
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of urea-bridged cyclic dominant negative pneumococcus competence-stimulating peptide analogs†
Mona
Mehrani
 ,
Muralikrishna
Lella
,
Katherine A.
Graham
,
Nicholas B.
Borotto
and
Yftah
Tal-Gan
*
,
Muralikrishna
Lella
,
Katherine A.
Graham
,
Nicholas B.
Borotto
and
Yftah
Tal-Gan
*
Department of Chemistry, University of Nevada, Reno, 1664 North Virginia Street, Reno, NV 89557, USA. E-mail: ytalgan@unr.edu
First published on 18th December 2024
Abstract
Cyclization is a widely used approach to exert conformational restraint on linear peptide sequences. Herein, urea bridge chemistry was deployed to achieve side chain-to-side chain peptide cyclization on the Streptococcus pneumoniae CSP1-E1A peptide scaffold. To determine the effects of ring size and bridge position on the overall peptide conformation and find the ideal area within the CSP sequence for cyclization, we performed biological evaluation as well as secondary structure analysis on all the cyclic analogs. Biological evaluation results exhibited that even minor modifications to cyclic analogs for each of the cyclization positions could significantly alter the interaction between the peptide and its target receptor, ComD. Furthermore, structural analysis using circular dichroism (CD) and Trapped Ion Mobility Spectrometry (TIMS) emphasized the significance of incorporating the bridge position as a parameter to be modified, in addition to the traditional ring position and ring size parameters. Overall, our results showcase the importance of comprehensive conformational screening in fine-tuning the secondary structure of cyclic peptide analogs. This knowledge could be very useful for future studies aimed at optimizing peptide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :protein interactions.
:protein interactions.
Introduction
Despite the successful efficacy of peptide drugs in treating certain human diseases, there are still obstacles to overcome to attain commercial drug status and strengthen the peptide drug market.1–3 Conformational modification and stabilization have become viable strategies for improving the pharmacological properties of peptides, as conformation is now considered one of the principal factors influencing peptide bioactivity and bioavailability.4,5 The α-helix conformation has gained significant attention as a potential target for conformational stabilization since it is a widely observed secondary structure in peptides and proteins.6 In this respect, numerous research groups are investigating synthetic methods for modifying peptides in the pursuit of reinforcing and stabilizing the native α-helical conformation of peptides and thus improving their binding affinity to specific protein targets.7,8Cyclization is one of the methods that can help constrain the conformation of peptide secondary structures and is the focus of this study.9–11 The structural rigidity in cyclic peptides minimizes the entropic penalty when binding, which translates to higher affinity and more selective interactions with target proteins compared to linear peptides.9,12 Cyclic peptides are used as therapeutic agents in a wide range of pharmacological treatments including antibiotics, anticancers, antifungals, and immunosuppressants.3,13,14 Superior pharmacokinetic and pharmacodynamic properties of the cyclic structure over linear peptides led to a rising interest in these molecules. As of today, more than 50 cyclic peptides have been approved as therapeutics and many more are being evaluated in various stages of clinical development for treating diverse conditions.15–18 Identifying the optimal region for cyclization is imperative and can mainly fall into four categories based on the types of amino acids involved in the structure: side chain-to-side chain, head-to-tail, head-to-side chain, and side chain-to-tail.12,19 Peptide cyclization can be carried out utilizing different synthetic approaches,20 including Native Chemical Ligation,21,22 Ser/Thr ligation,23,24 KAHA ligation,25 aldehyde-based ligations,26 bioorthogonal reactions,27 disulfide formation,28 and CyClick cyclization.20,29 Alternatively, peptide cyclization can also be accomplished through enzymatic processes such as: Non-Ribosomal Peptide Synthetases (NRPS),30 Ribosomally Synthesized and Post-Translationally Modified Peptides (RiPPs),31 Macrocyclases,32 Asparaginyl Endopeptidases (AEPs),33 Transglutaminases,34 Sortases,35 or Subtiligases.36–40
Herein, we employed urea bridge chemistry to perform side chain-to-side chain peptide cyclization on the Streptococcus pneumoniae CSP1-E1A peptide scaffold, which is a synthetic analog of the native competence stimulating peptide 1 (CSP1) that exhibits potent inhibitory activity of the S. pneumoniae competence regulon quorum sensing (QS) circuitry.4,41–43 QS is an intercellular communication system that many bacteria utilize to synchronize group behaviors based on cell density.44–46S. pneumoniae employs the competence regulon QS circuit to modulate the competence state through the release of a chemical cue called CSP.47 Furthermore, S. pneumoniae strains can be divided into two main specificity groups based on the signal they produce (CSP1 or CSP2).48,49 The competence regulon QS circuitry in S. pneumoniae consists of 5 components: upon expression of comC within the cell, a precursor for the CSP signal is generated, termed ComC, which is then cleaved, matured, and transported outside of the cell by the transmembrane ComAB transporter.47,50 As the exported, mature CSP reaches a particular threshold concentration outside of the cell, it binds and activates the transmembrane histidine kinase receptor, ComD. ComD goes on to phosphorylate an intracellular response regulator, ComE, causing the positive autoinduction of comAB and comCDE. Finally, ComE triggers the transcription of comX, the master regulator of many group behaviors associated with pathogenicity in this bacterium (Fig. 1).51–53
 | ||
| Fig. 1 Illustration of the competence regulon QS circuit regulating competence in S. pneumoniae. | ||
As such, designing potent peptide inhibitors that block the QS circuitry provides a powerful strategy to combat pathogenicity in S. pneumoniae.54–56 Previously, Yang and coworkers reported that peptide cyclization could lead to highly potent and metabolically stable CSP-based QS inhibitors.57 In Yang's study, a lactam bridge was created between positions 6 and 10 on CSP1-E1A. By altering the ring size and bridge position, the authors identified that the CSP1-E1A-cyc(Dap6E10) analog is an effective pan-group inhibitor capable of attenuating pneumococcal infections caused by both a CSP1 producing strain (D39) and a CSP2 producing strain (TIGR4).57 Later, Lella and colleagues extended the investigation of lead pan-group inhibitors by altering the bridge chemistry between positions 6 and 10 from a lactam to a urea bond.41 Even though the E1A modification has been previously described as a critical step in transforming CSPs into competitive inhibitors, Lella's studies revealed that some cyclic CSP1-E1A analogs could also activate noncognate ComD receptors, leading to a few analogs that exhibit dual-action where they inhibit the ComD1 receptor while activating the ComD2 receptor. Understanding the factors influencing the conformation and structure of these signaling peptides is imperative to enhance the properties of the designed peptides and effectively block QS.
Herein, we set out to design and synthesize new libraries of structurally unique cyclic peptide analogs by incorporating a urea bridge at different regions on the CSP1-E1A scaffold (aka, ring position scan). Through the employment of ring position scan, we sought to determine the optimal region within the CSP sequence for cyclization. At the same time, we also aimed to interrogate the effects ring size and bridge position have on the overall peptide conformation, with an emphasis on α-helicity, as well as on the biological activity of the peptides against S. pneumoniae. By leveraging this information, in the future, we can rationally design cyclic CSP-based QS modulators that exhibit high potency as well as enhanced pharmacological properties.
Results and discussion
Design and synthesis of the urea-bridge cyclic modulators
Previous structural characterization studies revealed that an α-helix structure is critical for S. pneumoniae CSP and analogs activity.58,59 Structural evaluation of CSP1-E1A revealed that its central region possesses two faces: one hydrophobic face (residues L4, F7, F8, F11, and I12) and one hydrophilic face (residues S5, K6, R9, and D10).59 Furthermore, it has been determined that the hydrophobic face directly contributes to ComD binding, whereas the hydrophilic face does not.59,60 As such, alterations to the hydrophilic face can be applied to stabilize the required α-helix conformation and potentially optimize the hydrophobic face binding interactions with ComD.57Considering that CSP1-E1A adopts an α-helical conformation, side chain-to-side chain cyclization can be employed on the hydrophilic face of the helix to stabilize this conformation by linking residues at (i → i + 3), (i → i + 4), (i → i + 7) or (i → i + 8) positions.6,61,62 As such, we sought to examine urea bridge cyclization between the following residues in CSP1-E1A: (6–9), (5–9), (10–14), (6–13) and (6–14) to gain a deeper understanding of the best possible region within the CSP1-E1A scaffold for cyclization.41,57,63 To this end, we incorporated different amino acids in these positions bearing an amino side chain functionality but differing in the methylene (–CH2–) side-chain lengths, including Lysine (Lys), Ornithine (Orn), 2,4-diaminobutyric acid (Dab), and 2,3-diaminopropionic acid (Dap) (Scheme 1). This strategy allowed us to probe three different cyclization parameters concurrently: ring position, ring size, and bridge position (Fig. 2).
 | ||
| Scheme 1 Step-by-step workflow for triphosgene-based side chain-to-side chain urea cyclization: first, the linear peptide sequence is synthesized with alloc-protected amino acids at positions 10 and 14 to enable later cyclization. Next, the alloc protective groups are selectively removed. Finally, the exposed amines are reacted with triphosgene to form a urea bridge. For additional experimental details, see ESI.† | ||
 | ||
| Fig. 2 Ring position scan on the CSP1-E1A scaffold: this figure highlights cyclization in three distinct regions of the peptide: (5–9), (10–14), and (6–13). The figure zooms into the region between residues 10 and 14 to provide a detailed view of ring size and bridge positions. (A) 21-Atom ring size: this panel showcases two cyclic peptides with identical ring sizes but differing bridge positions. The bridge is placed either near residue 10 or closer to residue 14. (B) 20-Atom and 24-atom ring sizes (shortest and largest): this panel compares the smallest and largest ring sizes examined. It highlights the cyclization bridge in the 24-atom ring, positioned at the center of the ring, to contrast it with the more compact structure of the 20-atom ring. By examining different ring sizes and bridge positions, we aim to explore their impact on helical stability and biological activity. | ||
We generated a wide range of peptide macrocycles with ring size ranging from 19 to 24 atoms for the (5–9) and (10–14) libraries via the (i → i + 4) cyclization strategy. Furthermore, the cyclization between positions 6 and 13 was only possible with a combination of K6K13, Orn6K13 and K6Orn13, with a macrocycle ring size varying from 32 to 33 atoms. The other permutations, bearing Dab and Dap that would have resulted in shorter ring sizes could not be synthesized as the side chains were not long enough to form a urea bond between the two amine groups. Cyclization between positions 6 and 9 (i → i + 3) or 6 and 14 (i → i + 8) were produced in very low yields, or not at all, likely due to these side chains being positioned poorly for cyclization. We therefore do not discuss these last two ring position libraries any further (cyclic or linear analogs).
Secondary structure analysis using circular dichroism (CD)
In order to see the effect of bridge position and cyclization on the helicity of our synthesized analogs, we set out to provide a quantitative measure of the helical content of both the cyclic and linear (pre-cyclic) analogs and evaluate their overall structure using CD spectroscopy. We assessed all analogs under both aqueous (phosphate-buffered saline (PBS) buffer, pH 7.4) and membrane-mimicking conditions (20% trifluoroethanol (TFE) in PBS, pH 7.4).64 Both linear and cyclic analogs maintained a helical conformation under membrane-mimicking conditions with characteristic negative minima at 208 and 222 nm (Fig. S9, S11, S13, S15, S17, S18†). Most of the cyclic peptides retained α-helical conformation even in an aqueous solution, while all the linear analogs adopted a random coil conformation in these conditions.We first examined the impact of side chain length in linear analogs on peptide helicity. For linear analogs in the 6–13 library, when Lys was maintained in position 6 while the amino acids at position 13 were modified, our findings revealed that reducing the side chain length at position 13 promotes higher helicity, as demonstrated in (Fig. 3A). We further analyzed the percent helicity when Orn was kept constant at position 6, but the amino acids at position 13 were modified to have shorter side chain lengths. The same trend was observed, indicating that reducing the side chain length at position 13 improves helicity (Fig. 3B). For linear analogs in the 5–9 library, when Dap was maintained in position 9 while the amino acids at position 5 were modified, our findings indicated that increasing the side chain length at position 5 improves helicity, as illustrated in (Fig. 3C). For the linear analogs in the 10–14 library, we did not observe any correlation between side chain length and helicity.
 | ||
| Fig. 3 Line graph representation of % helicity in 20% TFE for CSP1-E1A 6–13 and 5–9 linear analogs. This graph exhibits that % helicity increases with a decrease in amino acid side chain length at the 13th position for 6–13 linear analogs and with an increase in amino acid side chain length at the 5th position for 5–9 linear analogs. Panel A refers to Lys at position 6, panel B refers to Orn at position 6, and panel C refers to Dap at position 9. | ||
Regarding the cyclic peptide scaffolds, we examined the impact of bridge position on the percent helicity in peptides featuring identical ring sizes but varying amino acid arrangements. Starting with the (5–9) library, the majority of the peptides exhibited relatively high helicity (17–33%), with similar values for peptides bearing the same ring size (Table 3). The one exception was observed for peptides with a ring size of 21 atoms where having the bridge close to either one of the positions has led to nearly double the percent helicity compared to having the bridge in the middle of the linker: (51–94) and (54–91) exhibiting 32.8 and 33.1% helicity, respectively, compared to (52–93) and (53–92) exhibiting 18.3 and 18.5% helicity, respectively (Table 3). In peptides with a ring size of 20 or 22 atoms, positioning the bridge in the middle of the linker resulted in increased helicity compared to placing the bridge near either end, although the difference in helicity was not significant: (51–93) and (53–91) exhibited helicities of 17.2% and 17%, respectively, compared to (52–92), which exhibited helicity of 18.9% for ring size of 20 atoms, and (52–94) and (54–92) displayed helicities of 28.3% and 24.4%, respectively, compared to (53–93), which exhibited helicity of 31.5% for ring size of 22 atoms (Table 3).
Moving to the (10–14) library, in peptides with a ring size of 20 atoms, placing the bridge close to either one of the positions resulted in nearly triple the percent helicity compared to having the bridge in the middle of the linker: (101–143) and (103–141) exhibiting 22.1% and 19.8% helicity, respectively, compared to (102–142) exhibiting 7.7% helicity (Table 3). In contrast to peptides with a ring size of 20, peptides with a ring size of 22 displayed higher helicity when the bridge was positioned in the middle of the linker. However, the difference in helicity was not as pronounced as in peptides with a 20-atom ring size: (102–144) and (104–142) exhibited 12.6% and 10.6% helicity, respectively, compared to (103–143) exhibiting 14.6% helicity (Table 3). When evaluating peptides possessing a ring size of 21 and 23 atoms, a different trend was observed, where peptides exhibited higher helicity when the bridge was situated in close proximity to the 14th position. Among four peptides with a ring size of 21 atoms, (102–143) with 4% helicity and (104–141) with 16.9% helicity demonstrated the greatest difference in helicity despite possessing the exact same molecular formula (a 4-fold difference in helicity; Table 3). Again, the differences in helicity are attributed to the varying bridge position within the macrocyclic ring (caused by alteration in the number of methylene groups between the alpha carbon and the side chain amino functional group; Fig. 4). Overall, our findings indicate that both the ring size and the bridge position play crucial roles in influencing the conformation of the peptide. The four peptides with a ring size of 21 atoms from the (10–14) library were selected for further analysis where the CD findings were validated with Trapped Ion Mobility Spectrometry (TIMS).
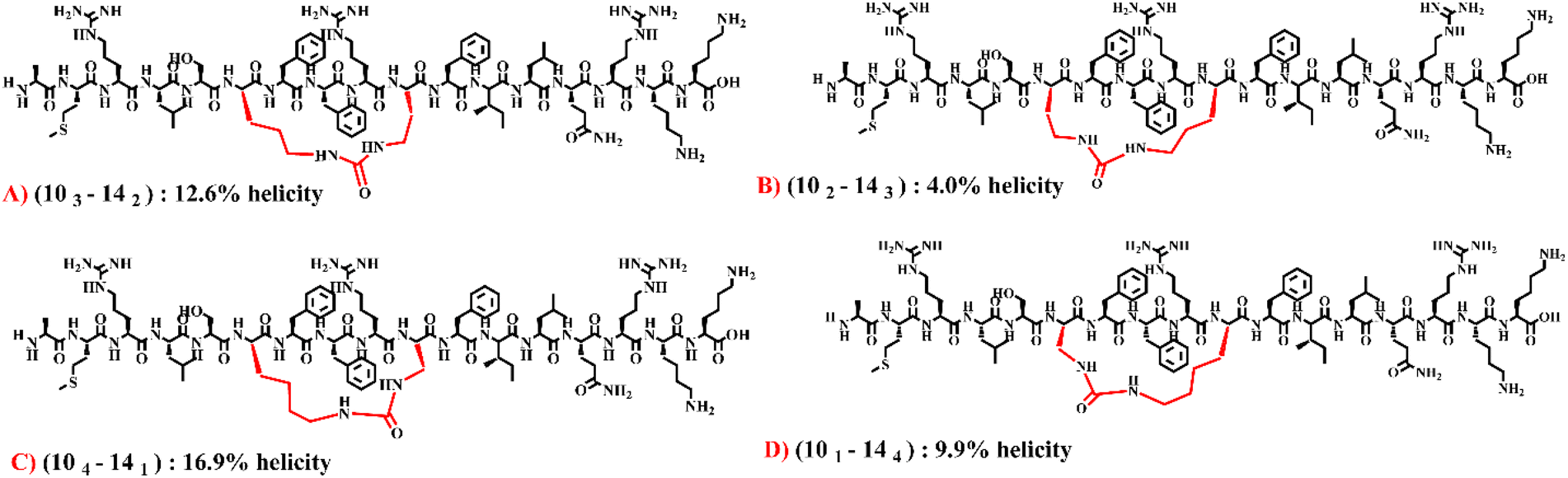 | ||
| Fig. 4 Depiction of the effect of bridge position on helicity of cyclic peptides in the (10–14) library. The four presented peptides have the exact same molecular formula and ring size. They only differ in the bridge position. | ||
Conformational properties analysis in cyclic peptides within (10–14) library utilizing trapped ion mobility spectrometry (TIMS)
TIMS measurements are gas-phase analyses that utilize an ion's mobility through a gas to assess the rotationally averaged collision cross section (CCS) and the size of an analyte.65,66 CCS thus provides valuable information about the ion's three-dimensional conformation in the gas phase and has been shown to be reflective of solution-phase peptide conformations.66–70 Higher helicity is generally associated with more organized and extended structures, whereas lower helicity can lead to flexible conformations capable of folding into compact random coils. Therefore, we expect that peptides with higher helicity will exhibit higher CCS values, as the more extended and defined conformations will lead to increased drag and lower gas phase mobilities.69,70 To corroborate our previously described results obtained via CD spectroscopy, we report the ion mobility spectra of the +4-charge state for each of the four selected peptides in Fig. 5. The TIMS results align well with the helicity trends observed in the CD spectra for the (10–14) analogs. The least helical analog, (102–143), exhibits a CCS value of 649.7 ± 0.2 Å2, the lowest CCS value measured. A CCS of 659.3 ± 0.6 Å2 was measured for (101–144), the second lowest helical analog; and the two analogs with the highest helicity, (103–142) and (104–141), exhibit CCS distributions centered around 694.9 ± 0.2 Å2 and 691.2 ± 0.8 Å2, respectively (Fig. 5). | ||
| Fig. 5 Ion mobility spectra of the +4 charge state of: (101–144) and (104–141) (top); and (102–143) and (103–142) (bottom). n = 3 technical replicates. | ||
Thermal denaturation experiment
Next, we carried out thermal denaturation and recovery measurements using CD spectroscopy to study the impact of the urea cyclization on the conformational stability of the four selected peptides, compared with the linear parent peptide analog, CSP1-E1A. The thermal stability of the peptides was determined by performing forward and reverse thermal denaturation experiments to determine the melting temperatures (Tm values) of the different peptide analogs. The melting temperature (Tm) is the temperature at which a peptide undergoes a conformational transition from a folded to an unfolded state.71 This parameter plays an important role in understanding the structural and thermodynamic aspects of peptides. Our experiments included CSP1-E1A, as well as the four selected cyclic peptide analogs in 2.5 mM DPC (dodecylphosphocholine). Thermal melting scans were recorded between 5 and 95 °C, and recovery from thermal denaturation was monitored from 95 to 5 °C between 200–260 nm wavelengths.The thermal denaturation results indicate that all four (10–14) cyclic peptide analogs have remarkable stability in DPC. This is indicated by the minimal loss of secondary structure at high temperatures and structure restoration upon cooling. The Tm value of each of the four cyclic peptides was determined and, in comparison with the Tm value of CSP1-E1A, the data reveal that cyclization enhanced the Tm value in all cases (Table 1). We analyzed the Tm values of all peptides and determined that (102–143) presents the highest Tm value of approximately 93.7 °C. We compared this value with the Tm values of CSP1-E1A and discovered that in this case cyclization elevated the Tm value significantly by roughly 36 °C. Additionally, when comparing (102–143) with (103–142), an analog with the exact same molecular formula that only differs by the position of the urea bridge, a substantially different Tm value was observed (93.7 °C and 66.2 °C, respectively). These results further highlight the effect the bridge position has on both thermodynamic stability and overall peptide structure in this case. In conclusion, our thermal denaturation experiments provided valuable insights into the conformational stability of the studied peptides and contributed to our understanding of peptide behavior under varying temperature conditions (Table 1).
| Peptide name | Melting temperatures (Tm) (°C) |
|---|---|
| CSP1-E1A | 57.1 |
| (101–144) | 65.9 |
| (104–141) | 60.9 |
| (102–143) | 93.7 |
| (103–142) | 66.2 |
Biological evaluation using QS cell-based reporters
S. pneumoniae D39 (D39pcomX::lacZ) and TIGR4 (TIGR4pcomX::lacZ) β-galactosidase reporter strains were used to perform all the biological assays and determine the structure–activity relationships (SARs) of all cyclic and linear CSP1-E1A-based analogs constructed in this study. To evaluate the effects of sequence modifications and the role of cyclization in receptor binding, specificity, and activation, we performed both activation and inhibition assays for all cyclic and linear analogs. None of the analogs exhibited significant activation (Fig. S1–S4†). Thus, we only discuss the inhibitory activity. According to the inhibition assay, the linear and cyclic peptides only displayed an inhibitory effect against the ComD1 receptor (Fig. S5 and S6†) and were inactive against the ComD2 receptor (Fig. S7 and S8†). Peptides that exhibited significant inhibitory activity in the initial inhibition assay (>50% inhibition) were further evaluated and their IC50 values determined.Biological evaluation of linear modulators
To better understand the importance of each residue used in cyclization on each site in the central region of CSP1-E1A and the effect of sequence modification for peptide cyclization on the activity of the CSP1-E1A analogs, we evaluated the biological activity of all the pre-cyclic analogs. The biological results for each pre-cyclic library are delineated and presented in detail below.5–9 linear library
Starting with the 5–9 library, all linear peptides exhibited inhibitory activity against the ComD1 receptor but were inactive against the ComD2 receptor. Substitutions at the 5th and 9th positions with the amine-containing amino acids have generally diminished the inhibitory activity of the linear peptides. The most potent inhibitor identified in this library was 53–94, with an IC50 value of 103 nM, similar to the parent peptide CSP1-E1A (Table 2). Minor changes, such as altering the side chain length and functional group, can be critical in optimizing protein–peptide binding interactions. Considering the data of substitutions at Ser5, changing the charge appears to be the most significant factor in optimizing CSP activity.| Peptide number | Peptide name | IC50b (nM) (95% CI)c |
% Helicity in 20% TFE (PBS) | Peptide sequence |
|---|---|---|---|---|
| ComD1 | ||||
| a See the ESI† for antagonism dose–response curves and CD data. All SAR assay data were averaged over three independent experiments. b IC50 values were calculated by testing peptides over a range of concentrations. c 95% confidence interval. d Previously reported value from ref. 58. e In 5n–9m, 6n–13m and 10n–14m, n and m indicate the carbon chain length at each position, corresponding to Lys (4), Orn (3), Dab (2), and Dap (1). f Due to low activity in the initial antagonism screening, the IC50 value was not determined. | ||||
| CSP1-E1Ad | 85.7 (50.7–145) | 18.8 (1.0) | AMRLSKFFRDFILQRKK | |
| 1 | 54–94e |
2500 (1630–3840) | 21.3 (0.4) | AMRLKKFFKDFILQRKK |
| 2 | 54–93 | >5000 | 21.1 (0.2) | AMRLKKFFOrnDFILQRKK |
| 3 | 54–92 | 1490 (1310–1700) | 23.8 (0.1) | AMRLKKFFDabDFILQRKK |
| 4 | 54–91 | >5000 | 30.1 (0.6) | AMRLKKFFDapDFILQRKK |
| 5 | 53–94 | 103 (54.9–194) | 24.9 (0.1) | AMRLOrnKFFKDFILQRKK |
| 6 | 53–93 | 1340 (1020–1760) | 18.5 (0.1) | AMRLOrnKFFOrnDFILQRKK |
| 7 | 53–92 | 1020 (910–1140) | 17.9 (0.1) | AMRLOrnKFFDabDFILQRKK |
| 8 | 53–91 | 2820 (2690–2960) | 23.6 (0.1) | AMRLOrnKFFDapDFILQRKK |
| 9 | 52–94 | 743 (625–881) | 15.4 (0.4) | AMRLDabKFFKDFILQRKK |
| 10 | 52–93 | 711 (637–795) | 15.2 (0.6) | AMRLDabKFFOrnDFILQRKK |
| 11 | 52–92 | 857 (703–1040) | 18.6 (0.9) | AMRLDabKFFDabDFILQRKK |
| 12 | 52–91 | 1610 (1290–2020) | 18.3 (0.4) | AMRLDabKFFDapDFILQRKK |
| 13 | 51–94 | 2520 (1800–3520) | 18.4 (0.1) | AMRLDapKFFKDFILQRKK |
| 14 | 51–93 | 2210 (1380–3530) | 19.1 (0.1) | AMRLDapKFFOrnDFILQRKK |
| 15 | 51–92 | 847 (634–1130) | 17.4 (0.1) | AMRLDapKFFDabDFILQRKK |
| 16 | 51–91 | >5000 | 14.7 (0.1) | AMRLDapKFFDapDFILQRKK |
| 17 | 104–144 | >5000 | 14.1 (0.6) | AMRLSKFFRKFILKRKK |
| 18 | 104–143 | 825 (670–1020) | 14.9 (0.3) | AMRLSKFFRKFILOrnRKK |
| 19 | 104–142 | 1380 (933–2050) | 11.9 (0.7) | AMRLSKFFRKFILDabRKK |
| 20 | 104–141 | 464 (259–829) | 16.4 (1.3) | AMRLSKFFRKFILDapRKK |
| 21 | 103–144 | >5000 | 14.4 (0.4) | AMRLSKFFROrnFILKRKK |
| 22 | 103–143 | >5000 | 15.7 (0) | AMRLSKFFROrnFILOrnRKK |
| 23 | 103–142 | 754 (366–1550) | 21.6 (0) | AMRLSKFFROrnFILDabRKK |
| 24 | 103–141 | 1910 (1100–3320) | 12.8 (0.2) | AMRLSKFFROrnFILDapRKK |
| 25 | 102–144 | >5000 | 9 (0) | AMRLSKFFRDabFILKRKK |
| 26 | 102–143 | 3510 (2020–6120) | 23.6 (0.6) | AMRLSKFFRDabFILOrnRKK |
| 27 | 102–142 | 1480 (886–2480) | 15.1 (0) | AMRLSKFFRDabFILDabRKK |
| 28 | 102–141 | >5000 | 5.1 (0.2) | AMRLSKFFRDabFILDapRKK |
| 29 | 101–144 | >5000 | 14.3 (0.4) | AMRLSKFFRDapFILKRKK |
| 30 | 101–143 | >5000 | 5.6 (0.4) | AMRLSKFFRDapFILOrnRKK |
| 31 | 101–142 | 1730 (879–3400) | 4.2 (0) | AMRLSKFFRDapFILDabRKK |
| 32 | 101–141 | 562 (282–1120) | 18.3 (1.2) | AMRLSKFFRDapFILDapRKK |
| 33 | 64–134 | —f | 15 (1.5) | AMRLSKFFRDFIKQRKK |
| 34 | 64–133 | —f | 16.6 (1.9) | AMRLSKFFRDFIOrnQRKK |
| 35 | 64–132 | >5000 | 17.5 (1.9) | AMRLSKFFRDFIDabQRKK |
| 36 | 64–131 | —f | 19 (2.4) | AMRLSKFFRDFIDapQRKK |
| 37 | 63–134 | >5000 | 17.4 (2.2) | AMRLSOrnFFRDFIKQRKK |
| 38 | 63–133 | >5000 | 18.1 (1.5) | AMRLSOrnFFRDFIOrnQRKK |
| 39 | 63–132 | >5000 | 21.0 (2.4) | AMRLSOrnFFRDFIDabQRKK |
| 40 | 63–131 | 3680 (1730–7820) | 24.6 (3.2) | AMRLSOrnFFRDFIDapQRKK |
| 41 | 62–134 | —f | 16.2 (1) | AMRLSDabFFRDFIKQRKK |
| 42 | 62–133 | —f | 16.9 (0.7) | AMRLSDabFFRDFIOrnQRKK |
| 43 | 62–132 | —f | 18.4 (1.3) | AMRLSDabFFRDFIDabQRKK |
| 44 | 62–131 | 3240 (945–11100) |
12.5 (1.3) | AMRLSDabFFRDFIDapQRKK |
| 45 | 61–134 | 705 (412–1210) | 16.6 (1.3) | AMRLSDapFFRDFIKQRKK |
| 46 | 61–133 | 575 (270–1230) | 15.1 (0.5) | AMRLSDapFFRDFIOrnQRKK |
| 47 | 61–132 | 148 (58.0–380) | 18.6 (0.8) | AMRLSDapFFRDFIDabQRKK |
| 48 | 61–131 | 121 (90.1–163) | 17.4 (1.3) | AMRLSDapFFRDFIDapQRKK |
To evaluate the impact of charge at position 5, we evaluated the pre-cyclic analogs containing Dap at position 5, as its side chain has a similar size to that of Ser. Upon substituting the neutral Ser with positively charged Dap, we observed a significant reduction in potency for all the pre-cyclic analogs with IC50 values ranging between 847–1490 nM, compared to CSP1-E1A with an IC50 value of 85.7 nM. Contrary, for substitutions at position 9, it appears that the length of the carbon chain has the more significant influence on the potency of the peptides. By evaluating the analogs while maintaining the same side chain residue at position 5 (either Lys, Orn, Dab, or Dap) and varying the side chain length at position 9, we observed a trend where the longer carbon chain (Lys) at position 9 generally exhibits the highest potency of the series, suggesting that this carbon length situates the positively-charged side-chain amino group in the most favorable position within the ComD binding pocket. For example, 53–94 exhibited an IC50 value of 103 nM, whereas the other three analogs in the series had markedly higher IC50 values (>1000 nM, Table 2).
10–14 linear library
The alanine scan and truncated CSP1 analog libraries initially reported by Yang and coworkers revealed that modifications to Gln14 significantly reduce the peptide activity.58 Furthermore, a recent report from Lella and colleagues identified that replacing Asp10 in CSP1-E1A with Lys, Orn, Dab, or Dap resulted in analogs with improved inhibitory activity against the S. pneumoniae ComD1 receptor. When evaluating the 10–14 pre-cyclic library, it appears that the dominant factor is the detrimental effect of modifying the 14th position, as the beneficial incorporation of positively charged residues at the 10th position cannot sufficiently compensate for the loss in affinity mediated by the modifications at the 14th position, resulting in an overall poor inhibitory activity of all the analogs (Table 2).6–13 linear library
Lastly, by evaluating the 6–13 linear analogs, we observed that at position 6, the incorporation of Dap results in a significant increase in inhibitory potency (Table 2). Furthermore, when comparing the series of analogs that contain Dap at position 6, a trend was observed where decreasing the length of the side chain at the 13th position leads to significantly improved activity (Table 2). Indeed, the most potent analog identified in this library was 61–131, with an IC50 value of 121 nM, similar to the IC50 of the parent peptide CSP1-E1A (85.7 nM).58Biological evaluation of macrocyclic modulators (5–9) cyclic library (i → i + 4)
Out of the 14 urea-bridged cyclic analogs in the (5–9) cyclic library, 5 analogs with macrocycle ring size between 21–24 atoms exhibited weak inhibitory activity against the ComD1 receptor (IC50 > 3000 nM, Table 3), while analogs with ring size between 19 to 20 atoms exhibited no activity against the ComD1 receptor. Despite having high helical content, which was previously shown to be important for biological activity, the cyclic modulators constructed through this library are functionally inactive. These results indicate that α-helix stabilization is not necessarily sufficient to maintain biological activity as individual side chain orientation could have a significant impact on the activity.| Peptide number | Peptide name | IC50b (nM) (95% CI)c |
% Helicity in 20% TFE (PBS) | Ring size | CCS (Å2) | T m (°C) | Peptide sequence |
|---|---|---|---|---|---|---|---|
| ComD1 | |||||||
| a See the ESI† for antagonism dose–response curves and CD data. All SAR assay data were averaged over three independent experiments. b IC50 values were calculated by testing peptides over a range of concentrations. c 95% confidence interval. d In (5n–9m), (6n–13m) and (10n–14m) n and m indicate the carbon chain length at each position, corresponding to Lys (4), Orn (3), Dab (2), and Dap (1). e Due to low activity in the initial antagonism screening, the IC50 value was not determined. | |||||||
| 49 | (54–94)d | 3050 (2590–3590) | 13.6 (3.1) | 24 | — | — | AMRL(KKFFK)DFILQRKK |
| 50 | (54–93) | >5000 | 30.2 (11.6) | 23 | — | — | AMRL(KKFFOrn)DFILQRKK |
| 51 | (53–94) | >5000 | 29.0 (5.6) | 22 | — | — | AMRL(OrnKFFK)DFILQRKK |
| 52 | (54–92) | >5000 | 24.4 (3.1) | 22 | — | — | AMRL(KKFFDab)DFILQRKK |
| 53 | (53–93) | —e | 31.5 (5.3) | 22 | — | — | AMRL(OrnKFFOrn)DFILQRKK |
| 54 | (52–94) | —e | 28.3 (7.1) | 22 | — | — | AMRL(DabKFFK)DFILQRKK |
| 55 | (54–91) | >5000 | 33.1 (4.4) | 21 | — | — | AMRL(KKFFDap)DFILQRKK |
| 56 | (53–92) | —e | 18.5 (1.3) | 21 | — | — | AMRL(OrnKFFDab)DFILQRKK |
| 57 | (52–93) | —e | 18.3 (2.7) | 21 | — | — | AMRL(DabKFFOrn)DFILQRKK |
| 58 | (51–94) | —e | 32.8 (11.5) | 21 | — | — | AMRL(DapKFFK)DFILQRKK |
| 59 | (53–91) | —e | 17.0 (6.1) | 20 | — | — | AMRL(OrnKFFDap)DFILQRKK |
| 60 | (52–92) | —e | 18.9 (1.5) | 20 | — | — | AMRL(DabKFFDab)DFILQRKK |
| 61 | (51–93) | —e | 17.2 (3.9) | 20 | — | — | AMRL(DapKFFOrn)DFILQRKK |
| 62 | (52–91) | —e | 8.2 (1.8) | 19 | — | — | AMRL(DabKFFDap)DFILQRKK |
| 63 | (104–144) | —e | 12.3 (6.0) | 24 | — | — | AMRLSKFFR(KFILK)RKK |
| 64 | (104–143) | >5000 | 12.1 (3.2) | 23 | — | — | AMRLSKFFR(KFILOrn)RKK |
| 65 | (103–144) | 3400 (2200–5250) | 10.2 (2.8) | 23 | — | — | AMRLSKFFR(OrnFILK)RKK |
| 66 | (104–142) | >5000 | 10.6 (2.6) | 22 | — | — | AMRLSKFFR(KFILDab)RKK |
| 67 | (103–143) | 1780 (881–3600) | 14.6 (1.8) | 22 | — | — | AMRLSKFFR(OrnFILOrn)RKK |
| 68 | (102–144) | 2260 (1350–3770) | 12.6 (3.4) | 22 | — | — | AMRLSKFFR(DabFILK)RKK |
| 69 | (104–141) | >5000 | 16.9 (2.1) | 21 | 691.2 ± 0.8 | 60.9 | AMRLSKFFR(KFILDap)RKK |
| 70 | (103–142) | >5000 | 12.6 (0.1) | 21 | 694.9 ± 0.2 | 66.2 | AMRLSKFFR(OrnFILDab)RKK |
| 71 | (102–143) | 2320 (2060–2620) | 4.0 (1.2) | 21 | 649.7 ± 0.2 | 93.7 | AMRLSKFFR(DabFILOrn)RKK |
| 72 | (101–144) | —e | 9.9 (3.9) | 21 | 659.3 ± 0.6 | 65.9 | AMRLSKFFR(DapFILK)RKK |
| 73 | (103–141) | >5000 | 19.8 (2.7) | 20 | — | — | AMRLSKFFR(OrnFILDap)RKK |
| 74 | (102–142) | 1370 (1090–1710) | 7.7 (0.0) | 20 | — | — | AMRLSKFFR(DabFILDab)RKK |
| 75 | (101–143) | —e | 22.1 (11.5) | 20 | — | — | AMRLSKFFR(DapFILOrn)RKK |
| 76 | (64–134) | —e | 6.0 (1.0) | 33 | — | — | AMRLS(KFFRDFIK)QRKK |
| 77 | (64–133) | —e | 21.6 (10.6) | 32 | — | — | AMRLS(KFFRDFIOrn)QRKK |
| 78 | (63–134) | 428 (330–556) | 4.4 (0.3) | 32 | — | — | AMRLS(OrnFFRDFIK)QRKK |
(10–14) cyclic library (i → i + 4)
SAR analysis of the (10–14) cyclic library revealed that nine analogs were weakly active against the ComD1 receptor with the best analog, (102–142), exhibiting an IC50 value of 1370 nM (Table 3). In this library, the low activity is generally correlated with relatively low helicity, suggesting that cyclization in this region of the peptide is not conducive to stabilizing the bioactive conformation.(6–13) cyclic library (i → i + 7)
As for the three (6–13) cyclic analogs, one exhibited a relatively high inhibitory potency: (63–134) with an IC50 value of 428 nM, the most potent cyclic analog identified in this study, only 5-fold less active than the parent scaffold CSP1-E1A. Interestingly, this analog exhibited very low helicity (4.4%, Table 3) whereas the other analog with the exact same molecular formula, (64-133), exhibited significantly higher helicity (21.6%, Table 3) yet was completely inactive. These results further highlight the importance of the individual side chain orientation to biological activity, rather than the global peptide conformation, and suggest that for these i to i + 7 analogs, the percent helicity obtained through the CD analysis may not be accurate due to the significant conformational constraint.Conclusions
S. pneumoniae is a notorious pathogen that poses serious health risks, especially when it develops resistance to many different antibiotics and vaccines. This pathogen utilizes the competence regulon QS circuitry to attack the host and promote its own virulence and pathogenesis. Developing novel methods to combat this pathogen by blocking its QS circuitry could thus be highly beneficial. In this study, we aimed to analyze the key parameters that influence the overall conformation of cyclic peptide analogs derived from the native signaling molecule of S. pneumoniae QS, CSP1. In this regard, we successfully constructed libraries of linear and cyclic analogs. Cyclic analogs were established via the formation of urea bridges between multiple amino acid side chains in positions (5–9), (10–14) and (6–13). Biological evaluation of the linear and cyclic analogs constructed in this study revealed that some analogs exhibited an inhibitory effect against the ComD1 receptor, however none of the analogs were active against the ComD2 receptor. Focusing on the cyclic peptides, our results indicate that minor modifications to cyclic analogs, for each of the cyclization positions, could significantly alter the peptide structure and consequently affect the interaction between the peptide and its target receptor, ComD. As for the pre-cyclic linear peptide analogs, our results revealed that changing the side chain charge at position 5 in the 5–9 library, as well as changing the side chain length at position 6 in the 6–13 library significantly influenced the inhibitory potency.Secondary structure analysis of the cyclic analogs revealed that even a relatively minor change, such as the bridge position within the macrocycle, can significantly alter the overall conformation of two (or more) peptides that share the exact same sequence and molecular formula. These results highlight the importance of including the bridge position as a parameter to be modified, in addition to the traditional ring position and ring size parameters, during conformational screening and optimization of cyclic peptides. Overall, by conducting this study, although we did not obtain analogs with higher inhibitory potencies than the parent CSP1-E1A analog, we were able to showcase the importance of comprehensive conformational screening in fine-tuning the secondary structure of cyclic peptide analogs. This knowledge could be very useful for future studies aimed at optimizing peptide:protein interactions.
Moving forward, the next parameter to be evaluated with regards to cyclization of the pneumococcus CSP is bridge chemistry, a parameter that was left constant in this study (urea). By applying different chemistries for cyclization, such as olefin metathesis, thioether, or azide–alkyne click chemistry, the interaction between the bridge and the peptide backbone or side-chain residues can be optimized to afford more stable and\or active scaffolds. Such methodology will likely require a full conformational screening to re-optimize the other parameters (ring position, ring size, and bridge position) for each new bridge chemistry applied. These studies are currently ongoing in our laboratory and will be reported in due course.
Experimental section
Materials and instrumentation
The chemical reagents and solvents used in this study were purchased from Sigma-Aldrich, Chem-Impex, and Fisher Scientific without further purification. Water (18 MΩ) was purified using Thermo Scientific Barnstead Smart2Pure Pro water purifying system. Peptide synthesis was performed on either a CEM Liberty1 microwave-assisted peptide synthesizer, or a CEM Liberty Prime microwave-assisted peptide synthesizer. Reversed-phase high-performance liquid chromatography (RP-HPLC) was performed using a Shimadzu system equipped with a CBM-20A communications bus module, two LC-20AT pumps, a SIL-20A autosampler, an SPD-20A UV/vis detector, a CTO-20A column oven, and an FRC-10A fraction collector. All RP-HPLC solvents (18 MΩ water and HPLC-grade acetonitrile (ACN)) contained 0.1% trifluoroacetic acid (TFA). Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) data were collected on a Bruker Microflex spectrometer using a nitrogen laser operating at 60 Hz and a reflectron. In reflectron positive ion mode, Ion Source 1 had a 19.01 kV acceleration voltage. Exact mass (EM) data were obtained using an Agilent Technologies 6230 TOF LC/MS spectrometer. The samples were sprayed with a capillary voltage of 3500 V, and the electrospray ionization (ESI) source parameters were as follows: gas temperature of 325 °C at a drying gas flow rate of 3 L min−1 and a pressure of 25 psi.Peptide synthesis and purification
All peptide analogs were synthesized by standard Fluorenylmethoxycarbonyl (Fmoc)-based solid-phase peptide synthesis (SPPS) procedures and purified using RP-HPLC. For full procedures please see the ESI.†Biological reagents and strain information
Standard biological reagents were purchased from Sigma-Aldrich and used according to the enclosed instructions. Donor horse serum (defibrinated) was purchased from Sigma-Aldrich and stored at 4 °C until use in bacterial growth conditions. To determine whether the synthesized CSP analogs modulate the competence regulon in S. pneumoniae, beta-galactosidase assays were performed using D39pcomX::lacZ (group I) and TIGR4pcomX::lacZ (group II) reporter strains.Bacterial growth conditions
Frozen stocks were prepared using 1.5 ml aliquots of pneumococcal bacteria (0.2 optical density [OD] 600 nm) in Todd Heitt Broth (THB) supplemented with 0.5% yeast extract (THY) and 0.5 ml of glycerol and kept at −80 °C. Assays were carried out upon frozen stocks streaked onto 5% serum-containing THY agar plates containing chloramphenicol at a final concentration of 4 μg mL−1. The plate was incubated for 8 to 9 h in a CO2 incubator (37 °C with 5% CO2). Freshly grown colonies were inoculated (single colony for D39pcomX::lacZ; 2–3 colonies for TIGR4pcomX::lacZ) into 5 mL of THY broth supplemented with 4 μg mL−1 final concentration of chloramphenicol, and the inoculated culture was incubated in a CO2 incubator overnight (15 h). Overnight cultures were then diluted (1:50 for D39pcomX::lacZ; 1:10 for TIGR4pcomX::lacZ) with THY, and incubated in a CO2 incubator for 3 to 4 h, until the bacteria reached exponential stage (0.30 to 0.35 for D39pcomX::lacZ; 0.25 to 0.30 for TIGR4pcomX::lacZ) as measured by a plate reader.
Beta-galactosidase assays
Beta-galactosidase reporter gene assays were used to determine the biological activity of all the CSP analogs. For full procedures please see the ESI.†Circular dichroism (CD) spectroscopy
CD spectra were obtained using either an Aviv Biomedical CD spectrometer (model 202-01) or a Jasco CD Spectrophotometer (model J-1500-150). All measurements were carried out using 200 μM peptide concentration in an aqueous environment, phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4; pH was adjusted to 7.4), and in membrane-mimicking milieu, 20% trifluoroethanol (TFE) in PBS. Spectra were recorded at 25 °C using a quartz cuvette of 0.1 cm in path length (Starna Scientific Ltd). Samples were scanned at wavelengths between 195 and 260 nm with the speed of 5 nm min−1, the bandwidth of 0.50 nm, and 16 S data integration time. The single scan was obtained, adjusted for solvent effects, and transformed to mean residue ellipticity (MRE) values using the following equation:where θ is the observed ellipticity in millidegrees, c is the peptide concentration in molarity, l is the path length in centimeters, and n is the length of the peptide sequence.

Percent helicity (fH) was calculated for peptides that exhibited a substantial helical pattern using the following equation:
 000 deg cm2 dmol−1), n is the number of residues in the potential helical region, and x is an empirical correction for end effects.
000 deg cm2 dmol−1), n is the number of residues in the potential helical region, and x is an empirical correction for end effects.
Thermal denaturation
All thermal denaturation CD experiments were carried out on a Jasco CD Spectrophotometer (model J-1500-150) equipped with a six-cell sample chamber and a temperature-controlled Peltier set-up. CD spectra were recorded using a quartz cuvette of 0.1 cm in path length (Starna Scientific Ltd) with a 100 nm min−1 scan rate, from 200 to 260 nm, with a 0.5 nm data pitch and a 1 s data integration time. The data were averaged over a minimum of three accumulations. The blank was subtracted, and the data were subsequently smoothed for better accuracy. Thermal melting scans were recorded between 5 and 95 °C for forward melting and 95 and 5 °C for reverse melting, at data interval of 5 °C with a temperature gradient of 1 °C min−1 and wait time of 30 seconds. Data are reported directly as ellipticity values in units of mdeg.72 To ensure that the detergent remained predominantly in the micellar form during spectroscopic measurements, we maintained the detergent concentrations above the critical micelle concentration (CMC) for DPC. Data processing was carried out using Interval Data Analysis Spectra Manager Jasco CD Spectrophotometer (model J-1500-150).Trapped ion mobility spectrometry
Ion mobility spectrometry experiments for all peptides were performed on either a Bruker timsTOF or timsTOF Pro 2 (Billerica, MA) trapped ion mobility spectrometry quadrupole-time-of-flight mass spectrometer. These instruments were equipped with electrospray ionization sources. The end plate offset, capillary, nebulizer, dry gas, and dry gas temperature for each instrument was set to or between 500 V, 4000 V, 1.5–2 bar, 2–4 L min−1, and 200 °C, respectively. Mobility separations were collected following instrument calibration and with a 1/K0 scan range of 0.7–1.7 V s cm−2, an accumulation time between 50 and 250 ms, an ion mobility ramp time of 250 ms, and a tunnel-in pressure between 2.6–3.0 mbar. Reduced mobility of an ion through a gas, K0, can be utilized to derive the collision cross section (Ω) of an ion through the Mason-Schamp equation:where KB is Boltzman's constant, T is temperature, N is the buffer gas density, μ is the reduced mass, z is the charge of an ion, and e is the elemental charge. CCS values were calculated by fitting the triplicate mobility spectra with the OriginPro 2022 Peak Analyzer tool and averaging the reported centroid values. All reported CCS values should be considered TIMSCCSN2 based on the recent recommended nomenclature.73 All peptide samples were diluted in 49/49/2% methanol/water/formic acid and directly infused into the instrument at a flow rate of 2 μL min−1.

Data availability
All data are presented in the article or the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by grants from the National Institutes of Health (R01HL142626) YT and (GM103440) NB. The S. pneumoniae D39pcomX::lacZ and TIGR4pcomX::lacZ reporter strains were a generous gift from G. W. Lau (University of Illinois at Urbana–Champaign).References
- A. A. Vinogradov, Y. Yin and H. Suga, Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges, J. Am. Chem. Soc., 2019, 141(10), 4167–4181, DOI:10.1021/jacs.8b13178.
- A. Zorzi, K. Deyle and C. Heinis, Cyclic Peptide Therapeutics: Past, Present and Future, Curr. Opin. Chem. Biol., 2017, 38, 24–29, DOI:10.1016/j.cbpa.2017.02.006.
- L. Costa, E. Sousa and C. Fernandes, Cyclic Peptides in Pipeline: What Future for These Great Molecules?, Pharmaceuticals, 2023, 16(7), 996, DOI:10.3390/ph16070996.
- M. Hurevich, Y. Tal-Gan, S. Klein, Y. Barda, A. Levitzki and C. Gilon, Novel Method for the Synthesis of Urea Backbone Cyclic Peptides Using New Alloc-Protected Glycine Building Units, J. Pept. Sci., 2010, 16(4), 178–185, DOI:10.1002/psc.1218.
- J. E. Bock, J. Gavenonis and J. A. Kritzer, Getting in Shape: Controlling Peptide Bioactivity and Bioavailability Using Conformational Constraints, ACS Chem. Biol., 2013, 8(3), 488–499, DOI:10.1021/cb300515u.
- F. Zhang, O. Sadovski, S. J. Xin and G. A. Woolley, Stabilization of Folded Peptide and Protein Structures via Distance Matching with a Long, Rigid Cross-Linker, J. Am. Chem. Soc., 2007, 129(46), 14154–14155, DOI:10.1021/ja075829t.
- Y. H. Lau, P. De Andrade, Y. Wu and D. R. Spring, Peptide Stapling Techniques Based on Different Macrocyclisation Chemistries, Chem. Soc. Rev., 2015, 44(1), 91–102, 10.1039/c4cs00246f.
- M. J. I. Andrews and A. B. Tabor, Forming Stable Helical Peptides Using Natural and Artificial Amino Acids, Tetrahedron, 1999, 55(40), 11711–11743, DOI:10.1016/S0040-4020(99)00678-X.
- N. H. Fischer, E. Fumi, M. T. Oliveira, P. W. Thulstrup and F. Diness, Tuning Peptide Structure and Function through Fluorobenzene Stapling, Chem. – Eur. J., 2022, 28(8), 1–7, DOI:10.1002/chem.202103788.
- S. Hess, O. Ovadia, D. E. Shalev, H. Senderovich, B. Qadri, T. Yehezkel, Y. Salitra, T. Sheynis, R. Jelinek, C. Gilon and A. Hoffman, Effect of Structural and Conformation Modifications, Including Backbone Cyclization, of Hydrophilic Hexapeptides on Their Intestinal Permeability and Enzymatic Stability, J. Med. Chem., 2007, 50(24), 6201–6211, DOI:10.1021/jm070836d.
- M. Serhan, M. Sprowls, D. Jackemeyer, M. Long, I. D. Perez, W. Maret, N. Tao and E. Forzani, Total Iron Measurement in Human Serum with a Smartphone, in AIChE Annu. Meet., Conf. Proc., 2019, vol. 2019-Novem, pp. 1–14. 10.1039/x0xx00000x.
- C. Bechtler and C. Lamers, Macrocyclization Strategies for Cyclic Peptides and Peptidomimetics, RSC Med. Chem., 2021, 12(8), 1325–1351, 10.1039/d1md00083g.
- H. Zhang and S. Chen, Cyclic Peptide Drugs Approved in the Last Two Decades (2001–2021), RSC Chem. Biol., 2022, 3(1), 18–31, 10.1039/d1cb00154j.
- T. S. Young, D. D. Young, I. Ahmad, J. M. Louis, S. J. Benkovic and P. G. Schultz, Evolution of Cyclic Peptide Protease Inhibitors, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(27), 11052–11056, DOI:10.1073/pnas.1108045108.
- M. A. Abdalla and L. J. McGaw, Natural Cyclic Peptides as an Attractive Modality for Therapeutics: A Mini Review, Molecules, 2018, 23(8), 2080, DOI:10.3390/molecules23082080.
- J. Matsoukas, V. Apostolopoulos, E. Lazoura, G. Deraos, M.-T. Matsoukas, M. Katsara, T. Tselios and S. Deraos, Round and Round We Go: Cyclic Peptides in Disease, Curr. Med. Chem., 2006, 13(19), 2221–2232, DOI:10.2174/092986706777935113.
- V. D'Aloisio, P. Dognini, G. A. Hutcheon and C. R. Coxon, PepTherDia: Database and Structural Composition Analysis of Approved Peptide Therapeutics and Diagnostics, Drug Discovery Today, 2021, 26(6), 1409–1419, DOI:10.1016/j.drudis.2021.02.019.
- Y. Y. Syed, Rezafungin: First Approval, Drugs, 2023, 83(9), 833–840, DOI:10.1007/s40265-023-01891-8.
- C. J. White and A. K. Yudin, Contemporary Strategies for Peptide Macrocyclization, Nat. Chem., 2011, 3(7), 509–524, DOI:10.1038/nchem.1062.
- H. C. Hayes, L. Y. P. Luk and Y. H. Tsai, Approaches for Peptide and Protein Cyclisation, Org. Biomol. Chem., 2021, 19(18), 3983–4001, 10.1039/d1ob00411e.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J. C. M. Monbaliu and O. Melnyk, Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations, Chem. Rev., 2019, 119(12), 7328–7443, DOI:10.1021/acs.chemrev.8b00712.
- A. C. Conibear, E. E. Watson, R. J. Payne and C. F. W. Becker, Native Chemical Ligation in Protein Synthesis and Semi-Synthesis, Chem. Soc. Rev., 2018, 47(24), 9046–9068, 10.1039/c8cs00573g.
- X. Li, H. Y. Lam, Y. Zhang and C. K. Chan, Salicylaldehyde Ester-Induced Chemoselective Peptide Ligations: Enabling Generation of Natural Peptidic Linkages at the Serine/Threonine Sites, Org. Lett., 2010, 12(8), 1724–1727, DOI:10.1021/ol1003109.
- H. Y. Lam, Y. Zhang, H. Liu, J. Xu, C. T. T. Wong, C. Xu and X. Li, Total Synthesis of Daptomycin by Cyclization via a Chemoselective Serine Ligation, J. Am. Chem. Soc., 2013, 135(16), 6272–6279, DOI:10.1021/ja4012468.
- J. W. Bode, R. M. Fox and K. D. Baucom, Chemoselective Amide Ligations by Decarboxylative Condensations of N-Alkylhydroxylamines and α-Ketoacids, Angew. Chem., Int. Ed., 2006, 45(8), 1248–1252, DOI:10.1002/anie.200503991.
- C. F. Liu and J. P. Tam, Peptide Segment Ligation Strategy without Use of Protecting Groups, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(14), 6584–6588, DOI:10.1073/pnas.91.14.6584.
- R. D. Row and J. A. Prescher, Constructing New Bioorthogonal Reagents and Reactions, Acc. Chem. Res., 2018, 51(5), 1073–1081, DOI:10.1021/acs.accounts.7b00606.
- F. Albericio, R. P. Hammer, C. García-Echeverría, M. A. Molins, J. L. Chang, M. C. Munson, M. Pons, E. Giralt and G. Barany, Cyclization of Disulfide–containing Peptides in Solid–phase Synthesis, Int. J. Pept. Protein Res., 1991, 37(5), 402–413, DOI:10.1111/j.1399-3011.1991.tb00755.x.
- R. Wills, V. Adebomi and M. Raj, Site-Selective Peptide Macrocyclization, ChemBioChem, 2021, 22(1), 52–62, DOI:10.1002/cbic.202000398.
- B. R. Miller and A. M. Gulick, Structural Biology of Nonribosomal Peptide Synthetases, Methods Mol. Biol., 2016, 1401, 3–29, DOI:10.1007/978-1-4939-3375-4_1.
- S. Wang, S. Lin, Q. Fang, R. Gyampoh, Z. Lu, Y. Gao, D. J. Clarke, K. Wu, L. Trembleau, Y. Yu, K. Kyeremeh, B. F. Milne, J. Tabudravu and H. Deng, A Ribosomally Synthesised and Post-Translationally Modified Peptide Containing a β-Enamino Acid and a Macrocyclic Motif, Nat. Commun., 2022, 13(1), 1–15, DOI:10.1038/s41467-022-32774-3.
- S. Sarkar, W. Gu and E. W. Schmidt, Expanding the Chemical Space of Synthetic Cyclic Peptides Using a Promiscuous Macrocyclase from Prenylagaramide Biosynthesis, ACS Catal., 2020, 10(13), 7146–7153, DOI:10.1021/acscatal.0c00623.
- T. M. S. Tang, D. Cardella, A. J. Lander, X. Li, J. S. Escudero, Y. H. Tsai and L. Y. P. Luk, Use of an Asparaginyl Endopeptidase for Chemo-Enzymatic Peptide and Protein Labeling, Chem. Sci., 2020, 11(23), 5881–5888, 10.1039/d0sc02023k.
- J. Touati, A. Angelini, M. J. Hinner and C. Heinis, Enzymatic Cyclisation of Peptides with a Transglutaminase, ChemBioChem, 2011, 12(1), 38–42, DOI:10.1002/cbic.201000451.
- J. M. Antos, G. L. Chew, C. P. Guimaraes, N. C. Yoder, G. M. Grotenbreg, M. W. L. Popp and H. L. Ploegh, Site-Specific N- and C-Terminal Labeling of a Single Polypeptide Using Sortases of Different Specificity, J. Am. Chem. Soc., 2009, 131(31), 10800–10801, DOI:10.1021/ja902681k.
- T. K. Chang, D. Y. Jackson, J. P. Burnier and J. A. Wells, Subtiligase: A Tool for Semisynthesis of Proteins, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(26), 12544–12548, DOI:10.1073/pnas.91.26.12544.
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Ligation Technologies for the Synthesis of Cyclic Peptides, Chem. Rev., 2019, 119(17), 9971–10001, DOI:10.1021/acs.chemrev.8b00657.
- X. Ji, A. L. Nielsen and C. Heinis, Cyclic Peptides for Drug Development, Angew. Chem., Int. Ed., 2024, 63(3), e202308251, DOI:10.1002/anie.202308251.
- J. S. Zheng, H. N. Chang, J. Shi and L. Liu, Chemical Synthesis of a Cyclotide via Intramolecular Cyclization of Peptide O-Esters, Sci. China: Chem., 2012, 55(1), 64–69, DOI:10.1007/s11426-011-4434-4.
- D. T. S. Rijkers, H. T. ten Brink, N. Ghalit, J. Tois and R. M. J. Liskamp, Synthesis of Cyclic Peptides via Transition Metal Catalyzed C-C Bond Formation, in Underst. Biol. Using Pept., 2006, pp. 50–52. DOI:10.1007/978-0-387-26575-9_11.
- M. Lella, M. W. Oh, S. H. Kuo, G. W. Lau and Y. Tal-Gan, Attenuating the Streptococcus Pneumoniae Competence Regulon Using Urea-Bridged Cyclic Dominant-Negative Competence-Stimulating Peptide Analogs, J. Med. Chem., 2022, 65(9), 6826–6839, DOI:10.1021/acs.jmedchem.2c00148.
- C. Duan, L. Zhu, Y. Xu and G. W. Lau, Saturated Alanine Scanning Mutagenesis of the Pneumococcus Competence Stimulating Peptide Identifies Analogs That Inhibit Genetic Transformation, PLoS One, 2012, 7(9), 1–7, DOI:10.1371/journal.pone.0044710.
- L. Zhu and G. W. Lau, Inhibition of Competence Development, Horizontal Gene Transfer and Virulence in Streptococcus Pneumoniae by a Modified Competence Stimulating Peptide, PLoS Pathog., 2011, 7(9), e1002241, DOI:10.1371/journal.ppat.1002241.
- S. T. Rutherford and B. L. Bassler, Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control, Cold Spring Harbor Perspect. Med., 2012, 2(11), 1–26, DOI:10.1101/cshperspect.a012427.
- S. B. Von Bodman, J. M. Willey and S. P. Diggle, Cell-Cell Communication in Bacteria: United We Stand, J. Bacteriol., 2008, 190(13), 4377–4391, DOI:10.1128/JB.00486-08.
- P. Williams, K. Winzer, W. C. Chan and M. Cámara, Look Who's Talking: Communication and Quorum Sensing in the Bacterial World, Philos. Trans. R. Soc., B, 2007, 362(1483), 1119–1134, DOI:10.1098/rstb.2007.2039.
- L. S. Håvarstein, G. Coomaraswamy and D. A. Morrison, An Unmodified Heptadecapeptide Pheromone Induces Competence for Genetic Transformation in Streptococcus Pneumoniae, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(24), 11140–11144, DOI:10.1073/pnas.92.24.11140.
- G. Pozzi, L. Masala, F. Iannelli, R. Manganelli, L. S. Havarstein, L. Piccoli, D. Simon and D. A. Morrison, Competence for Genetic Transformation in Encapsulated Strains of Streptococcus Pneumoniae: Two Allelic Variants of the Peptide Pheromone, J. Bacteriol., 1996, 178(20), 6087–6090 CrossRef CAS.
- A. M. Whatmore, V. A. Barcus and C. G. Dowson, Genetic Diversity of the Streptococcal Competence (Com) Gene Locus, J. Bacteriol., 1999, 181(10), 3144–3154 CrossRef CAS PubMed.
- F. M. Hui, L. Zhou and D. A. Morrison, Competence for Genetic Transformation in Streptococcus Pneumoniae: Organization of a Regulatory Locus with Homology to Two Lactococcin A Secretion Genes, Gene, 1995, 153(1), 25–31, DOI:10.1016/0378-1119(94)00841-F.
- O. Ween, P. Gaustad and L. S. Håvarstein, Identification of DNA Binding Sites for ComE, a Key Regulator of Natural Competence in Streptococcus Pneumoniae, Mol. Microbiol., 1999, 33(4), 817–827, DOI:10.1046/j.1365-2958.1999.01528.x.
- C. H. G. Johnston, A. L. Soulet, M. Bergé, M. Prudhomme, D. D. Lemos and P. Polard, The Alternative Sigma Factor Σx Mediates Competence Shut-off at the Cell Pole in Streptococcus Pneumoniae, eLife, 2020, 9, 1–27, DOI:10.7554/eLife.62907.
- R. A. Cuevas, R. Eutsey, A. Kadam, J. A. West-Roberts, C. A. Woolford, A. P. Mitchell, K. M. Mason and N. L. Hiller, A Novel Streptococcal Cell–Cell Communication Peptide Promotes Pneumococcal Virulence and Biofilm Formation, Mol. Microbiol., 2017, 105(4), 554–571, DOI:10.1111/mmi.13721.
- D. A. Rasko and V. Sperandio, Anti-Virulence Strategies to Combat Bacteria-Mediated Disease, Nat. Rev. Drug Discovery, 2010, 9(2), 117–128, DOI:10.1038/nrd3013.
- A. A. Brennan, M. Mehrani and Y. Tal-Gan, Modulating Streptococcal Phenotypes Using Signal Peptide Analogues, Open Biol., 2022, 12(8), 220143, DOI:10.1098/rsob.220143.
- T. A. Milly and Y. Tal-Gan, Targeting Peptide-Based Quorum Sensing Systems for the Treatment of Gram-positive Bacterial Infections, Pept. Sci., 2023, 115(2), 1–13, DOI:10.1002/pep2.24298.
- Y. Yang, J. Lin, A. Harrington, G. Cornilescu, G. W. Lau and Y. Tal-Gan, Designing Cyclic Competence-Stimulating Peptide (CSP) Analogs with Pan-Group Quorum-Sensing Inhibition Activity in Streptococcus Pneumoniae, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(3), 1689–1699, DOI:10.1073/pnas.1915812117.
- Y. Yang, B. Koirala, L. A. Sanchez, N. R. Phillips, S. R. Hamry and Y. Tal-Gan, Structure-Activity Relationships of the Competence Stimulating Peptides (CSPs) in Streptococcus Pneumoniae Reveal Motifs Critical for Intra-Group and Cross-Group ComD Receptor Activation, ACS Chem. Biol., 2017, 12(4), 1141–1151, DOI:10.1021/acschembio.7b00007.
- Y. Yang, G. Cornilescu and Y. Tal-Gan, Structural Characterization of Competence-Stimulating Peptide Analogues Reveals Key Features for ComD1 and ComD2 Receptor Binding in Streptococcus Pneumoniae, Biochemistry, 2018, 57(36), 5359–5369, DOI:10.1021/acs.biochem.8b00653.
- T. A. Milly, E. R. Engler, K. S. Chichura, A. R. Buttner, B. Koirala, Y. Tal-Gan and M. A. Bertucci, Harnessing Multiple, Nonproteogenic Substitutions to Optimize CSP:ComD Hydrophobic Interactions in Group 1 Streptococcus Pneumoniae, ChemBioChem, 2021, 22(11), 1940–1947, DOI:10.1002/cbic.202000876.
- J. Garner and M. M. Harding, Design and Synthesis of α-Helical Peptides and Mimetics, Org. Biomol. Chem., 2007, 5(22), 3577–3585, 10.1039/b710425a.
- Y. W. Kim, T. N. Grossmann and G. L. Verdine, Synthesis of All-Hydrocarbon Stapled ±-Helical Peptides by Ring-Closing Olefin Metathesis, Nat. Protoc., 2011, 6(6), 761–771, DOI:10.1038/nprot.2011.324.
- K. Bozovičar and T. Bratkovič, Small and Simple, yet Sturdy: Conformationally Constrained Peptides with Remarkable Properties, Int. J. Mol. Sci., 2021, 22(4), 1–25, DOI:10.3390/ijms22041611.
- A. J. Miles, R. W. Janes and B. A. Wallace, Tools and Methods for Circular Dichroism Spectroscopy of Proteins: A Tutorial Review, Chem. Soc. Rev., 2021, 50(15), 8400–8413, 10.1039/d0cs00558d.
- B. T. Ruotolo, C. C. Tate and D. H. Russell, Ion Mobility-Mass Spectrometry Applied to Cyclic Peptide Analysis: Conformational Preferences of Gramicidin S and Linear Analogs in the Gas Phase, J. Am. Soc. Mass Spectrom., 2004, 15(6), 870–878, DOI:10.1016/j.jasms.2004.02.006.
- M. E. Ridgeway, M. Lubeck, J. Jordens, M. Mann and M. A. Park, Trapped Ion Mobility Spectrometry: A Short Review, Int. J. Mass Spectrom., 2018, 425, 22–35, DOI:10.1016/j.ijms.2018.01.006.
- R. Wu, J. B. Metternich, A. S. Kamenik, P. Tiwari, J. A. Harrison, D. Kessen, H. Akay, L. R. Benzenberg, T. W. D. Chan, S. Riniker and R. Zenobi, Determining the Gas-Phase Structures of α-Helical Peptides from Shape, Microsolvation, and Intramolecular Distance Data, Nat. Commun., 2023, 14(1), 1–11, DOI:10.1038/s41467-023-38463-z.
- R. Wu, J. B. Metternich, P. Tiwari, L. R. Benzenberg, J. A. Harrison, Q. Liu and R. Zenobi, Structural Studies of a Stapled Peptide with Native Ion Mobility-Mass Spectrometry and Transition Metal Ion Förster Resonance Energy Transfer in the Gas Phase, J. Am. Chem. Soc., 2022, 144(32), 14441–14445, DOI:10.1021/jacs.2c02776.
- S. Hoyas, E. Halin, V. Lemaur, J. De Winter, P. Gerbaux and J. Cornil, Helicity of Peptoid Ions in the Gas Phase, Biomacromolecules, 2020, 21(2), 903–909, DOI:10.1021/acs.biomac.9b01567.
- A. K. Karanji, M. Khakinejad, S. G. Kondalaji, S. N. Majuta, K. Attanayake and S. J. Valentine, Comparison of Peptide Ion Conformers Arising from Non-Helical and Helical Peptides Using Ion Mobility Spectrometry and Gas-Phase Hydrogen/Deuterium Exchange, J. Am. Soc. Mass Spectrom., 2018, 29(12), 2402–2412, DOI:10.1007/s13361-018-2053-4.
- R. E. Austin, R. A. Maplestone, A. M. Sefler, K. Liu, W. N. Hruzewicz, C. W. Liu, H. S. Cho, D. E. Wemmer and P. A. Bartlett, A Template for Stabilizatgion of a Peptide α-Helix: Synthesis and Evaluation of Conformational Effects by Circular Dichroism and NMR, J. Am. Chem. Soc., 1997, 119(28), 6461–6472, DOI:10.1021/ja964231a.
- M. Lella, S. Kamilla, V. Jain and R. Mahalakshmi, Molecular Mechanism of Holin Transmembrane Domain i in Pore Formation and Bacterial Cell Death, ACS Chem. Biol., 2016, 11(4), 910–920, DOI:10.1021/acschembio.5b00875.
- V. Gabelica, A. A. Shvartsburg, C. Afonso, P. Barran, J. L. P. Benesch, C. Bleiholder, M. T. Bowers, A. Bilbao, M. F. Bush, J. L. Campbell, I. D. G. Campuzano, T. Causon, B. H. Clowers, C. S. Creaser, E. De Pauw, J. Far, F. Fernandez-Lima, J. C. Fjeldsted, K. Giles, M. Groessl, C. J. Hogan, S. Hann, H. I. Kim, R. T. Kurulugama, J. C. May, J. A. McLean, K. Pagel, K. Richardson, M. E. Ridgeway, F. Rosu, F. Sobott, K. Thalassinos, S. J. Valentine and T. Wyttenbach, Recommendations for Reporting Ion Mobility Mass Spectrometry Measurements, Mass Spectrom. Rev., 2019, 38(3), 291–320, DOI:10.1002/mas.21585.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ob01524j |
| This journal is © The Royal Society of Chemistry 2025 |