
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric ‘Clip-Cycle’ synthesis of 3-spiropiperidines†
Saikiran
Ravi
,
Christopher J.
Maddocks
,
Ian J. S.
Fairlamb
 *,
William P.
Unsworth
* and
Paul A.
Clarke‡
*,
William P.
Unsworth
* and
Paul A.
Clarke‡
Department of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: ian.fairlamb@york.ac.uk; william.unsworth@york.ac.uk
First published on 20th November 2024
Abstract
3-Spiropiperidines can be synthesized in up to 87% yield and 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 er using a two step ‘Clip-Cycle’ approach. The ‘Clip’ stage of this method is based on efficient and highly E-selective cross metathesis of N-Cbz-protected 1-amino-hex-5-enes with a thioacrylate. This is followed by the ‘Cycle’ step, in which an intramolecular asymmetric aza-Michael cyclization is promoted by a chiral phosphoric acid catalyst.
:4 er using a two step ‘Clip-Cycle’ approach. The ‘Clip’ stage of this method is based on efficient and highly E-selective cross metathesis of N-Cbz-protected 1-amino-hex-5-enes with a thioacrylate. This is followed by the ‘Cycle’ step, in which an intramolecular asymmetric aza-Michael cyclization is promoted by a chiral phosphoric acid catalyst.
Introduction
Nitrogen-containing heterocycles are privileged structures in medicinal chemistry and present in a high percentage of FDA-approved pharmaceuticals.1 Of these nitrogen-containing heterocycles, piperidine is the most abundant. Piperidines are common in biologically-active natural products and drugs,2 making them a regular target for synthesis.3 Spirocyclic piperidines are also of wide interest, being found in an array of natural product structures, and recognised as desirable motifs in medicinal chemistry (Fig. 1).4 This interest is part of a broader drive to explore biologically active molecules with greater 3-dimensionality, as molecules with high ‘3D character’ tend to occupy areas of chemical space that have been less well-explored (traditionally) in drug discovery.5,6 The advantages of such spirocyclic systems are that they are conformationally rigid and position hydrogen-bonding donor and acceptor atoms at well-defined positions for interaction with suitable protein receptors.5 They also offer the opportunity to develop novel intellectual property, through a richer diversification in chemical structure, beyond simple carbocyclic systems.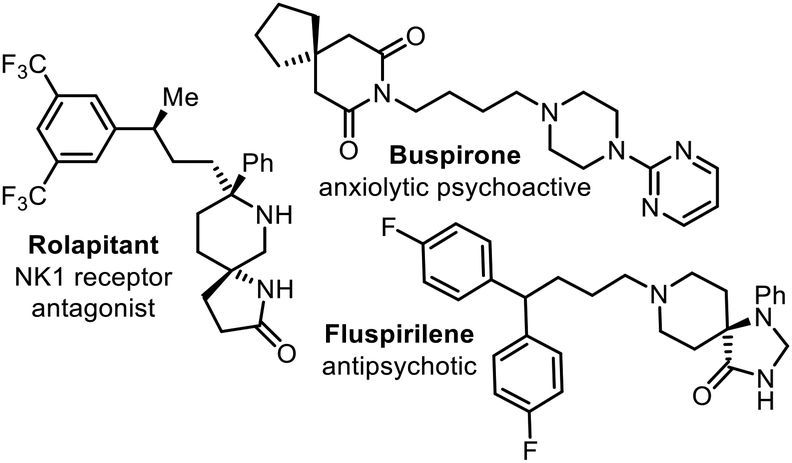 | ||
| Fig. 1 Marketed spiropiperidine-containing drugs. | ||
Contemporaneous with our work on the synthesis of racemic 2-spiropiperidines,7 a review article was published summarising methods for the synthesis of spiropiperidines and describing their use in pharmaceutical molecules.4a This review highlighted 3-spiropiperidines as being relatively underrepresented structures, while 4-spiropiperidines are the most frequently synthesized in medicinal chemistry research programs.4a
Thus, we decided to explore the development of a general strategy for the asymmetric synthesis of 3-spiropiperidines, based on our recently reported asymmetric ‘Clip-Cycle’ synthesis of pyrrolidines and tetrahydropyrans.8 The ‘Clip-Cycle’ approach had not been used to prepare piperidines prior to this study. We envisaged a synthesis based on “Clipping” a N-protected 1-amino-hex-5-ene with a thioacrylate via an alkene metathesis reaction, followed by the ‘Cycle’ step, where the N-protected amine undergoes aza-Michael cyclization catalysed by a chiral phosphoric acid (CPA) to yield enantioenriched piperidines (Scheme 1). Advantages of this ‘Clip-Cycle’ approach are that both the metathesis and the aza-Michael reactions are catalytic, it enables the straightforward synthesis of a range of functionalized piperidines by changing the aminoalkane reagent, and that the thioester can be transformed into a variety of other functional groups under mild conditions post cyclisation. The successful realisation of this strategy is described herein, demonstrated by the enantioselective synthesis of ten thioester-containing 3-spiropiperidines, formed in yields up to 87%, with enantiomeric ratios up to 97:3.
 | ||
| Scheme 1 Asymmetric ‘Clip-Cycle’ synthesis of 3-spiropiperidines. | ||
Results and discussion
Initial optimisation studies focused on the N-Cbz-2-spirocyclohexylhex-5-ene substrate 1a as a model for the formation of 3-spirocyclicpiperidines 4a–c (Table 1). The p-tolyl thioester 2a was chosen as the ‘Clip’-partner, as it had been applied successfully in pyrrolidine formation.8a,b For all of the optimisation studies, the ‘Clip’ product 3a–c was first prepared using an alkene cross-metathesis protocol, using the Hoveyda–Grubbs Catalyst™ 2nd generation (reaction conditions are described in detail later; vide infraScheme 3). The yields and er in Table 1 relate to the ‘cycle’ step, i.e. the conversion of alkenes 3a–c into piperidines 4a–c.|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | CPA | Solvent/Temp (°C) | Yield (%) | erb |
| The reactions were run for 24 h with 20 mol% of the specified CPA catalyst in the specified solvent (0.02 M) unless stated.a Run for 48 h.b Determined by chiral stationary phase HPLC (see ESI† for details). | |||||
| 1 | 3a | 5a | Cyclohexane/80 | 10 | 96:4 |
| 2a | 3a | 5a | Cyclohexane/80 | 21 | 96:4 |
| 3 | 3a | 5a | Octane/100 | 36 | 94:6 |
| 4 | 3b | 5a | Cyclohexane/80 | 20 | 92:8 |
| 5a | 3b | 5a | Cyclohexane/80 | 21 | 84:16 |
| 6 | 3b | 5a | Octane/100 | 55 | 89:11 |
| 7 | 3c | 5a | Octane/100 | 48 | 97:3 |
| 8 | 3c | 5a | Octane/110 | 63 | 95:5 |
| 9 | 3c | 5b | Octane/100 | 17 | 38:62 |
| 10 | 3c | 5c | Octane/100 | 67 | 92:8 |
| 11 | 3c | 5d | Octane/100 | 21 | 62:38 |
| 12 | 3a | 5c | Octane/100 | 80 | 93:7 |
| 13 | 3a | 5c | Cyclohexane/80 | 78 | 96:4 |

The reaction conditions identified as being successful for pyrrolidine synthesis previously8a were examined first (Table 1, entries 1 and 2). It was initially disappointing to discover that the yield of the piperidine 4a was low. Under these conditions, the cyclisation step was sluggish, limiting overall product formation due to low conversion. Increasing the temperature to 100 °C did not result in a substantial improvement (entry 3), although it was encouraging that high enantioselectivities were observed at these elevated temperatures. To help drive the cyclisation reaction to completion, the aryl group of the thioester was changed to a p-nitrophenyl group (2b), with the idea being to increase the electrophilicity of the Michael-accepting motif. However, this structural change made little difference at 80 °C (entry 4). Extending the reaction time to 48 h (entry 5) resulted in an erosion in the enantiomeric ratio of piperidine 4b and little change in product formation. When the reaction was run in octane at 100 °C, the reaction progressed further, and the yield of 4b increased to 55% at the expense of the enantioselectivity (entry 6). We reasoned that the reduction in enantioselectivity at higher temperatures could be mitigated by using a bulkier thioester. Mesityl thioester 2c was therefore investigated, and both the yield and the enantioselectivity increased using this substrate (cf. entries 3 and 7). Increasing the temperature further resulted in greater yields and only a slight drop in enantioselectivity (entry 8).
The next factor to be examined was the choice of chiral phosphoric acid (CPA) catalyst. Studies had initially employed only (R)-TRIP 5a as the catalyst. The use of (R)-TiPSY 5b, and (R)-phenanth 5d both resulted in a reduction in enantioselectivity and yields. Although interestingly, the use of 5b resulted in the opposite enantiomer being formed as the major product (entries 9 and 11). Catalyst (R)-anthra 5c, was the most effective, forming piperidine 4c in the highest yield so far of 67% and with excellent enantioselectivity 92:8 (entry 10). Subjecting the original test substrate 3a to these reaction conditions with 5c afforded 4a in 80% yield and with an enantiomeric ratio of 93:7. Finally, the temperature was reduced to 80 °C to see if the conversion/yield and enantioselectivity could be conserved under these milder reaction conditions; this was indeed the case, with 4a being formed in 78% yield with an enantiomeric ratio of 96:4. Note that Table 1 shows selected screening results only; for a more complete summary of the optimisation studies, see ESI Tables S1–S6.†
Next, attention turned to examining the scope of the reaction. To enable this, a series of N-Cbz-protected 1-amino-hex-5-ene substrates (1a,d–k) was prepared, starting from nitriles 6 using the three-step protocol summarised in Scheme 2. Full synthetic and characterisation details for all steps and products formed are described in the ESI.†
 | ||
| Scheme 2 Synthesis of N-Cbz-protected 1-amino-hex-5-ene starting materials. | ||
The resulting alkenes (1a,d–k) were then reacted with thioacrylate 2 in a cross metathesis reaction with the Hoveyda–Grubbs Catalyst™ 2nd generation, in the ‘Clip’ phase of the overall ‘Clip-Cycle’. Based on the optimisation results (Table 1) the p-toluene substituted thioester 2a was chosen to take forward into the substrate scoping studies; products 3b and 3c, used in the earlier optimisation study, are also shown for completeness. The expected products 3a–j were formed in 43–91% yields, as a single E-isomer in all cases (Scheme 3).
 | ||
| Scheme 3 ‘Clipping’ 1-amino-hex-5-enes 1 with thioacrylates 2 using cross metathesis. | ||
Attention then turned to the ‘Cycle’ phase, using the optimised conditions for cyclisation (Table 1, entry 13). Substrates containing a carbocyclic spirocycle all worked well; homologues 4a–d were each formed in good yield and er using the standard protocol (Scheme 4). To test whether the carbocycle provides a benefit to cyclisation, a dimethylated substrate 3g was also tested for comparison. In this case the isolated yield was much lower (29%), although notably the er (95:5) remained high. Larger carbocyclic product 4h and thioether spirocycle 4j were each formed in lower yield also, again with the er high. Cyclic ether containing spirocycle 4i was formed in much better yield, with the er also high. In the three lower yielding examples (4g,h,j) the majority of the mass balance was accounted for by unreacted starting material. As the er was high in all three cases, we expect that further optimisation would allow these products to be isolated in higher yield, by optimising the conditions to increase the reaction conversion (e.g. using conditions similar to those in Table 1, entry 12).§
 | ||
| Scheme 4 Asymmetric cyclisation of alkenes 3 to form enantioenriched piperidines 4. Enantiomeric ratios were determined by chiral stationary phase HPLC (see ESI† for details). | ||
To assign the absolute stereochemistry of the products formed, the ‘Clip-Cycle’ approach was used to prepare piperidine 4k without a spirocyclic moiety. The yield was low in this case, but the er was found to be in line with the spirocyclic derivatives; attempts to improve the outcome of this reaction using other CPA catalysts were unsuccessful (see ESI, Table S6†). Hydrolysis to carboxylic acid 8 enabled assignment of the S-stereochemistry shown, by measuring its optical rotation and comparing to literature data for the known R-enantiomer.9 This allowed for the assignment of the absolute stereochemistry of all other substrates described in this manuscript, by analogy (Scheme 5). The major enantiomer formed was the same as that produced in the preceding pyrrolidine- and tetrahydropyran-forming variants of the method, and hence the enantioselectivity can be explained using the models previously established.8a,c
 | ||
| Scheme 5 ‘Clip-Cycle’ synthesis of 4k and hydrolysis to 8 to confirm the absolute stereochemistry and the unsuccessful attempted synthesis of 2-spiropiperidine 4l. | ||
We also attempted the synthesis of 2-spiropiperidine 4l using the ‘Clip-Cycle’ approach. In this case, the ‘Clip’ step proceeded as expected, with alkenes 1l and 2a undergoing efficient cross metathesis to form 3l in good yield. However, 3l did not react under the optimised cyclisation conditions, likely due to the increase in steric bulk adjacent to the protected amine inhibiting the aza-Michael step; unreacted 3l was recovered.
Conclusion
In summary, the efficacy of the ‘Clip-Cycle’ approach for 3-spiropiperidine synthesis has been validated, with ten 3-spiropiperidine products generated in yields up to 87% yield. The ‘Clip’ step proceeds smoothly, with the Hoveyda–Grubbs Catalyst™ 2nd generation used to promote cross metathesis of N-Cbz-protected 1-amino-hex-5-enes with thioacrylates. The ‘Cycle’ step is best promoted by a chiral phosphoric acid catalyst (R)-anthra 5c, to afford the 3-spiropiperidine products in up to 97:3 er. The synthesis of a non-spirocyclic piperidine was also completed, to enable the assignment of absolute stereochemistry.
This study represents the first report of the ‘Clip-Cycle’ method being used to prepare piperidines. Having validated its efficacy for 3-spiropiperidine synthesis, we anticipate that similar ‘Clip-Cycle’ strategies will also allow the preparation of a much wider range of functionalised, biologically important piperidines.§
Author contributions
Synthetic studies were done by S.R and C.J.M. The project was conceived, designed and led by P.A.C. The paper was written by W.P.U. and I.J.S.F., with contributions from S.R.Data availability
The data supporting this article have been included as part of the Supplementary Information ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of York and the Wild Fund for supporting the PhD studentship of S. R. and the Royal Society for an Industry Fellowship (I. J. S. F. 2021–2025).References
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) M. Prashad, H.-Y. Kim, Y. Lu, D. Har, O. Repic, T. J. Blacklock and P. Giannousis, J. Org. Chem., 1999, 64, 1750–1753 CrossRef CAS PubMed; (b) N. Shankaraiah, R. A. Pilli and L. S. Santos, Tetrahedron Lett., 2008, 49, 5098–5100 CrossRef CAS; (c) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed; (d) X.-N. Cao, X.-M. Wan, F.-L. Yang, K. Li, X.-Q. Hao, T. Shao, X. Zhu and M.-P. Song, J. Org. Chem., 2018, 83, 3657–3668 CrossRef CAS PubMed; (e) S. P. Chavan, D. B. Kalbhor and R. G. Gonnade, Tetrahedron, 2021, 80, 131773 CrossRef CAS; (f) J. C. Anderson, E. Bouvier-Israel, C. D. Rundell and X. Zhang, Tetrahedron, 2021, 78, 131821 CrossRef CAS.
- (a) P. S. Watson, B. Jiang and B. Scott, Org. Lett., 2000, 2, 3679–3681 CrossRef CAS PubMed; (b) L. Zhou, D. W. Tay, J. Chen, G. Y. C. Leung and Y.-Y. Yeung, Chem. Commun., 2013, 49, 4412–4414 RSC; (c) P. A. Clarke, A. V. Zaytzev and A. C. Whitwood, Tetrahedron Lett., 2007, 48, 5209–5212 CrossRef CAS; (d) P. A. Clarke, A. V. Zaytsev, T. W. Morgan, A. C. Whitwood and C. Wilson, Org. Lett., 2008, 10, 2877–2880 CrossRef CAS PubMed; (e) M. A. Larsen, E. T. Hennessy, M. C. Deem, Y. H. Lam, J. Saurí and A. C. Sather, J. Am. Chem. Soc., 2020, 142, 726–732 CrossRef CAS PubMed; (f) J. Garcia, J. Eichwald, J. Zesiger and T. K. Beng, RSC Adv., 2022, 12, 309–318 RSC; (g) S. Fustero, S. Monteagudo, M. Sánchez-Roselló, S. Flores, P. Barrio and C. del Pozo, Chem. – Eur. J., 2010, 16, 9835–9845 CrossRef CAS PubMed; (h) R. R. Mittapalli, S. J. J. Guesné, R. J. Parker, W. T. Klooster, S. J. Coles, J. Skidmore and A. P. Dobbs, Org. Lett., 2019, 21, 350–355 CrossRef CAS PubMed; (i) H. Liu, D. Su, G. Cheng, J. Xu, X. Wang and Y. Hu, Org. Biomol. Chem., 2010, 8, 1899–1904 RSC; (j) S. G. Davies, A. M. Fletcher, J. A. Lee, P. M. Roberts, A. J. Russell, R. J. Taylor, A. D. Thomson and J. E. Thomson, Org. Lett., 2012, 14, 1672–1675 CrossRef CAS PubMed; (k) H. Kawabata, T. Hirama, T. Yanagisawa, K. Sato, N. Kogure, M. Kitajima and H. Takayama, Org. Lett., 2019, 21, 7982–7986 CrossRef CAS PubMed; (l) E. C. Carlson, L. K. Rathbone, H. Yang, N. D. Collett and R. G. Carter, J. Org. Chem., 2008, 73, 5155–5158 CrossRef CAS PubMed.
- (a) S. D. Griggs, N. Thompson, D. T. Tape and P. A. Clarke, Org. Biomol. Chem., 2018, 16, 6620 RSC; (b) T. M. McQueen and S. D. Griggs, Tetrahedron Lett., 2021, 65, 152752 CrossRef CAS; (c) L. Zhou, F. Yuan, Y. Zhou, W. Duan, M. Zhang, H. Deng and L. Song, Tetrahedron, 2018, 74, 3761–3769 CrossRef CAS; (d) A. A. Peshkov, A. A. Peshkov, A. Makhmet, O. Bakulina, E. Kanov, R. Gainetdinov, V. A. Peshkov, M. Krasavin and L. N. Gumilyov, Synthesis, 2022, 54, 2604–2615 CrossRef CAS; (e) A. A. Peshkov, O. Bakulina, D. Dar'in, G. Kantin, A. Bannykh, V. A. Peshkov and M. Krasavin, Eur. J. Org. Chem., 2021, 1726–1731 CrossRef CAS; (f) L. A. Martinez-Alsina, J. C. Murray, L. M. Buzon, M. W. Bundesmann, J. M. Young and B. T. O'Neill, J. Org. Chem., 2017, 82, 12246–12256 CrossRef CAS PubMed; (g) M.-C. Yang, C. Peng, H. Huang, L. Yang, X.-H. He, W. Huang, H.-L. Cui, G. He and B. Han, Org. Lett., 2017, 19, 6752–6755 CrossRef CAS PubMed; (h) H. E. Askey, J. D. Grayson, J. D. Tibbetts, J. C. Turner-Dore, J. M. Holmes, G. Kociok-Kohn, G. L. Wrigley and A. J. Cresswell, J. Am. Chem. Soc., 2021, 143, 15936–15945 CrossRef CAS PubMed.
- For perspective on the exploration of 3D space in medicinal chemistry, see: (a) E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257 CrossRef CAS PubMed; (b) A. W. Hung, A. Ramek, Y. Wang, T. Kaya, J. A. Wilson, P. A. Clemons and D. W. Young, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6799 CrossRef CAS PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed; (d) W.-Y. Siau and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 17726 CrossRef CAS PubMed; (e) K. B. Sippy, D. J. Anderson, W. H. Bunnelle, C. W. Hutchins and M. R. Schrimpf, Bioorg. Med. Chem. Lett., 2009, 19, 1682 CrossRef CAS PubMed.
- For the synthesis and properties of bioactive spirocycles, see: (a) K. Heisinger, D. Dar'in, E. Proschak and M. Krasavin, J. Med. Chem., 2021, 64, 150–183 CrossRef PubMed; (b) S. J. Chambers, G. Coulthard, W. P. Unsworth, P. O'Brien and R. J. K. Taylor, Chem. – Eur. J., 2016, 22, 6496 CrossRef CAS PubMed; (c) A. Ding, M. Meazza, H. Guo, J. W. Yang and R. Rios, Chem. Soc. Rev., 2018, 47, 5946–5996 RSC.
- S. D. Griggs, N. Thompson, D. T. Tape, M. Fabre and P. A. Clarke, Org. Biomol. Chem., 2018, 16, 6663–6674 RSC.
- (a) C. J. Maddocks, K. Ermanis and P. A. Clarke, Org. Lett., 2020, 22, 8116–8121 CrossRef CAS PubMed; (b) C. J. Maddocks and P. A. Clarke, Tetrahedron, 2021, 78, 131789 CrossRef CAS; (c) K. Alomari, N. S. P. Chakravarthy, B. Duchadeau, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2022, 20, 1181–1185 RSC.
- E. C. Carlson, L. K. Rathbone, H. Yang, N. D. Collett and R. G. Carter, J. Org. Chem., 2008, 73, 5155–5158 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01608d |
| ‡ Deceased. |
| § Unfortunately, the development of the ‘Clip-Cycle’ approach will not continue in York as Prof Paul A. Clarke passed away in November 2023. This manuscript is therefore presented for the scientific record, and to disclose value of this new piperidine forming method. Other researchers interested in continuing to study ‘Clip-Cycle’ reactivity based on the results described herein are strongly encouraged. |
| This journal is © The Royal Society of Chemistry 2025 |