DOI:
10.1039/D4OB01589D
(Review Article)
Org. Biomol. Chem., 2025,
23, 1479-1532
Nitrogen-bridgehead compounds: overview, synthesis, and outlook on applications
Received
1st October 2024
, Accepted 11th November 2024
First published on 3rd December 2024
Abstract
The nitrogen-bridgehead is a common structural motif present in a multitude of natural products. As many of these abundant compounds exhibit biological activities, e.g. against cancer or bacteria, these derivatives are of high interest. While natural products are often associated with problematic characteristics, such as elaborate separation processes, high molecular complexity and limited room for derivatization, purely synthetic approaches can overcome these challenges. Many synthetic procedures have been reported for preparation of artificial nitrogen bridgehead compounds, however, to our surprise only a fraction of these has been tested for their bioactivity. This review is therefore meant to give an overview of existing synthetic methods that provide scaffolds containing bridgehead nitrogen atoms, covering the period from 2000 to 2023. Reviews which cover subunits of this topic are referenced as well.
1 Introduction
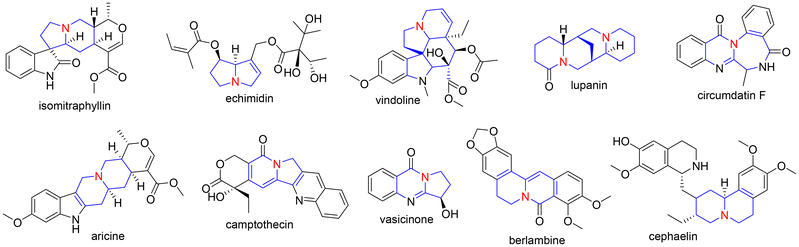
Natural products, the first medicines known to man, have been used to treat many different diseases.1 Despite advances in organic synthesis methodology, drug discovery research continues to focus on natural products for, as examples, antiresorptive activity,2 hypertension,3 and, currently, treatment of coronavirus infection.4 Therefore, it is not surprising that almost half of drugs used today are based on natural products,5 a selection of which are shown in Fig. 1. While this compound pool offers a broad variety of structures, natural products remain challenging to extract and purify,6–8 structurally complex (which can make identification of active substructures difficult)9 and difficult to modify.10,11 Total synthesis or synthesis of core structures can address these limitations,12 especially as a step-by-step build of the desired motif can give access to additional simplified derivatives. Nitrogen atoms are common in natural products and their derivatives (Fig. 1, marked in red). In fact, 59% of FDA approved drugs contain at least one nitrogen atom.13 Many of the compounds depicted in Fig. 1 bear another interesting feature in their scaffold: a bridgehead nitrogen, two rings connected by a nitrogen atom (Fig. 1, marked in blue). As all of these compounds exhibit bioactivity, the N-bridgehead, which all have in common, can be considered a key feature for their activity.14,15 A synthetic chemist would rightly assume that the compounds shown in Fig. 1 are natural alkaloids or semi-synthetic derivatives thereof, because installation of a bridgehead nitrogen is synthetically challenging. For this reason, to access this important source of structural complexity, chemists have had to rely on naturally occurring structures.
 |
| | Fig. 1 Natural products that have a bridgehead nitrogen and are used as drugs. | |
Synthetic access to the full array of complexity associated with the bridgehead nitrogen would offer an expanded structural ‘pool’ from which new molecules could be drawn and would allow researchers to overcome previously listed drawbacks of the natural pool. The ability to build N-bridgehead scaffolds from scratch would allow synthesis of compound families around new structural motifs, which would be crucial for structure relationship investigations, including, for example, optimization of bioactivity, toxicity and ADME properties. Optimization of these properties will require robust synthetic methods, which will give access to both broad structural diversity and the selectivity required to isolate material with high isomeric purity and high ee and dr values. Ready access to additional structural diversity will be beneficial in areas of drug discovery research, such as cancer treatment, where there is an urgent need for new molecules with enhanced bioactivity, especially against special cancer types and resistant forms and with decreased side effects.
Keeping these challenges in mind, we summarized the synthetic work toward compounds bearing a nitrogen bridgehead during the period of 2000–2022. As the scope of this review is extensive, in our schemes, we summarized the key step for N-bridgehead formation for each reaction. Many elegant approaches toward N-bridgehead compounds have been developed and reported, but to our surprise relatively few of these have included evaluation of bioactivity. We hope that this review will serve as both an overview of the many exciting methods available for the synthesis of a vast array of N-bridgehead compounds and as an opportunity for researchers to identify promising scaffolds that have yet to be investigated for their potential bioactivity (Scheme 1).
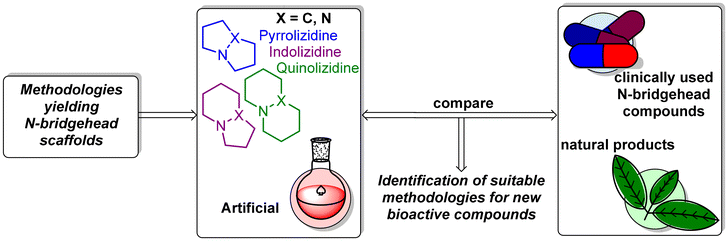 |
| | Scheme 1 Scope and goals of the review. | |
2. Pyrrolizidines (5,5 ring; 1-azabicyclo[3.3.0]-octane)
In the first part of the review, pyrrolizidines are discussed. The pyrrolizidine motif is a commonly found element in natural products, such as pyrrolizidine alkaloids, which are secondary metabolites of plants.16 The biological activities of many different alkaloids of this type have been investigated, and their importance is underlined by the numerous different approaches to their synthesis.17–20 It should be noted that many pyrrolizidines, especially 1,2-unsaturated pyrrolizidines, are toxic to humans, therefore much research has focused on their exclusion from food.21,22
Pyrrolizidines have a multitude of different applications as bioactive compounds.23,24 Examples are shown in Fig. 2. The N-oxide of indicine was tested for its activity against Leukemia in two different publications.25 In the first study, indicine N-oxide was used for the treatment of twenty-two patients with acute lymphoblastic leukemia, while the second study describes a phase II treatment of thirty-one patients. Retronecin and three other natural products were tested against Escherichia coli and Penicillium chrysogenum.26 Retronecin exhibited pronounced activity against both microorganisms. 7,7a-diepialexine is a natural product isolated from Alexa leiopetala.27 It was tested for its inhibition of human immunodeficiency virus (HIV) growth, where it restricted viral replication with an IC50 value of 0.38 mM. This correlates with inhibitory activity against purified pig kidney-glucosidase 1 of the glycoprotein processing enzymes and the reduced cleavage of the precursor HIV-1 glycoprotein gp160. Pyrrolizidines like heliotrine, which can be isolated from the seeds of Heliotropium indicum, exhibited ganglion blocking activity.28 As one of four different natural products, europine inhibited the production of nitric oxide in lipopolysaccharide induced murine macrophages RAW 264.7 cells with an IC50 value of 7.9 μM.29 Australine is a polyhydroxylated pyrrolizidine with anti-HIV activity.30 The activity of glycosidases, particularly the nitrogen-linked glycosylation process of HIV, was greatly inhibited at concentrations between 0.1 and 10 mM. Heliosupine is an acetylcholinesterase (AchE) inhibitor with an IC50 value below 0.60 mM. AchE is an enzyme that catalyses the hydrolysis of esters that act as neurotransmitters.31,32 While overstimulation of acetylcholine receptors is related to disorders like depression, under stimulation can cause other diseases, such as Alzheimer's and Myasthenia gravis.
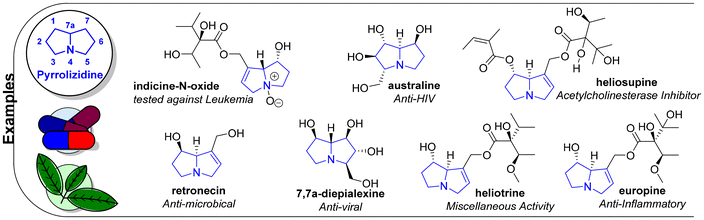 |
| | Fig. 2 Examples of pyrrolizidine-derived compounds. | |
Over the last 20 years, many synthetic pyrrolizidines have been prepared. To our surprise and to the best of our knowledge, relatively few of the reported compounds have been tested for biological activity. Because many of these derivatives are not 1,2-unsaturated, there may be a potential non-toxic bioactive application.
2.1. Free radical and photochemical reactions
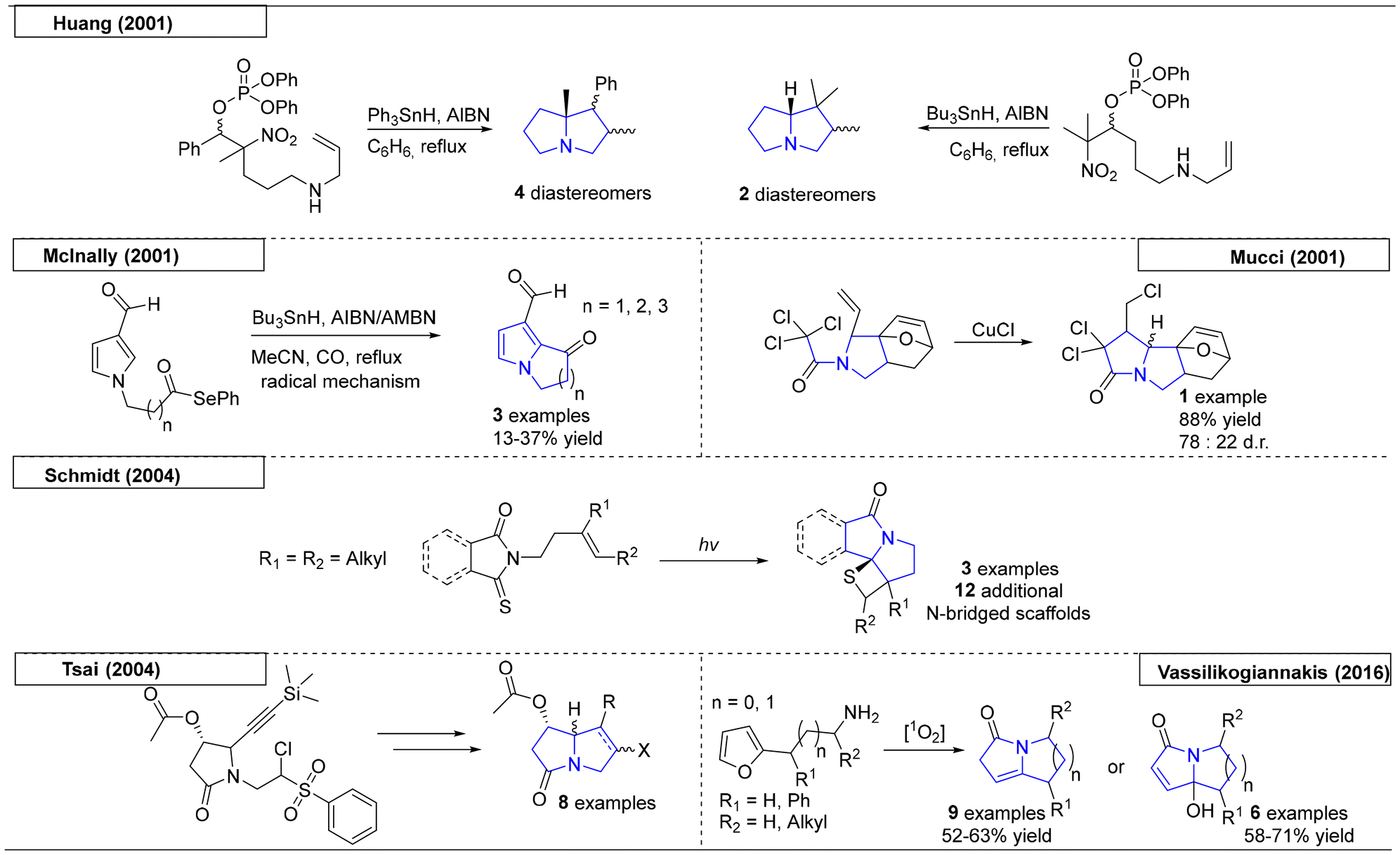
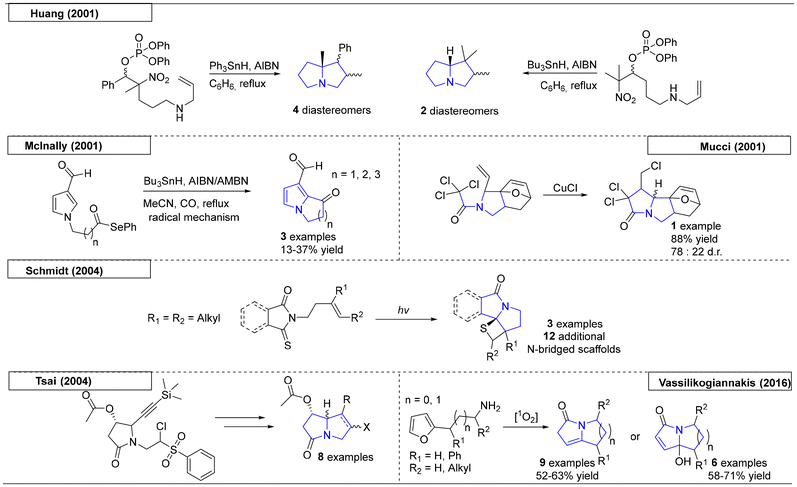
In the early 2000s, several different free radical approaches were reported to yield the desired pyrrolizidine scaffolds (Scheme 2). The first example, which was reported by the Huang research group utilized a phosphorous-derived homolytic cleavage.33 They disclosed the synthesis of substituted N-bridgehead heterocycles from β-phosphatoxy nitroalkanes on treatment with triphenyltin hydride (Ph3SnH) or tributyltin hydride (Bu3SnH) and azobisisobutyronitrile (AIBN). The tributyltin hydride reaction took advantage of the Thorpe–Ingold effect34 to provide the bicyclic product. The first bridgehead compound was isolated as a mixture of four diastereomers (ratio 2.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.6:1:1), with the major diastereomer being in trans-configuration about the newly formed bond. The second pyrrolizidine has only two diastereomers (isolated in 1:1 ratio) but has limited utility for drug discovery, because of the constraints of further derivatisation.
:1.6:1:1), with the major diastereomer being in trans-configuration about the newly formed bond. The second pyrrolizidine has only two diastereomers (isolated in 1:1 ratio) but has limited utility for drug discovery, because of the constraints of further derivatisation.
 |
| | Scheme 2 Free radical and photochemical approaches to pyrrolizidines. | |
Another example, published by McInally and coworkers,35 utilized an acyl selenium moiety to provide the initial free radical via homolytic cleavage (Scheme 2). Reaction with AIBN or azobismethylisobutyronitrile (AMBN) and Bu3SnH afforded a pyrrole ring fused to a 5-, 6- or 7-membered ring. Mechanistic studies indicated that the AIBN and AMBN act as radical initiators as well as oxidants of intermediate π-radicals to provide the final aromatic product. In the course of exploring a novel [4 + 2] cycloaddition reaction, the Mucci research group exploited a copper chloride-initiated free radical reaction to form a pyrrolizidine scaffold. Because the scope of this study was limited, only one pyrrolizidine derivative was reported.36
An elegant free radical approach reported by the Schmidt research group used UV light to form the di-radical that was involved in the subsequent cyclization (Scheme 2).37 Variations of the starting materials and post-cyclization modifications gave access to twelve additional N-bridgehead scaffolds that spanned from indolizidines to 5,7-fused ring systems. In 2004, another free radical approach was reported by the research group of Tsai.38 Starting from a silyl-protected alkyne precursor, AIBN and Bu3SnH were used to obtain the desired pyrrolizidine scaffold with excellent stereoselectivity. The paper contains eight pyrrolizidine scaffolds, including the naturally occurring derivatives (+)-heliotridine and (−)-retronecine. The last example of a free radical pyrrolizidine synthesis was reported in 2016 by Vassilikogiannakis and coworkers.39 This methodology employed readily accessible alkyl furan precursors with photooxygenation conditions, which transformed the starting materials into pyrrolizidine and indolizidine scaffolds. The first step involved the addition of singlet oxygen, which facilitated the furan ring opening and conversion toward pyrrolizidines.
2.2. Cycloaddition reactions
2.2.1 1,3-Dipolar cycloadditions.
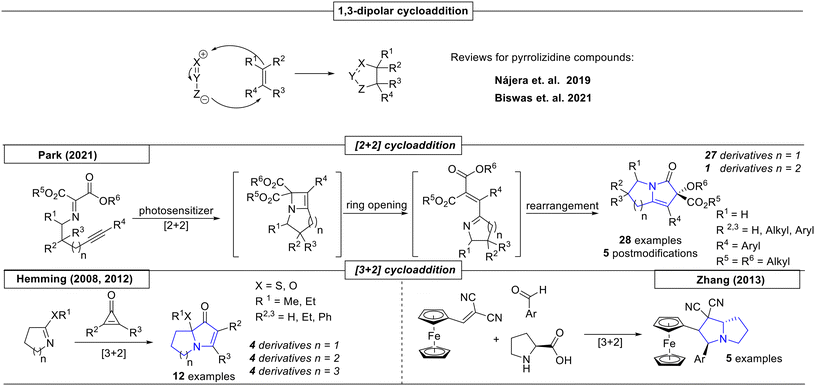
One of the most common ways to synthesize artificial pyrrolizidines is the 1,3-dipolar cycloaddition reaction (Scheme 3). Since these approaches have already been summarized in excellent recent reviews by Nájera et al.40 and Biswas et al.,41 they are not reviewed here.
 |
| | Scheme 3 Cycloaddition reactions that produce pyrrolizidines. | |
2.2.2 [2 + 2] cycloadditions.
In 2021, Park and coworkers reported a reaction sequence involving an imine alkyne [2 + 2] cycloaddition as a key-step (Scheme 3),42 which forms a four membered ring that undergoes ring opening. Visible light photochemical conditions were used to facilitate the following rearrangement, which yielded twenty-seven different pyrrolizidine derivatives. Additionally, a [4 + 2] cycloaddition was reported, which gave access to twenty different dihydro-1,4-oxazine derivatives.
2.2.3 [3 + 2] cycloadditions.
The Hemming research group reported a novel formal [3 + 2] cycloaddition reaction toward highly functionalized pyrrolizidines, indolizidines and 5,7-fused ring systems, depending on the ring size associated with the imine precursor (Scheme 3).43 In a subsequent publication, nine additional derivatives were reported by the same research group.44 In 2013, Zhang and coworkers reported a novel [3 + 2] cycloaddition of a ferrocene-derived dicyanoolefin with benzaldehyde-proline condensates, utilizing L-proline as catalyist.45 All five examples were pyrrolizidine-ferrocene hybrid compounds.
2.3 Palladium-catalysed reactions
Palladium catalysed carbon–nitrogen cross coupling reactions, among them the Buchwald–Hartwig reaction, are useful tools for the synthesis of highly substituted amines.46 A number of approaches have exploited this technique to obtain pyrrolizidine structures. In addition, various intramolecular 1,2-aminoalkylation reactions have been developed, which will be discussed in the following section. In 2003, Doye and coworkers reported the synthesis of six different pyrrolizidine derivatives starting from readily available di-halogenated benzenes (Scheme 4).47 The amine-derived precursors used for the palladium-catalysed cyclization were prepared from the corresponding alkyne derivatives via a procedure that involved hydroamination followed by in situ reduction.
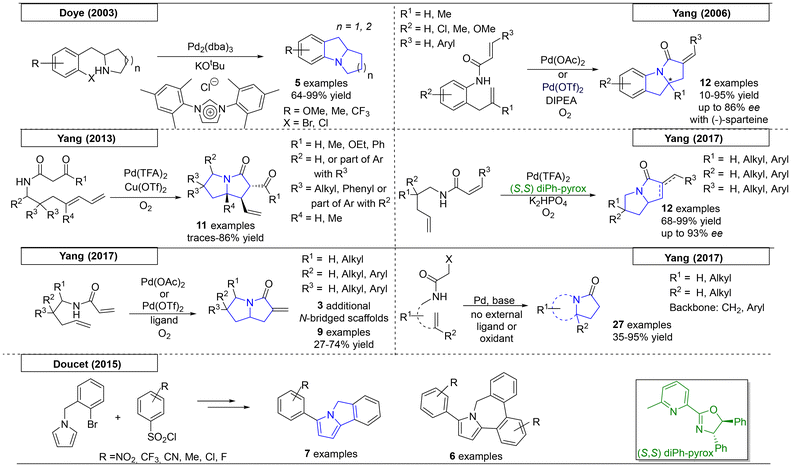 |
| | Scheme 4 Palladium-catalysed reactions that produce pyrrolizidines. | |
The Yang research group has been responsible for much research in this area. In 2006, they reported a novel oxidative tandem reaction that employed palladium acetate or palladium triflate as a catalyst and provided twelve examples of pyrrolizidines (Scheme 4).48 The formation of both rings was enabled by the palladium catalyst. By adding a chiral ligand to the system, (−)-sparteine was synthesized using this approach with an enantiomeric excess of up to 86%. In 2013, a 1,3-diketo-pyrrolizidine scaffold example was published by the same research group, which utilized a 1,3-diketone and addition of copper triflate to the palladium catalyst system.49 In the course of this work, eleven examples of highly substituted pyrrolizidine derivatives were synthesized. In 2017, the same research group added an additional variation of the palladium-catalysed cyclization reaction.50 By omitting the benzene backbone present in their 2006 publication, Yang and coworkers were able to synthesize twelve novel derivatives with varying substitution patterns and no fused benzene ring. In the same year, the Yang research team published an additional variation of the reaction using (S,S)-diPh-pyrox as a ligand, which resulted in the preparation of the desired pyrrolizidine compounds in high enantiomeric excess (up to 93% ee).51 Additionally, by modifying the starting material, three additional N-bridgehead heterocycles were obtained. In a fifth publication, the Yang research group demonstrated that α-halo-acetamides can also be utilized as starting materials for their palladium-catalysed cyclization reaction.52 This change allowed for the synthesis of an additional twenty-seven scaffolds without the need for a vinyl moiety in the starting material.
The last example of a palladium-catalysed procedure was reported in 2015 by Doucet and coworkers, who described a novel two-step procedure for the preparation of pyrrolizidines (Scheme 4).53 In this process, a desulfitative addition of a substituted benzene ring to the pyrrole system was followed by C–H bond arylation to yield the closed ring system. By using an excess of the sulfonyl chloride derivative (2.5 equivalents), two rings were added to the pyrrole. As the α-position was now occupied, the C–H bond arylation took place on one of the aryl rings, forming a 5,7-fused ring system with a bridgehead nitrogen.
2.4 Metathesis reactions
A well-established tool for the synthesis of ring systems is the metathesis reaction.54 In 2007, Mori and coworkers utilized this powerful methodology to prepare pyrrolizidines, indolizidines and one quinolizidine using a successive ring-opening metathesis (ROM)/ring-closing metathesis (RCM) approach (Scheme 5). Depending on the starting material, different ring sizes were provided.55
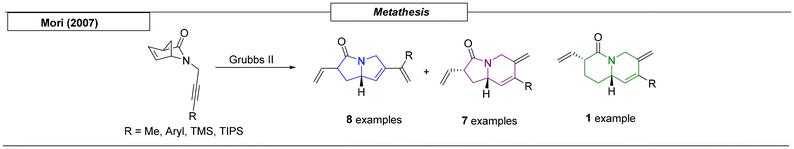 |
| | Scheme 5 Pyrrolizidine scaffolds obtained via metathesis reactions. | |
2.5 Cascade and multicomponent reactions
Cascade reactions, procedures with successively triggered reactions in a single approach, were found to be versatile tools capable of providing a multitude of complex molecules. Cascade reactions are of additional interest, because they are often considered ‘green’ chemistry.56 In this section, we highlight cascade reactions and one-pot processes, as both combine multiple reaction steps in a single reaction flask, toward pyrrolizidines. Reissig and coworkers synthesized various 1,2-oxazines, which were used in reductive cascade reactions (Scheme 6).57 Ring cleavage at the N–O bond followed by subsequent cyclization yielded three different pyrrolizidinone compounds. Modification of these compounds provided three additional pyrrolizidine scaffolds. In the same year, the Delgado research group published two novel routes of synthesis toward pyrrolizidines starting from pyrrole-aldehydes.58 Knoevenagel condensation of a pyrrole aldehyde with an activated methylene compound yielded the corresponding precursors, which were used in a Michael addition of dimethylmalonate and a subsequent lactamization. In total, seven different pyrrolizidine scaffolds were obtained.
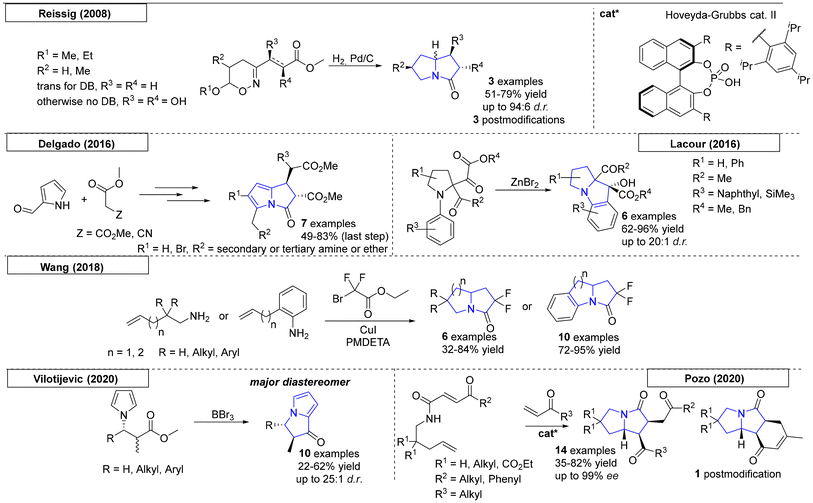 |
| | Scheme 6 Cascade and multicomponent reactions that produce pyrrolizidines. | |
In 2016, Lacour and coworkers developed a novel method for the synthesis of pyrrolidines bearing both a carbonyl and an ester moiety (Scheme 6).59 A Lewis acid-induced intramolecular nucleophilic reaction with a ketone provides an alcohol-functionalized pyrrolizidine scaffold. In total, six examples were reported with diastereomeric ratios as high as 20:1. Wang and coworkers reported a new cascade approach utilizing amine-functionalized olefins and ethyl bromodifluoroacetate.60 Catalysed by copper(I), difluorinated nitrogen-containing polycycles were obtained in up to 95% yield via a radical cascade annulation. In 2020, the Vilotijevic research group reported an enantioselective N-allylation of N-silyl pyrrole latent nucleophiles with allylic fluorides followed by hydrogenation. The resulting diastereomeric mixtures were used as starting material in a diastereoselective Friedel–Crafts-type cyclization sequence.61 This procedure yielded ten examples of enantioenriched, substituted 2,3-dihydro-1H-pyrrolizin-1-ones (enrichment due to steric hindrance).
The last protocol, published by Pozo and coworkers in 2020, described a triple-tandem reaction for the synthesis of pyrrolizidinones (Scheme 6).62 The mechanism can be described as a cross metathesis–intramolecular aza-Michael reaction–intramolecular Michael addition tandem sequence utilizing N-pentenyl-4-oxo-2-alkenamides and conjugated ketones. As the products bore two ketone groups, post-modification via Robinson annulation was possible and was demonstrated in one example.
2.6 One-pot procedures
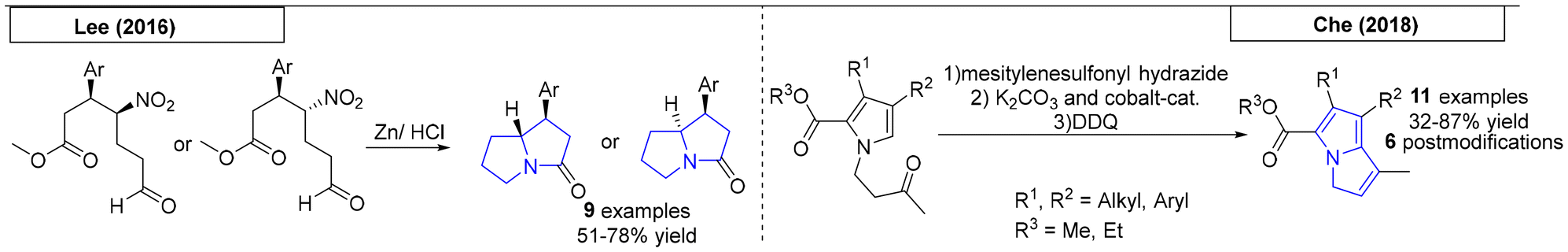
One-pot procedures are synthetic preparations involving multiple reactions that occur in a single reaction vessel. In 2016, Lee and coworkers synthesized valuable pyrrolizidine precursors from α,β-unsaturated aldehydes (Scheme 7).63 Subsequently, a one-pot nitro reduction/reductive amination/lactamization sequence was conducted using zinc powder to give access to nine different pyrrolizidine derivatives. Che and coworkers reported a one-pot synthesis of pyrrolizidines in 2018.64 This preparation of poly-substituted 3H-pyrrolizines and pyrrolizidines employed a cobalt(II) porphyrin catalyst. The reaction sequence, which consisted of an intramolecular cyclopropanation/ring-opening cascade reaction of hydrazones derived from pyrrolyl ketones, provided eleven examples. Six additional pyrrolizidines were obtained via post modification.
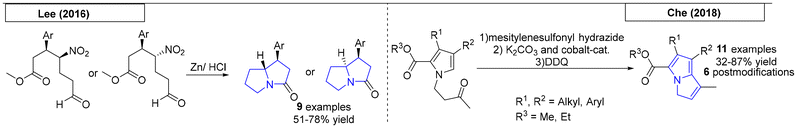 |
| | Scheme 7 One-pot procedures that provided pyrrolizidines. | |
2.7 Multistep procedures
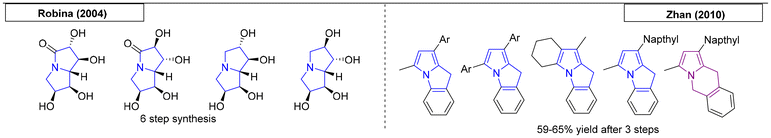
Two research groups have published multistep procedures that gave access to pyrrolizidines. Robina and coworkers developed a stereoselective route toward two enantiopure tetrahydroxy pyrrolizidin-5-ones and tetrahydroxy pyrrolizidines (Scheme 8).65 The starting material, D-allitol, was transformed into the desired compounds over six reaction steps. In 2010, the Zhan research group reported a zinc-catalysed, regioselective sequence of propargylation, amination and cyclo-isomerization reactions of propargylic acetates, enoxysilanes and primary amines.66 In this work, five new nitrogen bridgehead compounds were synthesized, including four pyrrolizidines and one indolizidine.
 |
| | Scheme 8 Multistep procedures that gave access to pyrrolizidines. | |
3 Indolizidine (5,6)
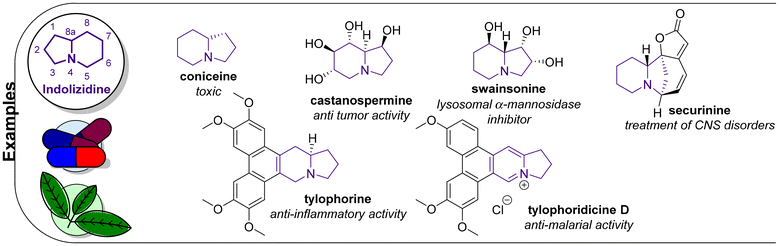
Indolizidines and various unsaturated analogs, including indolizines,67 are privileged structures that are present in many natural products (Fig. 3).68,69 Reported biological activities include antitumor, antimalarial, antibacterial, antifungal, antiparasitic, antiviral and anti-inflammatory.70 Coniceine, a well-known example of a bioactive indolizidine, was used to execute criminals in ancient Greece.71 Castanospermine (1,6,7,8-tetrahydroxyoctahydroindolizine) exerts diverse biological activities and has been investigated for its antitumor activities, specifically for use against multidrug-resistant breast cancer cell lines.72,73 The data suggested that this compound may be beneficial for the treatment of cancer, and, as a result, its synthesis has been reported many times. Tylophorine exerted anti-inflammatory activity by enhancing the phosphorylation of Akt and downregulating AP1, thereby suppressing nitric oxide (NO) production in lipopolysaccharide (LPS)/interferon-γ (IFN-γ)-stimulated RAW264.7 cells.74 When administered to animals, swainsonine, a specific directional inhibitor of the active site of lysosomal α-mannosidase, caused a phenocopy of mannosidosis, which is a genetic lysosomal storage disease.70 Securinine has been established clinically to treat CNS disorders, e.g. poliomyelitis and amyotrophic lateral sclerosis.75 Investigation of its mechanism of action in microglia and astrocytic cultures revealed the inhibition of NO formation and activation of the inflammatory mediator NF-κB and mitogen-activated protein kinases in BV2 cells stimulated by LPS. Securinine was also found to be capable of inhibition of both iNOS mRNA expression and the level of NO-induced by IFN-γ. Furthermore, securinine significantly reduced the neurotoxicity of midbrain dopamine compared to conditioned medium (CM) from LPS-stimulated microglia. Tylophoridicine D was tested for its in vitro antiplasmodial activity against a chloroquine-sensitive strain of Plasmodium falciparum. With an IC50 of 0.028 μM, its potency was comparable to artemisinin and chloroquine.76 Due to rising interest in the investigation of this compound class, efforts were made to develop stereo-controlled strategies toward naturally occurring derivatives.77–80 Even though numerous patents have been filed connected to this compound class, an indolizidine-based drug has not yet been approved.81
 |
| | Fig. 3 Examples of indolizidine compounds. | |
3.1 Reviews
In 2015, Burtoloso et al. reviewed the synthesis of indolizidines and quinolizidines starting from diazo derivatives.82
3.2 Free radical reactions
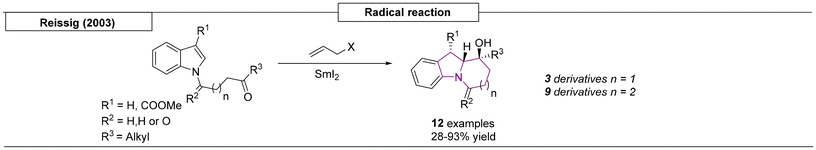
One method has been reported for the synthesis of pyrrolizidines and indolizidines using free radical chemistry. In 2003, the Reissig research group developed a samarium iodide-initiated reaction and used it to produce twelve different variants (Scheme 9).83 The diastereoselectivity can be explained by samarium alcoholate favouring an equatorial position for steric and electronic reasons.
 |
| | Scheme 9 Synthesis of pyrrolizidines and indolizidines using free radical chemistry. | |
3.3 Cycloadditions
3.3.1 1,3-Dipolar cycloadditions.
Two research groups have reported methods for the synthesis of indolizidines using 1,3-dipolar cycloaddition reactions. In 2001, the Georgescu research group described the 1,3-dipolar cycloaddition of activated, quaternary pyridinium or naphthalene salts with alkynes, yielding carbamoyl-substituted indolizines and benzoindolizines (Scheme 10).84 The salts were activated using propenoxide. In total, twenty-seven novel compounds were synthesized. A second 1,3-dipolar cycloaddition reaction example was reported by the Kakehi research group.85 The lanthanide-based, Lewis acid-catalysed reactions were highly enantioselective and utilized 3-(2-alkenoyl)-2-oxazolidinones and carbonyl ylides generated from N-diazoacetyl lactams. Quinolizidines and 7-membered ring systems were also synthesized.
 |
| | Scheme 10 Synthesis of indolizidines via cycloaddition reactions. | |
3.3.2 [4 + 2] cycloadditions.
In 2016, Rychnovsky and coworkers described the intramolecular [4 + 2] cycloaddition of the in situ prepared (via Strecker reaction) 2-cyano-1-azadiene (Scheme 10).86 The reaction, which yielded five different indolizidine derivatives and one quinolizidine derivative, showed moderate stereoselectivity (up to 1:4 dr) of the endo and exo reaction pathways. The reaction is enabled by direct reaction of the intermediate N-methylcarbamoyl pyridinium salts with activated alkynes in an epoxide, as acid acceptor and reaction solvent.
3.4 SN2 reactions
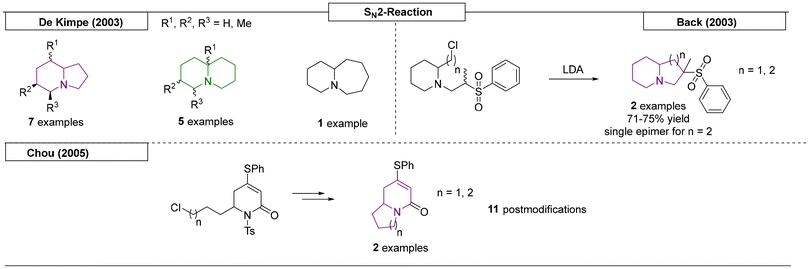
Several research groups have published methods for the synthesis of indolizidines using SN2 reactions. A very effective synthetic route for the construction of simple N-bridgehead scaffolds was reported by De Kimpe and coworkers in 2003 (Scheme 11).87 Starting with a kinetic deprotonation of 2-methyl-1-pyrroline (indolizidine) or 6-methyl-2,3,4,5-tetra-hydropyridine (quinolizidine), addition of a dihalogenated alkyl chain resulted in the desired core structures after two subsequent SN2-reactions. Another example of an SN2 reaction yielding an indolizidine scaffold was reported by the Back research group.88 LDA was utilized as a strong base to deprotonate the most acidic proton next to the sulfone group. The free electron pair then attacked the halogenated carbon via SN2-reaction to form the N-bridgehead motif. In one example, an indolizine scaffold occurred as an epimeric mixture, while in another example a quinolizidine scaffold was prepared as a single epimer. A similar reaction was reported by the Chou research group in 2005.89 After removal of a nitrogen protecting group, NaH was used as a strong base to deprotonate a secondary amide. The electron pair then reacted with the halogenated carbon to form the second ring of the N-bridgehead scaffold. Oxidative and reductive post modifications were used to produce nine additional derivatives.
 |
| | Scheme 11 Synthesis of indolizidines via SN2 reactions. | |
3.5 Metathesis reactions
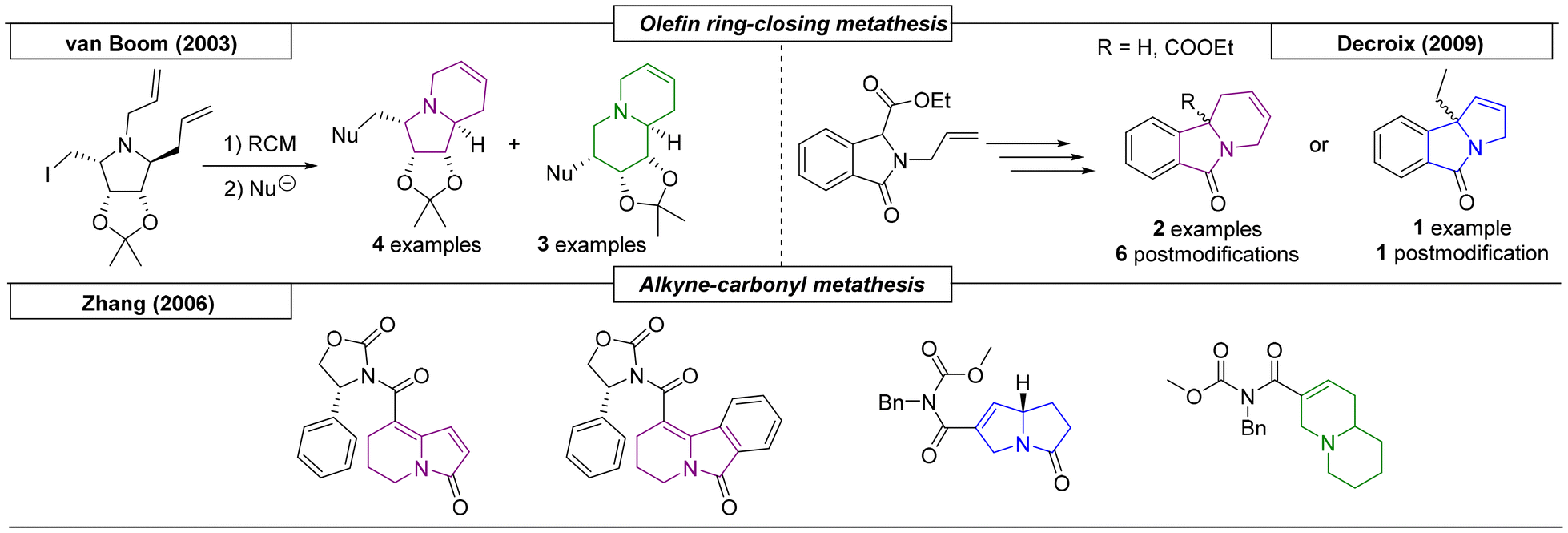
Several research groups have published methods for the preparation of indolizidines and quinolizidines using metathesis reactions. Van Boom and coworkers reported a synthesis of novel indolizidine and quinolizidine derivatives via a three step sequence starting from (3aS,4S,6R,6aR)-4,5-diallyl-6-(iodomethyl)-2,2-dimethyltetrahydro-4H-[1,3]dioxolo[4,5-c]pyrrole (Scheme 12).90 The ring closure was achieved by a ring-closing metathesis reaction. The resulting indolizidine was in equilibrium with the corresponding quinolizidine, and, by exploiting this equilibrium, additional derivatives were synthesized via nucleophilic substitution. In 2006, the Zhang research group described a boron trifluoride etherate-catalysed alkyne-olefin metathesis reaction that yielded two indolizidine scaffolds.91 By variation of the starting materials and reaction conditions, pyrrolizidine and quinolizidine derivatives were also prepared. The last metathesis reaction example was reported by Decroix and coworkers.92 A bicyclic lactam precursor was modified by SN2 reaction with an allyl-halogen reagent to introduce a second double bond, which participated in a RCM reaction to yield indolizidines and one pyrrolizidine. The obtained N-bridgehead compounds were then postmodified by oxidation of the resulting double bonds with OsO4 to introduce 1,2-dihydroxy functionalities, which allowed for further modification.
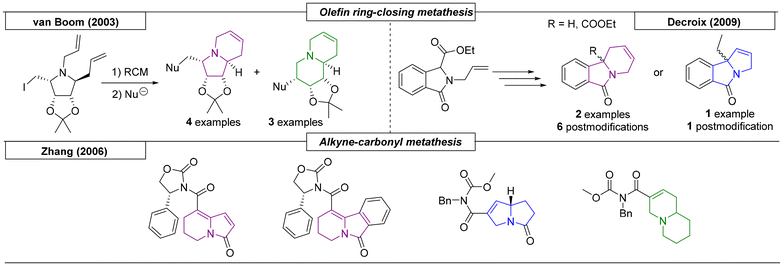 |
| | Scheme 12 Methods for the preparation of indolizidines and quinolizidines utilizing metathesis reactions. | |
3.6 Cascade and multicomponent reactions
Many methods for the preparation of indolizidine-type compounds using cascade and multicomponent reactions have been published. The Molander research group employed various amino-di-olefin compounds in an organo-lanthanide catalysed intramolecular amination/cyclization sequence, which gave access to different N-bridgehead scaffolds, including indolizidines, pyrrolizidines and quinolizidines (Scheme 13).93 The procedure allowed the straightforward synthesis of simple core structures and, for some indolizidines and quinolizidines, gave high diastereoselectivities. Babaev and coworkers discovered an unusual oxazole-to-pyrrole recyclization reaction.94 The oxazole was synthesized from a tetrahydroquinolone via a three-step transformation. By employing piperidine, morpholine or methanol as nucleophiles, three different indolizine compounds were synthesized. In 2005, the Ma research group developed a sequential SN2/Michael addition/SN2/SN2 reaction process, which involved the combination of iodo alkyne compounds and δ-chloropropylamines or β-amino esters (not shown).95 In addition to quinolizidines and a 6:7 ring-fused N-bridgehead system, natural occurring alkaloid indolizidine 223A was synthesized in a 12-step linear sequence with an overall yield of 14.5%. One year later, the same group exploited this reaction to synthesize twelve additional N-bridgehead scaffolds from different starting materials.96 During the course of this investigation, two pyrrolizidine, five indolizidine and five quinolizidine derivatives were obtained.
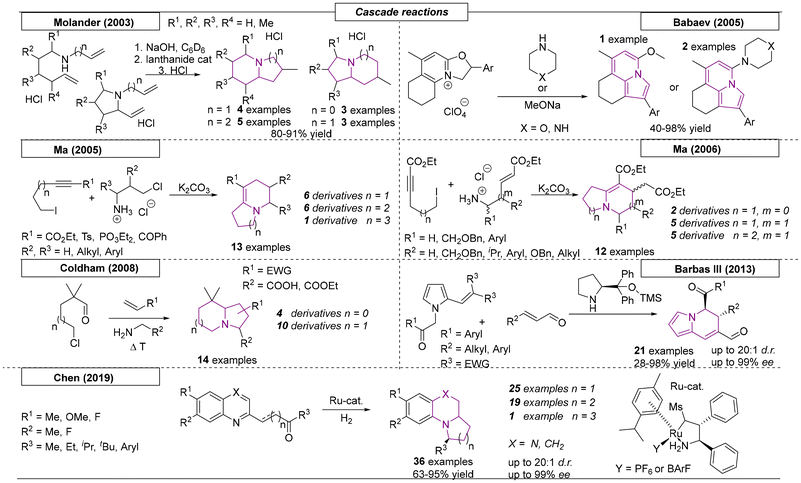 |
| | Scheme 13 Methods for the preparation of indolizidine-type compounds using cascade and multicomponent reactions. | |
In 2008, the Coldham research group developed a three-component coupling reaction, which utilized a primary amine (amino acid, aminoester or hydroxylamine), an alkene or alkyne dipolarophile, and an aldehyde bearing a halogen leaving group (Scheme 13).97 The reaction sequence, which involved a condensation and subsequent cyclization followed by a dipolar cycloaddition, was used to produce four pyrrolizidine and nine indolizidine derivatives. In 2013, Barbas III and coworkers reported a novel organocascade reaction toward indolizidine derivatives.98 Pyrrole-derived substrates were reacted with α,β-unsaturated aldehydes via iminium enolate activation, which enabled a Michael–Michael bond formation. After elimination of the condensed C(R3)2 units, the desired indolizidine derivatives were obtained in excellent yields and with excellent stereoselectivities. The last method reviewed here, a ruthenium-catalysed asymmetric cascade reaction, was published by Chen and coworkers in 2019.99 Quinolinyl- and quinoxalinyl-containing ketones underwent a hydrogenation/reductive amination sequence, which was catalysed by an enantiopure ruthenium complex. The indolizidine and quinolizidine derivatives were obtained with high stereoselectivities, diastereomeric ratios exceeding 20:1 and enantiomeric excesses of up to 99%. A set of sixteen compounds with an additional nitrogen in the core structure were also synthesized. Additionally, this methodology was used in the formal synthesis of (+)-gephyrotoxin.
3.7 One-pot syntheses
Many one-pot procedures have been used to produce indolizidines and quinolizidines. In 2009, the Charette research group reported a two-step, one-pot process that consisted of a pyridine activation step and a subsequent Grignard reaction (Scheme 14).100 In the first step, the cyclization resulted in a pyridinium salt, which was then attacked by the nucleophilic Grignard reagent. This resulted in the reduction of the ring system and introduction of an R3 substituent. The regioselectivity and stereoselectivity was explained by coordination of the Aux functionality to the Mg at the position α to the amide nitrogen. Furman and coworkers published a one-pot strategy toward indolizidines and quinolizidines in 2014.101 In the first step, a sugar-derived lactam was reduced with Schwartz's reagent. Subsequently, a Lewis acid-catalysed diastereoselective Mannich/Michael tandem reaction of the resulting sugar imine with Danishefsky's diene was carried out. The desired N-bridgehead scaffolds were obtained in yields as high as 81% and with diastereomeric ratios as high as 98:2.
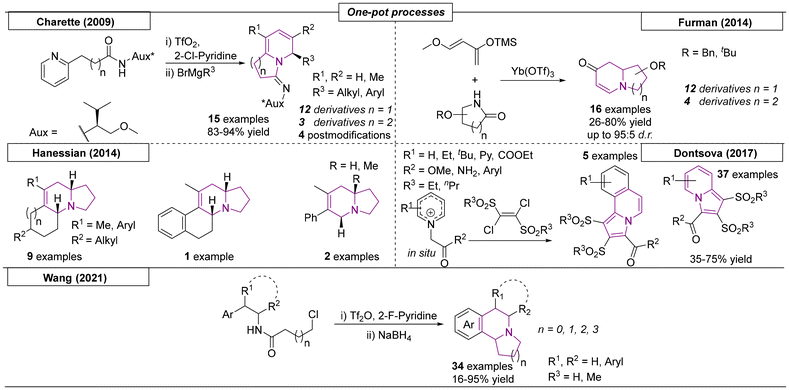 |
| | Scheme 14 One-pot procedures used to produce indolizidines and quinolizidines. | |
The Hanessian research group developed a novel one-pot method starting from L-proline and L-pipecolic acid, which consisted of a two component iminium ion/enamine sequence and employed mild reaction conditions (Scheme 14).102 In addition to indolizidines, azasteroids and other complex alkaloids were obtained by variation of the starting materials. Dontsova and coworkers developed a novel one pot reaction toward a multitude of indolizidine scaffolds.103 In this work, in situ-generated pyridinium ylides were reacted with E-1,2-di(alkyl-sulfonyl)-1,2-dichloroethene, yielding a total of forty-two novel indolizidine compounds. The last and most recent one-pot process reviewed here was developed by Wang and coworkers in 2021.104 In this work, fused indolizidine and quinolizidine frameworks were constructed by a one-pot tandem route starting from amides. In the first step, triflic anhydride (Tf2O) enabled the ring formation toward a secondary amide, which, after reduction with NaBH4, attacked the halogenated carbon via an SN2 reaction. The novel procedure was also employed as a key step in the total synthesis of (I)-xylopinine.
3.8 Metal-catalysed methods
Two research groups have published metal-catalysed methods that produce indolizidines and quinolizidines. Baudoin and coworkers reported a C(sp3)–H alkenylation, which was used to synthesize N-bridgehead compounds from tertiary cyclic amides (Scheme 15).105 In this work, one of the indolizidine compounds was further converted to the naturally-occurring alkaloid (±)-coniceine via two additional steps. In 2017, the Widenhoefer research group developed an Au(I)-catalysed intramolecular hydroamination reaction of alkenes attached to 2-pyridones.106 For substances with two enantiomeric centers, the reaction gave diastereoselectivities of up to 25:1 dr. In addition to the ten indolizidine and four quinolizidine derivatives produced, compounds with an additional nitrogen in the core structure were obtained.
 |
| | Scheme 15 Metal-catalysed methods that have been used to produce indolizidines and quinolizidines. | |
3.9 Multistep syntheses
Since 2009, four research groups have published multistep procedures that provide indolizidines and quinolizidines. In 2009, the Micalizio research group developed an efficient multistep synthetic strategy toward indolizidines and quinolizidines (Scheme 16).107 The first step yielded highly-substituted and stereo-defined allylsilanes through chemoselective coupling of functionalized allylsilanes. These were further transformed via acid-promoted, cationic annulation toward the desired scaffolds with diastereomeric ratios greater than 20:1. Caprio and coworkers exploited the stereoselective synthesis of nitrones as a starting point toward the construction of indolizines and quinolizidines.108 The nitrones were reacted with oxygen-functionalized lithium acetylides. Reduction of the resulting hydroxylamines and alkynes resulted in the corresponding secondary amines and cis-alkenes or alkyl chains, respectively. Conversion of the oxygen functionality to a leaving group followed by SN2 cyclization with the secondary amine provided the nitrogen bridgehead. This strategy was used to enable the synthesis of (+)-swainsonine. Another example of the synthesis of indolizidines was described by the Pohmakotr research group in 2018.109 The approach focused on the oxidative difluoromethylation of tetrahydroisoquinolines. Subsequent reductive desulfenylation resulted in free radical cyclization toward the indolizidine and quinolizidine scaffolds. In total, the procedure yielded seventeen N-bridgehead derivatives. The final multistep synthetic strategy toward indolizines and quinolizidines reviewed here was developed by the Gueyrard research group in 2020.110 Methyl- and benzyl-substituted succinimide and glutarimide derivatives were converted to the corresponding benzothiazolyl sulfones in four steps. The resulting cyclization precursors were then used in a regioselective olefination reaction to yield eighteen examples of the desired indolizine and quinolizidine compounds. This route was also used to synthesize Pundalizine A.
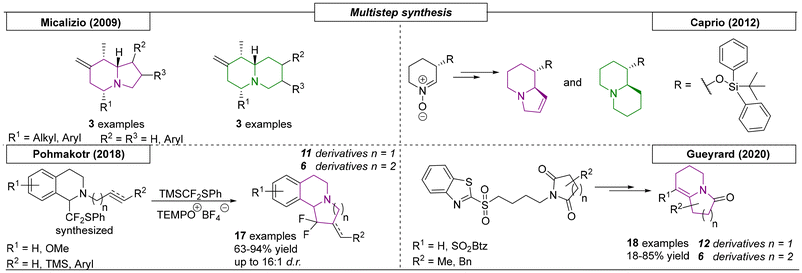 |
| | Scheme 16 Multistep procedures that provide indolizidines and quinolizidines. | |
3.10 Annulation reactions
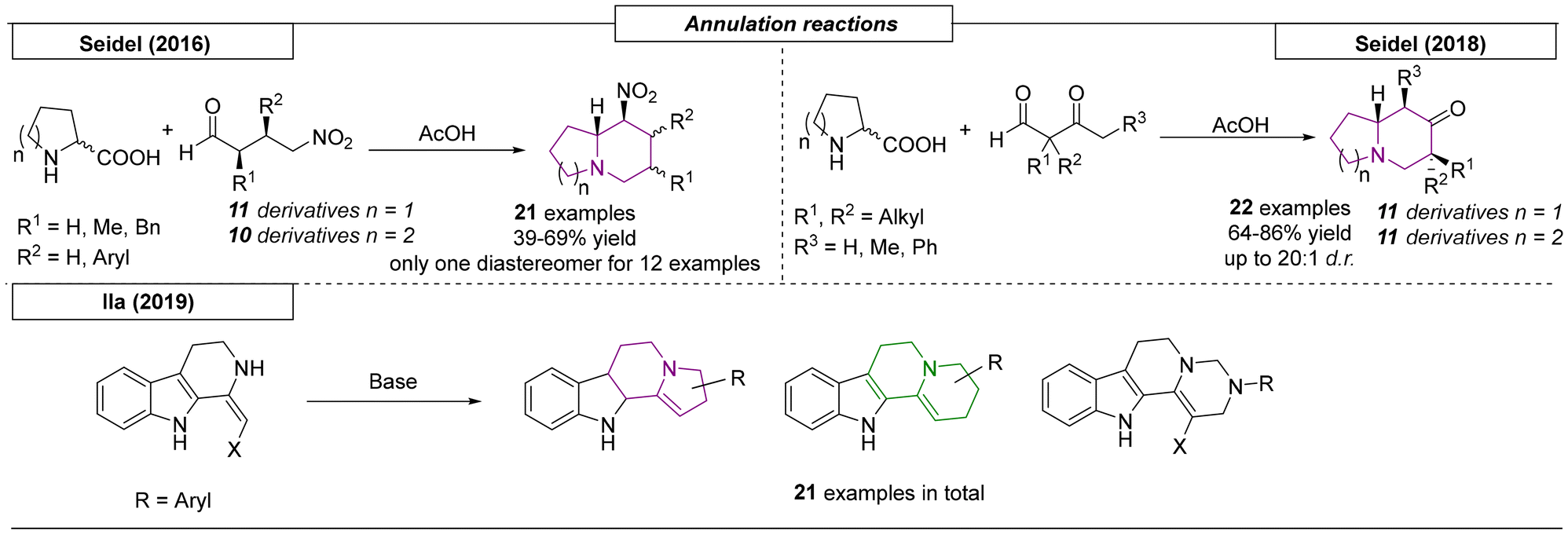
Several research groups have published the synthesis of indolizidines and quinolizidines using annulation reactions. The first example of an annulation reaction yielding nitrogen bridgehead scaffolds was described by Seidel and coworkers in 2016 (Scheme 17).111 In this approach proline or pipecolic acid were condensed with a γ-nitrobutyraldehyde. Acetic acid-promoted decarboxylation formed an azomethine ylide, which underwent nucleophilic annulation to form the final product. In addition to twenty-one examples of indolizidines and quinolizidines, an epiquinamide analog was synthesized. Two years later, the same research group published a decarboxylative annulation reaction of α-amino acids with β-ketoaldehydes.112 In this work, twenty-two additional indolizidine and quinolizidine derivatives were reported with excellent diastereoselectivities (up to 20:1 dr). The last annulation process reviewed here was described by Ila and coworkers.113 This approach investigated the aza-annulation of novel 1,2,3,4-tetrahydro-β-carboline-derived enaminones and nitroenamines, which were reacted with various 1,2- and 1,3-bis electrophiles. This simple reaction enabled the construction of highly functionalized tetrahydro-β-carboline and 1,2-fused five- and six-membered heterocyclic frameworks including indolizidine and quinolizidine scaffolds.
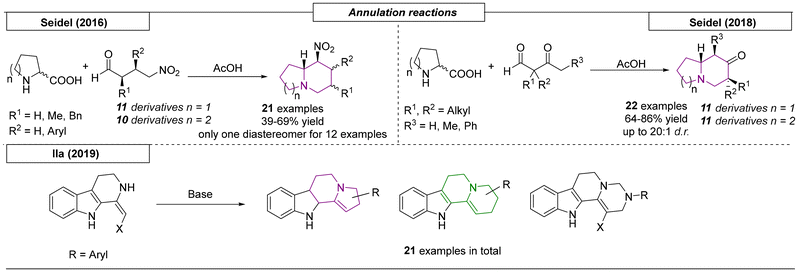 |
| | Scheme 17 Indolizidines and quinolizidines via annulation reactions. | |
4 Quinolizidines
The quinolizidine motif is present in various naturally occurring alkaloids, including berberin and spartein, as shown in Fig. 4.114 Many of these interesting scaffolds are present in plants, such as Lupinus albus, and possess a broad range of biological applications.115–117 An example of a quinolizidine subgroup is the 1-hydroxy-substituted derivatives, which are in some cases poisonous to herbivores.118 Zou and coworkers isolated a multitude of quinolizidine compounds from Sophora tonkinensis that showed anti-tobacco mosaic virus (TMV) properties.116 In other cases, compounds of this type exhibited activity against leukemia.119 Another example is the benzo[a]quinolizidines, which have been subjected to a multitude of physiological activity investigations.120 The last example of a bioactive quinolizidine subgroup is the naturally occurring phenanthroquinolizidine scaffold, which has anticancer (due to cytotoxicity), anti-inflammatory and antiviral applications.121 Many synthetic approaches toward quinolizidines have been reported. However, few of these compounds have been evaluated for biological activity.
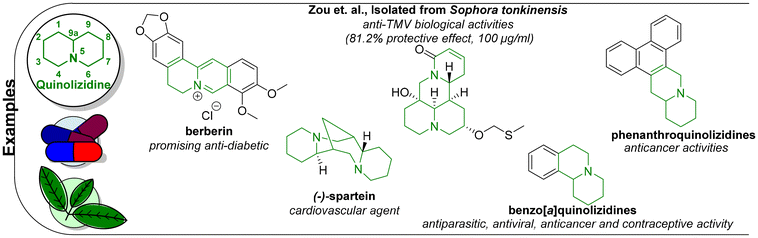 |
| | Fig. 4 Examples of various quinolizidine-derived compounds. | |
4.1 Free radical reactions
In 2017, the Zhou research group developed a light-induced tandem substitution reaction of two fluorine atoms toward novel fluorinated benzo[a]quinolizidines (Scheme 18).122 This radical-initiated sequence was facilitated by an iridium photocatalyst and provided access to sixteen different compounds with yields as high as 89% and diastereomeric ratios of up to 20:1.
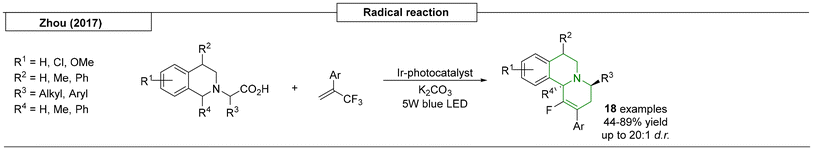 |
| | Scheme 18 Radical reactions that have been used to produce quinolizidines. | |
4.2 Cycloadditions
Several research groups have published cycloaddition reactions that give access to quinolizidines. In 2013, a highly enantio- and diastereoselective procedure toward indolo- and benzoquinolizidine scaffolds was developed by Jacobsen and coworkers (Scheme 19).123 The formal aza-Diels–Alder reaction was catalysed by a bifunctional primary aminothiourea, which was capable of activating both starting materials, cyclic imines and enones. In total, twenty-two examples were presented in the publication with up to quantitative yields and 99% ee. A second aza-Diels–Alder process was reported by Schneider and coworkers in 2018.124 A chiral Brønsted acid catalysed the intramolecular cycloaddition reaction of an ortho-quinone methide imine with a dienophile. The resulting quinolizidines and oxazinoquinolines were obtained in yields as high as 93% and diastereomeric ratios of up to 91:9. In 2013, the Doyle research group published a [3 + 3] cycloaddition reaction catalysed by a chiral dirhodium reagent.125 In this transformation, isoquinolinium or pyridinium methylides reacted with enol diazoacetates. By using enough catalyst, the concurrent [3 + 2] side reaction was suppressed. In addition to yields as high as 90%, enantiomeric excesses of up to 96% were also achieved.
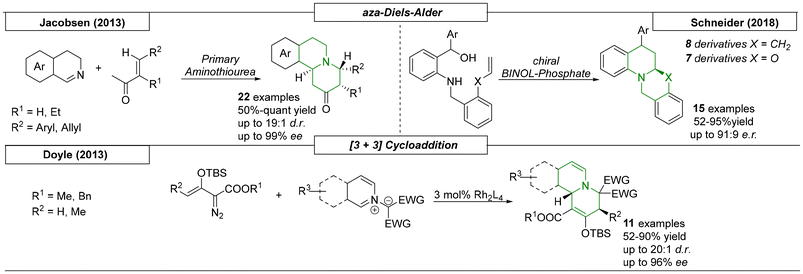 |
| | Scheme 19 Cycloaddition reactions that provide quinolizidines. | |
4.3 Metal-catalysed reactions
Many examples of the synthesis of quinolizidines via metal-catalysed reactions have been published. In 2013, Wu and coworkers developed a novel cascade sequence toward indoloquinolizidines that started with imino diesters and featured an asymmetric catalytic hydrogenation/lactamization step (Scheme 20).126 By employing a chiral ruthenium catalyst for the stereoselective hydrogenation, high optical purities of up to 99% were obtained with yields as high as 92%. 2-Epideviyl anthirine, Isogeissoschizol and other natural products were synthesized using this approach. Guinchard and coworkers developed a palladium(0)-catalysed, two-step cascade reaction toward the quinolizidine core structure.127 Precursors were synthesized from N-allyl tryptamine and 1,5-allenals using an asymmetric Pictet–Spengler reaction. Quinolizidines were used in subsequent deprotection and cyclization sequences to form the corresponding products in yields as high as 82% and with diastereomeric ratios of up to 93:7.
 |
| | Scheme 20 Quinolizidines via metal-catalysed reactions. | |
In 2016, the Unsworth research group investigated a silver-catalysed cyclization reaction that gave access to pyridine-, isoquinoline-, and pyrazine-ynone scaffolds (Scheme 20).128 The high yielding reaction (up to quantitative yield) employed pyridine-ynone starting materials, which were readily available from 2-picoline. In addition to the twelve quinolizidine examples, this new approach was used to prepare lasubine II, a naturally occurring alkaloid, in five steps. In the same year, Tokuyama and coworkers published a novel gold-catalysed reaction sequence toward highly substituted quinolizidines.129 The acationic gold complex facilitated a double cyclization cascade sequence of yn-amides, which were converted to enamides in the first cyclization and to the final quinolizidines in the second cyclization. The novel methodology was also used in the total synthesis of the naturally occurring alkaloid lupinine. In another example, the Castarlenas research group reported a rhodium-catalysed hydroalkenylation reaction of 2-vinylpyridine that yielded a quinolizidine.130 By changing 2-vinylpyridine to 2-vinylpyridazine, an additional nitrogen atom was introduced into the quinolizidine scaffold. In the last approach reviewed here, published by Fan and coworkers in 2020, 2-(quinolin-8-yl)ethyl ketones were transformed into their corresponding julolidines, which bear the quinolizidine structure with an additional aromatic ring.131 The employed ruthenium complex catalysed both the hydrogenation and subsequent reductive amination reactions with yields as high as 94%, diastereomeric ratios exceeding 20:1 and ee values of up to 99%. Three post modifications were reported, including the preparation of two chiral compounds and a molecular motor.
4.4 Metathesis reactions
Four research groups have reported the synthesis of N-bridgehead compounds using metathesis reactions. A straightforward synthetic procedure toward N-bridgehead scaffolds with different ring sizes was published by the Barluenga research group in 2004 (Scheme 21).132 RCM precursors were prepared by aza Diels–Alder reaction of amino trienes with imines. The subsequently RCM reaction formed the N-bridgehead system. In total, seven examples with ring sizes of up to nine atoms were synthesized. The Chou research group published four different papers about the use of thiophene-derived precursors to synthesize a multitude of different N-bridgehead heterocycles.133–136 Using this approach, the precursors were transformed into diallyl compounds, which were then used in RCM reactions to access various bicyclic scaffolds. In a different approach, Malhotra and coworkers reported the use of N-Boc β-lactams to construct N-bridgehead bicyclic compounds with varying ring sizes in a stereoselective fashion.137 The β-lactams were opened with an ethynyl Grignard reagent, and the resulting ynones were cyclized to form dihydropyridones. N-Alkylation of the pyridone nitrogen provided di-olefin intermediates, which were used in subsequent RCM reactions. In total, eight N-bridgehead compounds were synthesized. The last reviewed procedure for the RCM synthesis of quinolizidines was developed by Amat and coworkers in 2012.138 A chiral bicyclic lactam was stereoselectively opened with allyltrimethylsilane to provide an N-substituted pyridone. Removal of the N-substituent, followed by N-allylation provided the di-olefin that was used in the subsequent RCM reaction to prepare the target quinolizidine. Eight additional compounds were provided via postmodification.
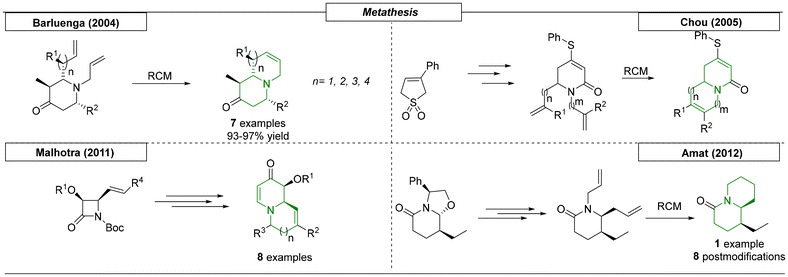 |
| | Scheme 21 Metathesis reactions toward N-bridgehead compounds. | |
4.5 Cascade reactions
Many research groups have published methods for the preparation of quinolizidines via cascade and multicomponent reactions. Khalafalla and coworkers developed a cascade reaction strategy for the preparation of nineteen N-bridgehead compounds (Scheme 22).139 The precursors used in the work were readily available from 1,4-naphthoquinone and a suitable alkylidyne malononitrile reagent. The intermediates obtained from the initial cycloaddition were reacted with Knoevenagl condensates of benzaldehyde to yield the corresponding quinolizidine scaffolds. In 2005, the Mikhal'chuk research group discovered a reaction sequence of an iso-quinoline Schiff base with α,β-keto esters that the authors described as a ‘cyclo-condensation’.140 This straightforward method gave access to four derivatives with yields as high as 93%. Another interesting approach was published by Babaev and coworkers in 2010,141 further expanding their work from 2005.94 Here, lithium cyclopentadienylide was reacted with oxazolo[1,2-a]pyridinium to obtain a novel ring-expanded pyrrolizidine scaffold. The yields of the four reported examples spanned from 11 to 44%. In the same year, the Zhao research group developed a Pictet–Spengler/lactamization cascade sequence toward a set of twenty-two indoloquinolizidines.142 The aldehydes used for this sequence were reacted with an S-proline-derived catalyst, which gave access to the enantiopure starting material. After the TFA-catalysed cascade sequence, the corresponding products retained high enantiopurities (up to 99% ee) with yields as high as 90%.
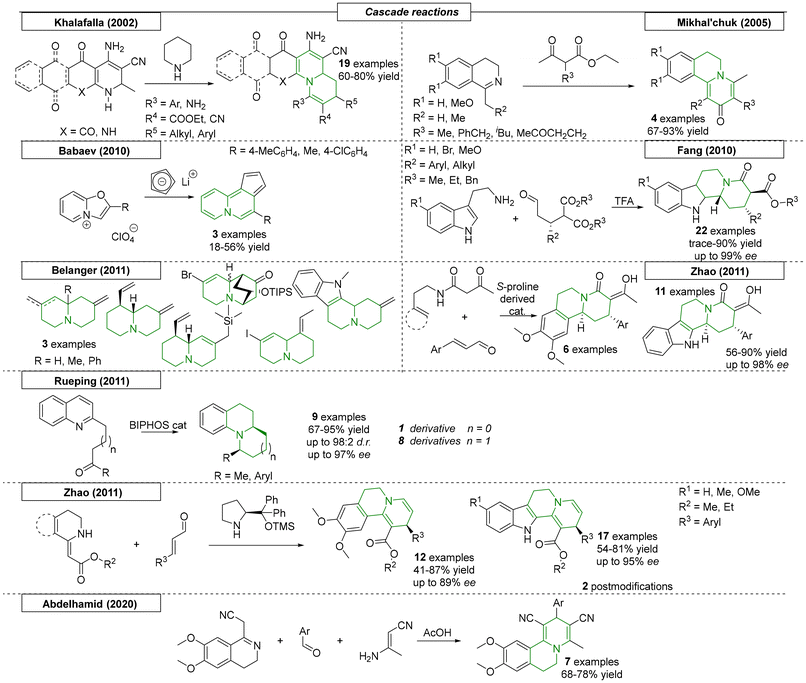 |
| | Scheme 22 Quinolizidines via cascade and multicomponent reactions. | |
In 2011, Belanger and coworkers synthesized a set of novel quinolizidine derivatives using a cyclization reaction cascade (Scheme 22).143 A tertiary amide, which bore two nucleophilic groups on the two alkyl chains, was chosen as the starting material. First, a Vilsmeier–Haack reaction occurred, resulting in a highly reactive imine intermediate, which was then attacked by the second nucleophile to yield the desired quinolizidine motifs with yields as high as 70%. In total, eight derivatives were synthesized. In the same year, the Zhao research group reported an enantioselective organocatalysed cascade reaction toward highly functionalized quinolizidines.144 In this process, catalysed by an S-proline-derived chiral catalyst, cinnamic aldehyde was reacted with β-ketoamide. As in their 2010 publication,142 the sequence was described as a Michael addition/Pictet Spengler cascade. During the investigation, seventeen examples were reported with yields as high as 90% and ee values of up to 98%. The Zhao research group continued their organocatalytic cascade investigations by changing their starting material to cyclic enamine esters, which were used in a novel sequence toward highly substituted indole[2,3-a]quinolizidines and benzo[a]quinolizidines.145 α,β-Unsaturated aldehydes were reacted with cyclic enamine esters using an enantiopure S-proline-derived catalyst. The activated LUMO of the aldehyde was attacked by the nucleophilic enamine derivative. After cyclization and dehydration, the corresponding N-bridgehead derivatives were obtained in yields as high as 87% and with up to 95% ee.
In 2011, an innovative approach toward quinolizidines and indolizidines was reported by Rueping et al., who enabled an enantioselective synthesis via a catalytic asymmetric hydrogenation cascade (Scheme 22).146 Starting from 5-(2-quinolinyl)-2-pentanone, a catalytic system of Hantzsch ester and BINOL-phosphate was optimized to provide nine examples of N-bridgehead derivatives. Depending on substitution and ring size, yields ranged from 67–95% and diastereomeric ratios of up to 98:2 and optical purities of up to 97% ee were obtained. The last quinolizidine cascade reaction reviewed here was reported by Abdelhamid and coworkers in 2020.147 In this Hantzsch ester synthesis inspired work, 3-aminocrotononitrile, benzaldehyde and 2-(6,7-dimethoxy-3,4-dihydroisoquinolin-1-yl)acetonitrile were reacted in acetic acid to yield the desired benzo-quinolizidine scaffolds. By varying the benzaldehyde, this novel sequence gave access to seven examples in yields as high as 78%.
4.6 One-pot syntheses
Many one-pot procedures have been used to produce quinolizidines. In 2007, Zou reported a one-pot, stereoselective synthesis toward polyhydroxylated quinolizidines (Scheme 23).148 The procedure used C-glycoside sugars from the natural pool in a one-pot, double-conjugate-addition procedure. Two quinolizidine derivatives were synthesized via a double-Michael addition. The hydroxy and ketone functional groups on the synthesized molecules were used in further modifications. Another one-pot procedure that gave access to novel quinolizidine derivatives was described by Franzén and coworkers 2009.149 The authors developed a novel procedure for the addition of amides to α,β-unsaturated aldehydes catalysed by optically pure proline derivatives. In total, ten new compounds were synthesized with stereoselectivities of up to 9:1 dr and 98% ee. Additional work pertaining to these scaffolds was published one year later.150 The Zhao research group reported an enantioselective, three-component, one-pot procedure that yielded indoloquinolizidines.151 The first part of the process was a conjugate addition enabled by an S-proline-derived optically pure catalyst. In the second part, tryptamine was added to allow a Pictet–Spengler cyclization to occur, which provided the desired compounds in yields of 56 to 95% and with enantiomeric excesses of up to 96%.
 |
| | Scheme 23 One-pot procedures used to produce quinolizidines. | |
In an innovative approach, Takemoto and coworkers synthesized oxidized quinolizidine derivatives via reduction of acylpyridinium cations (Scheme 23).152 The first reaction of the two-step, one-pot sequence was the acylation, which was enabled by Ghosez's reagent, while the second reaction was a Hantzsch ester reduction. In addition to nine new quinolizidines, 207I and 1-epi-207I were synthesized in five steps. In 2019, the Min research group reported a novel, one-pot synthesis of benzo[a]quinolizidines.153 The procedure consisted of a copper(I) bromide-catalysed, aza Michael addition reaction followed by a DDQ-oxidized Mannich process. In total, fourteen substrates were synthesized using the one-pot procedure or by performing each step separately. In addition, the authors showed that it was possible to combine the reactions into one-pot procedures in four more examples. The last quinolizidine one-pot sequence reviewed here was reported by Bélanger and coworkers, who described a sequential one-pot Vilsmeier–Haack and organocatalysed Mannich cyclization,154 which was inspired by their previous work.143 To overcome the reactivity issues of the second cyclization in the previous approach, an in situ-prepared enamine was used. In total, fourteen derivatives were synthesized with yields as high as 95% and stereoselectivities of up to 19:1 dr.
4.7 Multistep syntheses
Many research groups have published syntheses of quinolizidines using multistep procedures. In 2005, Bosch and coworkers developed a biogenetically inspired enantioselective synthesis of indolo[2,3-a]- and benzo[a]quinolizidines (Scheme 24).155 In the first step, they synthesized an aglycone of naturally occurring secologanin. This compound was subjected to a tandem kinetic resolution/desymmetrization sequence and then employed in a stereoselective cyclo-condensation with two different (S)-tryptanols. In total, four quinolizidine derivatives were synthesized. In 2007, this procedure was further developed to produce an additional seven examples.156 In 2005, Clive and coworkers described a synthetic procedure toward N-bridgehead compounds with various ring sizes.157 Methyl acrylate was reacted with cyclic Boc-protected β-amino aldehydes with ring sizes ranging from 5 to 8. Removal of the protecting group allowed cyclization to occur via an intramolecular Michael addition reaction. In a subsequent publication, the Clive research group expanded the scope to include additional sizes of both rings.158 Overall, eight additional examples were reported.
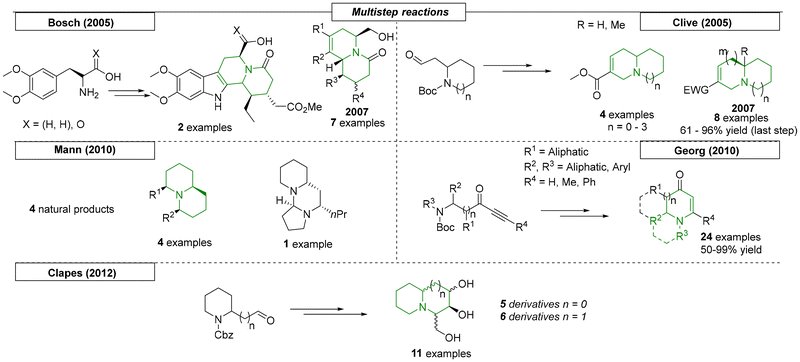 |
| | Scheme 24 Quinolizidines via multistep procedures. | |
In 2010, Mann and coworkers developed a hydroformylation of alkenylamines, which was used as a key step in the construction of quinolizidine compounds (Scheme 24).159 The protected cyclic amines, obtained by the aza-Sakurai–Hosomi hydroformylation, were stereoselectively converted to either an aldehyde or an α,β-unsaturated ketone, which were used to form the second quinolizidine ring. They also synthesized four natural products using this reaction sequence. In the same year, the Georg research group developed a novel multistep synthetic strategy toward enaminones.160 In the first step, various ynones, prepared from β-amino acids, were transformed to their corresponding heterocycles. If cyclic amines were used, quinolizidine and indolizidine derivatives were obtained, twenty-four examples in all. The last multistep quinolizidine synthesis reviewed here was reported by Clapés and coworkers.161 Using a two-step sequence, eleven novel quinolizidine and indolizidine derivatives were synthesized. All compounds were tested for their ability to inhibit α-L-rhamnosidase from P. decumbens; four indolizidines and two quinolizidines showed inhibitory activity.
4.8 Annulation reactions
Du and coworkers developed an N-heterocyclic carbene-catalysed annulation reaction of cyclic β-enamino esters with enals (Scheme 25). This approach gave entry to functionalized indolo[2,3-a]quinolizidines, which are present in Latifoliamide A, Corynantheol and Geissoschizine.162 The methodology was used to synthesize nineteen different derivatives.
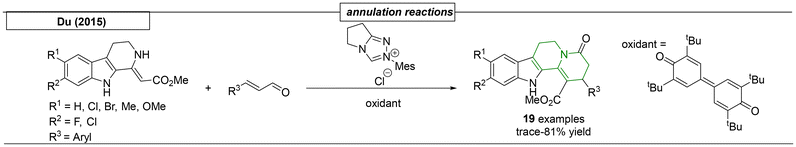 |
| | Scheme 25 Annulation reactions that have been used to prepare quinolizidines. | |
4.9 Other
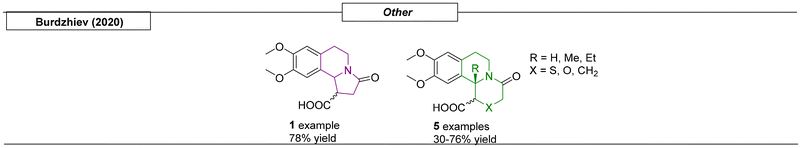
In 2020, the Burdzhiev research group synthesized a set of quinolizidines while developing a one-step route toward isoquinoline derivatives (Scheme 26).163 This straightforward method used dihydro-isoquinoline precursors, which were reacted with cyclic anhydrides to yield the corresponding N-bridgehead compounds.
 |
| | Scheme 26 Other reactions that have been used to obtain quinolizidines. | |
5 Other N-bridgehead compounds
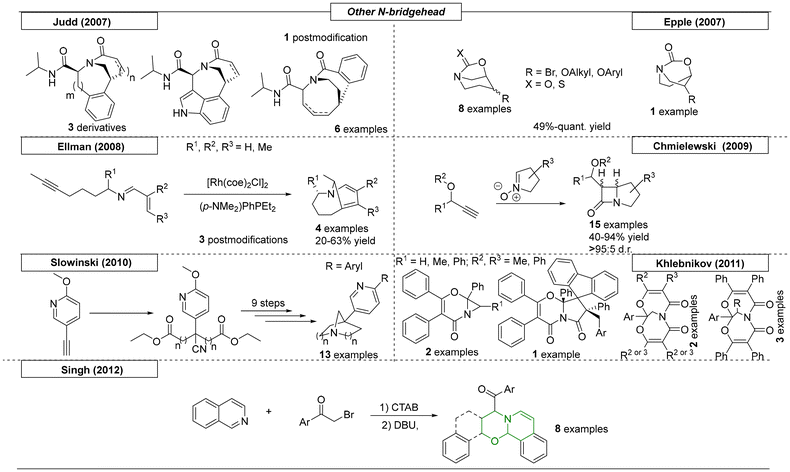
Other than the main scaffolds, pyrrolizidine, indolizidine and quinolizidine, a multitude of additional compounds classes exist bearing a bridgehead nitrogen atom. This section summarizes publications of N-bridged heterocycles that cannot be assigned to one of the main compound classes. In 2007, the Judd research group reported a concise construction of various bicyclic bridged lactam compounds (Scheme 27).164 In this publication, a three-step sequence utilizing simple starting materials was developed. The first reaction was an Ugi four component coupling reaction. The resulting diallyl compound was then used in an RCM and a Heck reaction to provide six novel bridged lactam compounds. In the same year, Epple and coworkers synthesized a set of bicyclic carbamates as non-peptide based inhibitors of papain-like cathepsin proteases.165 The core structure was identified during a high throughput screening based on monitoring cleavage rate of a coumarin-labelled synthetic tetrapeptide highly reactive to Cathepsin S. A set of compounds was synthesized starting from tetrahydropyridine, which was converted to its corresponding nitrogen-protected epoxide. After hydrolysis, the carbamate was formed.
 |
| | Scheme 27 Synthesis of other related N-bridgehead compounds. | |
A rhodium-catalysed, tandem reaction toward bicyclic enamines with bridgehead nitrogen was developed by Ellman and coworkers in 2008 (Scheme 27).166 The stereoselective transformation comprised activation, alkylation and electrocyclization steps. During this investigation, four examples and two post-modification examples were synthesized. By reacting cyclic nitrones with nonracemic acetylenes, the Chmielewski research group was able to synthesize fourteen novel N-bridgehead lactams with fused four- and five-membered rings.167 The asymmetric Kinugasa reaction was catalysed by copper(I) and gave access to the lactams in yields as high as 94% and diastereomeric ratios of up to 95:5.
Slowinski et al. synthesized novel azabicyclo-[2.2.1]heptane and -[3.3.1]nonane derivatives, which contained a pyridinyl substituent at the bridgehead position, via a ten step reaction sequence starting from commercially available (6-methoxy-pyridin-3-yl)acetonitrile (Scheme 27).168 The synthesized derivatives were subsequently evaluated in vitro for their affinity for α7-nicotinic receptors. The compounds were both highly active, with nanomolar EC50s, and selective, without significant affinity for the α4β2-nicotinic receptor subtype. Khlebnikov and coworkers published a non-concerted cycloaddition of 3-aryl, 2H-azirines and acylketenes that gave access to a set of N-bridgehead heterocycles.169 A cycloaddition reaction provided 3-membered ring systems, which were reacted with an additional acylketene to form complex bicyclic scaffolds. Structures were verified by X-ray analysis. A novel synthetic route toward fused heterocyclic oxa-aza-phenanthrene and -anthracene scaffolds was developed by the Singh research group in 2012.170 In this one-pot sequence, isoquinoline was reacted with various phenacyl bromides, and the resulting intermediates were subsequently reacted with styrene oxides. This process gave excellent yields, ranging from 90–98%, and is considered ‘green’ chemistry, because water is used as a solvent and DBU as an organic base. Additional structures and synthetic methodologies have been reviewed recently in separate works. As they have a small overlap with our review, we added these in the table below (Table 1).
Table 1 related reviews published from 2000–2023
| Year |
Title |
Ref. |
| 2017 |
Benzopyrroloxazines containing a bridgehead nitrogen atom as promising scaffolds for the achievement of biologically active agents |
171
|
| 2018 |
Synthetic approaches to non-tropane, bridged, azapolycyclic ring systems containing seven-membered carbocycles |
172
|
| 2018 |
A review on recent advances in nitrogen-containing molecules and their biological applications |
173
|
| 2018 |
Recent advances in the synthesis of bridgehead (or ring-junction) nitrogen heterocycles via transition metal-catalysed C–H bond activation and functionalization |
174
|
| 2019 |
Chemistry of bridged lactams: recent developments |
175
|
| 2021 |
Construction of heterocyclic bridgehead via de-aromatisation strategies |
176
|
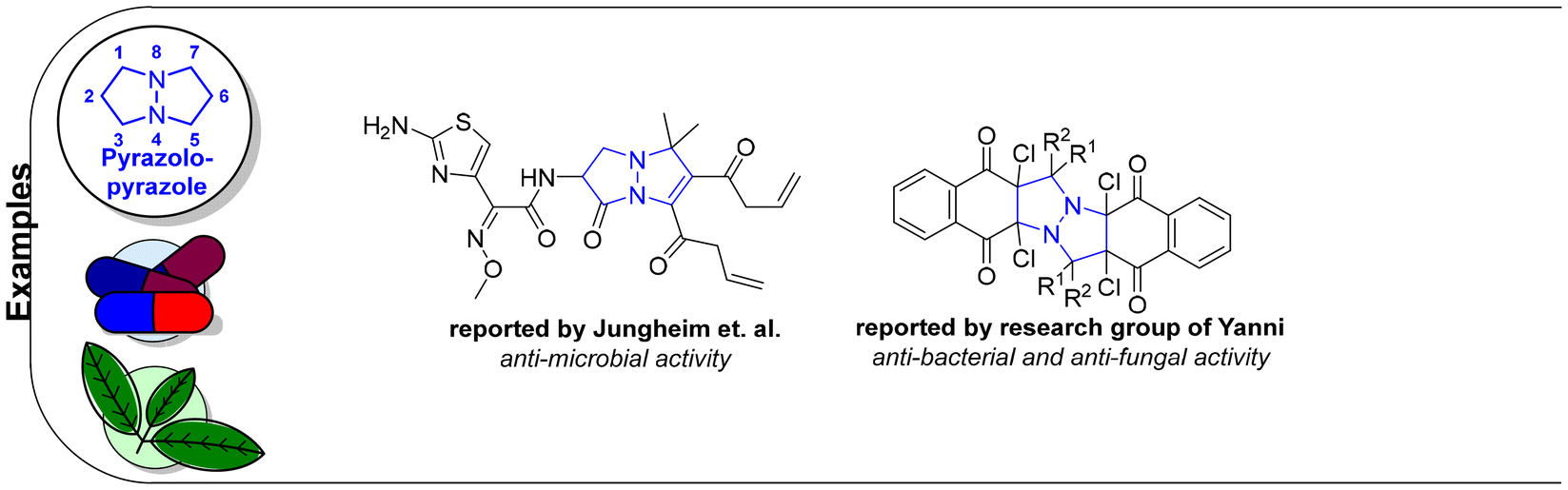
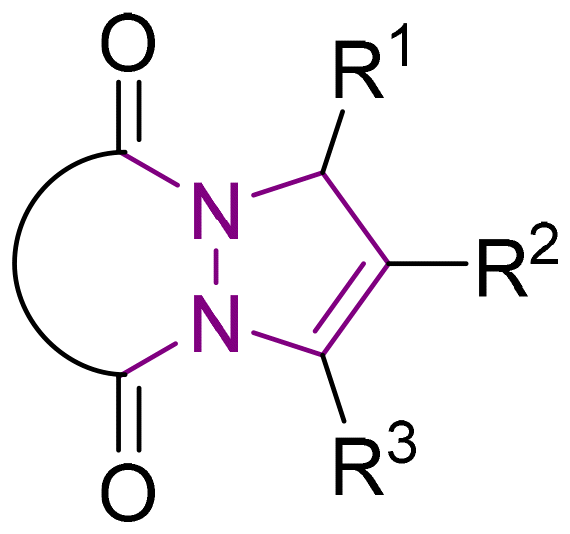
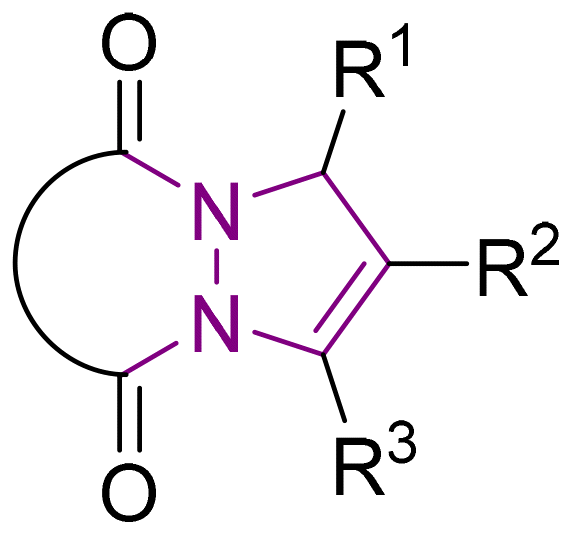
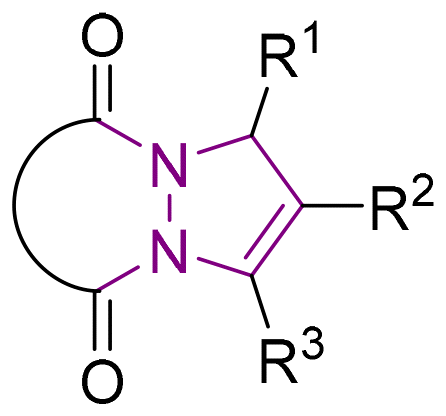
6 Tetrahydro-1H,5H-pyrazolo[1,2-a]pyrazoles
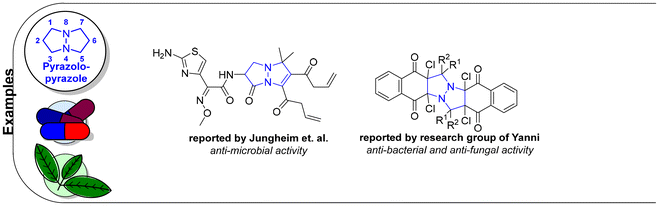
Relative to their single bridgehead nitrogen analogs, fewer studies have been published that feature pyrazolo-pyrazoles. However, they are also known to exhibit important bioactivity.177 For example, Jungheim et al. have reported synthetic efforts to access lactam analogs, which are known antibacterial agents (Fig. 5).178 In another example, the Yanni research group reported the antibacterial and antifungal activity of the pyrazolo-pyrazole scaffold. Here we have reviewed all synthetically available analogs.
 |
| | Fig. 5 Examples of pyrazolo-pyrazole-derived compounds. | |
6.1 Multistep procedures
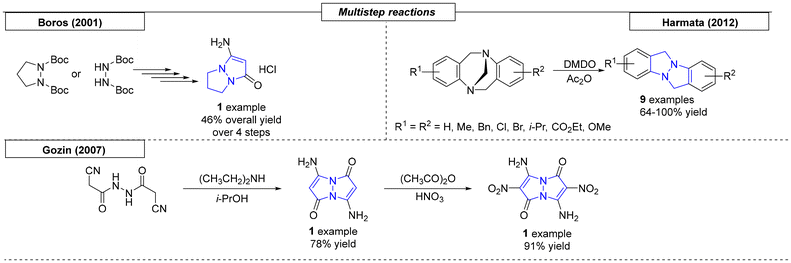
Three research groups have published multistep procedures that produce pyrazolo-pyrazoles. The first multistep approach reviewed here was described by Boros and coworkers in 2001 (Scheme 28).179 Starting from Boc-protected hydrazine, the authors were able to obtain the target pyrazole compound in four steps with a 46% overall yield. The Harmata research group employed a set of Trögers bases as starting materials to access nine examples of pyrazolo-pyrazole compounds.180 The two-step sequence started with the oxidation of one bridgehead nitrogen atom with dimethyldioxirane and concluded with a rearrangement in acetic anhydride to provide the desired scaffolds. In the last example reviewed here, Gozin and coworkers synthesized an ‘energetic butterfly’ in a four-step sequence.181 The authors started from the condensation of 2-cyano-N′-(2-cyanoacetyl)acetohydrazide and acetylacetone. The condensation product was converted into 3,7-diamino-1H-5H-pyrazolo[1,2-a]-pyrazole-1,5-dione bimane. Treatment of the pyrazolo-pyrazole derivative with concentrated nitric acid in acetic anhydride yielded the target compound. Because the molecule was both explosive and resembled a butterfly, it was referred to as an ‘energetic butterfly’ in the publication. However, it has been included here, because it adopts its explosive character only after the final nitration reaction.
 |
| | Scheme 28 Various multistep procedures have been used to prepare pyrazolo-pyrazole. | |
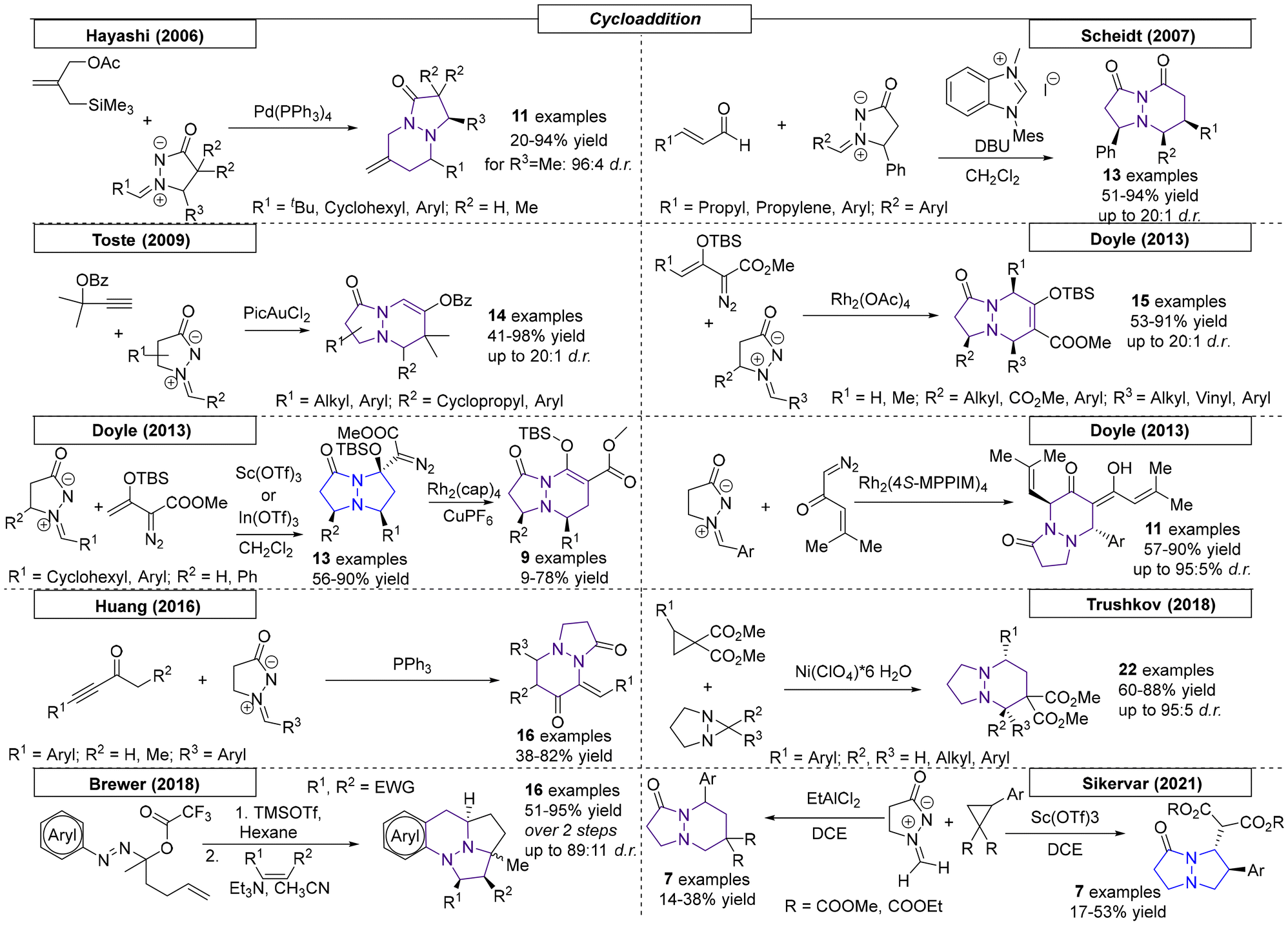
6.2 1,3-Dipolar cycloaddition reactions
By far the most developed synthetic methodology toward the pyrazolo-pyrazole motif is the 1,3-dipolar cycloaddition. Most of the reported approaches employ (E)-2-benzylidene-5-oxopyrazolidin-2-ium-1-ide in combination with a diene as shown in Scheme 29.
 |
| | Scheme 29 Generalized 1,3-dipolar cycloaddition reaction toward pyrazolo-pyrazoles. | |
Over the past twenty years over seventy publications have employed a 1,3-dipolar cycloaddition reaction to prepare a multitude of pyrazolo-pyrazole compounds. The first 1,3-dipolar cycloaddition toward multiple pyrazole scaffolds was described by the Sharpless research group in 2000 (Scheme 30).182 Azomethine precursors were synthesized in a four-step sequence and provided the corresponding twenty cycloadducts after reaction with various dipolarophiles. Products that were formed by attack on the less hindered site of the azomethine were favored by this reaction. In 2001, Svete and coworkers synthesized a novel (2Z)-2-(1,3-dimethyl-2,5-dioxoimidazolidin-4-ylidene)acetonitrile compound, which was converted to its 1-acetyl and 1-benzoyl analogs.183 By using azomethine, two pyrazolo-pyrazole-derived spiro compounds were obtained. Another 1,3-dipolar cycloaddition was published by Svete and coworkers in 2005.184 Various dipolarophiles were employed to provide twenty-three examples of pyrazolo-pyrazole derivatives in yields as high as 88%. In the same year, Svete and coworkers reacted β-ketoester enol dipolarophiles with various azomethines.185 Twenty-six diastereomerically pure compounds were synthesized in yields as high as 86%. In the same year, by swapping N-arylmaleimides for the β-keto esters, the Svete research group was able to synthesize additional examples of N-bridgehead compounds in yields as high as 89%.186 In some follow-up work in 2009, Svete and coworkers reported the reaction of alkynes and alkenes with the previously synthesized azomethine precursors.187 At no higher than 32%, the yields were relatively low. In the same year, Svete and coworkers reacted cis- or trans-dimethyl maleate with various benzyl-protected azomethine derivatives.188 The reaction selectively gave one diastereomer for six of the fourteen examples. In 2011, Svete and coworkers published a 1,3-dipolar cycloaddition reaction that provided five examples.189 In the course of their investigation, the structures of two representative compounds were unambiguously determined by NMR spectroscopy and X-ray diffraction. In a similar reaction published by the Chmielewski research group, alkynes substituted with electron withdrawing groups were used as dipolarophiles.190 By using various azomethines, ten derivatives were synthesized.
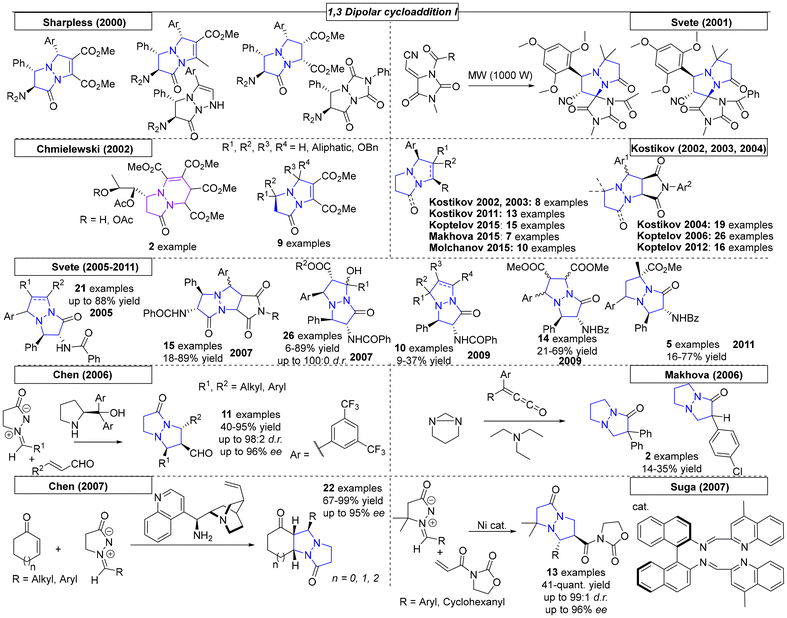 |
| | Scheme 30 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part I. | |
In three publications from 2002–2004, Kostikov and coworkers demonstrated that thermolysis of 6-aryl-1,5-diazabicyclo[3.1.0]hexanes resulted in azomethines, which could be utilized in 1,3-dipolar cycloadditions (Scheme 30).191–193 In the first report, N-arylmaleimides, alkenes and diphenylcyclopropenones were employed to provide thirty-two N-bridgehead compounds. To obtain insight into the diastereoselectivity of the 1,3-dipolar cycloaddition of azomethines and N-arylmaleimides, additional derivatives were synthesized by Kopetlov and coworkers. NMR studies were conducted on the twenty-six pyrazolo-pyrazoles in this and a subsequent study, but chemical yields were not reported.194,195 Koptelov and coworkers reported additional compounds provided by the reaction of stable azomethine imines and N-arylmaleimides with yields as high as 78% and stereoselectivities of up to 76:24.196 Following their procedure from 2011, an additional sixteen examples with dimethyl substitution on the azomethine imine backbone were prepared with yields as high as 82%.197 In the final Koptelov paper reviewed here, various N-arylmaleimides were employed to investigate the effects of steric hindrance in dipolar cycloadditions with azomethine imines.198 The authors found that the trans adduct is formed as the major product due to the favorability of the endo approach.
Makhova and coworkers described the dipolar cycloaddition of azomethine imines with various (arylmethylidene)malononitriles in dichloroethane (Scheme 30).199 The reaction showed complete regio- and stereoselectivity following the classical Michael mechanism, resulting in cis-positioned aryl groups. Molchanov and coworkers reported N-vinylpyrroles as highly efficient reactants in [3 + 2] cycloadditions.200 Heating various nitrile oxides and azomethine imines in a microwave reactor provided ten different N,N-bridgehead compounds in yields as high as 85% and with moderate cis–trans stereoselectivity. In 2006, the Chen research group reported a 1,3-dipolar cycloaddition of azomethine with various α,β-unsaturated aldehydes.201 By employing a chiral, secondary amine-derived organocatalyst to activate the aldehydes, diastereoselectivities of up to 98:2 and ee values of up to 96% were achieved. In the same year, the research group of Makhova investigated the reactivity of arylketenes with in situ-formed azomethines.202 In the course of this investigation, two novel pyrazolo-pyrazole derivatives were obtained, though with relatively low yields. Another organocatalytic approach toward pyrazolo-pyrazoles was published by Chen and coworkers in 2007.203 An organocatalyst bearing primary amine, tertiary amine and phenol functional groups enabled a reaction with various cyclic α,β-unsaturated ketones that provided twenty-two novel N,N-bridgehead compounds in yields as high as 99% and with up to 95% ee.
Suga and coworkers reported a highly enantioselective 1,3-dipolar cycloaddition reaction between azomethine imines and 3-acryloyl-2-oxazolidinone catalysed by a nickel(II) complex (Scheme 30).204 The oxygen atoms of 3-acryloyl-2-oxazolidinone interact nicely with the Ni catalyst. The reaction, which was carried out at room temperature, yielded the desired pyrazolo-pyrazoles in up to quantitative yield. Diastereomeric ratios of up to 99:1 and enantiomeric excesses of up to 97% were reported.
In 2008, the Sibi research group published a dipolar cycloaddition using dipolarophiles similar to those described by Suga (Scheme 31).205 In this work, a chiral copper(II) catalyst enabled the reaction at room temperature with yields as high as 90% and stereoselectivities of up to 96:4 dr and 98% ee. In 2009, Yamamoto and coworkers generated a set of benzynes in situ that were employed in a dipolar cycloaddition reaction with azomethine dipolarophiles.206 The benzynes were generated by potassium fluoride/18-crown-6 ether in THF and gave access to ten examples in yields as high as 68%. Similar to the work of Yamamoto, Larock and coworkers synthesized a set of novel pyrazolo-pyrazoles via the 1,3-cycloaddition of an azomethine with various in situ-generated benzynes.207 The novel derivatives were substituted on the backbone of the azomethine and were obtained in 19:1 regioisomeric mixtures with yields as high as 85%. A different approach for the 1,3-dipolar cycloaddition was enabled by Kobayashi and coworkers by employing a NOPF6 catalyst.208 By using various azomethines and vinyl ethers, fifteen N,N-bridgehead compounds were synthesized in yields as high as 82% and diastereoselectivities of up to 93:7.
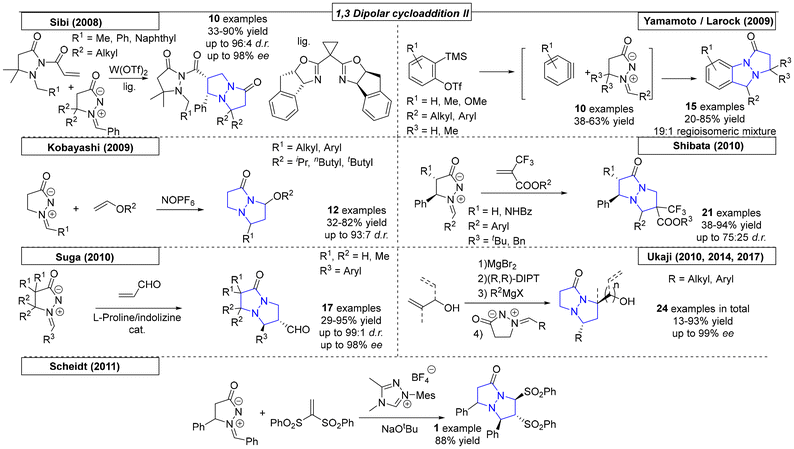 |
| | Scheme 31 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part II. | |
To access trifluormethylated heterocycles, Shibata and coworkers developed a noncatalytic methodology employing azomethine imines and (trifluoromethyl)prop-2-enoates as extremely electron deficient reagents (Scheme 31).209 Twenty-one examples were obtained in yields as high as 94% and with up to 75:25 dr. The regiochemistry was found to be opposite to that observed by Svete, which indicated that the electronic effect could be utilized to influence the resulting regiochemistry.188 In 2010, Suga and coworkers used S-proline or proline-derived compounds as catalysts to activate acrolein for a 1,3-dipolar cycloaddition reaction with various azomethine imine compounds.210 The investigations provided seventeen derivatives with yields as high as 95% and excellent selectivities, up to 99:1 dr and up to 98% ee. Another elegant approach toward pyrazoline-pyrazoles was published by the Ukaji research group in the same year.211 For the dipolar cycloaddition, a vinyl alcohol was activated by a Grignard reagent in the presence of a chelating chiral ligand; a precise mechanism was not proposed. Six compounds were prepared in yields as high as 74% and with excellent selectivities, up to 93% ee. In yet another example from 2010, a similar reaction was published by the same group that utilized homoallylic alcohols as dipolarophiles, which gave access to seven N,N-bridgehead compounds in yields as high as 93% and with up to 95% ee.212
Ukaji and coworkers utilized the asymmetric 1,3-dipolar cycloaddition reaction to de-symmetrize 1,4-pentadien-3-ol (Scheme 31).213 The magnesium-mediated, multi-nucleating chiral reaction system transferred chiral information from di-isopropyl (R,R)-tartrate, resulting in ee values of up to 98%. By further developing their highly enantioselective approach, the Ukaji research group synthesized five additional examples of N,N-bridgehead compounds, one of which was used as the starting point for the formal synthesis of Manzacidin C.214 An organocatalytic approach toward pyrazolo-pyrazoles was published by Scheidt and coworkers in 2011.215 In this publication, the use of vinyl sulfones in 1,3-dipolar cycloaddition reactions was investigated. One N-bridgehead compound was synthesized in 88% yield.
In 2011, Kwon and coworkers synthesized a multitude of N,N-bridgehead compounds via phosphine-catalysed [3 + N] reactions (Scheme 32).216 In addition to thirty-three pyrazolo-pyrazole compounds, 5,6-, 5,7- and 5,8-fused ring systems were synthesized. To access linear diaza-triquinanes, the Li research group developed a methodology employing an azomethine imine and sugar-derived hex-5-enals.217 The stereocontrolled cycloaddition was catalysed by triethylamine and yielded the corresponding diaza-triquinanes in up to 90% yield. In the same year, a metal-catalysed, 1,3-dipolar cycloaddition approach was published by Yu and coworkers.218 The authors employed chromium or tungsten Fisher carbene complexes, which were reacted with azomethine imines. After treatment of the intermediates with pyridine N-oxide, sixteen N,N-bridgehead compounds were obtained with yields as high as 89%. In 2012, Matsuyama and coworkers published an asymmetric conjugate addition–cyclization reaction.219 Deprotonation of pyrazolidine with n-BuLi enabled the 1,3-dipolar cycloaddition reaction. The authors synthesized an additional example of a 5,6-fused ring system by using hexahydropyridazine as the starting material.
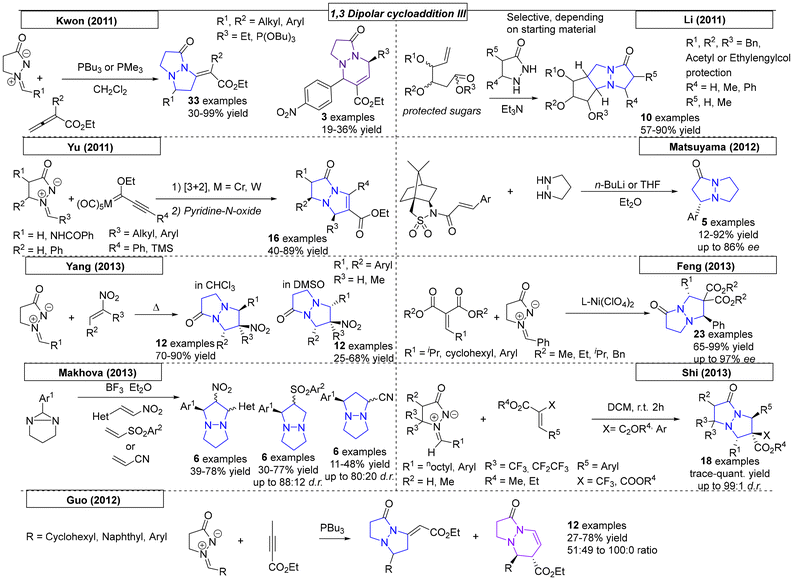 |
| | Scheme 32 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part III. | |
Many examples were published in 2013. Yang and coworkers employed various nitrostyrene compounds as dipolarophiles in a 1,3-dipolar cycloaddition reaction with azomethine imines (Scheme 32).220 The formation of the corresponding diastereomer was found to depend on the solvent choice; both diastereomers were selectively synthesized in up to 78% yield and 100:0 dr. In the same year, the Feng research group utilized a nickel complex as a catalyst for a 1,3-dipolar cycloaddition reaction between various alkylidene malonate derivatives and azomethine imines.221 Twenty-three pyrazolo-pyrazole compounds were prepared with excellent yields (up to 99%) and ee values as high as 97%. Investigation of the relationship between the enantiomeric excesses of the ligand and the resulting product revealed a slightly positive, nonlinear effect. Makhova and coworkers reported a novel, 1,3-dipolar cycloaddition reaction between in situ generated azomethine imines and six different nitrostyrene derivatives.222 The diastereoselective reaction, which tolerated different heterocycles as nitrostyrene substituents, was carried out in acetonitrile or [bmim][PF6] solvents with yields as high as 78% for five examples. The same group used similar reaction conditions in a dipolar cycloaddition reaction between various azomethine derivatives and acrylonitrile or 4-nitrophenyl vinyl sulfones.223 These reactions were carried out in ionic liquid solvent and gave opposite diastereoselectivities for the two substrates, which was rationalized by quantum chemistry calculations. Shi and coworkers developed a novel methodology toward trifluoromethylated heterocycles.224 The reaction, which employed azomethine imine and trifluoromethyl-functionalized olefins, yielded eighteen examples in up to quantitative yield. The electron-deficient olefins had either CF3 or CF2CF3 substituents on R5. The Guo research group reported phosphine-catalysed [3 + 2] and [3 + 3] annulations similar to those published by Kwon in 2011 that lead to various N,N-bridgehead compounds. Depending on the starting material, 5,5- and/or 5,6-fused ring systems were prepared in yields as high as 78% and of up to 100:0 dr.225
While studying the synthesis of dual conformation tricycles, Wulff and coworkers developed a dipolar cycloaddition reaction of azomethine imines and various vinyl sulfones (Scheme 33).226 Bicyclic vinyl sulfones can be used to synthesize, for example, an inhibitor of viral neuraminidase. NMR was used to confirm the fluxional behavior of synthesized compounds in solution at room temperature. The Guo research group used a phosphor-derived catalyst for the synthesis of various dinitrogen fused heterocycles.227 In the course of this work, twenty-four examples were synthesized in yields as high as 93%. Three cyclic systems with one bridgehead nitrogen were also prepared.
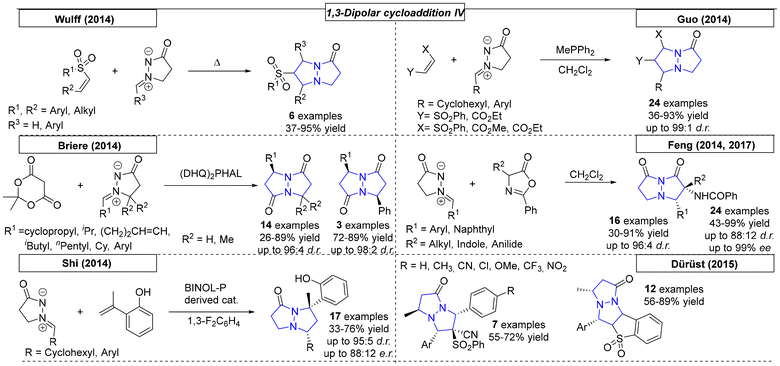 |
| | Scheme 33 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part IV. | |
An organocatalytic approach was developed by Briere and coworkers (Scheme 33).228 Interestingly, R1 was confirmed to be opposite of the expected site of the derived N,N-bridgehead compound, as confirmed by X-ray analysis. This multicomponent domino Knoevenagl-aza-Michael cyclo-condensation process used (DHQ)2PHAL as a multifunctional catalyst (except in three cases, where DIPEA was used) to provide the desired compounds in yields as high as 89% and with up to 98:2 dr. A catalyst-free variation of the 1,3-dipolar cycloaddition of azomethine imines was published by the Feng research group.229 The authors prepared N-(1,7-dioxotetrahydropyrazolo[1,2-a]pyrazol-2-yl)benzamide derivatives in dichloromethane at room temperature. The proposed mechanism included a rearrangement step after the cycloaddition. In total, sixteen examples were reported. Later, Feng and coworkers reported a reaction of azlactones with azomethine imines via an asymmetric formal [3 + 2]-cycloaddition.230 The reaction was catalysed by a chiral bifunctional bisguanidinium hemisalt and provided twenty-four compounds with high diastereomeric ratios (up to 20:1 dr) and high enantioselectivities (up to 99% ee). Another organocatalytic approach was reported by the Shi research group in 2014.231 In this work, a chiral BINOL-phosphate catalyst was used in the reaction of azomethine imines and o-hydroxystyrenes that provided eighteen examples of N,N-bridgehead compounds in yields as high as 76% and with selectivities of up to 95:5 dr and 88:12 er.
In 2015, Dürüst and coworkers reacted various azomethane imines with benzothiophene 1,2-dioxide under microwave conditions to obtain twelve N,N-bridgehead compounds (Scheme 33).232 Regio- and stereochemistry were confirmed by X-ray analysis. In a second publication, the same research group published a dipolar cycloaddition reaction between various azomethane imines and 2-phenylsulfonyl acrylonitrile derivatives.233 They showed that the resulting seven compounds can also be obtained in lower yields by using (E)-3-(2-hydroxyphenyl)-2-(phenyl-sulfonyl)acrylonitrile, however, two equivalents of the corresponding azomethane imines were required.
In 2016, Damljanović and coworkers used aluminum(III) chloride and zirconium(IV) chloride as Lewis acids to enable the [3 + 2]-cycloaddition reaction of enones and azomethine imines (Scheme 34).234 The two diastereomers formed in the reaction were easily separable, and the configurations of the diastereomers were confirmed by X-ray analysis. The Svete research group published a method for the synthesis of N,N-bridgehead compounds that was inspired by the Huisgen or ‘click’ reaction.235 Similar to the [3 + 2]-dipolar azide–alkyne cycloaddition, copper was used to enable the cycloaddition of azomethine imines and alkynes in high yields. The scope of this novel approach was also demonstrated in various model reactions, the most recent having been reported in 2022.177 The syntheses of six of the ten compounds prepared have already been described in literature. Fluorescent compounds were prepared that also possessed promising properties, including a remarkably large Stokes shift (150–268 nm). The authors showed that the emission wavelengths and intensities of these compounds were influenced by solvent polarity and pH.
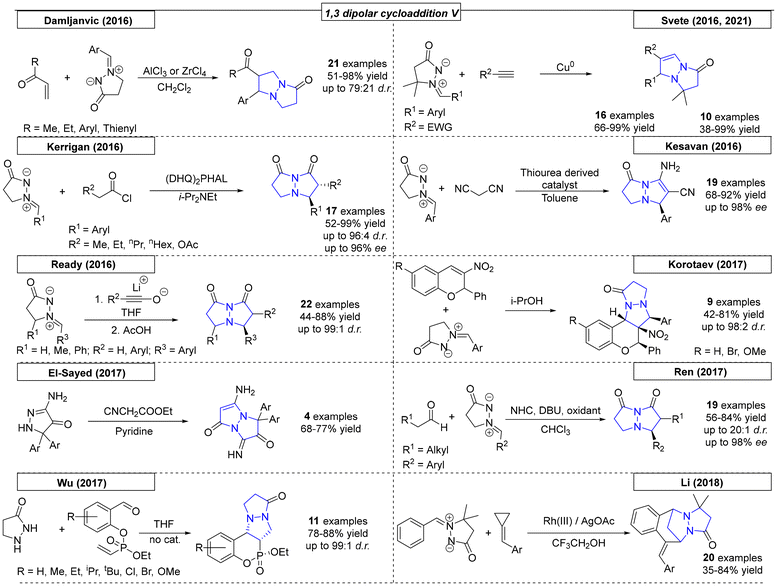 |
| | Scheme 34 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part V. | |
Kerrigan and coworkers reported an alkaloid-catalysed, enantioselective approach to the reaction of azomethine imines with in situ-formed ketenes (Scheme 34).236 When an organocatalyst ((DHQ)2PHAL) and Hünig's base (i-Pr2NEt) were used to form the ketene, yields as high as 99% and stereoselectivities of up to 96:4 dr and 96% ee were obtained. In another organocatalytic approach, a bifunctional thiourea-derived catalyst enabled the synthesis of N,N-bridgehead compounds.237 The procedure, reported by Kesavan and coworkers, used malonitrile, which can undergo dipolar cycloaddition after deprotonation. The thiourea catalyst both deprotonates the malonitrile and transfers stereo information, resulting in ee values as high as 98%.
In 2016, Ready and coworkers reported the reaction of azomethines with various ynolate dipolarophiles (Scheme 34).238 The ynolates were generated in situ from the corresponding alkynes using t-BuOOH and LiHMDS. In total, twenty-two examples with yields as high as 88% and selectivities of up to 99:1 dr were obtained. In 2017, the Korotaev research group reacted 3-nitro-2-phenyl-2H-chromenes with various azomethine imines in a highly stereoselective fashion.239 The reaction proceeded over two days and gave access to nine N,N-bridgehead compounds. The configurations of the stereoisomers were confirmed via X-ray analysis. In the same year, El-Sayed and coworkers reported the synthesis of novel pyrazole derivatives for use as antineoplastic agents.240 In addition to various nitrogen-containing scaffolds, four pyrazolo-pyrazole examples were synthesized. Unfortunately, the authors evaluated the bioactivity of only eleven compounds, and no N,N-bridgehead compounds were included.
The Ren research group reported the oxidative, asymmetric [2 + 3] annulation of aldehydes with azomethine imines (Scheme 34).241 For this reaction, an N-heterocyclic carbene bearing stereochemical information was used as a catalyst, while DBU was used as both a base and an oxidizing agent. Nineteen examples were produced with high enantioselectivities, up to 98% ee. A one-pot domino condensation, intramolecular 1,3-dipolar cycloaddition of 2-(vinylphosphoryloxy)benzaldehydes with azomethines was reported by Wu and coworkers.242 The reaction sequence does not require a catalyst and yielded the phosphadihydrocoumarin-fused N,N-bicyclic pyrazolidin-3-ones in yields as high as 88% and with diastereomeric ratios of up to 99:1. The Li research group reported a protocol for the C–H activation of alkylidenecyclopropanes with a rhodium catalyst, which enabled their reaction with azomethine imines.243 Twenty compounds were afforded by the broad substrate scope of this protocol, which can also be used for the reaction of nitrones.
In two separate publications in 2018, Damljanović and coworkers reported the synthesis of some new scaffolds that are likely to draw interest from biological investigators (Scheme 35).244 The reaction utilized aluminium chloride as a Lewis acid. In the course of this work, it was shown that these compounds exhibit strong antioxidant properties and good antimicrobial activities. The Kang research group reported a variation of the 1,3-dipolar cycloaddition with a chiral rhodium catalyst that employed α,β-unsaturated 2-acyl imidazoles and azomethine imines as reagents.245 The methodology was notable for its broad substrate scope, high yields (up to 99%) and excellent diastereomeric ratios (up to 20:1) and enantiomeric excesses (up to 99%). Ramachary and coworkers reported the synthesis of various N,N-bicyclic pyrazolidine derivatives starting from enolizable aldehydes and azomethine imines.246 The enolates were generated using KOtBu or DBU as a base. Reduction of the obtained compounds removed the hydroxyl group and resulted in eighteen additional derivatives.
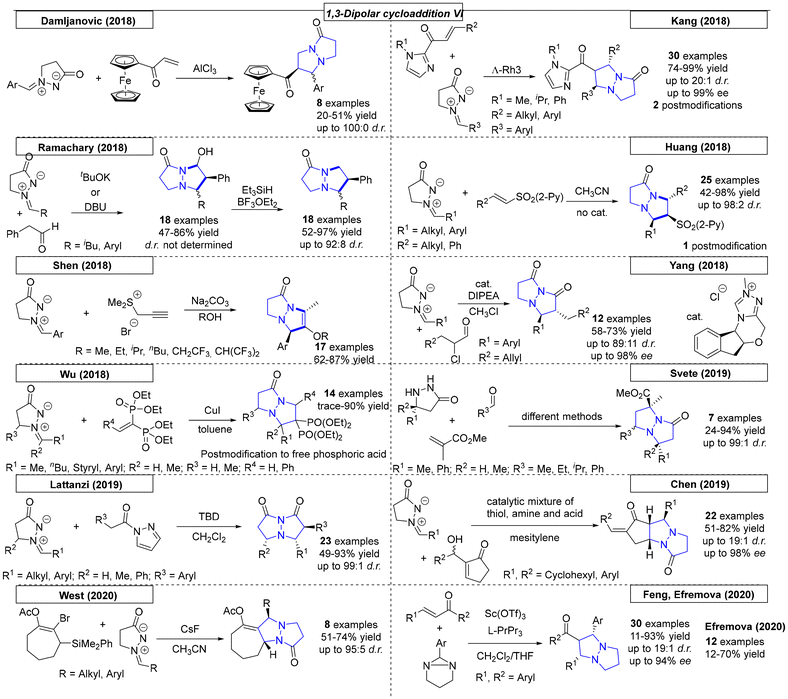 |
| | Scheme 35 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part VI. | |
In 2018, the Huang research group described the synthesis of pyrazolidinones and isoxazolidines using fluorinated α,β-unsaturated 2-pyridylsulfones as electron deficient dipolarophiles (Scheme 35).247 The catalyst- and additive-free procedure provided twenty-five examples in yields as high as 98% and diastereoselectivities of up to 98:2. The observed stereoselectivity was further investigated via DFT calculations. As a postmodification, it was possible to remove the SO2(2-Py) group from the ring system. By generating novel allelic sulfonium salts by deprotonation of propargyl sulfur ylides, Shen and coworkers were able to synthesize a set of novel N,N-bicyclic pyrazolidinone derivatives.248 The alcohol solvent takes part in this reaction as a nucleophile, substituting –OR for –SMe2. In 2018, Yang and coworkers published a chiral N-heterocyclic carbene-catalysed dipolar cycloaddition of α-chloroaldehydes with various azomethine imines.249 The enolates, which act as dipolarophiles, were generated in situ by carbine-catalysed formal elimination of HCl from the α-chloroaldehydes. The dipolar cycloaddition products were obtained in yields as high as 73% and selectivities of up to 89:11 dr and 98% ee. The Wu research group used copper(I) iodide to catalyze the [3 + 2] cycloaddition of hindered vinylidenebisphosphonates with azomethine imines toward novel phosphate-substituted pyrazolo-pyrazoles.250 The fourteen examples were converted to their corresponding free phosphoric acids by reaction with Me3SiBr.
Svete and coworkers used a one-pot method to synthesize a series of tetrahydro-1H,5H-pyrazolo[1,2-a]pyrazole-1-carboxylates and showed that these compounds selectively inhibit the dihydroorotate dehydrogenase (DHODH) mitochondrial enzyme of Plasmodium falciparum, a parasite that causes malaria (Scheme 35).251 The most active derivatives had an anti-configuration and a methyl ester functional group and showed selective inhibition of the parasite enzyme relative to the human enzyme. Free carboxylic acids, carboxamides and derivatives with a syn-configuration showed no activity. In 2019, Lattanzi and coworkers investigated the cycloaddition of pyrazoleamides to azomethine imines catalysed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene.252 The pyrazoleamides were enolizable with the pyrazole moiety functioning as a leaving group. Depending on the reactants, yields as high as 93% and diastereomeric ratios of up to 99:1 were obtained. In the same year, a formal [5 + 3] cycloaddition of azomethine imines with unmodified Morita–Baylis–Hillman alcohols was published by the Chen research group.253 The usage of a double catalytic system was reported to be crucial for the observed chemo- and enantioselectivities. Products were obtained in yields as high as 82% and with selectivity of up to 19:1 dr and 98% ee. The authors proposed a reaction mechanism and identified key intermediates via mass spectrometry. West and coworkers developed a methodology for the generation of 1,2-cycloheptadienes, which were reacted with various 1,3-dipoles, including azomethine imines.254 The resulting pyrazolo-pyrazoles were obtained in yields as high as 74% and diastereomeric ratios of up to 95:5.
In 2020, the Feng research group reported a highly enantioselective method for the synthesis of pyrazolo-pyrazoles that employed a chiral scandium(III) N,N′-dioxide catalyst (Scheme 35).255 The 1,3-dipole azomethine imine was generated in situ from meso-diaziridines and reacted with a chalcone, which was activated by ligand exchange from the scandium complex. The reaction provided selectivity of up to 19:1 dr and up to 94% ee. The Efremova research group used microwave irradiation to react similar chalcone substrates with azomethine imines.256 In this work, no second diastereomers were reported, and the configurations of the products were supported by X-ray analysis. In the same year, a similar approach was reported by Shibasaki and coworkers.257 In this case, an indium(III) complex with a chiral ligand was used to transfer stereo information and activate the chalcone-derived substrates (Scheme 36).
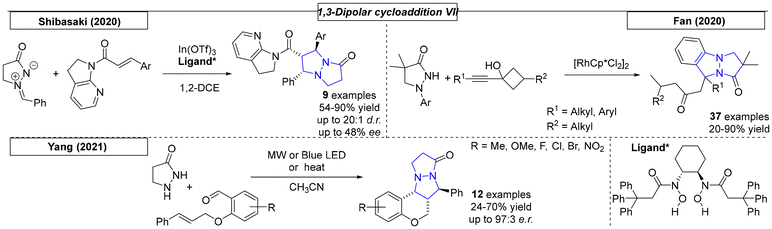 |
| | Scheme 36 Pyrazolo-pyrazoles via 1,3-dipolar cycloaddition reactions, part VII. | |
Fan and coworkers developed a Rh(III)-catalysed reaction toward pyrazolo[1,2-a]pyrazolones and 2-acylindoles, dependent on the chosen reaction conditions (Scheme 36).258 The optimized conditions, sodium acetate and dichloromethane, gave selective access to N,N-bridgehead compounds in high yields (up to 90%) and with a broad substrate scope (thirty-nine derivatives). Absolute configurations of the products were not explicitly determined in this work. The synthesis of similar derivatives was reported by the Yu research group in a publication from the same year.259 The Yang research group accessed tetrahydrochromeno[4,3-c]pyrazolo[1,2-a]pyrazol-9-ones via a novel dipolar cycloaddition of in situ-formed azomethines to 2-(cinnamyloxy)benzaldehyde.260 The reaction was performed using three different sources of energy (heating, microwave or 3 W blue LED irradiation) and yielded only one diastereomer in all cases. The absolute configurations of the products were determined by X-ray analysis.
6.3 Cascade reactions
There are many examples of cascade and multicomponent reactions that have been used to produce pyrazolo-pyrazoles. In 2001, Samyn and coworkers reported the unexpected reaction of various α,β-unsaturated esters with hydrazine hydrate (Scheme 37).261 Reaction temperatures of greater than 200 °C were required to favour the cycloaddition reaction over the competing Michael addition reaction. In the same year, the Mohamed research group reported the synthesis of a series of novel polyfunctional group-substituted pyrazole azo dyes; four core structures and sixteen dyes were obtained.262 The dyes were also investigated for their ability to provide new shades of color on nylon and acetate fabrics. In a very similar work, the El-Kharadly research group reported the synthesis of three additional, novel pyrazolo-pyrazole dyes.263 The azomethine precursors were synthesized by reacting 5-methyl-2,4-dihydro-3H-pyrazol-3-one with various diazo reagents. Treatment of the resulting derivatives with ethylcyanoacetate gave the corresponding N,N-bridgehead compounds as yellow or orange dyes.
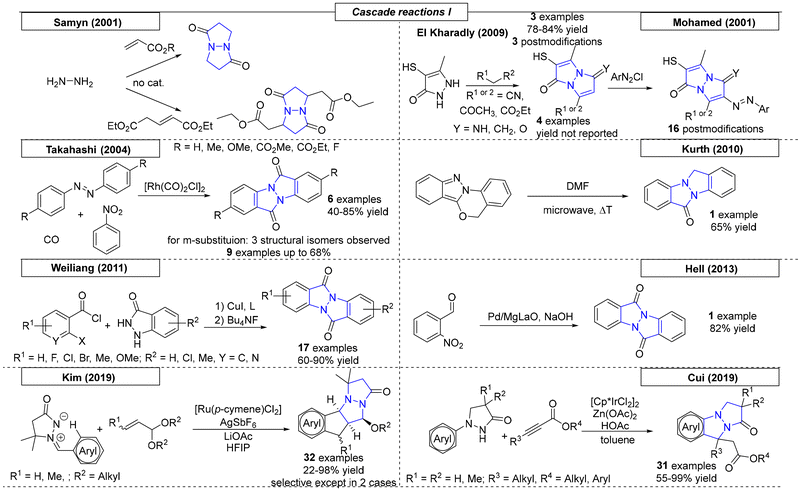 |
| | Scheme 37 Cascade and multicomponent reactions that have been used to produce pyrazolo-pyrazoles. | |
In 2004, Takahashi and coworkers reported the formation of various pyrazolo-pyrazole compounds via the reaction of azobenzene derivatives, carbon monoxide and nitrobenzene catalysed by a rhodium complex (Scheme 37).264meta-Substituted regio-isomers resulted from reaction at the two positions ortho to the diazo functional group. Kurth and coworkers reported the isolation of an N,N-bridgehead compound by microwave irradiation of indazole in DMF.265 In a later publication, the same rearrangement was observed, but it was catalysed by a nucleophile.266 In 2011, a copper-catalysed cascade process was reported by the Weiliang research group.267 The reaction utilized various 2-halobenzoyl chlorides and 1,2-dihydroindazol-3-ones in an N-acylation/intramolecular C–N coupling cyclization process sequence and provided seventeen derivatives in yields as high as 90%. The Hell research group observed the formation of a pyrazolo-pyrazole derivative while investigating the palladium-catalysed chain elongation of aromatic nitro compounds.268 The reported example was isolated in 82% yield.
Kim and coworkers developed a reaction sequence that consisted of an acrolein oxonium formation/tandem C–H allylation/[3 + 2] dipolar cycloaddition for the synthesis of a set of thirty-two novel pyrazolo-pyrazole compounds (Scheme 37).269 Post modifications, including late stage C–H functionalization and diazocine formation of complex bioactive molecules, demonstrated the potential applicability of this method to medicinal chemistry. Cui and coworkers reported an iridium-catalysed annulation of pyrazolidinones with propiolates.270 In the subsequent C–H bond activation/[4 + 1] cyclization that proceeds under mild reaction conditions the propiolates act as C1-synthons. In total, thirty-one examples were reported with yields as high as 99%.
In 2020, an indazolone-assisted sequential ortho-alkenylation-oxidative aza-Michael addition was developed by the Sakhuja research group (Scheme 38).271 In this work, the Rh(II)-catalysed reaction of acrylates and 1-arylindazolones provided a set of twenty-four indazolo[1,2-a]indazolylidenes in up to 89% yield. The imido group acted as a directing group for the C–H activation. The next example of a cascade sequence that lead to the formation of the pyrazolo-pyrazole scaffold was reported by Fan and coworkers in 2021.272 In this publication, a catalytic system consisting of a ruthenium complex and an antimony salt provided the reactive species that was necessary for the 1,3-dipolar cycloaddition. Anticancer testing of the resulting compounds revealed significant activity; some compounds were superior to the positive control, 5-fluoro uracil. In the same year, a very similar rhodium-catalysed approach was published by Zhou and coworkers.273 The procedure, which included a C–H coupling/intramolecular cyclization step, provided twenty-five examples in yields as high as 70% and with up to 97:3 er. The reaction pathways were also investigated using Gibbs free energy calculations. Gogoi and coworkers developed an unprecedented procedure toward all-carbon quaternary carbon-centered indolo[1,2-a]quinazolinones and indazolo[1,2-a]indazolones.274 The latter can be obtained by reacting dialkyl alkynes with phenylindazolone after Csp2–H activation, alkyne insertion and a 1,2-phenylshift. In total, eight examples were reported.
 |
| | Scheme 38 cascade and multicomponent reactions. | |
6.4 Criss-cross cycloaddition reactions
The first example of a ‘criss-cross’ cycloaddition reaction approach toward pyrazolo-pyrazoles was published by Domingues and coworkers in 2005 (Scheme 39).275 Various diaryl azines were reacted with maleic acid derivatives, such as maleic anhydride, maleimide, and N-alkyl and N-arylmaleimides. In total, twenty-five examples with yields as high as 94% were reported. In the same year, the Potácek research group was able to synthesize 3-substituted homo allenylaldazines, which were used in an intramolecular ‘criss-cross’ reaction.276 Nitrogen-containing branched and heterocyclic aliphatics and phenyl were introduced as R-groups. Snider and coworkers reported a ‘criss-cross’ reaction of (R)-citronellal-derived azine that provided a C2-symmetric pyrazolo-pyrazole and its diastereomer.277 These compounds were subsequently converted into eight-membered heteroatom-containing ring systems. The last example of a ‘criss-cross’ reaction toward the pyrazolo-pyrazole scaffold was reported by Yanni and coworkers in 2020. Substituted 2,3-diaz-1,3-butadienes were reacted with 2,3-dichloro-1,4-naphthoquinone in benzene to obtain bisnaphth[2,3-c]pyrazolidinetetrones in yields as high as 65%.278 These compounds were shown to exhibit antifungal and antibacterial activity.
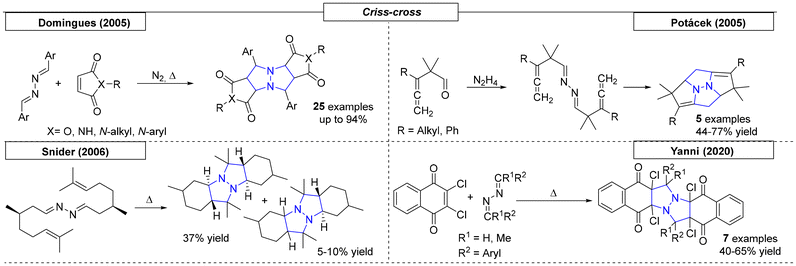 |
| | Scheme 39 Synthesis of pyrazolo pyrazoles using ‘criss-cross’ cycloaddition reactions. | |
7 5,6-Ring systems
The pyrazolo-pyridazine structure is similar to that of indolizidine, except that it contains two bridgehead nitrogen atoms (Fig. 6). This compound class is known to exhibit biological activity. For example, antiexudative activity with low toxicity has been reported.279 In a very well-explored one-pot pathway toward the pyrazolo-pyridazine scaffold, the reaction of a cyclic anhydride derivatives and hydrazine resulted in an intermediate that reacts with a condensate of a CH2-acidic compound and an aldehyde. In over 150 publications, the reaction has been tested with various catalytic systems, resulting in a multitude of different derivatives. A general reaction equation and mechanism is shown in Scheme 40.
 |
| | Fig. 6 Examples of various pyrazolo-pyridazine-derived compounds. | |
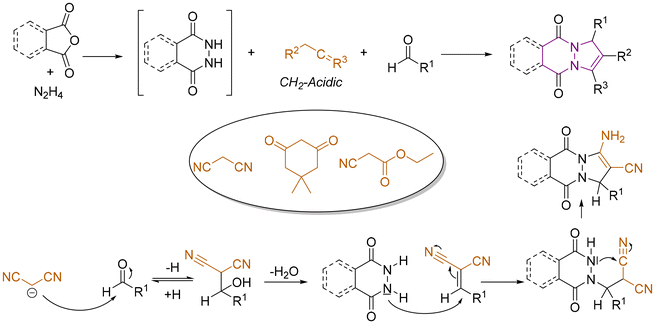 |
| | Scheme 40 General reaction equation and mechanism for synthesis of pyrazolo-pyridazines via 1,3-dipolar cycloaddition reactions. | |
7.1 1,3-Dipolar cycloaddition reactions
As stated before, many different methodologies exist for the synthesis of pyrazolo-pyridazines using a 1,3-dipolar cycloaddition reaction, as shown in Scheme 41.
 |
| | Scheme 41 Overview of 1,3-dipolar cycloaddition reaction approaches leading to the pyrazolo-pyridazine scaffold. | |
The first examples of 1,3-dipolar cycloaddition reactions used to produce pyrazolo-pyridazines reviewed here involve polymeric catalysts. In total, seven different approaches were reported (Table 2). While Shaterian,280 Hasaninejed,281 Deshmukh282 and Mouradzadegun283 used polymers functionalized with sulphur-based groups, Abedini284 and Schirini285 employed PVP and PVP-based polymers. The last example, reported by Mohamadpour,286 used a cellulose polymer functionalized with carboxymethyl groups.
Table 2 Pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions catalysed by polymeric catalysts
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2011 |
Starch sulfate |
20 |
72–92% |
280
|
| 2011 |
Sulfuric acid-modified PEG-6000 |
23 |
80–93% |
281
|
| 2015 |
Polymer-supported sulfonic acid |
12 |
80–92% |
282
|
| 2018 |
Sulfamic acid functionalized nano-catalyst |
11 |
0–98% |
283
|
| 2019 |
[PVPH]ClO4 |
20 |
90–97% |
284
|
| 2020 |
[PVP-SO3H]Cl |
22 |
47–96% |
285
|
| 2020 |
Carboxymethyl cellulose |
20 |
77–94% |
286
|
The 1,3-dipolar cycloaddition reaction can also be conducted without the benefit of a catalyst (Table 3). While the Patel287 and Davoodnia288 research groups used ethanol as a solvent, Khalafi-Nezhad289 and Sadek (with micro wave irradiation)290 used glycerol.
Table 3 Pyrazolo-pyridazine-yielding catalyst-free 1,3-dipolar cycloaddition reactions
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2012 |
EtOH |
16 |
69–83% |
287
|
| 2015 |
Glycerol |
23 |
89–94% |
288
|
| 2019 |
Glycerol, MW irradiation |
14 |
80–92% |
289
|
| 2019 |
EtOH, reflux |
6 |
70–91% |
290
|
There are examples of 1,3-dipolar cycloaddition reactions toward pyrazolo-pyridazines that use combinations of catalytic systems and irradiation, either microwave or ultrasonic (Table 4). Bazgir and coworkers used triethylamine in EtOH under ultrasonic irradiation conditions.291 A microwave irradiation example using montmorillonite as a catalyst was published by the Jeong research group.292 Rostamnia,293 Homayoon294 and Helal295 developed three additional ultrasonic irradiation reactions. A final microwave example, published by Kamanna and coworkers, was catalysed by burned mango peel.296
Table 4 Pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions supported by ultrasonic or microwave irradiation
| |
Year |
Condition |
Examples |
Yield |
Ref. |
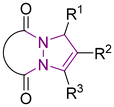
|
2010 |
US, Et3N, EtOH |
21 |
85–98% |
291
|
| 2012 |
MW, montmorillonite K-10, solv. free |
20 |
80–96% |
292
|
| 2013 |
US, SBA-15 sulfonic acid nanoreactor, EtOH |
9 |
92–96% |
293
|
| 2016 |
US, TBAF, H2O |
12 |
86–95% |
294
|
| 2017 |
US, Et3N, EtOH |
4 |
71–88% |
295
|
| 2020 |
MW, burned mango peel, EtOH |
12 |
83–89% |
296
|
Over the last two decades, interesting examples of 1,3-dipolar cycloaddition reactions using heterogeneous catalysis were reported (Table 5). The majority of heterogeneous systems feature catalysts supported on silica, as published by the research groups of Shaterian,297 Sabitha,298 Rostamnia,299 Reddy,300 Javanshir,301 Doustkhah,302 and Jeong.303 Other solid materials include solid acid catalysts as published by the research groups of Shaterian,304 Shaterian,305 Das,306 Shirini,307 and Massah.308 It is important to note that many of the resulting compounds have been converted to their corresponding sulfonamides and tested for antibacterial activity against S. aureus and E. coli. Tayebee309 and Nikoorazm310 reported the use of alumina- and copper metal-based catalytic systems. A final example, reported by the Jonnalagadda research group, was catalysed by powdered eggshells.311
Table 5 Pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions performed in a heterogeneous catalytic system
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2008 |
Silica-SO3H, solvent free |
8 |
80–91% |
297
|
| 2010 |
Phosphomolybdic acid-SiO2 |
19 |
80–92% |
298
|
| 2013 |
Phosphoric acid supported on alumina |
15 |
76–93% |
305
|
| 2013 |
Cellulose-SO3H |
14 |
0–91% |
304
|
| 2014 |
Carbon-based solid acid catalyst |
18 |
80–94% |
306
|
| 2014 |
Mesoporous/trifluoroethanol |
8 |
89–94% |
299
|
| 2014 |
Silica gel-supported tungstic acid |
20 |
85–93% |
300
|
| 2014 |
SO3H-functionalized mesoporous silica |
16 |
90–95% |
301
|
| 2015 |
Al-SBA-15-TPI/H6P2W18O62 |
21 |
45–84% |
309
|
| 2016 |
[PVP-SO3H]HSO4, solv. free |
18 |
88–95% |
307
|
| 2017 |
Diatomite-SO3H |
15 |
73–83% |
308
|
| 2018 |
Cu(II)-adenine-MCM-41 |
9 |
85–95% |
310
|
| 2019 |
Mesoporous SBA-15/PIDA |
8 |
85–95% |
302
|
| 2019 |
Silica-supported tungstic acid, solv. free |
16 |
87–94% |
303
|
| 2020 |
Eggshell powder |
16 |
93–98% |
311
|
Many examples of 1,3-dipolar cycloaddition reactions toward pyrazolo-pyridazines using organocatalytic systems have been reported (Table 6). While the research groups of Roy (L-proline),312 Bouraiou (DMAP),313 Lalitha,314 (2-aminopyridne) and Mohamadpour (Theophylline)315 catalysed the reaction using organic bases, the groups of Singh,316 Farahmand,317 Jeong,318 Saghanezhad,319 and Karhale320 established acid-catalysed reaction procedures. Similar approaches have been reported with organic salts as catalysts by the research groups of Li,321 Wu,322 Guo,323 Shirini,324 and Mamaghani.325 The research groups of Mohamadpour,326 Gill327 and Dalal328 reported the use of various sugars catalysts.
Table 6 Pyrazolo-pyridazine-yielding organocatalytic 1,3-dipolar cycloaddition reactions
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2011 |
(S)-Camphorsulforic acid |
20 |
90–94% |
316
|
| 2012 |
Citric acid |
12 |
81–92% |
317
|
| 2012 |
Proline triflate, solv. free |
10 |
87–94% |
321
|
| 2014 |
Methylimidazolium-3-yl-1-ethyl sulfate |
26 |
80–93% |
322
|
| 2015 |
β-Cyclodextrine-SO3H |
26 |
85–95% |
318
|
| 2016 |
Saccarin |
18 |
82–93% |
326
|
| 2016 |
L-Proline |
12 |
87–90% |
312
|
| 2016 |
Caffeine-H2SO4 |
10 |
45–96% |
319
|
| 2017 |
β-Cyclodextrin |
19 |
77–94% |
328
|
| 2017 |
DMAP |
12 |
85–97% |
313
|
| 2017 |
β-Cyclodextrin |
15 |
82–93% |
327
|
| 2018 |
Hydroxyethylammonium acetate |
13 |
61–96% |
323
|
| 2018 |
NS-[C4(DABCO-SO3H)2]·4Cl, solv. free |
23 |
85–96% |
324
|
| 2019 |
[cmdmim]I, EtOH |
15 |
71–89% |
325
|
| 2020 |
5-Sulphosalicylic acid |
14 |
0–92% |
320
|
| 2020 |
2-Aminopyridine |
14 |
90–98% |
314
|
| 2020 |
Theophylline |
23 |
75–93% |
315
|
The only example of an electrocatalytic approach toward pyrazolo-pyridazines was reported by Kefayati and coworkers in 2014 (Table 7).329
Table 7 Pyrazolo-pyridazine-yielding electrocatalytic 1,3-dipolar cycloaddition reactions
| |
Year |
Condition |
examples |
Yield |
Ref. |
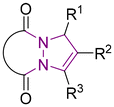
|
2014 |
Electrolysis, NaBr, PrOH |
11 |
85–98% |
329
|
Procedures toward pyrazolo-pyridazine scaffolds that use inorganic catalysts to enable a 1,3-dipolar cycloaddition reaction are summarized in the following paragraph (Table 8). In the cases of procedures reported by Guan,330 Reddy,331 Karimi-Jaberi,332 Khan,333 Foroumadi,334 Turgut,335 Varala,336 Ethiraj,337 Türkan338 and Taslimi,339 inorganic salts were used in combination with ethanol as a solvent. The Türkan research group tested the synthesized derivatives in assays that implicate Alzheimer's disease and diabetes mellitus. Kidwai and coworkers established a procedure that employed ceric ammonium nitrate and PEG-400 to access over fifty derivatives in two publications.340,341 Nickel-based complexes in acetic acid were used by Ebrahimipour and coworkers in two publications.342,343 A final example, reported by the Shajari research group, used zirconium nitrate as a catalyst and water as a ‘green’ solvent.336
Table 8 Pyrazolo-pyridazine-yielding metal-/metal salt-catalysed 1,3-dipolar cycloaddition reactions
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2012 |
(NH4)2[Ce(NO3)6], PEG 400 |
19 |
62–94% |
340
|
| 2012 |
NiCl2, EtOH |
17 |
71–94% |
330
|
| 2014 |
(NH4)2[Ce(NO3)6], PEG 400 |
40 |
84–92% |
341
|
| 2014 |
InCl3, EtOH |
24 |
70–85% |
331
|
| 2015 |
K2CO3, EtOH |
10 |
85–96% |
332
|
| 2015 |
[M + II(pzca)2(H2O)2]; M = (Ni, Co, Cu), AcOH |
7 |
80–94% |
342
|
| 2015 |
[Fe2(SO4)3H2O], EtOH |
21 |
79–93% |
333
|
| 2017 |
NiCl2·6H2O, EtOH |
21 |
70–95% |
334
|
| 2018 |
Mixed-ligand Ni(II) schiff base complex, AcOH |
6 |
84–94% |
343
|
| 2019 |
Y(OTf)3, EtOH |
5 |
76–86% |
335
|
| 2020 |
Cs2CO3, TBAB, EtOH |
16 |
80–95% |
336
|
| 2020 |
[ZrO(NO3)2·2H2O], H2O |
3 |
75–83% |
344
|
| 2020 |
(NH4)2[Ce(NO3)6] |
|
2–95% |
337
|
| 2020 |
Cu(OTf)2, EtOH |
6 |
—/— |
338
|
| 2020 |
Y(OTf)3, EtOH |
4 |
76–83% |
339
|
Over the last twenty years, various solvent-free approaches toward pyrazolo-pyridazines have been reported (Table 9). The majority of these procedures feature metal salts or metal-derived compounds as catalysts, as reported by the research groups of Zhang (H4SiW12O40),345 Mosaddegh (Ce(SO4)2),346 Azarifar (Cu(CH3CN)4PF6),347 Bazgir (ZrOCl2·8H2O),348 Jeong (InCl3),349,350 and Mohamadpour (Cu(OAc)2·H2O).351 Organocatalysts were featured in the procedures developed by the research groups of Bazgir,352 Wu,353 Ghorbani-Vaghei,354,355 Kidwai,356 Wang357 and Kiasat.358 Other solvent free methodologies include: iodine as reported by Varghese and coworkers,359 HFIP as described by Maleki and coworkers,360 B(OSO3H)3 as reported by the Soheilizad research group361 and simple inorganic compounds as described by the research groups of Davoodnia,362 Gomha363 and Habibi.364
Table 9 Solvent free 1,3-dipolar cycloaddition reactions that provide pyrazolo-pyridazines
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2008 |
p-PTSA |
13 |
73–97% |
352
|
| 2010 |
H4SiW12O40 |
17 |
60–95% |
345
|
| 2010 |
TCT |
11 |
89–97% |
353
|
| 2011 |
TBBDA, PBBS |
20 |
51–91% |
354
|
| 2011 |
Ce(SO4)2 |
10 |
71–95% |
346
|
| 2012 |
Cu(CH3CN)4PF6 |
14 |
76–92% |
347
|
| 2012 |
DPA |
16 |
79–96% |
356
|
| 2013 |
p-TSA |
12 |
83–92% |
357
|
| 2013 |
PSA |
11 |
75–98% |
358
|
| 2013 |
I2 |
13 |
87–95% |
359
|
| 2013 |
ZrOCl2 |
7 |
80–89% |
348
|
| 2013 |
InCl3 |
25 |
83–94% |
349
|
| 2014 |
TBBDA or PBBS |
14 |
63–90% |
355
|
| 2014 |
HFIP |
16 |
75–92% |
360
|
| 2014 |
B(OSO3H)3 |
12 |
65–94% |
361
|
| 2015 |
NaHCO3 |
13 |
85–92% |
362
|
| 2016 |
[Cu(OAc)2H2O] |
15 |
73–89% |
351
|
| 2018 |
NaOH, grinding |
10 |
73–92% |
363
|
| 2019 |
InCl3 |
35 |
85–94% |
350
|
| 2021 |
KHSO4 or CF3SO3H |
11 |
27–86% |
364
|
Ionic liquids can be employed in the synthesis of pyrazolo-pyridazines (Table 10). These versatile reagents can be used as a solvent, a catalyst or for both purposes. Bazgir,365 Khurana,366 and Fazaeli,367 reported the use of [bmim]-based ionic liquids as solvents in combination with various acid catalysts. Interestingly, many systems have been reported that use an ionic liquid as both a solvent and a catalyst. Singh,368 Xu,369 Shaterian,370 Shaterian,371 He,372 Habibi,373 Abedini,374 Shirini,375 Tayebee,376 Godajddr,377 Kermani,378 Kermani,379 Masihpour,380 Shirini,381 Raghavendra,382 Shaikh,383 Amirmahani,384 Rad-Moghadam,385 Shirini,386 and Keshavarz387 reported the use of inorganic ionic liquids, while Mombani Godajdar388 used an imidazole dimer-based ionic liquid in combination with FeCl4− as a counter ion. In a final example, the Nakhaei research group389 reported a reaction using an ionic liquid containing SnCl3 as a counter ion. Three research groups showed that an ionic liquid could be used as catalyst in combination with various polar solvents. Deshmukh and coworkers reported an example where water was used as a ‘green’ solvent.390 The Hongyun research group published twelve examples where ethanol was used as a solvent,391 and He and coworkers performed an ionic liquid-catalysed reaction using methanol as a solvent.392
Table 10 Pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions that employ ionic liquids
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2008 |
[bmim]Br, PTSA |
13 |
73–97% |
365
|
| 2009 |
[bmim]BF4, H2SO4 |
22 |
83–94% |
366
|
| 2010 |
[bmim]BF4, H3PW12O40 |
6 |
82–92% |
367
|
| 2011 |
[bmim]OH |
26 |
89–98% |
368
|
| 2011 |
[(CH2)4SO3HMIM]HSO4 |
10 |
55–86% |
369
|
| 2012 |
([Hnhp][HSO4]) and other ionic liquids |
15 |
70–95% |
370
|
| 2012 |
DBU[CH3COO], [Pyrr][HCOO] |
28 |
0–94% |
371
|
| 2012 |
[BSO3HmIm]HSO4 |
19 |
70–96% |
372
|
| 2013 |
[C4(MIm)2](FeCl4)2 |
12 |
85–90% |
388
|
| 2014 |
[DMSBP][HSO4] |
14 |
0–94% |
373
|
| 2015 |
[SuSA-H] HSO4 |
19 |
88–97% |
374
|
| 2015 |
DABCO(HSO3)2(HSO4)2 |
23 |
83–95% |
375
|
| 2015 |
[Simp]HSO4 |
17 |
33–88% |
376
|
| 2015 |
PEG-MIDL |
12 |
85–90% |
377
|
| 2016 |
[Et3N–SO3H]Cl |
17 |
80–95% |
378
|
| 2016 |
[bmim]OH |
10 |
80–93% |
379
|
| 2016 |
[Et3N–SO3H]HSO4 |
14 |
85–98% |
380
|
| 2017 |
[H-Suc]HSO4 |
2 |
89–97% |
381
|
| 2018 |
[DBUH][OAc] |
8 |
30–83% |
382
|
| 2018 |
[Bu3NH][HSO4] |
13 |
85–94% |
383
|
| 2020 |
[TBP]2SO4 |
28 |
65–95% |
384
|
| 2020 |
[Bn-DBU][TFA] |
26 |
76–93% |
385
|
| 2020 |
[[C4(H-DABCO)2]HSO4]4 |
17 |
87–96% |
386
|
| 2021 |
Alkylated phenanthroline and piperazine ClO4 IL |
13 |
86–96% |
387
|
| 2021 |
[DMDSP]Cl, [DMDSP]SnCl3 |
13 |
88–98% |
389
|
| 2013 |
[C8DABCO]Br, H2O |
12 |
85–92% |
390
|
| 2016 |
[bmim]OH, EtOH |
12 |
84–97% |
391
|
| 2019 |
ChCl/p-TsOH, MeOH |
11 |
83–93% |
392
|
Nanoparticles can be used as catalysts for 1,3-dipolar cycloaddition reactions toward pyrazolo-pyridazines (Table 11). Metal-based nanoparticles, such as iron or iron oxide-based nanostructures, are the most commonly used nanomaterials.393 Metal nanomaterials were successfully employed in the synthesis of pyrazolo-pyridazines by Azarifar (ZnO),394 Kiasat (Al),395 Pal (Ni),396 Safaei-Ghomi (CuI),397 Mosaddegh (Ca/Cu based),398 Albadi (Co/Al2O3-based),399 and Maleki (Cu/ZnO-based).400 These procedures do not require an additional solvent.
Table 11 Pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions that employ nanomaterials as catalysts
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2012 |
Nano-ZnO, solv. free |
14 |
86–92% |
394
|
| 2013 |
Nano-alumina, solv. free |
12 |
70–98% |
395
|
| 2013 |
Ni-NP, solv. free |
14 |
88–94% |
396
|
| 2014 |
CuI-NP, solv. free |
12 |
82–93% |
397
|
| 2015 |
Ca2CuO3/CaCu2O3/CaO, solv. free |
10 |
80–94% |
398
|
| 2017 |
Co/Al2O3, solv. free |
15 |
88–94% |
399
|
| 2020 |
Cu-doped ZnO hollow spheres |
17 |
81–93% |
400
|
| 2012 |
Fe3O4-NP, (3-aminopropyl)-Si(OEt)3 coat., solv. free |
28 |
0–93% |
401
|
| 2013 |
Fe3O4-NP supported guanidine, solv. free |
21 |
80–93% |
402
|
| 2013 |
Fe3O4-NP, silica coated@sulforic acid, solv. free |
15 |
84–94% |
403
|
| 2013 |
Fe3O4-NP, N-propylsulfamic acid |
20 |
82–93% |
404
|
| 2014 |
Fe3O4@ SiO2-NP, solv. free |
17 |
85–96% |
405
|
| 2014 |
CoFe2O4-CS-SO3H-NP, solv. free |
22 |
83–95% |
421
|
| 2015 |
Ni0.5Zn0.5Fe2O4@HAP-Cs2CO3, solv. free |
16 |
76–98% |
416
|
| 2016 |
Urea-functional. silica-coat. Fe3−xTixO4 NP, solv. free |
10 |
87–98% |
417
|
| 2016 |
Nano-FGT, solv. free |
19 |
87–97% |
422
|
| 2016 |
Fe3O4@GO-Pr-SO3H, EtOH |
8 |
90–95% |
406
|
| 2016 |
Fe3O4@SiO2-imid-PMAn, solv. free |
35 |
73–96% |
407
|
| 2016 |
PrxCoFe2−xO4 (x = 0.1), EtOH |
12 |
84–94% |
418
|
| 2017 |
STA-amine-Si-magnetite, CH3OH |
27 |
76–99% |
419
|
| 2017 |
H3PW12O40@Fe3O4/EN-MIL-101, solv. free |
4 |
31–94% |
408
|
| 2017 |
Fe3O4@SiO2–ZrCl2-MNPs |
18 |
85–95% |
409
|
| 2019 |
Nano-NiFe2O4@TiO2-ILPip, PEG 400 |
13 |
85–97% |
420
|
| 2019 |
Cu@Fe3O4 MNP, solv. free |
16 |
81–96% |
410
|
| 2020 |
Fe3O4@GOQDs-N-(β-alanine) |
9 |
90–98% |
411
|
| 2020 |
g-Al2O3/BFn/Fe3O4, solv. free |
8 |
90–98% |
412
|

|
2020 |
g-Al2O3/BF3/Fe3O4, solv. free |
10 |
50–98% |
413
|
| 2021 |
Fe3O4@SiO2-imine/phenoxy-Cu(II), solv. free |
8 |
88–96% |
414
|
| 2022 |
PAmPDA@Fe3O4 nanocomposite, solv. free |
—/— |
—/— |
415
|
| 2014 |
SBA-Pr-NH2, solv. free |
10 |
70–92% |
423
|
| 2014 |
SBA-Pr-SO3H, solv. free |
11 |
70–90% |
424 and 425 |
| 2015 |
SBA@BiPy2 + Cl− |
10 |
86–96% |
426
|
| 2015 |
RH@[SiPrDABCO@BuSO3H]HSO4, EtOH |
12 |
86–94% |
427
|
| 2015 |
SBA-15-Ph-SO3H, solv. free |
9 |
75–96% |
428
|
| 2015 |
Nanosilica supported dedritic polymer, solv. free |
27 |
74–95% |
429
|
| 2020 |
RH@DABCO, EtOH |
15 |
80–95% |
430
|
| 2020 |
SiO2–H2SO4 NP, EtOH |
10 |
85–95% |
431
|
Many examples of this reaction using iron-derived nanomaterials have been published, most commonly with Fe3O4 nanoparticles: Shaterian,401 Rostami,402 Kiasat,403 Rostami,404 Ghasemzadeh,405 Rostamnia,406 Esmaeilpour,407 Lee,408 Shirini,409 Druzhinin,410 Khaleghi Abbasabadi,411 Bamoniri,412 Taherpour,413 Ebrahimiasl414 and Pourmousavi (tested for anti-bacterial activity)415 (Table 11). Other systems feature iron-containing metal composite materials such as those established by Maleki,416 Azarifar,417 Zaheer,418 Arora419 and Hamidinasab.420 Zhang prepared CoFe2O4 sulfonic acid-derived nanoparticles, which were employed under solvent free conditions.421 Pal and coworkers synthesized nineteen derivatives with nano-FGT.422
Functionalized silica-based nanoporous materials were established by three different research groups (Table 11). The first two procedures, developed by Ziarani and coworkers, employed SBA-Pr-NS-functionalized nanomaterials with additional amine and sulphate functionalization.423–425 Kiasat and coworkers developed a method using a silica-based nanocomposite and a nano-Brønsted solid acid to access the desired derivatives.426,427 The final example reviewed here, published by the Veisi research group, employed a sulfonic acid-functionalized SBA under solvent free conditions.428
In addition to silica-based nanoporous materials, functionalized silica nanoparticles can be used in the synthesis of pyrazolo-pyridazines (Table 11). The Esmaeilpour research group used a nanosilica-supported dendrimer as a catalyst.429 Other examples used nanomaterials that were either base-functionalized as published by Rezaee and coworkers (DABCO)430 or acid-functionalized as reported by the Sadeghi research group.431
As discussed in previous sections, nanoparticles and ionic liquids are effective catalytic systems for this reaction. It is therefore not surprising that additional procedures have been developed that combine these approaches (Table 12). Mohammadpoor-Baltork,432 Rezaee,433 Mohammadpoor-Baltork434,435 and Davoodnia436 used silica-derived materials for ionic liquid functionalization. Other examples used metal-derived nanoparticles as reported by the research groups of Ghorbani-Vaghei (Fe3O4),437 Paul (Ti),438 Tayebee ([Simp]3PW12O40)439 and Kalhor (Mn@zeolite-Y).440
Table 12 Pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions that employ nanopartical and ionic liquids
| |
Year |
Condition |
Examples |
Yield |
Ref. |
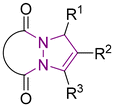
|
2013 |
IL@nano-SiO2, solvent free |
12 |
77–96% |
432
|
| 2015 |
[SBA-Im]HSO4, EtOH |
13 |
83–90% |
433
|
| 2017 |
Piperidinium benzene-1,3-disulfonate nanomagnetic ionic liquid, solvent free |
26 |
60–94% |
437
|
| 2011 |
Basic ionic liquid ([SiPMIM]OH@MNPs), solvent free |
9 |
82–96% |
434
|
| 2017 |
Phosphotungstic acid-containing ionic liquid immobilized on CuFe2O4@SiO2, solvent free |
10 |
88–95% |
436
|
| 2017 |
Ionic liquid coated sulfonated carbon@titania composites, water |
12 |
80–96% |
438
|
| 2017 |
Heteropolyacid-based ionic liquid [Simp]3PW12O40 nanoparticle, solvent free |
19 |
0–92% |
439
|
| 2018 |
[MSPP]HSO4@nSiO2, solv. free |
18 |
63–96% |
435
|
| 2020 |
4-Methylpyridinium chloride ionic liquid grafted on Mn@zeolite-Y, EtOH/H2O |
16 |
88–98% |
440
|
In the last section, special cases are summarized (Table 13). The research groups of Perumal,441 Zhang,442 Nabid,443 Wang,444 and Shi445 reported pyrazolo-pyridazine derived spiro-compounds. A review summarized additional reactions.446 The Salehi447 and MaGee448,449 research groups expanded the one-pot synthesis of pyrazolo-pyridazines with a ‘click’ reaction. By exchanging an alkyne-derived compound for malononitrile, a subsequent dipolar cycloaddition reaction was conducted. In a very similar one-pot procedure, published by both the Shaabani450 and Yoosefian451 research groups, alkyne and iso-cyano compounds were reacted with maleic acid and hydrazine. The Fu research group used various vinylketones and phthalhydrazide in the synthesis of highly functionalized pyrazolo-pyridazines.452 It was possible to run this domino reaction at gram scale. Bouraiou and coworkers developed a method for the synthesis of carbazole-fused heterocycles.453 Instead of isolating the pyrazolo-pyridazine, phenylhydrazine was added to the crude mixture, resulting in pyrazolo-pyridazine carbazole-fused structures. In two publications, the Gencer research group used nitro-pyridazolo-pyridazines as starting points for various post modification reactions.454,455 After reduction of the nitro group, various lactate and urea derivatives were synthesized. Mohebat and coworkers reacted benzo[a]phenazin-5-oles with phthalhydrazide and benzaldehydes toward quinoxaline-fused compounds.456 The one-pot procedure was catalysed by Fe3O4 nanoparticles and provided fused heterocyclic compounds in yields as high as 86%.
Table 13 Special case examples of pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2010 |
L-Proline, EtOH |
10 |
86–92% |
441
|
| 2011 |
NiCl2, PEG 600 |
26 |
80–95% |
442
|
| 2012 |
NEt3, EtOH |
3 |
93–96% |
443
|
| 2012 |
Piperidine, EtOH |
14 |
42–93% |
444
|
| 2013 |
Piperidine, CH3CN |
11 |
74–89% |
445
|
| 2013 |
Dipyridine/Fe3O4, review |
REVIEW |
|
446
|
|
2011 |
Cu(OAc)2, Na L-ascorbate, [Hmim](CF3COO) |
14 |
75–92% |
447
|
| 2012 |
Cu(OAc)2, Na L-ascorbate, p-TSA, EtOH |
19 |
79–95% |
448
|
| 2014 |
Cu(OAc)2, Na L-ascorbate, [Hmim](CF3COO) |
16 |
65–82% |
449
|
| 2012 |
EtOH/acetone |
15 |
54–73% |
450
|
| 2012 |
Acetone |
5 |
42–59% |
451
|

|
2017 |
Phosphotungstic acid, PhCN |
19 |
0–95% |
452
|
| 2017 |
1. TFAA/HOAc, 2. PhNH-NH2 |
9 |
56–87% |
453
|
| 2013 |
—/— |
16 |
48–84% |
454
|
| 2015 |
—/— |
10 |
26–53% |
455
|
| 2017 |
Fe3O4-MNP, PEG-400 |
10 |
75–86% |
456
|
The last three examples use catalytic systems that do not fit into the previous sections (Table 14). Nagarapu and coworkers employed trimethylsilyl chloride as a catalyst and acetonitrile as a solvent.457 The Ma research group developed a methodology that employed elemental iodine as a catalyst in ethanol.458 Finally, sodium hydroxide was established as a readily available and inexpensive catalyst by the Sangani research group.459
Table 14 Other pyrazolo-pyridazine-yielding 1,3-dipolar cycloaddition reactions
| |
Year |
Condition |
Examples |
Yield |
Ref. |

|
2009 |
TMSCl, CH3CN |
16 |
86–95% |
457
|
| 2011 |
I2, EtOH |
11 |
86–96% |
458
|
| 2015 |
NaOH, EtOH |
16 |
70–89% |
459
|
7.2 Metal-catalysed reactions
There are many examples of metal-catalysed reactions that have been used to produce pyrazolo-pyridazines. In 2002, Amarasekara and coworkers reported the reaction of two furfural derivatives with 1,4-phthalazinedione to form two pyrazolo-pyridazine compounds in yields as high as 64% (Scheme 42).460 This [4 + 2] dipolar cycloaddition reaction was catalysed by lead acetate. In 2013, the Li research group developed a novel silver-catalysed tandem addition/cyclization reaction of alkyne azomethines with various soft nucleophiles.461 In this work, ketones, nitroalkanes, water, and terminal alkynes were used to form forty-two derivatives in yields as high as 90%. Though the chemoselectivities were high, diastereoselectivities were moderate to low. Huang and coworkers reported a rhodium-catalysed C–H activation reaction that provided novel N,N-bicyclic pyrazolo-pyridazine derivatives.462 The ortho C–H activation was found to be directed by the pyrazolidine moiety, which initiated the formation of the desired benzopyridazine analogs in yields as high as 92%. A ruthenium-catalysed synthetic approach was reported by the Kianmehr research group in 2018.463 The reaction sequence included an alkenylation/annulation step, was initiated by a C–H activation and was carried out in water. In total, twenty derivatives were synthesized in yields as high as 99% and with up to 95:5 dr.
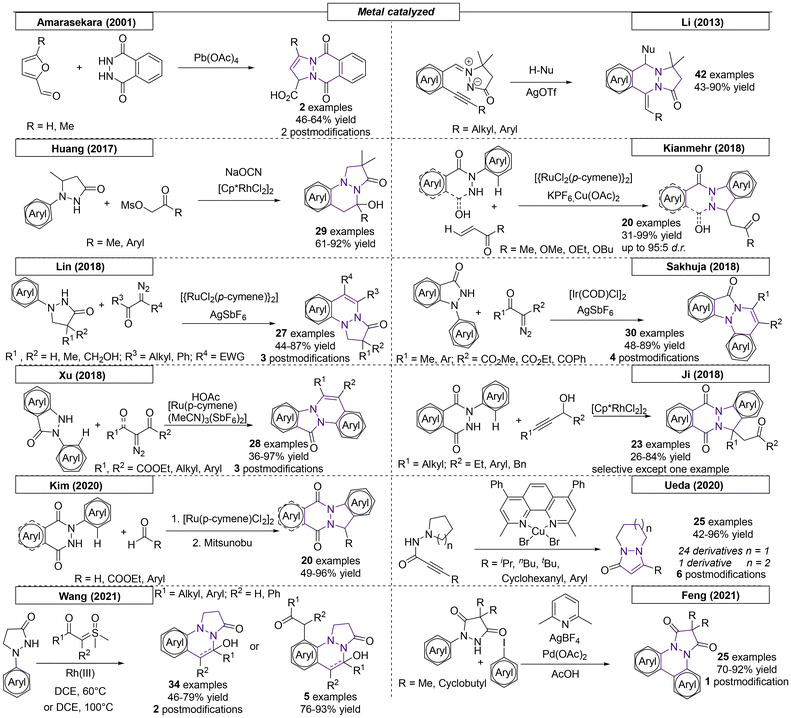 |
| | Scheme 42 Metal-catalysed reactions that produce pyrazolo-pyridazines. | |
Lin and coworkers reported a redox neutral annulation of pyrazolidinones with diazo compounds that provided twenty-seven derivatives that had the pyrazolo-pyridazine core structure (Scheme 42).464 The reaction sequence, which was catalysed by a ruthenium complex, was described as a C–H activation/cyclization/dehydration process and gave only N2 and H2O as byproducts. In addition to a broad substrate scope, three postmodifications were reported. In the same year, a similar approach was published by Sakhuja and coworkers, who used an iridium complex instead of a ruthenium complex.465 In total, thirty examples and four postmodifications were reported. The Xu research group reported a related reaction in which diazo reagents were reacted with 2-phenylindazol-3-ones.466 In this sequence, C–H ruthenation, Ru(II)-carbene formation, migratory insertion, and condensation take place in a sequential fashion. The scope included twenty-eight examples with yields as high as 97% and three postmodifications. Additional derivatives were reported in a follow-up publication in which the bioactivities of the novel molecules were also investigated.467 Ji and coworkers reported a rhodium-catalysed [4 + 1] cyclization via C–H activation that was used to synthesize various quaternary carbon-containing heterocycles.468 The reaction, which featured various heterocyclic and propargyl alcohol starting materials, provided twenty-three pyrazolo-pyridazine examples in yields as high as 84% and with excellent stereoselectivities in almost all cases.
In 2020, the Kim research group developed a C–H hydroxyalkylation/Mitsunobu/cyclization sequence starting from N-aryl phthalazinones (Scheme 42).469 The ruthenium-catalysed procedure, which involved aldehydes or activated ketones, provided twenty indazolophthalazinone examples in yields as high as 96%. In the same year, Ueda and coworkers reported a copper-catalysed sequential cyclization/migration reaction toward pyrazolo-pyridazines.470 Control experiments revealed the importance of the copper catalyst in the reaction of phenylalkynyl hydrazide. In total, twenty-five examples with two different ring sizes were synthesized in yields as high as 96%. Wang and coworkers developed a sequence that included a rhodium(III)-catalysed C–H activation of pyrazolidinones followed by a [4 + 2] sulfoxonium ylide annulation toward pyrazolo[1,2-a]cinnolines.471 Depending on the reaction temperature, two different substitution patterns were obtained. It was also reported that the synthesis could be performed at gram scale. In 2021, the Feng research group published a palladium-catalysed dual C–H activation that enabled the construction of C–C and C–N bonds between pyrazolones and aryl iodides leading to phenylpyrazolidine-3,5-diones.472 The derivatives were obtained in yields as high as 94% and were readily converted into benzo[c]cinnoline analogs by reaction with NaOH in ethanol.
7.3 Condensation reactions
El-Abhar and coworkers synthesized a set of pyrazolo and pyridazino compounds by condensation of 1,2-dihydro-3H-indazol-3-one with various dicarboxylic acids (Scheme 43).473 The anti-inflammatory activity of these compounds was determined, and one pyrazolo-pyridazine exhibited significant anti-inflammatory potential.
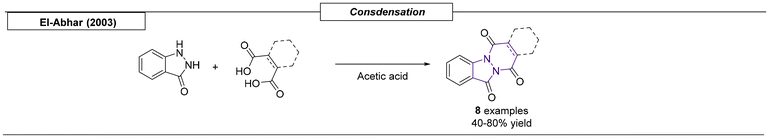 |
| | Scheme 43 Pyrazolo-pyridazine via condensation reactions. | |
7.4 Multistep syntheses
Several examples of multistep synthetic procedures that produce pyrazolo-pyridazines have been published. Abell and coworkers described the eight-step, enantioselective synthesis of a peptidic tetrahydropyridazinone starting from (S)-phenylalanine (Scheme 44).474 The derivative was further converted into a bicyclic peptidomimetic, which is an important class of β-strand mimetics. In 2003, the Moeller research group developed a synthetic method for the incorporation of pyrazoline derivatives into substituted bicyclic lactam peptidomimetics.475 The bicyclic products have either a six- or seven-membered lactam ring in their core structure. In 2008, Wang and coworkers constructed three different aza-phenanthroindo(quino)lizidine alkaloid analogs.476 The reaction sequence started with the four-step synthesis of a biacylhydrazine. The N,N-bridgehead scaffold was then obtained using an aldol condensation reaction. The authors also demonstrated that the two aryl rings could be oxidatively coupled. Some of the compounds demonstrated anticancer potential in an in vitro assay.
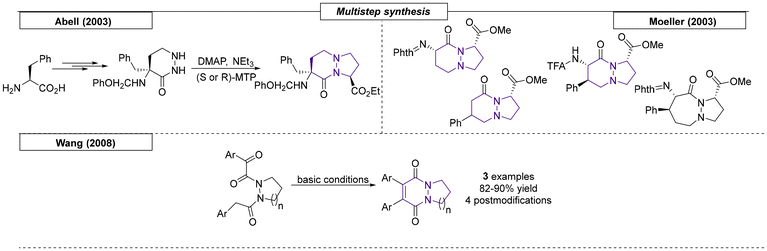 |
| | Scheme 44 Multistep synthetic procedures that produce pyrazolo-pyridazines. | |
7.5 Domino reactions
Two research groups have published cascade or multicomponent reactions that produce pyrazolo-pyridazines. Kostikov and coworkers reported the reaction of thermolized 6-aryl-1,5-diazabicyclo[3.1.0]hexanes with 2-alkyl-3-phenylcyclopropenones (Scheme 45).477 The fused polycyclic systems, 4a,7b-diazacyclopenta[cd]inden-7-ones, were found to be the result of two cyclopropenone molecules added with extrusion of a CO molecule. In total, six substrates were synthesized in yields as high as 63%. In 2021, Li and coworkers developed a rhodium-catalysed cascade reaction consisting of C–H bond activation/intramolecular cyclization toward pyrazolo[1,2-a]cinnoline derivatives.478 Starting from pyrazolidiones and an iodonium ylide, which served as carbene precursor, twenty examples bearing various functional groups were obtained in yields as high as 77%.
 |
| | Scheme 45 Cascade and multicomponent reactions that produce pyrazolo-pyridazines. | |
7.6 Cycloaddition reactions
There are many examples of cycloaddition reactions that have been used to produce pyrazolo-pyridazines. The first example was published by Hayashi and coworkers in 2006 (Scheme 46).479 In this palladium-catalysed [3 + 3] cycloaddition, reaction of azomethine imines with trimethylenemethane provided eleven pyrazolo-pyridazine compounds in yields as high as 94%. An absolute configuration was postulated and confirmed by X-ray analysis. Another approach that exploited azomethine imines as starting materials was developed by the Scheidt research group.480 Using NHC catalysis, the authors created homo-enolates, which underwent subsequent [3 + 3] reactions to produce the desired N,N-bridgehead compounds. The thirteen examples were obtained in yields as high as 94% and with diastereoselectivities of up to 20:1 dr. Toste and coworkers reported a [3 + 3] cycloaddition reaction between an azomethine imine and propargyl esters.481 The mechanism represented the first example of a formal cycloaddition of alkenyl-gold-carbenoids and 1,3-dipoles. Fourteen examples were reported with yields as high as 98% yield and diastereoselectivities of up to 20:1 dr.
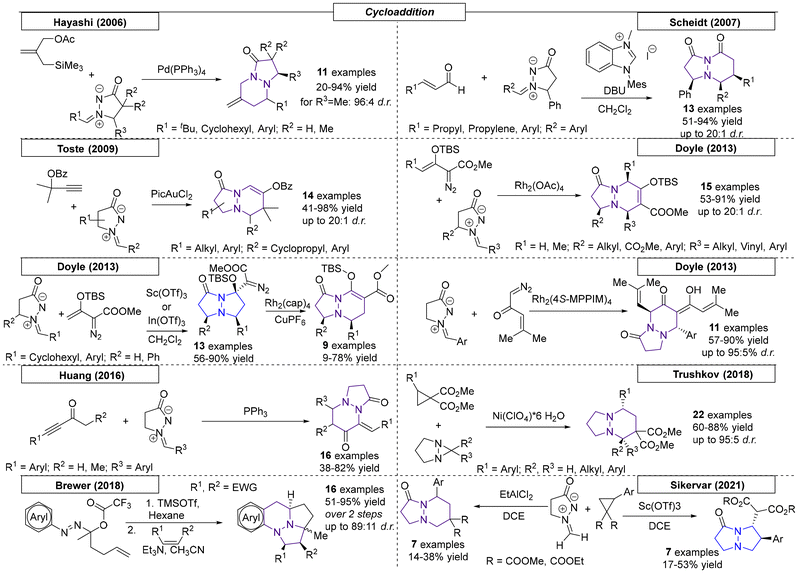 |
| | Scheme 46 Cycloaddition reactions that provide pyrazolo-pyridazines. | |
The Doyle research group synthesized pyrazolo-pyridazines by reacting vinylcarbene intermediates, obtained from enoldiazoacetates, with azomethine imines using a rhodium catalyst (Scheme 46).482 The absolute configuration of the major diastereomer was identified by X-ray analysis. All examples were obtained by this [3 + 3] cycloaddition process in yields as high as 91% and with diastereoselectivities of up to 20:1 dr. In the same year, Doyle and coworkers published the [3 + 2] cycloaddition reaction of enoldiazoacetates and azomethine imines using gold or copper triflate catalysts.483 The obtained pyrazolo-pyrazole derivatives were then used for selective 1,2-C → C and –N → C migrations that provided the target pyrazolo-pyridazine compounds in yields as high as 78%. The Doyle research group also published a [3 + 2 + 1] annulation of azomethine imines with two diazo ketone molecules.484 The reaction provided the desired N,N-bridgehead compounds in yields of up to 90% and diastereoselectivities of up to 95:5 dr.
In 2016, Huang and coworkers developed a phosphine-catalysed [3 + 3] domino cycloaddition of ynones and azomethine imines toward pyrazolo-pyridazine derivatives (Scheme 46).485 Triphenylphosphine was used to generate a zwitterionic species, which enabled the formation of the desired products. The authors obtained nineteen different derivatives with high stereoselectivities and yields as high as 82%. The Trushkov research group reported the [3 + 3] annulation of donor–acceptor cyclopropanes with diaziridines.486 The reaction, which was catalysed by a Lewis acid nickel catalyst, provided the desired pyrazolo-pyridazines in yields of up to 88% and with high diastereoselectivities, up to 95:5 dr. Brewer and coworkers described a novel two-step sequence of cycloadditions toward tetracyclic 1,2,3,4-tetrahydrocinnolines.487 The structurally complex derivatives were constructed by consecutive [4 + 2] and [3 + 2] cycloadditions in yields as high as 95% over 2 steps and with up to 89:11 dr. Another Lewis acid-mediated cycloaddition reaction was reported by the Sikervar research group.488 In this work, pyrazolo-pyrazoles or pyrazolo-pyridazines were obtained by [3 + 2] or [3 + 3] cycloadditions, respectively, depending on the catalyst employed, Sc(OTf)3 or EtAlCl2, respectively. Seven examples were produced for each scaffold, and their absolute configurations were confirmed using X-ray analysis. Additionally, three tetrahydrofuran-fused systems were reported.
7.7 Organocatalysed reactions
In 2021, Briere and coworkers reported an organocatalytic approach toward the pyrazolo-pyridazine core structure (Scheme 47).489 In this multicomponent Knoevenagel/aza-Michael/cyclocondensation approach, pyridazinones were added to alkylidene Meldrum's acid intermediates using the organic base DBU as a catalyst. The procedure provided twenty-one examples in yields as high as 87% and with high regio- and diastereoselectivities.
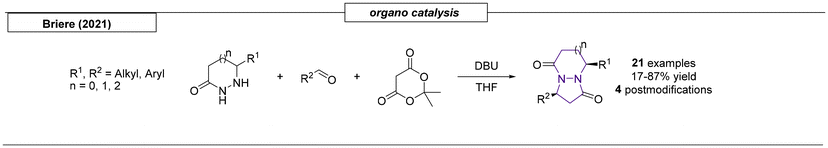 |
| | Scheme 47 Pyrazolo-pyridazines via organocatalytic reactions. | |
8 Octahydro pyridazino[1,2-a]pyridazine
The final compound class to be discussed in this review is the pyridazino[1,2-a]pyridazines. Since the 1970s, partially or fully saturated derivatives of the pyridazino[1,2-a]pyridazine ring system have been considered as potential drug molecules (Fig. 7).490
 |
| | Fig. 7 Examples of pyridazino-pyridazine-derived compounds. | |
8.1 Metal-catalysed reactions
As the synthesis of these pyridazino-pyridazines is very difficult, relatively few approaches that yield expanded scaffolds have been published. In 2018, the Shang research group developed a ruthenium- or iridium-catalysed procedure for the synthesis of 8H-isoquinolino[1,2-b]quinazolin-8-ones and phthalazino[2,3-a]cinnoline-8,13-diones (Scheme 48).491 The latter were obtained using various 2-phenyl-2,3-dihydrophthalazine-1,4-diones, cyclic diazo-1,3-diketones and an iridium catalyst. Twenty-four examples were obtained in yields as high as 95%. Kim and coworkers reported a rhodium-catalysed C–H annulation of N-aryl heterocycles toward cinnolines.492N-Aryl phthalazinones were reacted with vinylene carbonate to obtain the pyridazolo-pyridazine or pyridazino-pyridazine core structures, depending on starting materials and reaction conditions. In total, twenty-eight derivatives were synthesized in yields as high as 98%.
 |
| | Scheme 48 Pyridazino-pyridazines via metal-catalysed reactions. | |
8.2 Condensation reactions
Four methods for the synthesis of pyridazino-pyridazines using condensation reactions have been published. In 2006, Orlov and coworkers reported the synthesis of a pyridazino[1,2-a]pyridazine derivative by reaction of 3-nitrophthalic acid with hydrazine in fuming sulfuric acid. The product was isolated in 19% yield (Scheme 49).493 Another approach, yielding the same core structure, was reported by Habibi and coworkers one year later.494 Instead of the free acids, anhydrides were brought to reaction with semi carbazides or thiosemicarbazides under solvent free conditions with K-10 clay under microwave irradiation. In total, ten examples were obtained in yields as high as 82%. The Sakhuja research group developed an additive-driven, rhodium-catalysed annulation reaction of N-arylphthalazine-1,4-diones and α-diazo carbonyl compounds.495 A [4 + 1] or a [4 + 2] annulation took place, depending on the additive, CsOAc or AgSbF6, respectively. Twenty examples of both motifs were prepared in yields as high as 88%. One year later, the same research group published a C–H acylmethylation of N-arylphthalazine-1,4-diones with α-carbonyl sulfoxonium ylides.496 The direct ruthenium-catalysed, ortho-Csp2–H acylmethylation with an α-carbonyl sulfoxonium ylide featured high functional group tolerance. The product was used as a key intermediate for the synthesis of various pyridazino-pyridazine scaffolds in yields ranging from 61 to 93%.
 |
| | Scheme 49 Condensation reactions that produce pyridazino-pyridazines. | |
9 Conclusion
We have reviewed the available structures and synthetic methods for N-bridgehead compounds published since 2000. These compounds, which have been reported in over 450 publications, offer the potential to serve as first-generation bioactive compounds or building blocks for novel pharmaceutics. We believe that these compounds have great potential, as the bioactivity of a broad majority of them has yet to be determined. Given the broad spectrum of synthetic methods reviewed here, historical drawbacks associated with these structures, such as availability or limited possibility of modification, can now be almost completely avoided.
As the de novo synthesis of these compounds is well-documented, we hope that this review is useful for drug design and discovery in multiple ways (Scheme 50): first, exemplary N-bridgehead compounds can be added to high-throughput compound libraries to cover the chemical space of these scaffolds. Compounds that are identified as potential drug candidates can be easily optimized by variation of the corresponding starting materials and, therefore, tailored to the desired purposes. Second, methodologies used for the construction of the N-bridgehead core can be utilized in the design of total synthesis procedures of natural products. In addition, research around pseudo natural products can profit from the multiple possibilities of constructing the complex core structure. Finally, this review can serve as a collection of N-bridgehead building blocks for drug design in general.
 |
| | Scheme 50 Numbers of publications and outlook. | |
Author contributions
B. W. G. and S. V. M. developed the general concept of the review. B. W. G. developed outlay, conducted literature research, writing of the manuscript and design of schemes, figures, and tables. P. K. and A. N. refined the concept and improved schemes, figures, and tables of the review. All authors contributed to editing, proofreading and finalizing the article.
Data availability
This is a review article on nitrogen bridgehead compounds, summarizing 20 hears of literature, that has not been done in two decades.
There is no data to be transferred.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
SVM would like to acknowledge OHSU Knight Cancer Institute for partial support of the work described in this article.
References
- B. A. P. Wilson, C. C. Thornburg, C. J. Henrich, T. Grkovic and B. R. O'Keefe, Nat. Prod. Rep., 2020, 37, 893–918 Search PubMed.
- A. L. Q. Franca, H. V. Chaves, J. M. O. Freire, L. H. T. de Sousa, A. T. A. Pimenta, M. A. S. Lima, B. R. de Oliveira, M. C. de Mattos, V. P. T. Pinto, A. Portela, K. M. A. Pereira, J. Costa, P. Goes, R. J. B. Jorge, J. A. M. Silveira, H. L. B. Braz, M. E. A. de Moraes and M. M. Bezerra, Clin. Oral Investig., 2022, 26, 1701–1711 Search PubMed.
- M. G. da Silva, S. L. F. Barbosa, D. S. Silva, I. B. M. Bezerra, E. A. Bezerra, A. G. Coelho, I. C. P. da Silva Morais, L. M. Rezende-Junior, I. S. do Carmo, J. S. Lima-Neto, S. G. Comerma-Steffensen, A. Cito and D. D. R. Arcanjo, J. Evidence-Based Complementary Altern. Med., 2022, 2022, 8499625 Search PubMed.
- Y. D. Singh, B. Jena, R. Ningthoujam, S. Panda, P. Priyadarsini, S. Pattanayak, M. K. Panda, M. C. Singh and K. B. Satapathy, Adv. Tradit. Med., 2020, 22, 259–270 Search PubMed.
- D. L. Chen, B. W. Wang, Z. C. Sun, J. S. Yang, X. D. Xu and G. X. Ma, Molecules, 2020, 25, 2485 Search PubMed.
- X.-Y. Zong, D.-C. Xu, J.-Y. Yin, S.-P. Nie and M.-Y. Xie, Bioact. Carbohydr. Diet. Fibre, 2022, 27, 100284 Search PubMed.
- E. Karrar, I. A. M. Ahmed, M. F. Manzoor, W. Wei, F. Sarpong and X. Wang, Food Chem., 2022, 373, 131436 Search PubMed.
- M. Kumar, A. Dahuja, S. Tiwari, S. Punia, Y. Tak, R. Amarowicz, A. G. Bhoite, S. Singh, S. Joshi, P. S. Panesar, R. Prakash Saini, A. Pihlanto, M. Tomar, J. Sharifi-Rad and C. Kaur, Food Chem., 2021, 353, 129431 Search PubMed.
- C. Wawrosch and S. B. Zotchev, Appl. Microbiol. Biotechnol., 2021, 105, 6649–6668 Search PubMed.
- M. Oliverio, M. Nardi, M. L. Di Gioia, P. Costanzo, S. Bonacci, S. Mancuso and A. Procopio, Nat. Prod. Rep., 2021, 38, 444–469 Search PubMed.
- V. Sharma, T. Sharma, S. Kaul, K. K. Kapoor and M. K. Dhar, Phytochem. Rev., 2016, 16, 513–526 Search PubMed.
- C. Kibayashi, Chem. Pharm. Bull., 2005, 53, 1375–1386 Search PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 Search PubMed.
- T. A. Crabb, R. F. Newton and D. Jackson, Chem. Rev., 1970, 71, 109–126 Search PubMed.
- J. T. Kim and V. Gevorgyan, J. Org. Chem., 2005, 70, 2054–2059 Search PubMed.
- P. Brugnerotto, S. K. T. Seraglio, M. Schulz, L. V. Gonzaga, R. Fett and A. C. O. Costa, Food Chem., 2021, 342, 128384 Search PubMed.
- S. V. Kauloorkar, V. Jha, G. Jogdand and P. Kumar, Org. Biomol. Chem., 2014, 12, 4454–4460 Search PubMed.
- C. Bhat and S. G. Tilve, RSC Adv., 2014, 4, 5405 Search PubMed.
- Y. Quevedo-Acosta, I. D. Jurberg and D. Gamba-Sanchez, Org. Lett., 2020, 22, 239–243 Search PubMed.
- J. Robertson and K. Stevens, Nat. Prod. Rep., 2014, 31, 1721–1788 Search PubMed.
- J. R. Liddell, Nat. Prod. Rep., 2002, 19, 773–781 Search PubMed.
- T. Kopp, M. Abdel-Tawab and B. Mizaikoff, Toxins, 2020, 12, 320 Search PubMed.
- X. Wei, W. Ruan and K. Vrieling, Molecules, 2021, 26, 1970 Search PubMed.
- R. Moreira, D. M. Pereira, P. Valentao and P. B. Andrade, Int. J. Mol. Sci., 2018, 19, 1668 Search PubMed.
- J. S. Miser, W. A. Smithson, W. Krivit, C. H. Hughes, D. Davis, M. D. Krailo and G. D. Hammond, Am. J. Clin. Oncol., 1992, 15, 135 Search PubMed.
- B. Singh, P. M. Sahu and S. Singh, Fitoterapia, 2002, 73, 153–155 Search PubMed.
- D. L. Taylor, R. Nash, L. E. Fellows, M. S. Kang and A. S. Tyms, Antiviral Chem. Chemother., 2016, 3, 273–277 Search PubMed.
- V. B. Pandey, J. P. Singh, Y. V. Rao and S. B. Acharya, Planta Med., 1982, 45, 229–233 Search PubMed.
- M. Aboelmagd, K. Elokely, M. A. Zaki, A. Said, E. G. Haggag and S. A. Ross, Med. Chem. Res., 2018, 27, 1066–1073 Search PubMed.
- P. Cos, D. Berghe, T. Bruyne and A. Vlietinck, Curr. Org. Chem., 2003, 7, 1163–1180 Search PubMed.
- H. Khan, Marya, S. Amin, M. A. Kamal and S. Patel, Biomed. Pharmacother., 2018, 101, 860–870 Search PubMed.
- H. Benamar, L. Tomassini, A. Venditti, A. Marouf, M. Bennaceur and M. Nicoletti, Nat. Prod. Res., 2016, 30, 2567–2574 Search PubMed.
- D. Crich, K. Ranganathan and X. Huang, Org. Lett., 2001, 3, 1917–1919 Search PubMed.
- R. M. Beesley, C. K. Ingold and J. F. Thorpe, J. Chem. Soc., Trans., 1915, 107, 1080–1106 Search PubMed.
- S. M. Allin, W. R. S. Barton, W. R. Bowman and T. McInally, Tetrahedron Lett., 2001, 42, 7887–7890 Search PubMed.
- F. Ghelfi, A. F. Parsons, D. Tommasini and A. Mucci, Eur. J. Org. Chem., 2001, 2001, 1845–1852 Search PubMed.
- A. Padwa, M. N. Jacquez and A. Schmidt, J. Org. Chem., 2004, 69, 33–45 Search PubMed.
- J.-M. Huang, S.-C. Hong, K.-L. Wu and Y.-M. Tsai, Tetrahedron Lett., 2004, 45, 3047–3050 Search PubMed.
- D. Kalaitzakis, M. Triantafyllakis, M. Sofiadis, D. Noutsias and G. Vassilikogiannakis, Angew. Chem., Int. Ed., 2016, 55, 4605–4609 Search PubMed.
- C. Nájera and J. M. Sansano, Pure Appl. Chem., 2019, 91, 575–596 Search PubMed.
- R. G. Biswas and V. K. Singh, Tetrahedron Lett., 2021, 69, 152954 Search PubMed.
- H. Oh, B. Ryou, J. Park, M. Kim, J.-H. Choi and C.-M. Park, ACS Catal., 2021, 11, 13670–13679 Search PubMed.
- P. A. O'Gorman, T. Chen, H. E. Cross, S. Naeem, A. Pitard, M. I. Qamar and K. Hemming, Tetrahedron Lett., 2008, 49, 6316–6319 Search PubMed.
- V. V. R. Kondakal, M. Ilyas Qamar and K. Hemming, Tetrahedron Lett., 2012, 53, 4100–4103 Search PubMed.
- Y.-M. Zhang, P. Liu and H.-L. Zhang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 340–342 Search PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 Search PubMed.
- I. Bytschkov, H. Siebeneicher and S. Doye, Eur. J. Org. Chem., 2003, 2888–2902 Search PubMed.
- K. T. Yip, M. Yang, K. L. Law, N. Y. Zhu and D. Yang, J. Am. Chem. Soc., 2006, 128, 3130–3131 Search PubMed.
- D. Xing and D. Yang, Org. Lett., 2013, 15, 4370–4373 Search PubMed.
- W. Du, Q. Gu, Y. Li, Z. Lin and D. Yang, Org. Lett., 2017, 19, 316–319 Search PubMed.
- D. Yang, K.-Y. Lo and L. Ye, Synlett, 2017, 1570–1575 Search PubMed.
- L. Ye, K. Y. Lo, Q. Gu and D. Yang, Org. Lett., 2017, 19, 308–311 Search PubMed.
- W. Hagui, K. Yuan, N. Besbes, E. Srasra, J.-F. Soulé and H. Doucet, ChemCatChem, 2015, 7, 3544–3554 Search PubMed.
- M. Schuster and S. Blechert, Angew. Chem., Int. Ed. Engl., 1997, 36, 2036–2056 Search PubMed.
- H. Wakamatsu, Y. Sato, R. Fujita and M. Mori, Adv. Synth. Catal., 2007, 349, 1231–1246 Search PubMed.
- R. A. Batey, J. Am. Chem. Soc., 2007, 129, 7476–7476 Search PubMed.
- R. Zimmer, M. Collas, R. Czerwonka, U. Hain and H.-U. Reissig, Synthesis, 2008, 237–244 Search PubMed.
- J. Tamariz, E. Martínez-Mora, M. Caracas, C. Escalante, C. Espinoza-Hicks, H. Quiroz-Florentino and F. Delgado, Synthesis, 2016, 1055–1068 Search PubMed.
- S. Goudedranche, C. Besnard, L. Egger and J. Lacour, Angew. Chem., Int. Ed., 2016, 55, 13775–13779 Search PubMed.
- X. Wang, M. Li, Y. Yang, M. Guo, X. Tang and G. Wang, Adv. Synth. Catal., 2018, 360, 2151–2156 Search PubMed.
- M. Lange, Y. Zi and I. Vilotijevic, J. Org. Chem., 2020, 85, 1259–1269 Search PubMed.
- M. Escolano, J. T. Fernández, F. Rabasa-Alcaniz, M. Sanchez-Rosello and C. D. Pozo, Org. Lett., 2020, 22, 9433–9438 Search PubMed.
- C.-H. Lin, B.-C. Hong and G.-H. Lee, RSC Adv., 2016, 6, 8243–8247 Search PubMed.
- F. Hao, A. R. Reddy, C.-Y. Zhou and C.-M. Che, Adv. Synth. Catal., 2018, 360, 1433–1438 Search PubMed.
- A. T. Carmona, J. Fuentes, P. Vogel and I. Robina, Tetrahedron: Asymmetry, 2004, 15, 323–333 Search PubMed.
- X. T. Liu, L. Hao, M. Lin, L. Chen and Z. P. Zhan, Org. Biomol. Chem., 2010, 8, 3064–3072 Search PubMed.
- V. Sharma and V. Kumar, Med. Chem. Res., 2014, 23, 3593–3606 Search PubMed.
- J. P. Michael, Alkaloids: Chem. Biol., 2016, 75, 1–498 Search PubMed.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556–1575 Search PubMed.
- J. Zhang, S. L. Morris-Natschke, D. Ma, X. F. Shang, C. J. Yang, Y. Q. Liu and K. H. Lee, Med. Res. Rev., 2021, 41, 928–960 Search PubMed.
- B. Reddy, D. Chaya, J. Yadav and R. Grée, Synthesis, 2011, 297–303 Search PubMed.
- K. Whitby, T. C. Pierson, B. Geiss, K. Lane, M. Engle, Y. Zhou, R. W. Doms and M. S. Diamond, J. Virol., 2005, 79, 8698–8706 Search PubMed.
- K. Wojtowicz, R. Januchowski, P. Sosinska, M. Nowicki and M. Zabel, Oncol. Rep., 2016, 35, 2896–2906 Search PubMed.
- C. W. Yang, W. L. Chen, P. L. Wu, H. Y. Tseng and S. J. Lee, Mol. Pharmacol., 2006, 69, 749–758 Search PubMed.
- D. Leonoudakis, A. Rane, S. Angeli, G. J. Lithgow, J. K. Andersen and S. J. Chinta, Mediators Inflammation, 2017, 2017, 8302636 Search PubMed.
- M. Kubo, W. Yatsuzuka, S. Matsushima, K. Harada, Y. Inoue, H. Miyamoto, M. Matsumoto and Y. Fukuyama, Chem. Pharm. Bull., 2016, 64, 957–960 Search PubMed.
- S. Veeraswamy, A. Anjaiah, S. Yennam and A. Jayashree, Asian J. Chem., 2015, 27, 1667–1670 Search PubMed.
- Y. H. Jung and I. S. Kim, Heterocycles, 2011, 83, 2489 Search PubMed.
- A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chemistry, 2009, 15, 7808–7821 Search PubMed.
- F. Abels, C. Lindemann and C. Schneider, Chemistry, 2014, 20, 1964–1979 Search PubMed.
- K. M. Dawood and A. A. Abbas, Expert Opin. Ther. Pat., 2020, 30, 695–714 Search PubMed.
- A. C. Burtoloso, R. M. Dias and B. Bernardim, Acc. Chem. Res., 2015, 48, 921–934 Search PubMed.
- S. Gross and H. U. Reissig, Org. Lett., 2003, 5, 4305–4307 Search PubMed.
- E. Georgescu, F. Georgescu, M. G. Danila, P. I. Filip, C. Drăghici and M. T. Căproiu, J. Chem. Soc., Perkin Trans. 1, 2001, 3382–3392, 10.1039/b106832f.
- H. Suga, Y. Hashimoto, S. Yasumura, R. Takezawa, K. Itoh and A. Kakehi, J. Org. Chem., 2013, 78, 10840–10852 Search PubMed.
- G. C. Tay, N. Sizemore and S. D. Rychnovsky, Org. Lett., 2016, 18, 3050–3053 Search PubMed.
- K. A. Tehrani, M. D'Hooghe and N. De Kimpe, Tetrahedron, 2003, 59, 3099–3108 Search PubMed.
- T. G. Back, M. Parvez and H. Zhai, J. Org. Chem., 2003, 68, 9389–9393 Search PubMed.
- S.-S. P. Chou and C.-W. Ho, Tetrahedron Lett., 2005, 46, 8551–8554 Search PubMed.
- S. H. Verhelst, B. Paez Martinez, M. S. Timmer, G. Lodder, G. A. van der Marel, H. S. Overkleeft and J. H. van Boom, J. Org. Chem., 2003, 68, 9598–9603 Search PubMed.
- K. C. Kurtz, R. P. Hsung and Y. Zhang, Org. Lett., 2006, 8, 231–234 Search PubMed.
- R. Ben-Othman, M. Othman, K. Ciamala, M. Knorr, C. Strohmann and B. Decroix, Tetrahedron, 2009, 65, 4846–4854 Search PubMed.
- G. A. Molander and S. K. Pack, J. Org. Chem., 2003, 68, 9214–9220 Search PubMed.
- E. V. Babaev, A. A. Tsisevich, D. V. Al'bov, V. B. Rybakov and L. A. Aslanov, Russ. Chem. Bull., 2005, 54, 259–261 Search PubMed.
- W. Zhu, D. Dong, X. Pu and D. Ma, Org. Lett., 2005, 7, 705–708 Search PubMed.
- D. Ma and W. Zhu, Synlett, 2006, 1181–1184 Search PubMed.
- I. Coldham, S. Jana, L. Watson and C. D. Pilgram, Tetrahedron Lett., 2008, 49, 5408–5410 Search PubMed.
- X. Jiang, B. Tan and C. F. Barbas 3rd, Angew. Chem., Int. Ed., 2013, 52, 9261–9265 Search PubMed.
- Y. Chen, Y. M. He, S. Zhang, T. Miao and Q. H. Fan, Angew. Chem., Int. Ed., 2019, 58, 3809–3813 Search PubMed.
- G. Barbe, G. Pelletier and A. B. Charette, Org. Lett., 2009, 11, 3398–3401 Search PubMed.
- P. Szczesniak, S. Stecko, E. Maziarz, O. Staszewska-Krajewska and B. Furman, J. Org. Chem., 2014, 79, 10487–10503 Search PubMed.
- S. Hanessian and A. K. Chattopadhyay, Org. Lett., 2014, 16, 232–235 Search PubMed.
- N. E. Dontsova and A. M. Shestopalov, Russ. Chem. Bull., 2017, 66, 1030–1043 Search PubMed.
- Q. Song, Y. Liu, L. Cai, X. Cao, S. Qian and Z. Wang, Chin. Chem. Lett., 2021, 32, 1713–1716 Search PubMed.
- P. M. Holstein, D. Dailler, J. Vantourout, J. Shaya, A. Millet and O. Baudoin, Angew. Chem., Int. Ed., 2016, 55, 2805–2809 Search PubMed.
- J. C. Timmerman, S. Laulhe and R. A. Widenhoefer, Org. Lett., 2017, 19, 1466–1469 Search PubMed.
- D. Yang and G. C. Micalizio, J. Am. Chem. Soc., 2009, 131, 17548–17549 Search PubMed.
- G. Archibald, C. P. Lin, P. Boyd, D. Barker and V. Caprio, J. Org. Chem., 2012, 77, 7968–7980 Search PubMed.
- T. Punirun, D. Soorukram, C. Kuhakarn, V. Reutrakul and M. Pohmakotr, J. Org. Chem., 2018, 83, 765–782 Search PubMed.
- B. Raison, N. Dussart, L. Levy, P. G. Goekjian and D. Gueyrard, J. Org. Chem., 2020, 85, 864–875 Search PubMed.
- Y. Kang and D. Seidel, Org. Lett., 2016, 18, 4277–4279 Search PubMed.
- A. Paul, N. R. Thimmegowda, T. G. Cruz and D. Seidel, Org. Lett., 2018, 20, 602–604 Search PubMed.
- A. Avadhani, P. Iniyavan, A. Acharya, V. Gautam, S. Chakrabarti and H. Ila, ACS Omega, 2019, 4, 17910–17922 Search PubMed.
- N. Veerasamy and R. G. Carter, Tetrahedron, 2016, 72, 4989–5001 Search PubMed.
- J. C. Li, W. F. Dai, D. Liu, Z. J. Zhang, M. Y. Jiang, K. R. Rao, R. T. Li and H. M. Li, Bioorg. Chem., 2021, 110, 104781 Search PubMed.
- J. Zou, L. Zhao, P. Yi, Q. An, L. He, Y. Li, H. Lou, C. Yuan, W. Gu, L. Huang, Z. Hu and X. Hao, J. Agric. Food Chem., 2020, 68, 15015–15026 Search PubMed.
- D. Mancinotti, K. M. Frick and F. Geu-Flores, Nat. Prod. Rep., 2022, 39, 1423–1437 Search PubMed.
- M. Wink, J. Chem. Ecol., 2019, 45, 109–115 Search PubMed.
- S. Davies, A. Fletcher, P. Roberts and J. Thomson, Synlett, 2017, 2697–2706 Search PubMed.
- A. S. Pashev, N. T. Burdzhiev and E. R. Stanoeva, Org. Prep. Proced. Int., 2016, 48, 425–467 Search PubMed.
- X. H. Jia, H. X. Zhao, C. L. Du, W. Z. Tang and X. J. Wang, Phytochem. Rev., 2020, 20, 845 Search PubMed.
- H. Chen, T. Xiao, L. Li, D. Anand, Y. He and L. Zhou, Adv. Synth. Catal., 2017, 359, 3642–3647 Search PubMed.
- M. P. Lalonde, M. A. McGowan, N. S. Rajapaksa and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 1891–1894 Search PubMed.
- M. Kretzschmar, F. Hofmann, D. Moock and C. Schneider, Angew. Chem., Int. Ed., 2018, 57, 4774–4778 Search PubMed.
- X. Xu, P. Y. Zavalij and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 12439–12447 Search PubMed.
- Y. Liu, Q. Wang, Y. Zhang, J. Huang, L. Nie, J. Chen, W. Cao and X. Wu, J. Org. Chem., 2013, 78, 12009–12017 Search PubMed.
- V. Gobe and X. Guinchard, Org. Lett., 2014, 16, 1924–1927 Search PubMed.
- M. J. James, N. D. Grant, P. O'Brien, R. J. Taylor and W. P. Unsworth, Org. Lett., 2016, 18, 6256–6259 Search PubMed.
- S. Nonaka, K. Sugimoto, H. Ueda and H. Tokuyama, Adv. Synth. Catal., 2016, 358, 380–385 Search PubMed.
- R. Azpíroz, A. Di Giuseppe, V. Passarelli, J. J. Pérez-Torrente, L. A. Oro and R. Castarlenas, Organometallics, 2018, 37, 1695–1707 Search PubMed.
- L. R. Wang, D. Chang, Y. Feng, Y. M. He, G. J. Deng and Q. H. Fan, Org. Lett., 2020, 22, 2251–2255 Search PubMed.
- J. Barluenga, C. Mateos, F. Aznar and C. Valdes, J. Org. Chem., 2004, 69, 7114–7122 Search PubMed.
- S.-S. P. Chou, C.-F. Liang, T.-M. Lee and C.-F. Liu, Tetrahedron, 2007, 63, 8267–8273 Search PubMed.
- S.-S. P. Chou and C.-F. Liu, J. Chin. Chem. Soc., 2010, 57, 811–819 Search PubMed.
- S.-S. P. Chou, C.-W. Huang and C.-C. Chang, Tetrahedron, 2011, 67, 4505–4513 Search PubMed.
- S.-S. P. Chou, C.-L. Lu and Y.-H. Hsu, J. Chin. Chem. Soc., 2012, 59, 365–372 Search PubMed.
- A. Pepe, M. Pamment, G. I. Georg and S. V. Malhotra, J. Org. Chem., 2011, 76, 3527–3530 Search PubMed.
- M. Amat, V. Semak, C. Escolano, E. Molins and J. Bosch, Org. Biomol. Chem., 2012, 10, 6866–6875 Search PubMed.
- A. K. Khalafallah, R. M. Abd Elal and N. A. A. El Kanzi, Heterocycl. Commun., 2002, 8, 397 Search PubMed.
- O. V. Gulyakevich, P. V. Kurman and A. L. Mikhal'chuk, Russ. J. Org. Chem., 2005, 41, 1806–1809 Search PubMed.
- P. V. Gormay, V. B. Rybakov and E. V. Babaev, Eur. J. Org. Chem., 2010, 5364–5368 Search PubMed.
- H. Fang, X. Wu, L. Nie, X. Dai, J. Chen, W. Cao and G. Zhao, Org. Lett., 2010, 12, 5366–5369 Search PubMed.
- G. Belanger, G. O'Brien and R. Larouche-Gauthier, Org. Lett., 2011, 13, 4268–4271 Search PubMed.
- X. Dai, X. Wu, H. Fang, L. Nie, J. Chen, H. Deng, W. Cao and G. Zhao, Tetrahedron, 2011, 67, 3034–3040 Search PubMed.
- X. Wu, L. Nie, H. Fang, J. Chen, W. Cao and G. Zhao, Eur. J. Org. Chem., 2011, 6755–6763 Search PubMed.
- M. Rueping and L. Hubener, Synlett, 2011, 1243–1246 Search PubMed.
- I. A. Abdelhamid, F. M. Saleh and H. M. Hassaneen, Synlett, 2020, 1126–1128 Search PubMed.
- W. Zou, M. Sandbhor and M. Bhasin, J. Org. Chem., 2007, 72, 1226–1234 Search PubMed.
- J. Franzén and A. Fisher, Angew. Chem., Int. Ed., 2009, 48, 787–791 Search PubMed.
- W. Zhang and J. Franzén, Adv. Synth. Catal., 2010, 352, 499–518 Search PubMed.
- X. Wu, X. Dai, L. Nie, H. Fang, J. Chen, Z. Ren, W. Cao and G. Zhao, Chem. Commun., 2010, 46, 2733–2735 Search PubMed.
- C. Tsukano, A. Oimura, I. Enkhtaivan and Y. Takemoto, Org. Lett., 2012, 14, 1902–1905 Search PubMed.
- A. Jung and S. J. Min, Asian J. Org. Chem., 2019, 8, 1617–1620 Search PubMed.
- J. Outin, P. Quellier and G. Bélanger, J. Org. Chem., 2020, 85, 4712–4729 Search PubMed.
- O. Bassas, N. Llor, M. M. Santos, R. Griera, E. Molins, M. Amat and J. Bosch, Org. Lett., 2005, 7, 2817–2820 Search PubMed.
- M. Amat, M. M. Santos, O. Bassas, N. Llor, C. Escolano, A. Gomez-Esque, E. Molins, S. M. Allin, V. McKee and J. Bosch, J. Org. Chem., 2007, 72, 5193–5201 Search PubMed.
- D. L. Clive, M. Yu and Z. Li, Chem. Commun., 2005, 906–908, 10.1039/b413481h.
- D. L. Clive, Z. Li and M. Yu, J. Org. Chem., 2007, 72, 5608–5617 Search PubMed.
- E. Airiau, N. Girard, M. Pizzeti, J. Salvadori, M. Taddei and A. Mann, J. Org. Chem., 2010, 75, 8670–8673 Search PubMed.
- M. J. Niphakis, B. J. Turunen and G. I. Georg, J. Org. Chem., 2010, 75, 6793–6805 Search PubMed.
- L. Gomez, X. Garrabou, J. Joglar, J. Bujons, T. Parella, C. Vilaplana, P. J. Cardona and P. Clapés, Org. Biomol. Chem., 2012, 10, 6309–6321 Search PubMed.
- S. Hu, B. Wang, Y. Zhang, W. Tang, M. Fang, T. Lu and D. Du, Org. Biomol. Chem., 2015, 13, 4661–4667 Search PubMed.
- A. Pashev, N. Burdzhiev and E. Stanoeva, Beilstein J. Org. Chem., 2020, 16, 1456–1464 Search PubMed.
- T. P. Ribelin, A. S. Judd, I. Akritopoulou-Zanze, R. F. Henry, J. L. Cross, D. N. Whittern and S. W. Djuric, Org. Lett., 2007, 9, 5119–5122 Search PubMed.
- R. Epple, H. D. Urbina, R. Russo, H. Liu, D. Mason, B. Bursulaya, C. Tumanut, J. Li and J. L. Harris, Bioorg. Med. Chem. Lett., 2007, 17, 1254–1259 Search PubMed.
- S. Yotphan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 2452–2453 Search PubMed.
- S. Stecko, A. Mames, B. Furman and M. Chmielewski, J. Org. Chem., 2009, 74, 3094–3100 Search PubMed.
- F. Slowinski, O. B. Ayad, J. Vache, M. Saady, O. Leclerc and A. Lochead, Org. Lett., 2010, 12, 5004–5007 Search PubMed.
- A. F. Khlebnikov, M. S. Novikov, V. V. Pakalnis and D. S. Yufit, J. Org. Chem., 2011, 76, 9344–9352 Search PubMed.
- M. Srivastava, J. Singh, S. B. Singh, K. Tiwari, V. K. Pathak and J. Singh, Green Chem., 2012, 14, 901 Search PubMed.
- F. Grande, M. A. Occhiuzzi, G. Ioele, G. Ragno and A. Garofalo, Eur. J. Med. Chem., 2018, 151, 121–144 Search PubMed.
- D. R. Griffith and A. H. Shoemaker, Synthesis, 2020, 65–78 Search PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 3067 Search PubMed.
- B. Nie, W. Wu, Y. Zhang, H. Jiang and J. Zhang, Org. Chem. Front., 2020, 7, 3067–3099 Search PubMed.
- R. Szostak and M. Szostak, Molecules, 2019, 24, 274 Search PubMed.
- Z. Zhang, H. Han, L. Wang, Z. Bu, Y. Xie and Q. Wang, Org. Biomol. Chem., 2021, 19, 3960–3982 Search PubMed.
- N. Petek, B. Erjavec, D. Slapšak, A. Gaber, U. Grošelj, F. Požgan, S. Ričko, B. Štefane, M. Klemenčič and J. Svete, Dyes Pigm., 2022, 201, 110224 Search PubMed.
- L. N. Jungheim, S. K. Sigmund and J. W. Fisher, Tetrahedron Lett., 1987, 28, 285–288 Search PubMed.
- E. E. Boros, F. Bouvier, S. Randhawa and M. H. Rabinowitz, J. Heterocycl. Chem., 2001, 38, 613–616 Search PubMed.
- X. Gao, C. S. Hampton and M. Harmata, Eur. J. Org. Chem., 2012, 7053–7056 Search PubMed.
- P. Zhang, D. Kumar, L. Zhang, D. Shem-Tov, N. Petrutik, A. K. Chinnam, C. Yao, S. Pang and M. Gozin, Molecules, 2019, 24, 4324 Search PubMed.
- T.-H. Chuang and K. B. Sharpless, Helv. Chim. Acta, 2000, 83, 1734–1743 Search PubMed.
- U. Grošelj, A. Drobnič, S. Rečnik, J. Svete, B. Stanovnik, A. Golobič, N. Lah, I. Leban, A. Meden and S. Golič-Grdadolnik, Helv. Chim. Acta, 2001, 84, 3403–3417 Search PubMed.
- L. Pezdirc, V. Jovanovski, D. Bevk, R. Jakše, S. Pirc, A. Meden, B. Stanovnik and J. Svete, Tetrahedron, 2005, 61, 3977–3990 Search PubMed.
- L. Pezdirc, D. Bevk, U. Groselj, A. Meden, B. Stanovnik and J. Svete, J. Comb. Chem., 2007, 9, 717–723 Search PubMed.
- L. Pezdirc, J. Cerkovnik, S. Pirc, B. Stanovnik and J. Svete, Tetrahedron, 2007, 63, 991–999 Search PubMed.
- L. Pezdirc, U. Grošelj, A. Meden, B. Stanovnik and J. Svete, J. Heterocycl. Chem., 2009, 45, 181–188 Search PubMed.
- L. Pezdirc, B. Stanovnik and J. Svete, Collect. Czech. Chem. Commun., 2009, 74, 835–856 Search PubMed.
- A. Novak, J. Bezenšek, L. Pezdirc, U. Grošelj, M. Kasunič, Č. Podlipnik, B. Stanovnik, P. Šimůnek and J. Svete, Tetrahedron, 2011, 67, 9729–9735 Search PubMed.
- I. Panfil, Z. Urbańczyk-Lipkowska, K. Suwińska, J. Solecka and M. Chmielewski, Tetrahedron, 2002, 58, 1199–1212 Search PubMed.
- A. P. Molchanov, D. I. Sipkin, Y. B. Koptelov, J. Kopf and R. R. Kostikov, Russ. J. Org. Chem., 2004, 40, 67–78 Search PubMed.
- A. P. Molchanov, D. I. Sipkin, Y. B. Koptelov and R. R. Kostikov, Eur. J. Org. Chem., 2002, 453–456 Search PubMed.
- A. P. Molchanov, D. I. Sipkin, Y. B. Koptelov, J. Kopf and R. R. Kostikov, Russ. J. Org. Chem., 2003, 39, 1338–1345 Search PubMed.
- Y. B. Koptelov, Russ. J. Org. Chem., 2006, 42, 1510–1515 Search PubMed.
- Y. B. Koptelov, S. P. Saik, A. P. Molchanov and S. I. Selivanov, Russ. J. Org. Chem., 2011, 47, 421–432 Search PubMed.
- Y. B. Koptelov and M. V. Sednev, Russ. J. Org. Chem., 2011, 47, 547–555 Search PubMed.
- Y. B. Koptelov, M. V. Sednev and R. R. Kostikov, Russ. J. Org. Chem., 2012, 48, 804–814 Search PubMed.
- Y. B. Koptelov, D. O. Antuganov, A. P. Molchanov and R. R. Kostikov, Russ. J. Org. Chem., 2015, 51, 972–981 Search PubMed.
- M. I. Pleshchev, N. V. D. Gupta, M. I. Struchkova, A. S. Goloveshkin, I. S. Bushmarinov, D. V. Khakimov and N. N. Makhova, Mendeleev Commun., 2015, 25, 188–190 Search PubMed.
- M. M. Efremova, A. P. Molchanov, A. V. Stepakov, R. R. Kostikov, V. S. Shcherbakova and A. V. Ivanov, Tetrahedron, 2015, 71, 2071–2078 Search PubMed.
- W. Chen, X.-H. Yuan, R. Li, W. Du, Y. Wu, L.-S. Ding and Y.-C. Chen, Adv. Synth. Catal., 2006, 348, 1818–1822 Search PubMed.
- A. V. Shevtsov, V. V. Kuznetsov, A. A. Kislukhin, V. Y. Petukhova, Y. A. Strelenko, N. N. Makhova and K. A. Lyssenko, J. Heterocycl. Chem., 2006, 43, 881–888 Search PubMed.
- W. Chen, W. Du, Y. Z. Duan, Y. Wu, S. Y. Yang and Y. C. Chen, Angew. Chem., Int. Ed., 2007, 46, 7667–7670 Search PubMed.
- H. Suga, A. Funyu and A. Kakehi, Org. Lett., 2007, 9, 97–100 Search PubMed.
- M. P. Sibi, D. Rane, L. M. Stanley and T. Soeta, Org. Lett., 2008, 10, 2971–2974 Search PubMed.
- T. Jin, F. Yang and Y. Yamamoto, Collect. Czech. Chem. Commun., 2009, 74, 957–972 Search PubMed.
- F. Shi, R. Mancuso and R. C. Larock, Tetrahedron Lett., 2009, 50, 4067–4070 Search PubMed.
- Y. Yamashita and S. Kobayashi, Chem. Lett., 2009, 38, 678–679 Search PubMed.
- N. Shibata, S. Ogawa, T. Nishimine and E. Tokunaga, Synthesis, 2010, 3274–3281 Search PubMed.
- H. Suga, T. Arikawa, K. Itoh, Y. Okumura, A. Kakehi and M. Shiro, Heterocycles, 2010, 81, 1669 Search PubMed.
- Y. Ukaji, K. Inomata, K. Tanaka and T. Kato, Heterocycles, 2010, 80, 887 Search PubMed.
- K. Tanaka, T. Kato, S. Fujinami, Y. Ukaji and K. Inomata, Chem. Lett., 2010, 39, 1036–1038 Search PubMed.
- M. Yoshida, N. Sassa, T. Kato, S. Fujinami, T. Soeta, K. Inomata and Y. Ukaji, Chemistry, 2014, 20, 2058–2064 Search PubMed.
- T. M. Tong, T. Soeta, T. Suga, K. Kawamoto, Y. Hayashi and Y. Ukaji, J. Org. Chem., 2017, 82, 1969–1976 Search PubMed.
- R. L. Atienza, H. S. Roth and K. A. Scheidt, Chem. Sci., 2011, 2, 1772–1776 Search PubMed.
- R. Na, C. Jing, Q. Xu, H. Jiang, X. Wu, J. Shi, J. Zhong, M. Wang, D. Benitez, E. Tkatchouk, W. A. Goddard, H. Guo and O. Kwon, J. Am. Chem. Soc., 2011, 133, 13337–13348 Search PubMed.
- Y. Li, Y. Meng, X. Meng and Z. Li, Tetrahedron, 2011, 67, 4002–4008 Search PubMed.
- N. Luo, Z. Zheng and Z. Yu, Org. Lett., 2011, 13, 3384–3387 Search PubMed.
- H. Matsuyama, C. Seki, M. Hirama, T. Sato, S. Takeda, Y. Kohari, K. Ishigaki, M. Ohuchi, K. Yokoi, H. Nakano, K. Uwai, N. Takano and K. Umemura, Heterocycles, 2012, 85, 1045 Search PubMed.
- M. Fan, D. Yang, H. Zhu, Y. Guo and J. Guo, Synthesis, 2013, 1325–1332 Search PubMed.
- J. Li, X. Lian, X. Liu, L. Lin and X. Feng, Chemistry, 2013, 19, 5134–5140 Search PubMed.
- M. I. Pleshchev, M. A. Epishina, V. V. Kachala, V. V. Kuznetsov, A. S. Goloveshkin, I. S. Bushmarinov and N. N. Makhova, Mendeleev Commun., 2013, 23, 206–208 Search PubMed.
- M. I. Pleshchev, V. V. Kachala, A. S. Goloveshkin, I. S. Bushmarinov, V. V. Kuznetsov, D. V. Khakimov and N. N. Makhova, Mendeleev Commun., 2013, 23, 271–273 Search PubMed.
- D. Wang, H.-P. Deng, Y. Wei, Q. Xu and M. Shi, Eur. J. Org. Chem., 2013, 401–406 Search PubMed.
- J. Liu, H. Liu, R. Na, G. Wang, Z. Li, H. Yu, M. Wang, J. Zhong and H. Guo, Chem. Lett., 2012, 41, 218–220 Search PubMed.
- S. S. Wong, M. G. Brant, C. Barr, A. G. Oliver and J. E. Wulff, Beilstein J. Org. Chem., 2013, 9, 1419–1425 Search PubMed.
- Z. Li, H. Yu, H. Liu, L. Zhang, H. Jiang, B. Wang and H. Guo, Chemistry, 2014, 20, 1731–1736 Search PubMed.
- E. Pair, C. Berini, R. Noel, M. Sanselme, V. Levacher and J. F. Briere, Chem. Commun., 2014, 50, 10218–10221 Search PubMed.
- Y. Xu, W. Liu, X. Sun, D. Lu and L. Guo, Synlett, 2014, 1093–1096 Search PubMed.
- Q. Zhang, S. Guo, J. Yang, K. Yu, X. Feng, L. Lin and X. Liu, Org. Lett., 2017, 19, 5826–5829 Search PubMed.
- R. Y. Zhu, C. S. Wang, J. Zheng, F. Shi and S. J. Tu, J. Org. Chem., 2014, 79, 9305–9312 Search PubMed.
- Y. Dürüst, E. Sagirli and A. Sagirli, Turk. J. Chem., 2015, 39, 789–800 Search PubMed.
- Y. Dürüst, E. Sağırlı and A. Sağırlı, Tetrahedron, 2015, 71, 9307–9312 Search PubMed.
- I. Damljanović, J. Jovanović and G. Bogdanović, Synlett, 2016, 664–668 Search PubMed.
- E. P. Kirar, U. Groselj, G. Mirri, F. Pozgan, G. Strle, B. Stefane, V. Jovanovski and J. Svete, J. Org. Chem., 2016, 81, 5988–5997 Search PubMed.
- M. Mondal, K. A. Wheeler and N. J. Kerrigan, Org. Lett., 2016, 18, 4108–4111 Search PubMed.
- M. Vishwanath, K. Sivamuthuraman and V. Kesavan, Chem. Commun., 2016, 52, 12314–12317 Search PubMed.
- S. E. Winterton and J. M. Ready, Org. Lett., 2016, 18, 2608–2611 Search PubMed.
- A. Y. Barkov, N. S. Zimnitskiy, I. B. Kutyashev, V. Y. Korotaev and V. Y. Sosnovskikh, Tetrahedron Lett., 2017, 58, 3989–3992 Search PubMed.
- S. A. Rizk, A. A. El-Sayed and M. M. Mounier, J. Heterocycl. Chem., 2017, 54, 3358–3371 Search PubMed.
- S. Yuan, Y. Luo, J. Peng, M. Miao, J. Xu and H. Ren, Org. Lett., 2017, 19, 6100–6103 Search PubMed.
- M. Wu, J. Jiang, Z. Zhu, Q. Wang and D. Kong, Synthesis, 2017, 139–145 Search PubMed.
- D. Bai, T. Xu, C. Ma, X. Zheng, B. Liu, F. Xie and X. Li, ACS Catal., 2018, 8, 4194–4200 Search PubMed.
- J. P. Jovanović, S. B. Novaković, G. A. Bogdanović, A. Minić, A. Pejović, J. Katanić, V. Mihailović, B. Nastasijević, D. Stevanović and I. Damljanović, J. Organomet. Chem., 2018, 860, 85–97 Search PubMed.
- J. Gong, Q. Wan and Q. Kang, Org. Lett., 2018, 20, 3354–3357 Search PubMed.
- J. Gujral, T. P. Reddy, B. Gorachand and D. B. Ramachary, ChemistrySelect, 2018, 3, 7900–7905 Search PubMed.
- Y.-D. Kou, Z.-N. Zhao, X. Yang, S. J. Kalita, X.-J. Chen, Z.-Z. Xie, Y. Zhao and Y.-Y. Huang, Asian J. Org. Chem., 2018, 7, 1830–1834 Search PubMed.
- S. Shen, Y. Yang, J. Duan, Z. Jia and J. Liang, Org. Biomol. Chem., 2018, 16, 1068–1072 Search PubMed.
- L. Yang, Y. Lv, F. Wang and G. Zhong, Org. Biomol. Chem., 2018, 16, 4433–4438 Search PubMed.
- Z. Zhu, Q. Wang, D. Kong, T. Huang and M. Wu, Synthesis, 2018, 2601–2607 Search PubMed.
- N. Strasek, L. Lavrencic, A. Ostrek, D. Slapsak, U. Groselj, M. Klemencic, H. B. Žugelj, J. Wagger, M. Novinec and J. Svete, Bioorg. Chem., 2019, 89, 102982 Search PubMed.
- C. Volpe, S. Meninno, A. Capobianco, G. Vigliotta and A. Lattanzi, Adv. Synth. Catal., 2019, 361, 1018–1022 Search PubMed.
- Q.-Q. Yang, X. Yin, X.-L. He, W. Du and Y.-C. Chen, ACS Catal., 2019, 9, 1258–1263 Search PubMed.
- Y. A. Almehmadi and F. G. West, Org. Lett., 2020, 22, 6091–6095 Search PubMed.
- H. Hu, J. Xu, F. Wang, S. Dong, X. Liu and X. Feng, Org. Lett., 2020, 22, 93–97 Search PubMed.
- A. P. Molchanov, M. M. Efremova, M. A. Kryukova and M. A. Kuznetsov, Beilstein J. Org. Chem., 2020, 16, 2679–2686 Search PubMed.
- Z. Li, N. Kumagai and M. Shibasaki, Chem. Pharm. Bull., 2020, 68, 552–554 Search PubMed.
- Y. Xu, M. Shen, X. Zhang and X. Fan, Org. Lett., 2020, 22, 4697–4702 Search PubMed.
- J. Zhou, L. Zhang, J. Chen, J. Chen, C. Yin and C. Yu, Tetrahedron Lett., 2020, 61, 152350 Search PubMed.
- M.-C. Wu, P.-J. Xia, Y.-Z. Hu, Z.-P. Ye, K. Chen, H.-Y. Xiang and H. Yang, Tetrahedron, 2021, 83, 131992 Search PubMed.
- T. Kozlecki, C. Samyn, R. W. Alder and P. G. Green, J. Chem. Soc., Perkin Trans. 2, 2001, 243–246, 10.1039/b000828l.
- H. Z. Shams, M. H. Helal and F. A. Mohamed, Pigm. Resin Technol., 2001, 30, 99–108 Search PubMed.
- H. Z. Shams, Y. A. Youssef, F. A. Mohamed, M. M. El-Zawahry, M. H. Helal and E. A. El-Kharadly, Pigm. Resin Technol., 2009, 38, 372–379 Search PubMed.
- D.-Y. Zhou, T. Koike, S. Suetsugu, K. Onitsuka and S. Takahashi, Inorg. Chim. Acta, 2004, 357, 3057–3063 Search PubMed.
- M. B. Donald, W. E. Conrad, J. S. Oakdale, J. D. Butler, M. J. Haddadin and M. J. Kurth, Org. Lett., 2010, 12, 2524–2527 Search PubMed.
- W. E. Conrad, R. Fukazawa, M. J. Haddadin and M. J. Kurth, Org. Lett., 2011, 13, 3138–3141 Search PubMed.
- Z. Wang, B. Yu, Y. Cui, X. Sun and W. Bao, Chin. J. Chem., 2011, 29, 2769–2774 Search PubMed.
- J. Németh, Á. Kiss and Z. Hell, Tetrahedron Lett., 2013, 54, 6094–6096 Search PubMed.
- H. Lee, D. Kang, S. H. Han, R. Chun, A. K. Pandey, N. K. Mishra, S. Hong and I. S. Kim, Angew. Chem., Int. Ed., 2019, 58, 9470–9474 Search PubMed.
- Z. Yang, Z. Song, L. Jie, L. Wang and X. Cui, Chem. Commun., 2019, 55, 6094–6097 Search PubMed.
- C. K. Mahesha, S. K. Mandal and R. Sakhuja, Asian J. Org. Chem., 2020, 9, 1199–1204 Search PubMed.
- L. Zhang, J. Zhao, Y. Jiang, X. Zhang and X. Fan, Org. Chem. Front., 2021, 8, 3734–3739 Search PubMed.
- M. Wu, H. Gao, H. Xu, W. Yi and Z. Zhou, Chin. Chem. Lett., 2022, 33, 842–846 Search PubMed.
- K. Gogoi, B. R. Bora, G. Borah, B. Sarma and S. Gogoi, Chem. Commun., 2021, 57, 1388–1391 Search PubMed.
- A. C. Tomé, J. A. S. Cavaleiro, F. M. J. Domingues and R. J. Cremlyn, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 2617–2634 Search PubMed.
- H. Zachová, S. Man, M. Nečas and M. Potáček, Eur. J. Org. Chem., 2005, 2548–2557 Search PubMed.
- B. B. Snider, J. F. Grabowski, R. W. Alder, B. M. Foxman and L. Yang, Can. J. Chem., 2006, 84, 1242–1249 Search PubMed.
- E. F. Kirolos and A. S. Yanni, Int. J. Org. Chem., 2020, 10, 104–110 Search PubMed.
- F. Al’-Assar, K. N. Zelenin, E. E. Lesiovskaya, I. P. Bezhan and B. A. Chakchir, Pharm. Chem. J., 2002, 36, 598–603 Search PubMed.
- H. R. Shaterian and F. Rigi, Starch/Staerke, 2011, 63, 340–346 Search PubMed.
- A. Hasaninejed, M. R. Kazerooni and A. Zare, Catal. Today, 2012, 196, 148–155 Search PubMed.
- A. Mulik, D. Chandam, P. Patil, D. Patil, S. Jagdale, S. Sankpal and M. Deshmukh, J. Heterocycl. Chem., 2015, 52, 931–937 Search PubMed.
- A. Mouradzadegun, M. A. Mostafavi and M. R. Ganjali, React. Kinet., Mech. Catal., 2018, 124, 741–755 Search PubMed.
- M. Mousapour, F. Shirini and M. Abedini, Polycyclic Aromat. Compd., 2019, 41, 419–426 Search PubMed.
- S. Z. D. Heirati, F. Shirini and A. F. Shojaei, J. Iran. Chem. Soc., 2020, 17, 3217–3225 Search PubMed.
- F. Mohamadpour, Polycyclic Aromat. Compd., 2020, 1–12, DOI:10.1080/10406638.2020.1768412.
- N. M. Shah, M. P. Patel and R. G. Patel, J. Heterocycl. Chem., 2012, 49, 1310–1316 Search PubMed.
- M. Elmi-Mehr, A. Davoodnia and M. Pordel, Russ. J. Gen. Chem., 2019, 88, 2595–2600 Search PubMed.
- M. Shekouhy, A. M. Sarvestani, S. Khajeh and A. Khalafi-Nezhad, RSC Adv., 2015, 5, 63705–63710 Search PubMed.
- O. S. Zaky, M. A. Selim, M. M. Ebied and K. U. Sadek, J. Heterocycl. Chem., 2019, 56, 2796–2803 Search PubMed.
- M. R. Nabid, S. J. Rezaei, R. Ghahremanzadeh and A. Bazgir, Ultrason. Sonochem., 2010, 17, 159–161 Search PubMed.
- M. V. Reddy, G. C. S. Reddy and Y. T. Jeong, Tetrahedron, 2012, 68, 6820–6828 Search PubMed.
- S. Rostamnia, H. Xin, X. Liu and K. Lamei, J. Mol. Catal. A: Chem., 2013, 374–375, 85–93 Search PubMed.
- H. Kefayati, A. Delafrooz and S. Homayoon, Russ. J. Gen. Chem., 2016, 86, 1735–1740 Search PubMed.
- N. Ghareb, H. A. Elshihawy, M. M. Abdel-Daim and M. A. Helal, Bioorg. Med. Chem. Lett., 2017, 27, 2377–2383 Search PubMed.
- P. B. Hiremath and K. Kamanna, Polycyclic Aromat. Compd., 2020, 1–17, DOI:10.1080/10406638.2020.1830129.
- H. R. Shaterian, M. Ghashang and M. Feyzi, Appl. Catal., A, 2008, 345, 128–133 Search PubMed.
- G. Sabitha, C. Srinivas, A. Raghavendar and J. S. Yadav, Helv. Chim. Acta, 2010, 93, 1375–1380 Search PubMed.
- S. Rostamnia and E. Doustkhah, Tetrahedron Lett., 2014, 55, 2508–2512 Search PubMed.
- M. V. Reddy, P. C. R. Kumar, G. C. S. Reddy and C. S. Reddy, C. R. Chim., 2014, 17, 1250–1256 Search PubMed.
- A. A. Amiri, S. Javanshir, Z. Dolatkhah and M. G. Dekamin, New J. Chem., 2015, 39, 9665–9671 Search PubMed.
- E. Doustkhah, A. Baghban, M. H. N. Assadi, R. Luque and S. Rostamnia, Catal. Lett., 2019, 149, 591–600 Search PubMed.
- A. M. Jadhav, S. G. Balwe and Y. T. Jeong, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 195, 201–210 Search PubMed.
- H. R. Shaterian and F. Rigi, Res. Chem. Intermed., 2013, 40, 1989–1995 Search PubMed.
- H. R. Shaterian, N. Fahimi and K. Azizi, Res. Chem. Intermed., 2013, 40, 1879–1898 Search PubMed.
- K. Pradhan, S. Paul and A. R. Das, Monatsh. Chem., 2014, 145, 1343–1352 Search PubMed.
- O. Goli-Jolodar, F. Shirini and M. Seddighi, RSC Adv., 2016, 6, 44794–44806 Search PubMed.
- M. Abbasi, S. M. R. Nazifi, Z. S. Nazifi and A. R. Massah, J. Chem. Sci., 2017, 129, 1257–1266 Search PubMed.
- R. Tayebee, M. M. Amini, S. Pouyamanesh and A. Aliakbari, Dalton Trans., 2015, 44, 5888–5897 Search PubMed.
- M. Nikoorazm, A. Ghorbani-Choghamaranai, M. Khanmoradi and P. Moradi, J. Porous Mater., 2018, 25, 1831–1842 Search PubMed.
- N. Kerru, L. Gummidi, S. V. H. S. Bhaskaruni, S. N. Maddila and S. B. Jonnalagadda, Res. Chem. Intermed., 2020, 46, 3067–3083 Search PubMed.
- H. N. Roy, M. Rana, A. Z. A. Munsur, K.-I. Lee and A. K. Sarker, Synth. Commun., 2016, 46, 1370–1376 Search PubMed.
- E. Lamera, S. Bouacida, H. Merazig, A. Chibani, M. Le Borgne, Z. Bouaziz and A. Bouraiou, Z. Naturforsch., B: J. Chem. Sci., 2017, 72, 361–368 Search PubMed.
- C. S. Maheswari, V. Tamilselvi, R. Ramesh and A. Lalitha, Org. Prep. Proced. Int., 2020, 52, 22–36 Search PubMed.
- F. Mohamadpour, Org. Prep. Proced. Int., 2020, 52, 64–68 Search PubMed.
- G. Shukla, R. K. Verma, G. K. Verma and M. S. Singh, Tetrahedron Lett., 2011, 52, 7195–7198 Search PubMed.
- M. A. Zolfigol, M. Mokhlesi and S. Farahmand, J. Iran. Chem. Soc., 2012, 10, 577–581 Search PubMed.
- A. B. Atar, S. D. Lee, B. G. Cho, D. W. Cho and Y. T. Jeong, Res. Chem. Intermed., 2015, 42, 1707–1728 Search PubMed.
- S. J. Saghanezhad and S. Sayyahi, Res. Chem. Intermed., 2016, 43, 2491–2500 Search PubMed.
- S. Karhale and V. Helavi, SN Appl. Sci., 2020, 2, 1227 Search PubMed.
- X. Shi, J. Li, W. Zhong and J. Li, J. Chem. Res., 2012, 36, 17–20 Search PubMed.
- Y. Wan, C. Wang, X.-X. Zhang, J. Shi, S.-Y. Huang, G.-X. Liu, L.-F. Chen, L.-L. Zhao, H.-Y. Wang and H. Wu, J. Heterocycl. Chem., 2014, 51, E77–E83 Search PubMed.
- X. Shi, M. Ding, C. Li, W. Wang and H. Guo, J. Heterocycl. Chem., 2018, 55, 440–446 Search PubMed.
- O. Goli-Jolodar, F. Shirini and M. Seddighi, J. Nanosci. Nanotechnol., 2018, 18, 591–603 Search PubMed.
- P. Jahanshahi and M. Mamaghani, New J. Chem., 2019, 43, 8266–8278 Search PubMed.
- F. Mohamadpour, M. T. Maghsoodlou, R. Heydari and M. Lashkari, J. Iran. Chem. Soc., 2016, 13, 1549–1560 Search PubMed.
- A. V. Chate, P. K. Bhadke, M. A. Khande, J. N. Sangshetti and C. H. Gill, Chin. Chem. Lett., 2017, 28, 1577–1582 Search PubMed.
- Y. A. Tayade and D. S. Dalal, Catal. Lett., 2017, 147, 1411–1421 Search PubMed.
- H. Kefayati, S. H. Amlashi, R. Kazemi-Rad and A. Delafrooz, C. R. Chim., 2014, 17, 894–898 Search PubMed.
- S.-H. Song, J. Zhong, Y.-H. He and Z. Guan, Tetrahedron Lett., 2012, 53, 7075–7077 Search PubMed.
- Y. D. Reddy, B. S. Narayana, C. V. Ramana Reddy and P. K. Dubey, Synth. Commun., 2014, 44, 3037–3046 Search PubMed.
- M. Abdesheikhi and Z. Karimi-Jaberi, J. Chem. Res., 2015, 39, 482–483 Search PubMed.
- A. Choudhury, S. Ali and A. T. Khan, J. Korean Chem. Soc., 2015, 59, 280–283 Search PubMed.
- L. Jalili-Baleh, H. Nadri, A. Moradi, S. N. A. Bukhari, M. Shakibaie, M. Jafari, M. Golshani, F. H. Moghadam, L. Firoozpour, A. Asadipour, S. Emami, M. Khoobi and A. Foroumadi, Eur. J. Med. Chem., 2017, 139, 280–289 Search PubMed.
- K. Turhan and Z. Turgut, Russ. J. Org. Chem., 2019, 55, 250–253 Search PubMed.
- P. M. Pisal, V. T. Kamble, R. Varala and P. B. Gujarathi, Org. Commun., 2020, 13, 194–201 Search PubMed.
- D. Pavithra and K. R. Ethiraj, Polycyclic Aromat. Compd., 2020, 1–14, DOI:10.1080/10406638.2020.1732430.
- P. Taslimi, K. Turhan, F. Türkan, H. S. Karaman, Z. Turgut and I. Gulcin, Bioorg. Chem., 2020, 97, 103647 Search PubMed.
- P. Taslimi, F. Türkan, K. Turhan, H. S. Karaman, Z. Turgut and İ. Gulcin, J. Heterocycl. Chem., 2020, 57, 3116–3125 Search PubMed.
- K. Mazaahir, C. Ritika and J. Anwar, Chin. Sci. Bull., 2012, 57, 2273–2279 Search PubMed.
- M. Kidwai and R. Chauhan, J. Heterocycl. Chem., 2014, 51, 1689–1696 Search PubMed.
- B. Afzalian, J. T. Mague, M. Mohamadi, S. Y. Ebrahimipour, B. P. Amiri and E. T. Kermani, Chin. J. Catal., 2015, 36, 1101–1108 Search PubMed.
- F. K. Nejad, M. Khosravan, S. Y. Ebrahimipour and F. Bisceglie, Appl. Organomet. Chem., 2018, 32, e3907 Search PubMed.
- S. S. Biroon, N. Shajari and H. Yahyaei, J. Heterocycl. Chem., 2020, 57, 2433–2445 Search PubMed.
- H.-J. Wang, X.-N. Zhang and Z.-H. Zhang, Monatsh. Chem., 2010, 141, 425–430 Search PubMed.
- E. Mosaddegh and A. Hassankhani, Tetrahedron Lett., 2011, 52, 488–490 Search PubMed.
- D. Azarifar, R. Nejat-Yami, Z. Akrami, F. Sameri and S. Samadi, Lett. Org. Chem., 2012, 9, 128–132 Search PubMed.
- H. R. Tavakoli, S. M. Moosavi and A. Bazgir, J. Korean Chem. Soc., 2013, 57, 472–475 Search PubMed.
- M. V. Reddy and Y. T. Jeong, Tetrahedron Lett., 2013, 54, 3546–3549 Search PubMed.
- A. M. Jadhav, S. G. Balwe, J. S. Kim, K. T. Lim and Y. T. Jeong, Tetrahedron Lett., 2019, 60, 560–565 Search PubMed.
- F. Mohamadpour, M. T. Maghsoodlou, R. Heydari and M. Lashkari, Res. Chem. Intermed., 2016, 42, 7841–7853 Search PubMed.
- M. Sayyafi, M. Seyyedhamzeh, H. R. Khavasi and A. Bazgir, Tetrahedron, 2008, 64, 2375–2378 Search PubMed.
- X. Wang, W.-W. Ma, L.-Q. Wu and F.-L. Yan, J. Chin. Chem. Soc., 2010, 57, 1341–1345 Search PubMed.
- R. Ghorbani-Vaghei, R. Karimi-Nami, Z. Toghraei-Semiromi, M. Amiri and M. Ghavidel, Tetrahedron, 2011, 67, 1930–1937 Search PubMed.
- R. Ghorbani-Vaghei, S. Noori, Z. Toghraei-Semiromi and Z. Salimi, RSC Adv., 2014, 4, 47925–47928 Search PubMed.
- M. Kidwai, A. Jahan, R. Chauhan and N. K. Mishra, Tetrahedron Lett., 2012, 53, 1728–1731 Search PubMed.
- X.-C. Jia, J. Li, Y. Ding, B. Zhang, N. Wang and Y.-H. Wang, J. Chem., 2013, 2013, 1–5 Search PubMed.
- R. Kiasat, A. Mouradezadegun and J. Saghanezhad, J. Serb. Chem. Soc., 2013, 78, 469–476 Search PubMed.
- A. Varghese, A. Nizam, R. Kulkarni and L. George, Eur. J. Chem., 2013, 4, 132–137 Search PubMed.
- B. Maleki, S. S. Ashrafi and R. Tayebee, RSC Adv., 2014, 4, 41521–41528 Search PubMed.
- M. Soheilizad, M. Adib and S. Sajjadifar, Monatsh. Chem., 2014, 145, 1353–1356 Search PubMed.
- A. Vafaee, A. Davoodnia, M. Pordel and M. Bozorgmehr, Orient. J. Chem., 2015, 31, 2153–2158 Search PubMed.
- H. R. M. Rashdan, S. M. Gomha, M. S. El-Gendey, M. A. El-Hashash and A. M. M. Soliman, Green Chem. Lett. Rev., 2018, 11, 264–274 Search PubMed.
- D. Habibi, S. Shojaei and S. Heydari, Russ. J. Org. Chem., 2021, 57, 85–90 Search PubMed.
- A. Bazgir, R. Ghahremanzadeh and G. Shakibaei, Synlett, 2008, 1129–1132 Search PubMed.
- J. M. Khurana and D. Magoo, Tetrahedron Lett., 2009, 50, 7300–7303 Search PubMed.
- R. Fazaeli, H. Aliyan and N. Fazaeli, Open Catal. J., 2010, 3, 14–18 Search PubMed.
- D. S. Raghuvanshi and K. N. Singh, Tetrahedron Lett., 2011, 52, 5702–5705 Search PubMed.
- F. Yang, H. J. Zang, Q. K. Wang, B. W. Cheng, Y. L. Ren and X. L. Xu, Adv. Mater. Res., 2011, 332–334, 1884–1887 Search PubMed.
- H. R. Shaterian and M. Aghakhanizadeh, C. R. Chim., 2012, 15, 1060–1064 Search PubMed.
- H. R. Shaterian and M. Mohammadnia, J. Mol. Liq., 2012, 173, 55–61 Search PubMed.
- S. Song, X. Deng, Z. Guan and Y. He, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 717–724 Search PubMed.
- D. Habibi and A. Shamsian, Res. Chem. Intermed., 2014, 41, 6245–6255 Search PubMed.
- M. Abedini, F. Shirini and J. M.-A. Omran, J. Mol. Liq., 2015, 212, 405–412 Search PubMed.
- F. Shirini, M. S. N. Langarudi and O. Goli-Jolodar, Dyes Pigm., 2015, 123, 186–195 Search PubMed.
- R. Tayebee, M. Jomei, B. Maleki, M. K. Razi, H. Veisi and M. Bakherad, J. Mol. Liq., 2015, 206, 119–128 Search PubMed.
- B. Godajddr, M. Etamad and S. Soliemani, Orient. J. Chem., 2015, 31, 1231–1236 Search PubMed.
- B. Pouramiri and E. T. Kermani, Tetrahedron Lett., 2016, 57, 1006–1010 Search PubMed.
- B. Pouramiri and E. T. Kermani, J. Iran. Chem. Soc., 2016, 13, 1011–1017 Search PubMed.
- A. Zare and F. Masihpour, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 1160–1165 Search PubMed.
- O. Goli-Jolodar and F. Shirini, Theor. Exp. Chem., 2017, 52, 349–357 Search PubMed.
- G. Jeevan Raghavendra and V. Siddaiah, Russ. J. Gen. Chem., 2018, 88, 1892–1898 Search PubMed.
- M. A. Shaikh, M. Farooqui and S. Abed, Res. Chem. Intermed., 2018, 44, 5483–5500 Search PubMed.
- N. Amirmahani, N. O. Mahmoodi, M. Malakootian and A. Pardakhty, Mol. Divers., 2022, 26, 15–25 Search PubMed.
- S. Fallah-Ghasemi Gildeh, M. Mehrdad, H. Roohi, K. Ghauri, S. Fallah-Ghasemi Gildeh and K. Rad-Moghadam, New J. Chem., 2020, 44, 16594–16601 Search PubMed.
- N. Safari, F. Shirini and H. Tajik, J. Mol. Struct., 2020, 1201, 127143 Search PubMed.
- F. Arian, M. Keshavarz, H. Sanaeishoar and N. Hasanzadeh, J. Mol. Struct., 2021, 1229, 129599 Search PubMed.
- B. Mombani Godajdar, A. R. Kiasat and M. M. Hashemi, Heterocycles, 2013, 87, 559 Search PubMed.
- A. Vafaee, A. Davoodnia, A. Nakhaei, S. Yadegarian and M. Nejatianfar, Russ. J. Gen. Chem., 2021, 91, 273–278 Search PubMed.
- A. G. Mulik, D. R. Chandam, D. R. Patil, P. P. Patil, G. N. Mulik, S. T. Salunkhe and M. B. Deshmukh, Res. Chem. Intermed., 2015, 41, 10085–10096 Search PubMed.
- W. Wang, L. Cong-Hao, Y. Yi, L. Xiao-Jun and G. Hong-Yun, J. Chem. Res., 2016, 40, 354–357 Search PubMed.
- L. Wang, M. Zhou, Q. Chen and M.-Y. He, J. Chem. Res., 2019, 37, 598–600 Search PubMed.
- H. Wu, J. J. Yin, W. G. Wamer, M. Zeng and Y. M. Lo, J. Food Drug Anal., 2014, 22, 86–94 Search PubMed.
- A. Azarifar, R. Nejat-Yami and D. Azarifar, J. Iran. Chem. Soc., 2012, 10, 297–306 Search PubMed.
- A. R. Kiasat, S. Noorizadeh, M. Ghahremani and S. J. Saghanejad, J. Mol. Struct., 2013, 1036, 216–225 Search PubMed.
- M. Saha, S. Phukan, R. Jamatia, S. Mitra and A. K. Pal, RSC Adv., 2013, 3, 1714–1721 Search PubMed.
- J. Safaei-Ghomi, H. Shahbazi-Alavi, A. Ziarati, R. Teymuri and M. R. Saberi, Chin. Chem. Lett., 2014, 25, 401–405 Search PubMed.
- E. Mosaddegh and A. Hassankhani, Catal. Commun., 2015, 71, 65–69 Search PubMed.
- J. Albadi, M. Jalali and A. Momeni, Res. Chem. Intermed., 2017, 44, 2395–2404 Search PubMed.
- B. Maleki, R. Nejat, H. Alinezhad, S. M. Mousavi, B. Mahdavi and M. Delavari, Org. Prep. Proced. Int., 2020, 52, 328–339 Search PubMed.
- H. R. Shaterian and M. Mohammadnia, Res. Chem. Intermed., 2012, 40, 371–383 Search PubMed.
- B. Atashkar, A. Rostami, H. Gholami and B. Tahmasbi, Res. Chem. Intermed., 2013, 41, 3675–3681 Search PubMed.
- A. R. Kiasat and J. Davarpanah, J. Mol. Catal. A: Chem., 2013, 373, 46–54 Search PubMed.
- A. Rostami, B. Tahmasbi and A. Yari, Bull. Korean Chem. Soc., 2013, 34, 1521–1524 Search PubMed.
- B. Mirhosseini-Eshkevari, M. A. Ghasemzadeh and J. Safaei-Ghomi, Res. Chem. Intermed., 2014, 41, 7703–7714 Search PubMed.
- E. Doustkhah and S. Rostamnia, J. Colloid Interface Sci., 2016, 478, 280–287 Search PubMed.
- M. Esmaeilpour, J. Javidi, F. N. Dodeji and S. Zahmatkesh, Res. Chem. Intermed., 2016, 47, 2629–2652 Search PubMed.
- A. Hashemzadeh, M. M. Amini, R. Tayebee, A. Sadeghian, L. J. Durndell, M. A. Isaacs, A. Osatiashtiani, C. M. A. Parlett and A. F. Lee, Mol. Catal., 2017, 440, 96–106 Search PubMed.
- F. Kamali and F. Shirini, New J. Chem., 2017, 41, 11778–11791 Search PubMed.
- L. Jiang and Z. Druzhinin, RSC Adv., 2019, 9, 15061–15072 Search PubMed.
- M. Khaleghi Abbasabadi and D. Azarifar, Appl. Organomet. Chem., 2020, 34, e5872 Search PubMed.
- M. M. F. Chegeni, A. Bamoniri and B. B. F. Mirjalili, Polycyclic Aromat. Compd., 2020, 1–9, DOI:10.1080/10406638.2020.1735457.
- M. M. F. Chegeni, A. Bamoniri and A. A. Taherpour, J. Heterocycl. Chem., 2020, 57, 2801–2814 Search PubMed.
- M. Nesarvand, D. Azarifar and H. Ebrahimiasl, Res. Chem. Intermed., 2021, 47, 3629–3644 Search PubMed.
- S. M. Nezhad, S. A. Pourmousavi and E. N. Zare, Curr. Org. Synth., 2022, 19, 246–266 Search PubMed.
- B. Maleki, S. B. N. Chalaki, S. S. Ashrafi, E. R. Seresht, F. Moeinpour, A. Khojastehnezhad and R. Tayebee, Appl. Organomet. Chem., 2015, 29, 290–295 Search PubMed.
- D. Azarifar, O. Badalkhani, Y. Abbasi and M. Hasanabadi, J. Iran. Chem. Soc., 2016, 14, 403–418 Search PubMed.
- Z. Zaheer, F. A. Khan, J. N. Sangshetti, R. H. Patil and K. S. Lohar, Bioorg. Med. Chem. Lett., 2016, 26, 1696–1703 Search PubMed.
- P. Arora and J. K. Rajput, Appl. Organomet. Chem., 2017, 32, e4001 Search PubMed.
- M. Hamidinasab and A. Mobinikhaledi, J. Iran. Chem. Soc., 2019, 16, 1255–1263 Search PubMed.
- X.-N. Zhao, G.-F. Hu, M. Tang, T.-T. Shi, X.-L. Guo, T.-T. Li and Z.-H. Zhang, RSC Adv., 2014, 4, 51089–51097 Search PubMed.
- B. Dam, M. Saha, R. Jamatia and A. K. Pal, RSC Adv., 2016, 6, 54768–54776 Search PubMed.
- G. M. Ziarani, N. H. Mohtasham, N. Lashgari and A. Badiei, Res. Chem. Intermed., 2014, 41, 7581–7591 Search PubMed.
- G. M. Ziarani, N. H. Mohtasham, A. Badiei and N. Lashgari, J. Chil. Chem. Soc., 2014, 61, 990–994 Search PubMed.
- G. M. Ziarani, A. Badiei, N. H. Mohtasham and N. Lashgari, J. Chil. Chem. Soc., 2014, 59, 2271–2274 Search PubMed.
- A. Bashti, A. R. Kiasat and B. Mokhtari, RSC Adv., 2015, 5, 25816–25823 Search PubMed.
- J. Davarpanah and A. R. Kiasat, RSC Adv., 2015, 5, 7986–7993 Search PubMed.
- H. Veisi, A. Sedrpoushan, A. R. Faraji, M. Heydari, S. Hemmati and B. Fatahi, RSC Adv., 2015, 5, 68523–68530 Search PubMed.
- M. Esmaeilpour, J. Javidi and F. Dehghani, J. Iran. Chem. Soc., 2015, 13, 695–714 Search PubMed.
- M. Ghahremani, J. Davarpanah, P. Rezaee and G. Davoodi, Res. Chem. Intermed., 2020, 46, 2683–2704 Search PubMed.
- S. R. Shafe-Mehrabadi, B. Sadeghi and A. Hassanabadi, Polycyclic Aromat. Compd., 2020, 1–8, DOI:10.1080/10406638.2020.1747094.
- S. Safaei, I. Mohammadpoor-Baltork, A. R. Khosropour, M. Moghadam, S. Tangestaninejad and V. Mirkhani, Catal. Sci. Technol., 2013, 3, 2717 Search PubMed.
- J. Davarpanah, P. Rezaee and S. Elahi, Res. Chem. Intermed., 2015, 41, 9903–9915 Search PubMed.
- F. H. Sabour, M. Nasr-Esfahani, I. Mohammadpoor-Baltork, S. Tangestaninejad, M. Moghadam and V. Mirkhani, J. Iran. Chem. Soc., 2017, 15, 671–683 Search PubMed.
- A. Khalili, M. Nasr-Esfahani, I. Mohammadpoor-Baltork, S. Tangestaninejad, V. Mirkhani and M. Moghadam, J. Mol. Liq., 2018, 253, 1–10 Search PubMed.
- N. Hosseininasab, A. Davoodnia, F. Rostami-Charati and A. Khojastehnezhad, Russ. J. Gen. Chem., 2017, 87, 2436–2443 Search PubMed.
- R. Ghorbani-Vaghei, J. Mahmoodi and Y. Maghbooli, Appl. Organomet. Chem., 2017, 31, e3717 Search PubMed.
- M. Kour, M. Bhardwaj, H. Sharma, S. Paul and J. H. Clark, New J. Chem., 2017, 41, 5521–5532 Search PubMed.
- R. Tayebee, M. F. Abdizadeh, B. Maleki and E. Shahri, J. Mol. Liq., 2017, 241, 447–455 Search PubMed.
- M. Kalhor, Z. Orouji and M. Khalaj, Microporous Mesoporous Mater., 2022, 329, 111498 Search PubMed.
- G. Shanthi and P. T. Perumal, J. Chem. Sci., 2010, 122, 415–421 Search PubMed.
- X.-N. Zhang, Y.-X. Li and Z.-H. Zhang, Tetrahedron, 2011, 67, 7426–7430 Search PubMed.
- S. J. T. Rezaei, Y. Bide and M. R. Nabid, Tetrahedron Lett., 2012, 53, 5123–5126 Search PubMed.
- J. Wang, X. Bai, C. Xu, Y. Wang, W. Lin, Y. Zou and D. Shi, Molecules, 2012, 17, 8674–8686 Search PubMed.
- H. Chen and D.-Q. Shi, J. Heterocycl. Chem., 2013, 50, 56–60 Search PubMed.
- S. M. Sadeghzadeh and M. A. Nasseri, Catal. Today, 2013, 217, 80–85 Search PubMed.
- L. Torkian, M. Dabiri, P. Salehi and M. Bararjanian, Helv. Chim. Acta, 2011, 94, 1416–1425 Search PubMed.
- P. Salehi, D. I. MaGee, M. Dabiri, L. Torkian and J. Donahue, Mol. Divers., 2012, 16, 231–240 Search PubMed.
- M. Dabiri, D. MaGee, P. Salehi, L. Torkian, M. Fakharian and J. Donahue, Synth. Commun., 2014, 44, 2037–2044 Search PubMed.
- S. Keshipour, S. Shojaei and A. Shaabani, Tetrahedron, 2012, 68, 6141–6145 Search PubMed.
- M. Yoosefian, H. Raissi, E. Davamdar, A. A. Esmaeili and M. Azaroon, Chin. J. Chem., 2012, 30, 779–784 Search PubMed.
- Y.-L. Xu, J.-Y. Fu, C.-H. Liu and T. Ding, RSC Adv., 2017, 7, 38733–38736 Search PubMed.
- E. Lamera, S. Bouacida, M. Le Borgne, Z. Bouaziz and A. Bouraiou, Tetrahedron Lett., 2017, 58, 1305–1307 Search PubMed.
- N. Berber, M. Arslan, E. Yavuz, C. Bilen and N. Gencer, J. Chem., 2013, 2013, 1–8 Search PubMed.
- N. Berber, M. Arslan, Ç. Bilen, Z. Sackes, N. Gençer and O. Arslan, Russ. J. Bioorg. Chem., 2015, 41, 414–420 Search PubMed.
- A. Yazdani-Elah-Abadi, R. Mohebat and M. Lashkari, Polycyclic Aromat. Compd., 2017, 40, 268–279 Search PubMed.
- L. Nagarapu, R. Bantu and H. B. Mereyala, J. Heterocycl. Chem., 2009, 46, 728–731 Search PubMed.
- X. Wang, G. Lu, W. Ma and L. Wu, E–J. Chem., 2011, 8, 1000–1005 Search PubMed.
- C. B. Sangani, J. A. Makwana, Y.-T. Duan, N. J. Thumar, M.-Y. Zhao, Y. S. Patel and H.-L. Zhu, Res. Chem. Intermed., 2015, 42, 2101–2117 Search PubMed.
- A. S. Amarasekara and S. Chandrasekara, Org. Lett., 2002, 4, 773–775 Search PubMed.
- Y. Liu, W. Zhen, W. Dai, F. Wang and X. Li, Org. Lett., 2013, 15, 874–877 Search PubMed.
- C. Yang, F. Song, J. Chen and Y. Huang, Adv. Synth. Catal., 2017, 359, 3496–3502 Search PubMed.
- M. Gholamhosseyni and E. Kianmehr, Org. Biomol. Chem., 2018, 16, 5973–5978 Search PubMed.
- P. Li, X. Xu, J. Chen, H. Yao and A. Lin, Org. Chem. Front., 2018, 5, 1777–1781 Search PubMed.
- C. K. Mahesha, D. S. Agarwal, P. Karishma, D. Markad, S. K. Mandal and R. Sakhuja, Org. Biomol. Chem., 2018, 16, 8585–8595 Search PubMed.
- L. Su, Z. Yu, P. Ren, Z. Luo, W. Hou and H. Xu, Org. Biomol. Chem., 2018, 16, 7236–7244 Search PubMed.
- X. Zhang, R. Bai, H. Xiong, H. Xu and W. Hou, Bioorg. Med. Chem. Lett., 2020, 30, 126916 Search PubMed.
- X. Wu and H. Ji, J. Org. Chem., 2018, 83, 4650–4656 Search PubMed.
- K. Kim, S. H. Han, D. Jeoung, P. Ghosh, S. Kim, S. J. Kim, J. M. Ku, N. K. Mishra and I. S. Kim, J. Org. Chem., 2020, 85, 2520–2531 Search PubMed.
- K. Konishi, M. Yasui, H. Okuhira, N. Takeda and M. Ueda, Org. Lett., 2020, 22, 6852–6857 Search PubMed.
- S. Hu, X. Han, X. Xie, F. Fang, Y. Wang, A. Saidahmatov, H. Liu and J. Wang, Adv. Synth. Catal., 2021, 363, 3311–3317 Search PubMed.
- H. Li, J. Zhao, S. Yi, K. Hu and P. Feng, Organometallics, 2021, 40, 880–889 Search PubMed.
- K. A. Abouzid and H. S. El-Abhar, Arch. Pharmacal Res., 2003, 26, 1–8 Search PubMed.
- J. Gardiner and A. D. Abell, Tetrahedron Lett., 2003, 44, 4227–4230 Search PubMed.
- B. Liu, J. D. Brandt and K. D. Moeller, Tetrahedron, 2003, 59, 8515–8523 Search PubMed.
- K. Wang, W. Wang, Q. Wang and R. Huang, Lett. Org. Chem., 2008, 5, 383–390 Search PubMed.
- A. P. Molchanov, D. I. Sipkin, Y. B. Koptelov and R. R. Kostikov, Russ. J. Org. Chem., 2005, 41, 567–574 Search PubMed.
- Z. Yang, Y. Zhou, H. Li, J. Lei, P. Bing, B. He and Y. Li, Asian J. Org. Chem., 2021, 11, e202100656 Search PubMed.
- R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2006, 128, 6330–6331 Search PubMed.
- A. Chan and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 5334–5335 Search PubMed.
- N. D. Shapiro, Y. Shi and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 11654–11655 Search PubMed.
- Y. Qian, P. J. Zavalij, W. Hu and M. P. Doyle, Org. Lett., 2013, 15, 1564–1567 Search PubMed.
- X. Xu, Y. Qian, P. Y. Zavalij and M. P. Doyle, J. Am. Chem. Soc., 2013, 135, 1244–1247 Search PubMed.
- X. Xu, X. Xu, P. Y. Zavalij and M. P. Doyle, Chem. Commun., 2013, 49, 2762–2764 Search PubMed.
- L. Liang and Y. Huang, Org. Lett., 2016, 18, 2604–2607 Search PubMed.
- A. O. Chagarovskiy, V. S. Vasin, V. V. Kuznetsov, O. A. Ivanova, V. B. Rybakov, A. N. Shumsky, N. N. Makhova and I. V. Trushkov, Angew. Chem., Int. Ed., 2018, 57, 10338–10342 Search PubMed.
- R. Dhakal, M. Ivancic and M. Brewer, J. Org. Chem., 2018, 83, 6202–6209 Search PubMed.
- R. Sonawane, V. Sikervar, R. Mandadapu, A. S. Dehade, S. A. Shete and M. Montgomery, Synthesis, 2021, 2865–2873 Search PubMed.
- A. Lebrene, T. Martzel, L. Gouriou, M. Sanselme, V. Levacher, S. Oudeyer, C. Afonso, C. Loutelier-Bourhis and J. F. Briere, J. Org. Chem., 2021, 86, 8600–8609 Search PubMed.
-
N. Haider and W. Holzer, Product Class 11: Pyridazino[1,2-a]pyridazines, 2004, DOI:10.1055/sos-SD-016-00455.
- P. Cai, E. Zhang, Y. Wu, T. Fang, Q. Li, C. Yang, J. Wang and Y. Shang, ACS Omega, 2018, 3, 14575–14584 Search PubMed.
- S. Kim, S. B. Choi, J. Y. Kang, W. An, S. H. Lee, H. Oh, P. Ghosh, N. K. Mishra and I. S. Kim, Asian J. Org. Chem., 2021, 10, 3005–3014 Search PubMed.
- V. V. Ganzha, A. D. Kotov, T. V. Kesareva and V. Y. Orlov, Chem. Heterocycl. Compd., 2006, 42, 963–964 Search PubMed.
- D. Habibi, N. Mahmoudi and O. Marvi, Synth. Commun., 2007, 37, 3165–3171 Search PubMed.
- P. Karishma, C. K. Mahesha, D. S. Agarwal, S. K. Mandal and R. Sakhuja, J. Org. Chem., 2018, 83, 11661–11673 Search PubMed.
- P. Karishma, D. S. Agarwal, B. Laha, S. K. Mandal and R. Sakhuja, Chem. – Asian J., 2019, 14, 4274–4288 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Praveen
Kumar
ab,
Aaron
Nilsen
c and
Sanjay V.
Malhotra
ab,
Praveen
Kumar
ab,
Aaron
Nilsen
c and
Sanjay V.
Malhotra