) J. Llaveria, A. Beltrán, M. M. Díaz-Requejo, M. I. Matheu, S. Castillón and P. J. Pérez, Angew. Chem., Int. Ed., 2010, 49, 7092 CrossRef CAS; (f) J. A. Morales-Serna, Y. Díaz, M. I. Matheu and S. Castillón, Synthesis, 2009, 5, 710 Search PubMed; (g) J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillón, Org. Lett., 2009, 11, 205 CrossRef CAS; (h) J. A. Morales-Serna, J. Llaveria, Y. Díaz, M. I. Matheu and S. Castillón, Org. Biomol. Chem., 2008, 6, 4502 RSC; (i) P. Rivero, V. Ivanova, X. Barril, M. Casampere, J. Casas, G. Fabriàs, Y. Díaz and M. I. Matheu, Bioorg. Chem., 2024, 145, 107233 CrossRef CAS PubMed.
- (a) M. Escudero-Casao, A. Cardona, R. Beltrán-Debón, Y. Díaz, M. I. Matheu and S. Castillón, Org. Biomol. Chem., 2018, 16, 7230 RSC; (b) M. Corro-Morón, A. Granell, V. Ivanova, E. Domingo, R. Beltrán-Debón, X. Barril, M.-J. Sanz, M. I. Matheu, S. Castillón and Y. Díaz, Bioorg. Chem., 2022, 121, 105668 CrossRef.
- The dipole moment for 1,4-disubstituted-1,2,3-triazole is 4.18 D. See: J.-L. M. Abboud, C. Foces-Foces, R. Notario, R. E. Trifonov, A. P. Volovodenko, V. A. Ostrovskii, I. Alkorta and J. Elguero, Eur. J. Org. Chem., 2001, 3013 CrossRef CAS.
- (a) M. A. Miller and E. M. Sletten, ChemBioChem, 2020, 21, 3451 CrossRef CAS; (b) M. Bassetto, S. Ferla and F. Pertusati, Future Med. Chem., 2015, 7, 527 CrossRef CAS.
- In relation to the fluorine effect on compounds interacting with hydrophobic fragments of proteins, see: L. Liu, N. Jalili, A. Baergen, S. Ng, J. Bailey, R. Derda and J. S. Klassen, J. Am. Soc. Mass Spectrom., 2014, 25, 751 CrossRef CAS PubMed.
- Incorporating rigid elements into flexible ligands to enhance protein–ligand binding affinity directly influences the entropic component of Gibbs free energy. See: (a) M. Schauperl, M. Podewitz, B. J. Waldner and K. R. Liedl, J. Chem. Theory Comput., 2016, 12, 4600 CrossRef CAS; (b) G. Klebe, F. Dullweber and H.-J. Böhm, Thermodynamic models of drug-receptor interactions: a general introduction, in Drug-Receptor Thermodynamics: Introduction and Applications, John Wiley & Sons, Chichester, UK, 2001, 83–104 Search PubMed; (c) I. D. Kuntz, K. Chen, K. A. Sharp and P. A. Kollman, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9997 CrossRef CAS PubMed.
- M. T. Reetz, M. W. Drewes and A. Schmitz, Angew. Chem., 1987, 99, 1186 CrossRef CAS.
- (a) G. Silveira-Dorta, O. J. Donadel, V. S. Martín and J. M. Padrón, J. Org. Chem., 2014, 79, 6775 CrossRef CAS; (b) G. Silveira-Dorta, I. J. Sousa, M. X. Fernandes, V. S. Martín and J. M. Padrón, Eur. J. Med. Chem., 2015, 96, 308 CrossRef CAS PubMed.
- R. A. da Silva, I. H. S. Estevam and L. W. Bieber, Tetrahedron Lett., 2007, 48, 7680 CrossRef CAS.
- M. A. Ayoub, J. Trebaux, J. Vallaghe, F. Charrier-Savournin, K. Al-Hosaini, A. Gonzalez Moya, J.-P. Pin, K. D. G. Pfleger and E. Trinquet, Front. Endocrinol., 2014, 5, 1 Search PubMed.
- (a) T. A. Klink, K. M. Kleman-Leyer, A. Kopp, T. A. Westermeyer and R. G. Lowery, J. Biomol. Screening, 2008, 13, 476 CrossRef CAS PubMed; (b) Adapta Universal Kinase Assay User Guide. Protocol part PV5099. Rev. 14 February 2008. Invitrogen.
- D. Soler, Y. Westermaier and R. Soliva, J. Comput. Aided Mol. Des., 2019, 33, 613 CrossRef CAS.
- D. R. Adams, S. Tawati, G. Berretta, P. Lopez Rivas, J. Baiget, Z. Jiang, A. Alsfouk, S. P. Mackay, N. J. Pyne and S. Pyne, J. Med. Chem., 2019, 62, 3658 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, additional docking figures and inhibition curves. See DOI: https://doi.org/10.1039/d4ob01656d |
| ‡ These authors contributed equally. |
| § Relevant studies on this topic can be found in ref. 9b–d. |
| ¶ Although the bulkier nature of the terminal trifluoromethyl group compared to a methyl group could favour a priori a better accommodation of the end of the hydrophobic chain in the toe of the J-shaped channel of the enzyme (more expanded for SphK2 than for SphK1),30 the higher polarity of this group could promote interactions with the polar residues at the head of the active site, preventing the compound from entering the J-shaped channel. |
| This journal is © The Royal Society of Chemistry 2025 |