
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkyl groups in organic molecules are NOT inductively electron-releasing†
Mark C.
Elliott
 *,
Colan E.
Hughes
,
Peter J.
Knowles
and
Benjamin D.
Ward
*,
Colan E.
Hughes
,
Peter J.
Knowles
and
Benjamin D.
Ward
School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK. E-mail: elliottmc@cardiff.ac.uk
First published on 12th November 2024
Abstract
It is commonly stated that alkyl groups exert an inductive electron-releasing effect when compared to hydrogen. This information has been given in numerous organic chemistry textbooks over the last 75 years. The evidence for this position is weak, and does not withstand scrutiny, and there is some evidence for the contrary position. We provide a significant body of computational data that clearly show that alkyl groups exert an inductive electron-withdrawing (–I) effect when compared to hydrogen. This revised position is not in conflict with experimental data, since alkyl group inductive effects are small and are likely to be masked by hyperconjugation/polarizability effects (particularly in charged species), and also by solvent effects.
Introduction
Early in the development of organic chemistry, it was postulated that alkyl groups exert an inductive electron-donating effect (+I, in the terminology introduced by Ingold1) compared to hydrogen. By the 1960s, this supposed electronic nature of alkyl groups was entrenched in textbooks, and that continues to this day.2 The opposite position, an inductive electron-withdrawing effect, would be supported by the fact that carbon is more electronegative than hydrogen (2.52 versus 2.20 on the Pauling scale).The apparent electron-donating inductive nature of alkyl groups made its way into early organic chemistry textbooks, starting with that of Ingold.1 In a classic textbook popular in the UK from the 1960s–1980s, Sykes3 stated (p. 22) that “alkyl groups are electron-donating”, referring to the higher rate of nitration of toluene compared to benzene as partial evidence of this. This observation can be fully explained by hyperconjugative stabilization of the reaction intermediate, and indeed the fact that alkyl groups can stabilize carbocations is probably at least partly responsible for the problem. In early textbooks,4 prior to our present understanding of hyperconjugation, carbocation stabilization by alkyl groups was presented as an inductive effect, and this remains the case even in some modern textbooks.5 The effect is also quoted in some textbooks as the reason for trends in the acidity of alcohols and carboxylic acids, and the basicity of amines which we will discuss.6
The stabilization of carbanions by alkyl groups is complex,7 and relatively few textbooks give a trend for carbanion stability.8 Nevertheless, some textbooks (including one written by one of the present authors) do draw incorrect conclusions based on the perceived inductive electron-releasing ability of alkyl groups.9 In the case of the present author, this is largely a result of having learned the trends in inductive effects very early on and not, until recently, having questioned this position.
In fact, there is already compelling evidence that alkyl groups exert an inductive electron-withdrawing effect (–I), accompanied by an electron-releasing hyperconjugation effect (+R).10 It would appear that the evidence for this position is not widely known and, as a result, textbook authors and educators routinely invoke alkyl group inductive electron-donating effects to explain phenomena that are best explained in other ways.
To properly understand the relative importance of such factors (hyperconjugation and other stereoelectronic effects,11 polarizability, solvent effects, etc.) it is critical that the direction of the alkyl group inductive effect is corrected. In this work, we consider the evidence on which the current position is based and the already published evidence for alkyl groups being inductively electron-withdrawing. We then present the results of a computational study that clearly demonstrates that alkyl groups are inductively electron-withdrawing (–I) relative to hydrogen in all cases. This is true for all partitioning schemes used to define partial charges. We find no conflict between this new position and existing experimental data.
What is the evidence for alkyl groups being inductively electron-donating?
The genesis of the alkyl group inductive effect is rather elusive. In 1916, Lewis gave the “familiar” relative acidity of acetic acid and chloroacetic acid as an example of the impact of an inductive effect.12 In 1925, Lucas, Simpson and Carter noted that formic acid is more acidic than acetic acid, drawing the conclusion that “hydrogen exerts a stronger pull on electrons than methyl does.”13 This is the earliest mention of the alkyl group inductive effect that we are aware of, although it is now clear that relative acidity is unlikely to be due to an inductive effect.14 Ingold uses the same example,1 with the subtle difference that the lower acidity of acetic acid would be attributed to an inductomeric (i.e. polarizability) effect rather than to a purely inductive effect. In an influential 1935 review,15 which formed the basis of significant portions of his seminal textbook,1 Ingold classified alkyl groups as +I (inductively electron-donating). The order of basicity of amines in water follows the trend:| Me2NH > MeNH2 > Me3N > NH3 |
This has long been attributed to the electron-donating ability of alkyl groups increasing the negative charge, although the importance of solvation of the corresponding ammonium ion was recognized as a reason for the lower basicity of Me3N. Again, a more regular trend is observed in the gas phase,16,17 with the tertiary amine being more basic as a result of stabilization of the ammonium ion due to the polarizability of the butyl groups, rather than as an inductive electron-donating effect. Finally, the dipole moments for representative alkylbenzenes are as follows (Fig. 1):18
 | ||
| Fig. 1 Dipole moments of representative alkylbenzenes. | ||
The dipole vector is directed towards the benzene ring, which has been interpreted as an electron-donating effect. Libit and Hoffmann19 interpret such data as a perturbation of the π system by the alkyl substituent rather than as electron-donation from the alkyl substituent, and we will return to this point later in the manuscript.
In short, although there was initially good reason to suppose that alkyl groups are inductively electron-donating relative to hydrogen, the experimental data for this position are not compelling by modern standards.
What is the evidence for alkyl groups being inductively electron-withdrawing?
In 1966, Laurie and Muetner used elegant dipole moment measurements of deuterated compounds to show that a methyl group attached to saturated carbon exerted an electron-withdrawing inductive effect.20 Laurie and Muetner did not challenge the position of alkyl groups more generally, and indeed stated that “it is well established that a methyl group bonded to an unsaturated carbon acts as an electron donor”. Hehre and Pople presented the dipole moments for ammonia and methylamine, and calculated acidities/basicities for a small selection of amines and alcohols to support an inductive electron-withdrawing effect for methyl groups in 1970, although polarizability was also shown to be a factor.21 Texidor et al. showed that a methyl group attached to boron in carboranes is electron-withdrawing, which then hinders further methylation.22 Early work in this area was summarized by Sebastian.23 In 2013, de Freitas and Firme presented calculations that suggest an inductive electron-withdrawing effect for alkyl groups attached to alkenes.24Are alkyl groups inductively electron-donating?
Before proceeding further, it is worth reviewing the definition of the inductive effect, and perhaps refining it for the purposes of the present work. The IUPAC definition of an inductive effect is “an experimentally observable effect (on rates of reaction, etc.) of the transmission of charge through a chain of atoms by electrostatic induction.”25 The explicit mention of rates of reaction in this definition would tend to imply the inclusion of polarizability effects within transition states and other charged species. Widing and Levitt state that “the polar inductive effect of the group R comprises all those processes (our emphasis) whereby it can modify the electrostatic forces operating at the reaction center X relative to the reference group Ro acting in the molecule RoX”.26In the present work, we will apply a narrower definition of inductive effects, this being the ground state polarization of σ-bonds in neutral molecules as a result of the ability of groups at either end of the bond to draw electron-density towards themselves. It is important to restrict the definition to neutral molecules to avoid polarizability effects which are likely to be more significant in charged species. By attempting to isolate the inductive effects, this will provide greater clarity of the hyperconjugation and polarizability effects.
To eliminate complications from effects such as polarizability, we must only consider neutral molecules, ideally lacking electronegative elements. Structural changes inherent in the introduction of alkyl groups mean that experimental results will inevitably be accompanied by steric and solvent effects. Indeed, if the direction of alkyl group inductive effects was easy to probe experimentally, the present ambiguity would not remain. We have selected calculated charges as our point of comparison.
There are many different ways to calculate atomic charges, all of which are limited by the fact that the charge on an atom cannot be precisely defined. Wiberg and Rablen have recently critically reviewed the various methods used to calculate atomic charges.27 Cho et al. have undertaken population analysis to compare many of the available charge models.28 We have evaluated several commonly-used charge models (Mulliken,29 NBO,30 Hirshfeld,31 CM5,32 QTAIM33).
We used non-empirical Kohn–Sham Density-Functional Theory (DFT) with the PBEh1PBE34 functional and a flexible orbital basis set (aug-cc-pVTZ35), using the Gaussian 09 software,36 and initially considered the charges on carbon atom in a range of alkanes as shown in Fig. 2. Specifically, we replaced each hydrogen atom in methane with one to four methyl groups. Charges on carbon are shown using each of the charge models. The Mulliken charges are exaggerated, and are known to be problematic.37 All other charge models put this charge close to zero, which we find to be logical. A methyl group attached to a carbon atom should really exert little or no net inductive effect. If we consider Hirshfeld charges, methane has −0.159 on carbon, which represents the inductive electron-donating effect of four hydrogen atoms. Since hydrogen is electron-donating relative to a methyl group, it follows that a methyl group is electron-withdrawing relative to hydrogen.
 | ||
| Fig. 2 Calculated charges (e) on the central C atom in representative alkanes. Hirshfeld charges are shown in blue, CM5 in orange, Mulliken in green, NBO in red and QTAIM in purple. | ||
From this point, we will give only Hirshfeld charges in the manuscript, for reasons given in the next section.
Having established that methyl groups attached to sp3 hybridized carbon exert a net electron-withdrawing effect relative to hydrogen, we broadened the scope of the study. Five series of compounds were evaluated, giving the calculated charges on the underlined atoms as shown in Fig. 3.
 | ||
| Fig. 3 Calculated Hirshfeld atomic charges (e) on the black underlined carbon atom as a function of the group ‘R’. | ||
The charge on the underlined atom becomes more positive by a similar amount for all series when hydrogen is replaced by any of the alkyl groups. If we assume this is an inductive effect, then we are once again forced to conclude that alkyl groups are inductively electron-withdrawing (–I) when compared to hydrogen, and that this is the case for alkyl groups attached to sp3, sp2, and sp hybridized carbon. Identical conclusions are reached when the other charge models are used and corresponding figures using these charge models are also given in the ESI.†
In summary, the answer to the question posed in the title of this section is ‘no’. For systems designed to isolate inductive effects, an alkyl group is inductively electron-withdrawing compared to hydrogen. It may be more convenient to consider alkyl groups as having no inductive effect when attached to carbon in organic molecules, and hydrogen to be inductively electron-donating towards carbon.
It is clear from Fig. 3 that the difference between alkyl groups is much smaller than that between H and alkyl. Furthermore, the direction of any trend, as measured by the calculated Hirshfeld charges, depends on what the alkyl group is attached to, and as such may not be due to inductive effects. While alkyl group inductive effect trends have been reported in the early years,38 we find no evidence for a trend. Indeed, it would be almost impossible to attribute any experimental observation to a difference in inductive effect between alkyl groups given that the experiment would inevitably introduce steric effects, which will then also affect solvation.
Benchmarking of computational data
The results of this study depend on the calculated atomic charges being a helpful representation of the overall charge distribution. There are two aspects to this:1. The density obtained using DFT should be a good approximation to exact electron density;
2. The point charge distribution arising from the calculated atomic charges should generate an electrostatic potential that is similar to that obtained from the parent DFT calculation.
We have investigated both of these aspects by comparing computed dipole moments for several molecules with appropriate reference values.
Table 1 shows the dipole moments for propane and toluene obtained from the DFT density, together with values from more accurate coupled-cluster calculations, and empirical values.
Single and double excitation coupled-cluster with perturbative correction for triples (CCSD(T)) gives 0.0856 Debye for propane. The calculations presented show that the orbital basis set limit has been reached, and the small deviation from the empirical value can be associated with remaining electron correlation, and vibrational effects. PBEh1PBE is in good agreement, overestimating the dipole moment, which is itself a small value that is sensitive to density fluctuations, by about 6%. Also shown for propane is a calibration of Pair Natural Orbital Local CCSD(T) (PNO-LCCSD(T)), which underestimates the dipole very slightly. In the case of toluene, the strong resource demands of full CCSD(T) calculations are avoided by using PNO-LCCSD(T), again yielding a dipole that is close to the empirical value. The PBEh1PBE dipole is in reasonable agreement, overestimating by about 15%.
Within molecular orbital theory, total molecular dipole moments can be calculated exactly as the sum of contributions from individual bond orbitals, and to a good approximation these have been shown to be transferable.44 As validation of the charge partitioning, dipole moments can also be calculated directly from the atomic charges. These are shown in Table 2 for each of the charge models considered, colour-coded according to their level of agreement with the dipole moment from the overall density (green = within 0.1 D, yellow = within 0.5 D, amber ≧ 0.5 D error; a negative sign indicates that the dipole from the charges is in the opposite direction to that calculated from the density). The RMS error for each charge model is given. Mulliken charges give, as expected with extended basis sets, poor agreement, and warrant no further discussion. QTAIM charges give good agreement for alkanes and for alkylbenzenes, but are poor for alkenes and alkynes; however, there is no particular reason to suppose that the charge partitioning arising from the shape of the density will give rise to a point-charge model with similar electrostatics to the total density. In practice, we find that the QTAIM charges change significantly when obtaining the density from different density functionals, or with different orbital basis sets, indicating that they are sensitive to small changes in the density, and probably unsuitable for use in interpreting trends in chemical reactivity. NBO is constructed primarily to support qualitative analysis of bonding contributions from localised orbitals, and the resultant atomic charge partitioning may or may not turn out to give the best representation of the overall Coulomb field of the molecule; with the set of molecules studied, it performs reasonably in this respect.

|
CM5 is constructed to give improved overall dipole moments relative to Hirshfeld, and one might expect it to be the best approach here. For the molecules studied, the opposite appears to be true; overall, Hirshfeld charges give the best agreement with the dipole moment from the overall density. The dipole moments for propane and isobutane are underestimated by this method, but in both cases the overall value is small, and sensitive to changes in the density.
On the use of dipole moments as a measure of alkyl group inductive effects
The use of dipole moments as evidence of inductive effects in hydrocarbons is complex and warrants further discussion. The experimental43 dipole moments of propane and isobutane have been used to support the position that methyl groups are electron-withdrawing when compared to hydrogen, as was previously noted by Salvatella.10The problem with using dipole moments as evidence of inductive effects can be seen in the following simplified situation for methane and propane. If we assume that each hydrogen atom is inductively electron-donating to the extent of 0.1 electrons, and the carbon charges are a consequence of only this, the atomic charges will be as shown in Fig. 5. In this scheme, we have replaced two electron-donating hydrogen atoms (in methane) with methyl groups (in propane) that have no net electronic effect (the overall charge on the methyl groups is zero). We would therefore conclude that the methyl groups are electron-withdrawing compared to the hydrogen atoms they replaced. For the optimized geometry of propane, the calculated dipole moment of propane from these atomic charges is 0.012 D, in the opposite direction to that given in Fig. 4. These atomic charges are similar to the calculated CM5 charges for these compounds.
 | ||
| Fig. 4 Dipole moments in propane and isobutane. | ||
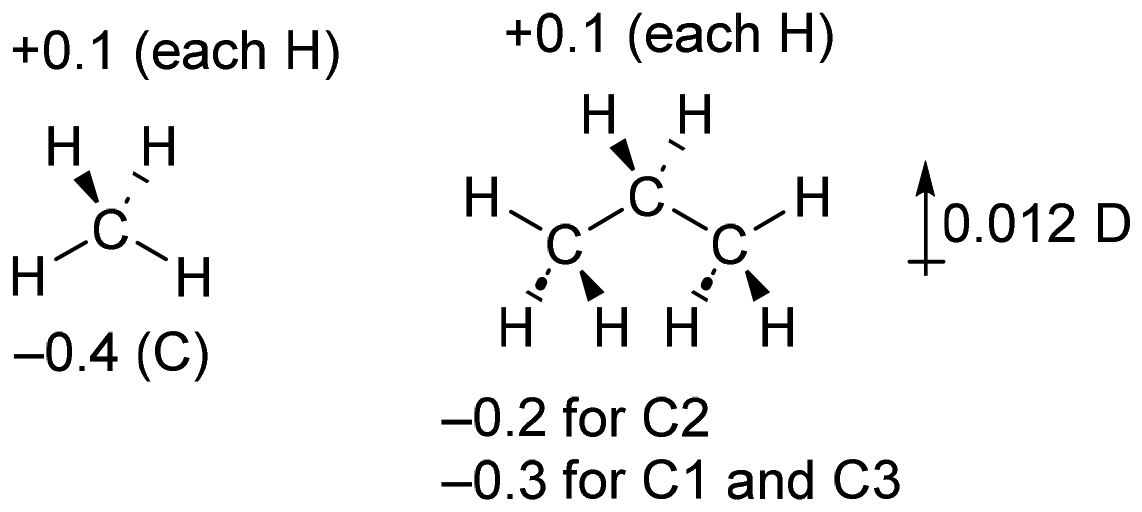 | ||
| Fig. 5 Hypothetical simplified charges for methane and propane and the calculated dipole moment of propane based on this data. | ||
That is, depending on the precise charge distribution, it is perfectly reasonable to have a methyl group exert an inductive electron-withdrawing (or donating) effect, and to find a dipole moment in either direction depending on small variations in atomic charge. This is rather counter-intuitive but follows directly from the calculated charges and atomic positions. We should be very cautious inferring alkyl group inductive effects from the dipole moment without more thorough consideration of the charge distribution.
The dipole moment in toluene is larger (0.37 D) and was originally used to support the position that alkyl groups donated electron-density towards the benzene ring.18 The overall effect was considered to be a combination of an electron-donating inductive (+I) effect in addition to a mesomeric electron-donating effect (+R, hyperconjugation). In fact, the dipole moment in toluene does not mean that the methyl group is electron-donating. It is perfectly reasonable for the methyl group to exert an electron-withdrawing effect relative to hydrogen, and to have the dipole in the direction shown in Fig. 6.18 The dipole moment in toluene is a result of perturbation of the benzene ring by the methyl group (and of the methyl group by the benzene ring), rather than donation of electron-density from the methyl group.19
 | ||
| Fig. 6 Experimental and calculated dipole moment for toluene. | ||
Impact on experimental data and the limits of inductive effects
We have seen that the evidence for alkyl groups being inductively electron-releasing was tenuous at best. The most compelling experimental evidence for alkyl groups attached to benzene rings being electron-donating effect is the Hammett linear free energy parameters.45 However, these represent a hyperconjugation effect, and changing the direction of the inductive effect of an alkyl group to electron-withdrawing does contradict these parameters. The inductive effect impacts the point of attachment of the alkyl group, whereas the Hammett parameters are used to describe effects at the meta and para positions. Such parameters are not used for the ortho position for steric reasons. On this basis it is not possible to determine the relative contribution of the two effects, but it seems likely that hyperconjugation is the smaller effect in alkylbenzenes.46,47Even if inductive effects are larger than hyperconjugation effects in neutral organic molecules, there is no reason to believe that this would be the case in charged species. In Fig. 7 we show the calculated Hirshfeld charges of alkanes (R-CH3), alcohols (R-OH) and alkoxide anions (R-O⊖). While the effects of alkyl groups compared to H and to each other is essentially identical for the alkanes and alcohols, the alkoxide charge is much more responsive to the change from H to alkyl as well as within the series of alkyl groups.
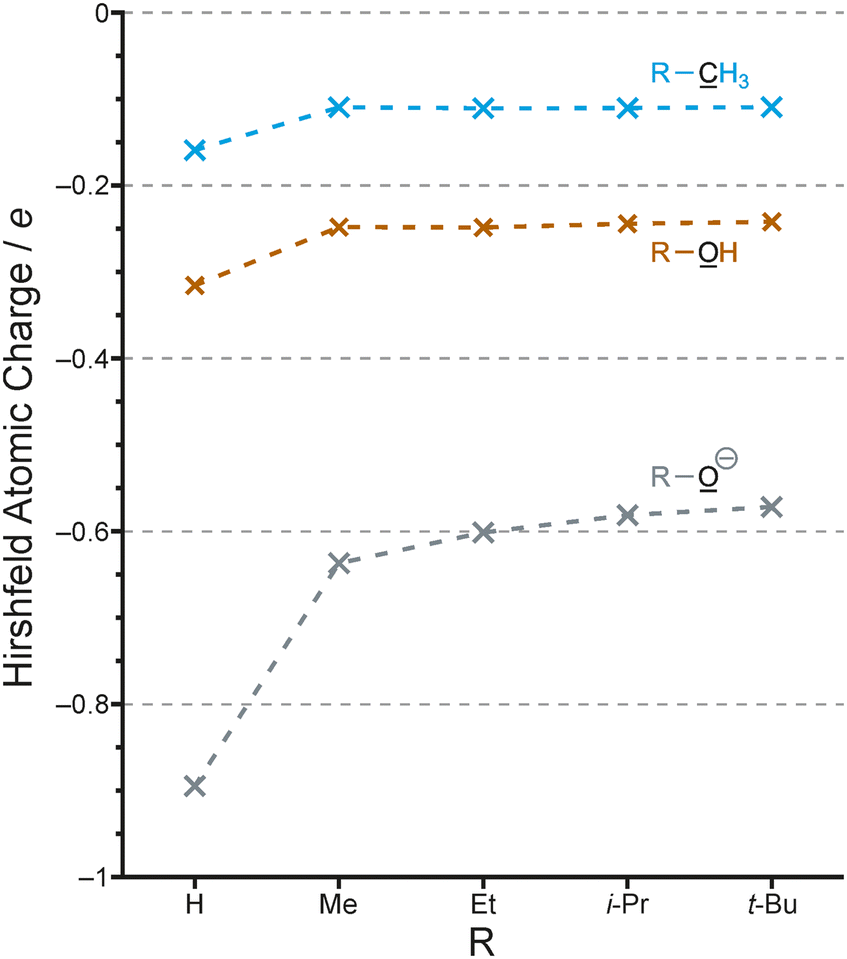 | ||
| Fig. 7 Calculated Hirshfeld charges on carbon and oxygen for R-X (R as above; X = CH3, OH and O⊖). | ||
The better dispersal of negative charge by the t-butyl group compared to methyl etc. results in the higher stability of the t-butoxide anion (compared to methoxide, etc.) in the gas phase, and it is well established that this is due to polarizability rather than being an inductive effect.48 Methanol is more acidic than t-butanol in aqueous solution,49 so that even this larger electronic effect can be masked by solvent effects. This further reinforces the point that there will be few, if any, measurable consequences of the alkyl group inductive effect.
Similarly, the stabilization of the t-butyl carbocation relative to the methyl carbocation can be attributed entirely to hyperconjugation, and in fact any inductive effect from the three methyl groups is now expected to be electron-withdrawing and therefore destabilizing. Since the electron-donating effect of hyperconjugation in the carbocation is so much larger in magnitude than the inductive effect, the latter is irrelevant.
Conclusions
All alkyl groups should be considered as inductively electron-withdrawing compared to hydrogen. This is found to be the case using various partitioning models for atomic charge when representative alkyl groups are attached to sp3, sp2 or sp hybridized carbon, to OH, and presumably to other groups. The previous position, that alkyl groups are inductively electron-releasing relative to hydrogen, is illogical and was reached based on data that are better interpreted in other ways. This position has been challenged intermittently over the years, most recently and comprehensively by Salvatella.10 To a very large extent, the problem is more pedagogic than practical. In reality, there are probably no directly observable consequences for the revised position. We would advise not mentioning alkyl group inductive effects beyond (possibly) a simple statement of their direction and that this makes little difference in reality, since the effect is small and will be masked by other effects. We would certainly recommend that alkyl group inductive effects no-longer be used to explain properties such as alcohol and carboxylic acid acidity and amine basicity, for which polarizability effects dominate in the gas phase and solvent effects in solution. Most importantly, alkyl group inductive effects should never be used as an explanation for carbocation stability. Hyperconjugation, with its corresponding molecular orbital representation, should be used exclusively at the explanation for the stabilization of carbocations by alkyl groups.Author contributions
MCE proposed the concept. MCE, BDW and PJK designed and carried out the computational study. All authors contributed to discussion and interpretation of the data, and to the writing and editing of the manuscript.Data availability
The computational data supporting the results presented in this article are freely available via the Cardiff University Data Catalogue (DOI: 10.17035/cardiff.27073675).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Professor Barry Carpenter, Professor Philip Davies, Dr Niek Buurma (all Cardiff University), Professor Keith Jones (Kingston University) and Professor Nick Greeves (University of Liverpool) for helpful discussions during preparation of this manuscript. We would like to thank Professor Mirosław Jabłoński (Warsaw University) for discussion of the propyne atomic charges. This research was undertaken using the supercomputing facilities at Cardiff University operated by Advanced Research Computing at Cardiff (ARCCA) on behalf of the Cardiff Supercomputing Facility and the HPC Wales and Supercomputing Wales (SCW) projects. We acknowledge the support of the latter, which is part-funded by the European Regional Development Fund (ERDF) via the Welsh Government.References
- C. K. Ingold, Structure and Mechanism in Organic Chemistry, G. Bell and Sons, London, 1953 Search PubMed.
- M. B. Smith and J. March, in March's Advanced Organic Chemistry, Wiley Interscience, New York, 7th edn, 2013, p. 21 Search PubMed; G. Solomons and C. Fryhle, Organic Chemistry, Wiley, New York, 7th edn, 2000, p. 247 Search PubMed; J. M. Hornback, in Organic Chemistry, Brooks/Cole, Pacific Grove, 1998, p. 121 Search PubMed; P. Vollhardt and N. Schore, in Organic Chemistry: Structure and Function, Freeman, New York, 7th edn, 2014, p. 696 Search PubMed; J. Karty, in Organic Chemistry: Principles and Mechanisms, Norton, New York, 2014, p. 325–327 Search PubMed.
- P. Sykes, in A Guidebook to Mechanism in Organic Chemistry, Longman, 6th edn, 1986 Search PubMed.
- R. T. Morrison and R. N. Boyd, in Organic Chemistry, Allyn and Bacon, New York, 2nd edn, 1966, p. 167 Search PubMed.
- F. A. Carey, in Organic Chemistry, McGraw Hill, Boston, 5th edn, 2003, p. 161 Search PubMed; J. Karty, in Organic Chemistry: Principles and Mechanisms, Norton, New York, 2014, p. 327 Search PubMed; L. G. Wade and J. W. Simek, in Organic Chemistry, Pearson, 10th Global edn, 2023, p. 231 Search PubMed; J. E. McMurry, in Organic Chemistry, Brooks/Cole, 8th edn, 2012, p. 245 Search PubMed.
- G. Solomons and C. Fryhle, Organic Chemistry, Wiley, New York, 7th edn, 2000, p. 947 Search PubMed; J. Karty, in Organic Chemistry: Principles and Mechanisms, Norton, New York, 2014, p. 325 Search PubMed; D. Klein, in Organic Chemistry as a Second Language, Wiley, 3rd edn, 2012, p. 62 Search PubMed; A. Streitwieser Jr. and C. H. Heathcock, in Introduction to Organic Chemistry, Collier MacMillan, 3rd edn, 1985, p. 197 Search PubMed; J. M. Hornback, in Organic Chemistry, Brooks/Cole, Pacific Grove, 1998, p. 121 Search PubMed; P. Sykes, in A Guidebook to Mechanism in Organic Chemistry, Longman, 6th edn, 1986, p. 66 Search PubMed; R. T. Morrison and R. N. Boyd, in Organic Chemistry, Allyn and Bacon, New York, 2nd edn, 1966, p. 745 Search PubMed.
- C. H. DePuy, S. Gronert, S. E. Barlow, V. M. Bierbaum and R. Damrauer, J. Am. Chem. Soc., 1989, 111, 1968–1973 CrossRef CAS.
- P. Sykes, in A Guidebook to Mechanism in Organic Chemistry, Longman, 6th edn, 1986, p. 273 Search PubMed; L. G. Wade and J. W. Simek, in Organic Chemistry, Pearson, 10th Global edn, 2023, p. 234 Search PubMed; M. C. Elliott, How to Succeed in Organic Chemistry, Oxford University Press, Oxford, 2020, p. 135 Search PubMed.
- P. Sykes, in A Guidebook to Mechanism in Organic Chemistry, Longman, 6th edn, 1986, p. 82–83 Search PubMed; J. Clayden, N. Greeves and S. Warren, Organic Chemistry, Oxford University Press, Oxford, 2012, pp. 399–491 Search PubMed; P. Vollhardt and N. Schore, in Organic Chemistry: Structure and Function, Freeman, New York, 7th edn, 2014, p. 696 Search PubMed; M. C. Elliott, How to Succeed in Organic Chemistry, Oxford University Press, Oxford, 2020, p. 203 Search PubMed.
- L. Salvatella, Educ. Quim., 2017, 28, 232–237 Search PubMed.
- I. Alabugin, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, Wiley, Chichester, 2016 Search PubMed.
- G. N. Lewis, J. Am. Chem. Soc., 1916, 38, 762–785 CrossRef CAS.
- H. J. Lucas, T. P. Simpson and J. M. Carter, J. Am. Chem. Soc., 1925, 47, 1462–1469 CrossRef CAS.
- M. F. R. Siggel and T. D. Thomas, J. Am. Chem. Soc., 1992, 114, 5795–5800 CrossRef CAS.
- C. K. Ingold, Chem. Rev., 1934, 15, 238–274 CrossRef.
- J. I. Brauman, J. M. Riveros and L. K. Blair, J. Am. Chem. Soc., 1971, 93, 3914–3916 CrossRef CAS.
- D. H. Aue, H. M. Webb and M. T. Bowers, J. Am. Chem. Soc., 1976, 98, 311–317 CrossRef CAS.
- T. L. Brown, J. Am. Chem. Soc., 1959, 81, 3232–3235 CrossRef CAS.
- L. Libit and R. Hoffmann, J. Am. Chem. Soc., 1974, 96, 1370–1383 CrossRef CAS.
- V. W. Laurie and J. S. Muetner, J. Am. Chem. Soc., 1966, 88, 2883–2884 CrossRef CAS.
- W. J. Hehre and J. A. Pople, Tetrahedron Lett., 1970, 11, 2959–2962 CrossRef.
- F. Texidor, G. Barberà, A. Vaca, R. Kivekäs, R. Sillanpää, J. Oliva and C. Viñas, J. Am. Chem. Soc., 2005, 127, 10158–10159 CrossRef PubMed.
- J. F. Sebastian, J. Chem. Educ., 1971, 48, 97–98 CrossRef CAS.
- G. R. S. de Freitas and C. L. Firme, J. Mol. Model., 2013, 19, 5267–5276 CrossRef.
- ‘Inductive effect’ in IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry, 3rd edn, 2006. Online version 3.0.1, 2019. DOI:10.1351/goldbook.I03021.
- H. F. Widing and L. S. Levitt, Z. Naturforsch., 1979, 34b, 321–326 CrossRef CAS.
- K. B. Wiberg and P. R. Rablen, J. Org. Chem., 2018, 83, 15463–15469 CrossRef CAS PubMed.
- M. Cho, N. Sylvetsky, S. Eshafi, G. Santra, I. Efremenko and J. M. L. Martin, ChemPhysChem, 2020, 21, 688–696 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527–541 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms In Molecules: A Quantum Theory, Clarendon Press, Oxford, 1990 Search PubMed.
- M. Ernzerhof and J. P. Perdew, J. Chem. Phys., 1998, 109, 3313–3320 CrossRef CAS.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS; D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1994, 100, 2975–2988 CrossRef; R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- B. Wang, S. L. Li and D. G. Truhlar, J. Chem. Theory Comput., 2014, 10, 5640–5650 CrossRef CAS PubMed.
- J. W. Baker and W. S. Nathan, J. Chem. Soc., 1935, 1844–1847 RSC.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip Rev. Comput. Mol. Sci., 2012, 242–253 CrossRef CAS; H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller III, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, J. Chem. Phys., 2020, 152, 144107 CrossRef PubMed; H.-J. Werner, P. J. Knowles, P. Celani, W. Györffy, A. Hesselmann, D. Kats, G. Knizia, A. Köhn, T. Korona, D. Kreplin, R. Lindh, Q. Ma, F. R. Manby, A. Mitrushenkov, G. Rauhut, M. Schütz, K. R. Shamasundar, T. B. Adler, R. D. Amos, J. Baker, S. J. Bennie, A. Bernhardsson, A. Berning, J. A. Black, P. J. Bygrave, R. Cimiraglia, D. L. Cooper, D. Coughtrie, M. J. O. Deegan, A. J. Dobbyn, K. Doll, M. Dornbach, F. Eckert, S. Erfort, E. Goll, C. Hampel, G. Hetzer, J. G. Hill, M. Hodges, T. Hrenar, G. Jansen, C. Köppl, C. Kollmar, S. J. R. Lee, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, B. Mussard, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. A. Peterson, K. Pflüger, R. Pitzer, I. Polyak, P. Pulay, M. Reiher, J. O. Richardson, J. B. Robinson, B. Schröder, M. Schwilk, T. Shiozaki, M. Sibaev, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, J. Toulouse, M. Wang, M. Welborn and B. Ziegler, MOLPRO, version 2024.2, a package of ab initio programs, see https://www.molpro.net Search PubMed.
- M. Schwilk, Q. Ma, C. Köppl and H.-J. Werner, J. Chem. Theory Comput., 2017, 13, 3650–3675 CrossRef CAS PubMed; Q. Ma, M. Schwilk, C. Köppl and H.-J. Werner, J. Chem. Theory Comput., 2017, 13, 4871–4896 CrossRef PubMed; Q. Ma and H.-J. Werner, J. Chem. Theory Comput., 2018, 14, 198–215 CrossRef.
- K. A. Peterson, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2008, 128, 084102 CrossRef.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide and H. P. R. Frederikse, CRC Press, Boc Raton, 76th edn, 1995 CrossRef CAS; G. Tasi, F. Mizukami and I. Palinko, J. Mol. Struct., 1997, 401, 21–27 CrossRef CAS.
- S. Böhm and O. Exner, Phys. Chem. Chem. Phys., 2004, 6, 510–514 RSC; O. Exner, Dipole Moments in Organic Chemistry, Georg Thieme, Stuttgart, 1975 Search PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. W. Baker and L. G. Groves, J. Chem. Soc., 1939, 1144–1150 Search PubMed.
- O. Exner and S. Böhm, J. Chem. Soc., Perkin Trans. 2, 1997, 1235–1240 RSC.
- H. Umeyama and K. Morokuma, J. Am. Chem. Soc., 1976, 98, 4400–4404 CrossRef CAS.
- J. I. Brauman and L. K. Blair, J. Am. Chem. Soc., 1968, 90, 6561–6562 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Diagrams and commentary relating to other charge models investigated. See DOI: https://doi.org/10.1039/d4ob01572j |
| This journal is © The Royal Society of Chemistry 2025 |