
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cross-bridged cyclam derivatives with bis(phosphinate) and phosphinate–phosphonate pendant arms (cb-BPC) as chelators for copper radioisotopes†
Peter
Urbanovský
 a,
Tomáš
David‡
ab,
Veronika
Hlinová
a,
Vojtěch
Kubíček
a,
Hans-Jürgen
Pietzsch
b and
Petr
Hermann
*a
a,
Tomáš
David‡
ab,
Veronika
Hlinová
a,
Vojtěch
Kubíček
a,
Hans-Jürgen
Pietzsch
b and
Petr
Hermann
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 128 40 Prague, Czech Republic. E-mail: petrh@natur.cuni.cz; Tel: +420221921263
bInstitute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, Bautzner Landstrasse 400, 01328 Dresden, Germany
First published on 12th November 2024
Abstract
Copper radioisotopes can be used for imaging as well as for therapy and, thus, can form ideal theranostic pairs. The Cu(II) complexes of cross-bridged cyclam (cb-cyclam) derivatives are considered to be highly stable in vivo. However, the complexes are mostly formed under harsh conditions not compatible with sensitive biomolecules. Here, a new class of cb-cyclam derivatives, cross-bridged bis(phosphinate)cyclams (“cb-BPC”), were investigated. Ligands with one or two methylene-bis(phosphinate) –CH2–PO2H–CH2–PO2H(R) (R = H, OH, substituted alkyl) pendant arms were synthesized. Bifunctionalization on the distant phosphorus atom was carried out by employing P-nitrobenzyl (R = CH2-Ph-4-NO2) precursors and/or, for cb-BPC with two bis(phosphinate) pendant arms, by reactions of silyl-phosphites obtained by silylation of their P(O)–H fragments. The reactive bifunctional groups include amine, carboxylate, azide, isothiocyanate, maleimide and/or tetrazine, and also their orthogonally reactive combination in a single molecule of chelator. The cb-BPCs with one bis(phosphinate) arm were not efficiently radiolabelled with 64Cu. The cb-BPCs with two pendant arms were radiolabelled even at room temperature and with only a small excess of chelator, leading to a high specific activity. Radiolabelling was fully comparable with that of analogous bis(phosphinate) derivatives of cyclam and identical radiolabelling of cyclam and cb-cyclam derivatives was observed for the first time. The cb-BPCs with two bis(phosphinate) pendant arms represent a new class of rigid chelators for copper radioisotopes that are easily synthetically modifiable, highly hydrophilic and radiolabelled under mild conditions.
Introduction
Personalized medicine has become an indispensable part of the medical arsenal used to identify and cure human diseases. This approach involves the right choice of molecular target based on the individual patient biochemistry, conjugation of a drug to a suitable targeting molecule that directs the conjugate to its molecular target, and the diagnostic/therapeutic action of the drug in diseased tissue. Among various ways, the utilization of “theranostic” agents combining diagnostic (imaging) probes and therapeutic drugs is an attractive approach. Nuclear medicine offers the theranostic combination of positron or gamma emitters for diagnostic PET/SPECT imaging with alpha- or beta-emitters useful for therapy. If the imaging and therapeutic radioisotopes are derived from the same element, such one-element theranostic agents are chemically identical and have the same properties in vivo.Radioisotopes of many metal elements have been used in, or have been suggested for, nuclear medicine applications as their features cover a wide range of properties.1–5 For safe applications, any metal radioisotope requires a suitable chelator, which tightly wraps ions of these elements into thermodynamically stable and, more importantly, kinetically inert complexes.6 The strong binding of the metal ions ensures the stability of these radiopharmaceuticals in vivo, i.e., no undesired leaching of the metal radioelements from the agents. So-called bifunctional ligands having a reactive group are then used for the conjugation of the complexes to bioactive vectors (e.g., small molecules, (oligo)peptides, antibodies, etc.) for targeted delivery. Besides the stability and inertness, the chelators/complexes have to meet some other requirements. For efficient radiolabelling, the ligands should be as selective as possible for the desired metal ion (i.e., radiolabelling is less affected by metal impurities), and complex formation should be very fast. Therefore, the search for suitable chelators is a vital branch of current coordination chemistry.
Among metal elements, copper radioisotopes are promising.7–961Cu (β+; t1/2 3.3 h) and 64Cu (17.9% β+; 39% β−, t1/2 12.7 h) can be used for PET diagnosis, and 64Cu and mainly 67Cu (β−; t1/2 61.8 h) are studied as radiotherapeutic isotopes. 64Cu is now available worldwide from various academic and commercial providers. 67Cu recently started to be regularly produced.10 The last two copper radioisotopes are mainly interesting from the theranostic point of view11 as they form an “ideal” (or “true”, or “matched”) metal radioisotope theranostic pair. Several tracers with copper radioisotopes are undergoing clinical trials.12–14
Despite a long history of the development of chelators for copper radioisotopes,4,7–9 the ideal ligand family (fast radiolabelling, kinetic inertness, selectivity for Cu(II) over other metal ions, stabilization of Cu(II) over Cu(I), bifunctionality etc.) has not been unambiguously defined. Almost all suggested ligands are derivatives of parent macrocyclic ligands, such as H3nota (tacn derivatives), H4dota (cyclen derivatives), or H4teta (cyclam derivatives); or cryptands, such as sarcophagines (Chart 1). Among them, sarcophagine derivatives are the most suitable ones, forming hexacoordinated Cu(II) complexes fully wrapping the metal ion, and radiolabelled sarcophagine conjugates have entered into several clinical trials.12
 | ||
| Chart 1 Ligands discussed in the text. | ||
Ligands used for Cu(II) complexation in early radiopharmaceuticals were based on cyclen or cyclam with fully substituted amine groups, and the complexes have been shown to be insufficiently stable in vivo. Currently, the most commonly utilized ligands are derivatives of tacn (1,4,7-triazacyclononane) but the Cu(II) complex of H3nota is not kinetically inert enough.15 Therefore, other chelators have been sought. As cyclam-based ligands are thermodynamically very selective for divalent copper, a lot of ligands investigated have been based on this macrocyclic skeleton. Complexes of cyclam-based ligands having two coordinating pendant arms were shown to be more kinetically inert and/or stable in vivo than those of fully substituted cyclams or H3nota derivatives.16–20 The most kinetically inert are complexes of compounds derived from cross-bridged cyclam (cb-cyclam).21–23 However, the diacetic acid derivative (H2cb-te2a, Chart 1) suffers from slow and inefficient radiolabelling.24 The introduction of phosphonate pendant arm(s) significantly improved the radiolabelling properties of cyclam (BPC ligands, Chart 1)25,26 as well as cb-cyclam (H4cb-te2p or H3cb-te1a1p, Chart 1)27–29 derivatives due to better coordination and proton-transfer abilities of the phosphonate groups (if compared to the carboxylate pendants).
We recently showed that copper(II) binding by cyclam derivatives could be highly improved by the utilization of one geminal methylene-bis(phosphinic acid) or methylene-(phosphinic–phosphonic acid) pendant arm (H2te1ppH or H3te1pp, respectively; Chart 1).17 It has also been confirmed for cb-cyclam derivatives H4cb-te2p and H4cb-te2ppH (Chart 1).30 Based on these convenient properties of the bis(phosphinate) coordinating unit(s), we introduced a new family of bifunctional ligands, “bis-phosphinate cyclams” (BPC ligands, Chart 1), for the binding of copper radioisotopes.25,26 The cyclam derivatives exhibit efficacious 64Cu radiolabelling and the radiolabelled tracers are highly stable in vivo. Their bifunctionality was introduced onto the distant phosphorus atom of the bis(phosphinic acid) moiety. Modification is far away from the macrocyclic metal binding site and preserves excellent demetallation stability of the complexes, and the conveniently high hydrophilicity of the phosphorus acid containing groups. We have shown that phosphorus atom modification is synthetically accessible, and common bifunctional reactive groups can be used for various coupling reactions.25,26 A biodistribution study of the radiolabelled BPC conjugate with an antibody against prostate cancer showed much better properties than those with a H3nota derivative.25
To date, only a handful of bifunctional cb-cyclam chelators have been proposed. The most commonly used conjugation method is amide coupling through the methylcarboxylate pendant arm (e.g., in H3cb-te1a1p) but it changes the coordination properties of such ligands, as carboxamides are weakly coordinating groups.31 A bifunctional reactive group (carboxylate, isothiocyanate) has also been introduced into the cyclam macrocyclic skeleton but this methodology is synthetically inconvenient as it requires the synthesis of the substituted macrocycle.32,33 In this work, we propose a new class of chelators, “cross-bridged bis(phosphinate) cyclams” (cb-BPC). These chelators offer a flexible approach for the attachment of bifunctional reactive group(s) that utilizes the phosphorus atom(s). It leads to the modification of the most distant phosphorus atom of the bis(phosphinic acid) group(s) and, therefore, the convenient chelation properties of the parent ligand, H4cb-te2ppH, are preserved. We also present 64Cu radiolabelling of the ligands.
Results and discussion
Methylene-bis(H-phosphinic acids) CH2[P(O)(OH)(H)]2 (A) and (4-NO2-Ph-CH2)(OH)(O)P–CH2–P(O)(OH)(H) (C) were obtained by following a published procedure.25 The solid-state structure of compound C was determined by X-ray diffraction (for details, see the ESI†). Methylene-(H-phosphinic–phosphonic acid) H2O3P–CH2–P(O)(OH)(H) (B) was obtained by a partial oxidation of methylene-bis(H-phosphinic acid) A by HgCl2 followed by the chromatographic removal of unreacted acid A and methylene-bis(phosphonic acid). Only very mild oxidation agents such as HgCl2 (suggested for such P–H oxidations previously)34 could be used to control the oxidation. Utilization of other common oxidation reagents such as Br2, I2 or peroxo compounds led mainly to methylene-bis(phosphonic acid) or decomposition of the compounds.Parent cb-BPC chelators
First, we had to find the conditions for the syntheses of the parent bis(H-phosphinic acid) derivatives of cb-cyclam. Thus, the simplest derivatives with bis(H-phosphinic acid) pendants, 1 and 2 (Scheme 1), were synthesized in a one-step procedure from methylene-bis(H-phosphinic acid) A and unprotected cb-cyclam using the phospha-Mannich reaction under conditions analogous to those previously reported.17,25,30 Mono- and di-substitutions were controlled by the reactant stoichiometry. To achieve selectively monosubstituted compound 1, paraformaldehyde was used as the reaction controlling reagent with an excess of both cb-cyclam and bis(H-phosphinic acid) A. The non-consumed reactants can be recovered during workup (depending on the purification method). The reaction proceeded well with the products being isolated in ∼60% yield on a multigram scale and with only small amounts of side products. The main by-products detected in the reaction mixture were partially oxidized and partially P-hydroxymethylated parent acid A, i.e., methylene-(H-phosphinic–phosphonic) acid B and H2O2P–CH2–PO2H–CH2OH, respectively, and a product of double substitution, i.e., compound 2 (∼5, ∼3, and ∼5%, respectively; based on cb-cyclam). The double-substituted compound 2 was prepared previously in moderate yield using an excess of both reactants, bis(H-phosphinic acid) A and paraformaldehyde, with respect to the amount of cb-cyclam.30 In attempts to optimize the yield, a lower reactivity of the other secondary amine was observed, leading to the more problematic attachment of the second bis(H-phosphinic acid) pendant arm. An interesting by-product was a ditopic ligand, bis-1 (Fig. S1†), identified by mass and NMR spectra of the reaction mixtures, containing two cb-cyclam fragments 1 bridged by the –CH2–PO2H–CH2–PO2H–CH2– moiety. It probably originates from the reaction of desired product 2 with another molecule of 1 and excess formaldehyde. If an excess of formaldehyde is used in phospha-Mannich reactions, P-hydroxymethylation is expected. Here, minimum amounts of P-hydroxymethylated derivatives of compounds 1 or 2 as well as only negligible P-hydroxymethylation of the starting acid A (<5%, see also below) were observed. This points to an unexpected resistance of the geminal bis(H-phosphinic acid) fragment towards this side reaction under the conditions used. It contrasts with the reactivity of H3PO2, H3PO3 and various H-phosphinic acids; they are readily P-hydroxymethylated if they are used as phosphorus components in the phospha-Mannich reactions.35 | ||
| Scheme 1 Syntheses of the parent bis(phosphinic acid) and phosphinic–phosphonic acid derivatives of cb-cyclam. (i) (A, 1 equiv.), paraformaldehyde (0.66 equiv.), 6 M aq. HCl, 60 °C, 2 d (60%, based on formaldehyde). (ii) A (6 equiv.), paraformaldehyde (20 equiv.), 6 M aq. HCl, 60 °C, 2 d (48%).30 (iii) 1. HgCl2 (1.5 equiv. per P–H group), H2O, 75 °C, 1 d; 2. H2S (>95%). (iv) B (3 equiv.), conc. aq. HCl, paraformaldehyde (2.2 equiv.), 60 °C, 24 h (41%). (v) A (2 equiv.), paraformaldehyde (∼1 equiv.), conc. aq. HCl, 60 °C, 3 d (9%). (vi) paraformaldehyde (>30 equiv.), ∼90% aq. TFA, 80 °C, 4 d (>90%). (vii) paraformaldehyde (12 equiv.), ∼90% aq. TFA, 80 °C, 2 d (>95%). | ||
The phosphonic acid pendant arms generally accelerate metal ion complexation and, thus, ligands with a phosphinate–phosphonate group were also prepared. Oxidation of the terminal H-phosphinate group(s) of 1 and 2 with HgCl2 analogously to that published17 led almost quantitatively to derivatives 3 and 4, respectively, with the geminal phosphinate–phosphonate group(s) (Scheme 1). In the latter case, minor by-products were removed by simple recrystallization. An alternative synthesis of 4 utilizing methylene-(H-phosphinic–phosphonic acid) B and cb-cyclam was also tested using typical conditions for the phospha-Mannich reaction. The reaction had to be run in a relatively concentrated solution of all components in a closed vial to avoid loss of gaseous formaldehyde. This direct synthesis of 4 using B and cb-cyclam did not bring any improvement over the above two-step approach (45% and 41% overall yields in the two- and one-step procedures, respectively).
Unsymmetrically disubstituted cb-cyclam, compound 5, was obtained in the reaction of compound 3 with methylene-bis(H-phosphinic acid) A under the same conditions as those used for compound 3 (Scheme 1). However, product 5 was isolated in a very low yield (<10%) as the reaction had to be run with a low conversion of up to only ∼15% and with a sub-stoichiometric amount of paraformaldehyde due to easy P-hydroxomethylation of compound 5 leading to compound 7 (see also below). It was surprising as P-hydroxomethylation was barely observed during the syntheses of compounds 1 and 2. The use of dilute aq. HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) instead of conc. aq. HCl significantly suppressed the P-hydroxymethylation reaction of compound 5. In this preparation, the formation of compound 7 had to be prevented as the purification of a mixture of compounds 5 and 7 was not possible.
:1) instead of conc. aq. HCl significantly suppressed the P-hydroxymethylation reaction of compound 5. In this preparation, the formation of compound 7 had to be prevented as the purification of a mixture of compounds 5 and 7 was not possible.
Aminomethyl-(H-phosphinic acids) are generally not fully stable as their P–H bond can spontaneously undergo further reactions such as oxidation in air or reactions with other functional groups in the same molecule. However, the reactivity of the P–H bond can be employed. The simplest way to modify the P–H group is P-hydroxymethylation. As given above, some methylene-(H-phosphinic acid) moieties react with formaldehyde much less efficiently than the P–H bond in simple H-phosphinic acids. This unusually low reactivity of, e.g., methylene-bis(H-phosphinic acid) A is synthetically useful as phospha-Mannich reactions involving this acid are faster than P-hydroxymethylation, and the formaldehyde stoichiometry can be used to control only partial N-substitution of azacycles, e.g., in the synthesis of 1. Commonly, P-hydroxymethylation of H-phosphinic acid was carried out in hot 1:1 aq. HCl with an excess of formaldehyde but these conditions were not satisfactory here (i.e., retro-phospha-Mannich reaction and/or oxidation of H-phosphinic acid group(s) were observed). We found that the utilization of hot aq. trifluoroacetic acid (TFA) generally led to better yields. The lower reactivity of the P–H bonds in 1 and 2 with formaldehyde and their “cleaner” phospha-Mannich reactions were further confirmed. Attempts to modify compound 1 with an excess of paraformaldehyde in hot aq. HCl or TFA led to a mixture of compounds where ditopic bis-1 (Fig. S1†), with a methylene-bis(phosphinic acid) bridge between the cb-cyclam rings of 1, was detected by MS and 31P NMR; however, isolation or the selective preparation of the compound failed.
The best P-hydroxymethylation of compound 2 (to prepare compound 6) was achieved in ∼90% aq. TFA solution at 80 °C (the maximal conversion ∼95%) with a high excess of paraformaldehyde (>30 equiv.) added in portions over a period of 4 d. Such harsh conditions led to a slight decomposition of starting compound 2 (mainly oxidation of the terminal H-phosphinic acid group(s) to the phosphonic acid group) and purification of this reaction mixture was difficult.
Aqueous TFA can also be used for the efficient preparation of compound 7 in a two-step procedure. In the first step, compound 3 reacted with an excess (2 equiv.) of compound A in the phospha-Mannich reaction in aq. HCl to give a mixture of compounds 5 and 7 (see also above). If the reaction was carried out in aq. TFA, it produced a complex mixture. Then, HCl was removed and the residue was dissolved in aq. TFA and more paraformaldehyde was added to produce 7. A prolonged heating of the mixture of compounds 5 and 7 in aq. HCl led to a partial decomposition of the compounds and to a rich reaction mixture. Due to the easy reaction of compound 5 with formaldehyde in aq. TFA, compound 7 was produced in a good yield (65% based on compound 3). Thus, we can suggest aq. TFA as a new solvent for the efficient P-hydroxymethylation of the P–H bond of terminal H-phosphinic acids.
Nitrobenzyl cb-BPC chelators
To obtain bifunctional ligands to utilize these chelators in targeted imaging or therapy, another reactive group(s) has to be introduced onto the ligand skeleton. Ideally, the bifunctional group should be located at a distant position to maintain the coordination properties of the parent ligand. Such a remote bifunctional reactive group can be attached to the geminal bis(phosphinic acid) moiety either through the central methylene carbon or, more conveniently, the distant phosphorus atom. These strategies were recently used for analogous BPC ligands (Chart 1) and it was confirmed that such substitutions did not decrease the radiolabelling efficacy of the BPC ligands.25Compounds with an aryl-nitro group are suitable precursors for bifunctional ligands with the corresponding aryl-amino and aryl-azido groups (Chart 1).25 Compound 8 (Scheme 2) was obtained by the reaction of P-(4-nitro-benzyl) methylene-bis(phosphinic acid) C, paraformaldehyde, and cb-cyclam under previously used conditions (6 M aq. HCl, moderate temperature) with a satisfactory yield (62%) as ion exchange purification led to some losses. Several procedures were tested for the preparation of disubstituted derivative 9. The direct reaction of cb-cyclam and compound C led to only a moderate yield (55%, using 6 equiv. of C); despite relatively high conversions (>80%), purification was complicated. The utilization of lower excesses of C led to significantly lower conversions/yields. Thus, to get 9, a two-step procedure, with two consecutive phospha-Mannich reactions (with preparation and isolation of 8), was used; this led to a similar yield (47% over two steps) to that of the direct “one-pot” reaction. The two-step synthesis was easily scaled up and required less compound C; however, the procedure overall required more time.
 | ||
| Scheme 2 (i) C (1 equiv.), paraformaldehyde (0.66 equiv.), 6 M aq. HCl, 60 °C, 3 d (62%). (ii) C (6 equiv.), paraformaldehyde (2.5 equiv.), 12 M aq. HCl, 80 °C, 2 d (55%). (iii) C (1.5 equiv.), paraformaldehyde (1.8 equiv.), 12 M aq. HCl, 80 °C, 2 d (76%). (iv) B (2.5 equiv.), paraformaldehyde (1.6 equiv.), 12 M aq. HCl, 80 °C, 2 d (70%). (v) C (1.5 equiv.), paraformaldehyde (3 equiv.), 12 M aq. HCl, 60 °C, 3 d (65%). (vi) A (4 equiv.), paraformaldehyde (3 equiv.), 6 M aq. HCl, 60 °C, 3 d (50%). (vii) Paraformaldehyde (12 equiv.), ∼90% aq. TFA, 80 °C, 3 d (>90%). | ||
The other secondary amine group in 8 was modified in a reaction with mixed phosphorus acid B giving phosphonate-containing ligand 10 in a satisfactory yield (70%; Scheme 2). As the mixed phosphorus acid B can be obtained only in a low yield (32%), an alternative route to ligand 10 was tested starting with compound 3 and acid C (Scheme 2). The reaction proceeded in conc. aq. HCl reasonably well, even on a multigram scale (∼60% isolated yield). Preparation of compound 11 from mono-substituted cb-cyclam 1 and acid C failed as the rich reaction mixture could not be purified (the main impurity was compound 12). Compound 11 was successfully prepared by the phospha-Mannich reaction of compound 8 with acid A in conc. aq. HCl at elevated temperature in a moderate yield (50%; Scheme 2) as, under these conditions, P-hydroxymethylation of 11 was not significant (<5%). In these reactions, compounds 1, 8 and/or 11 do not seem to be fully stable and they undergo a retro-phospha-Mannich reaction with re-formation of a secondary amine and the corresponding H-phosphinic acids, and these decomposition products can further react. It was confirmed by isolation of compound 9 (∼10%) from the reaction mixture formed in the reaction of 8 to give 11 where acid C released from 8 or 11 reacted with still unreacted 8 to produce compound 9, despite the presence of an excess of acid A.
The P-hydroxymethylation of acid C is much easier than that of acid A and this side process decreases the yields of reactions involving acid C. The observations point to the fact that the P–H bond in acid C is much more reactive than that in acid A. The P-hydroxymethylation of 11 with an excess of paraformaldehyde easily proceeded in aq. TFA solution to give 12 in a high isolated yield (>90%, Scheme 2) similarly to that for the other disubstituted compounds (see above).
In general, the reactions of compound 8 to give disubstituted derivatives 9, 10 or 11 proceeded, if compared with the reactions of compound 3, in relatively high yields. We can speculate that the presence of the bulky and electron-withdrawing nitro-benzyl moiety close to the ring skeleton might alter the conformations and/or intramolecular hydrogen bond system, and the resulting orientation of the remaining secondary amine group is more accessible for the second substitution.
P–H bond modification
Compound 11 could be further modified on the reactive terminal P–H group to add a higher variability on the ligand skeleton. To find the scope of possible reactions, common reactions of the H-phosphinic acid group were tested. The five-valent R–P(H)(O)(OH) moiety can be transformed into a very reactive trivalent R–P(OSiMe3)2 group, which can be further transformed.36,37 Thus, the utilization of a mixture of triethylamine or (N,N-diisopropyl)-ethylamine (DIPEA), with N,O-bis(trimethylsilyl)acetamide (BSA) and trimethylsilyl-chloride (TMS-Cl), both used in a large excess, was the most efficient way to transform the R–P(O)(OH)(H) group in 11 into the R–P(OSiMe3)2 moiety almost quantitatively (Scheme 3 and Fig. S2†). Other commonly used silylating agents (e.g., N-(trimethylsilyl)imidazole, hexamethyldisilazane, or TMS-Cl alone) with, or without, the amines (DIPEA or Et3N) did not lead to the quantitative formation of the P(III) derivative. Therefore, BSA proved to be necessary in this type of reaction as shown earlier.38,39,40 | ||
Scheme 3 (i) DIPEA (8 equiv.), BSA (16 equiv.), TMS-Cl (3 equiv.), anh. CHCl3, under Ar atm, 50 °C, 1 h (conversion >95% by 31P{1H} NMR). (ii) 1. H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCO2tBu/H2CCH–CN/paraformaldehyde/PhtCH2CHO (5 equiv.), anh. CHCl3, Ar atmosphere, 50 °C, overnight. 2. anh. CHCl3/MeOH (excess), Ar atmosphere, room temperature, 30 min (∼60% for 12, 13a, and 15a; and 35% for 14; over two steps based on 11). (iii) TFA, room temperature, overnight (>95%). (iv) N2H4 hydrate (20 equiv.), 50% aq. MeOH, room temperature, 2 d (70%). CHCO2tBu/H2CCH–CN/paraformaldehyde/PhtCH2CHO (5 equiv.), anh. CHCl3, Ar atmosphere, 50 °C, overnight. 2. anh. CHCl3/MeOH (excess), Ar atmosphere, room temperature, 30 min (∼60% for 12, 13a, and 15a; and 35% for 14; over two steps based on 11). (iii) TFA, room temperature, overnight (>95%). (iv) N2H4 hydrate (20 equiv.), 50% aq. MeOH, room temperature, 2 d (70%). | ||
The silylated intermediate derived from 11 was reacted first with activated double bonds,41,42 such as those in aldehydes or Michael acceptors (Scheme 3). The reactions were carried out with the in situ generated intermediate and were quenched by the addition of MeOH to hydrolyse all silyl-containing species. The ligands were extracted into water and, after HPLC purification, compounds 12, 13a, 14 and 15a were obtained in moderate yields (∼60% with exception of 14 with 35% yield) and high purity (>95%). The synthesis of compound 12 is less efficient than the direct reaction of 11 with paraformaldehyde in hot aq. TFA (see above) mainly due to purification losses. The t-butyl ester group of compound 13a was quantitatively cleaved in TFA to give compound 13, the nitro group of which could be reduced (see below). The phthalimide group of compound 15a was deprotected with hydrazine in aq. MeOH to obtain compound 15 (70% isolated yield) with an alkyl amine group. Deprotection of compound 15a in boiling aq. HCl (1:1) led to partial decomposition (caused by the retro-phospha-Mannich reaction) and to a lower isolated yield (30%). Another common reaction of phosphites is the Arbuzov reaction with activated alkylhalogenides.43 Thus, silylated H-phosphite 11 was also tested in the reaction with t-butyl bromoacetate, benzyl bromide and methyl iodide. These reactions were not selective and mixtures always formed. Besides the desired products, compounds with an alkylated ring amine group were detected and some cleavage of the pendant bis(phosphinic acid) group was also observed by MS after methanolysis of the reaction mixtures. Utilization of an excess of the alkyl halides led to even more complex mixtures. Side reactions significantly decreased the yields and made the purification of the reaction mixtures almost impossible. Thus, only the addition of silylated 11 to activated double bonds can be used to modify the distant H-phosphinic acid group. Such reactions can be used for the syntheses of orthogonally bifunctional derivatives of cb-cyclam (see below). It should be noted that silylation was not possible for the parent P–H containing compounds 1, 2 or 5 as these macrocycles were practically insoluble in any silylation mixture due to their high polarity.
Bifunctional cb-BPC chelators
Ligands with mixed phosphinic–phosphonic acid (e.g., 10) and those with two bis(phosphinic acid) pendant arms exhibited the best radiolabelling (see below) and, therefore, the synthesis of other bifunctional ligands was exemplified for these two parent motifs. Thus, the 4-nitro group(s) in 9, 10, and 13 were reduced by catalytic hydrogenation using Pd/C in water or aq. AcOH to give the corresponding amine group containing ligands 16, 19, and 22, respectively (Scheme 4). The reactions proceeded in aq. AcOH with almost quantitative conversion in a few hours while virtually no reaction was observed in EtOH. Reduction of 10 on a gram scale was performed in ∼75% aq. AcOH and ligand 19 was obtained in an isolated yield of 70%. Unfortunately, the anilines are not fully stable upon long-term storage (especially compound 16, which is also light sensitive) and it is better to use them directly in the next reactions. Therefore, amines 16, 19 and 22 were converted (Scheme 4) into the corresponding azides 17 and 20, respectively, through the Sandmeyer reaction by treatment with NaNO2 and NaN3 in aq. HCl with moderate overall yields (∼30–50% over two steps, based on the nitro derivatives). The yield of compound 23 (∼18%) was determined over four steps as compounds 13a, 13 and 22 could only be isolated as oils and were always used directly in the next step. Ligand 23, containing a carboxylic group for amide coupling and an aryl-azide group for alkyne click reactions, represents an orthogonally double bifunctional cb-cyclam-based chelator. The reaction of amines 16 and 19 with thiophosgene under common biphasic conditions (Scheme 4) led to bifunctional ligands 18 and 21, respectively, with isothiocyanate group(s) in moderate isolated yields (40–50% over two steps based on the nitro derivatives). Reduction of the nitrile group of 14 was problematic. As there is no fully selective reduction of nitriles in the presence of an aryl-nitro group, both groups have to be reduced. However, hydrogenolysis of 14 with Pd/C in aq. AcOH led to a mixture of compounds without any conversion to the desired product 24. Commonly used reduction of the nitrile group44 using freshly prepared Raney nickel was also attempted. Only reactions in alkaline aqueous solutions (14 is only soluble in water) led to the isolation of compound 24 in a very low yield (∼10% on a milligram scale) and the formation of Ni(II) complexes of compounds 24 and 24a was observed (more details are given in the ESI†). The formation of the Ni(II) complexes in aqueous solution was surprising as, to date, only Cu(II) complexes of cross-bridged cyclams could be prepared in aqueous media. It indirectly proves that methylene-bis(phosphinic acid) pendant arms facilitate the incorporation of metal ions into the cb-cyclam ligand cavity. | ||
| Scheme 4 (i-a) Pd/C (20% w/w), H2 (1 atm), ∼75% aq. AcOH, 50 °C, 1 d (>95% conversion). (i-b) Pd/C (10% w/w), H2 (1 atm), ∼75% aq. AcOH, 50 °C, 2 d (70%). (i-c) Pd/C (20% w/w), H2 (1 atm), ∼75% aq. AcOH, 50 °C, 4 h (>95% conversion). (ii) 1. NaNO2 (2.4, 1.5, or 1.5 equiv. for 17, 20, and 23, respectively), 1% aq. HCl, ∼0 °C, 5 min; 2. NaN3 (3.5, 2, or 2 equiv. for 17, 20, and 23, respectively), from ∼0 °C to room temperature, 3 h (yields: 9 → 17 57%, 10 → 20 45%, 11 → 23 18%). (iii) Thiophosgene (6 and 3 equiv. for 18 and 21, respectively), 1% aq. HCl/CCl4, room temperature, 24 h (yields: 9 → 18 55%, 10 → 21 43%). (iv) Ra–Ni (1 equiv.), NaBH4 (20 equiv.), water, room temperature, 2 d (∼10%). | ||
To extend the toolbox of bifunctional groups on cb-cyclam bis(phosphinates), a tetrazine moiety for the inverse electron-demand Diels–Alder click reaction was introduced into aniline compound 19 by the reaction with an active ester of a carboxylate tetrazine derivative (Scheme 5). Tetrazine-containing product 25 was obtained in a moderate yield (42%) after semi-preparative HPLC purification. Orthogonally reactive bifunctional ligand 26 (for reactions with thiols and amines) was obtained from the reaction of compound 18 with one equivalent of N-(2-aminoethyl)maleimide (Scheme 6). The reaction produced a statistical mixture of di-maleimide 26a (24%), mono-maleimide 26 (43%) and unreacted di-isothiocyanate 18. The components of the mixture were separated by semi-preparative HPLC after washing of the reaction mixture with AcOEt to remove excess iPr2NEt. These unsymmetrical compounds are the first doubly bifunctional derivatives of cross-bridged cyclam.
 | ||
| Scheme 5 (i) NHS-ester of 1,2,4,5-tetrazine derivative (1.1 equiv.), MES–NaOH buffer (1.0 M, pH 6.2, 25 equiv.) in ∼50% aq. MeCN, room temperature, 2 d (42%). | ||
 | ||
| Scheme 6 N-(2-Aminoethyl)maleimide (1 equiv.), DMF, iPr2NEt, room temperature, 5 h, (yields: 26a 24% and 26 43%). | ||
Radiolabelling with 64Cu
To evaluate the usefulness of the new chelator family for the preparation of copper-based radiopharmaceuticals, the radiolabelling efficiency of the selected ligands was assessed. Radiolabelling was carried out under conditions already used for 64Cu radiolabelling of the cyclam-based BPC chelator H2te1ppH (Chart 1) and its bifunctional P-substituted derivatives/conjugates.17,25 To compare the title ligands with the well-established chelators H3nota and H4dota and cross-bridged cyclams H2cb-te2a and H4cb-te2p (Chart 1), they were also radiolabelled using the same protocol. The results are shown in Fig. 1. To account for possible differences between various 64Cu batches (i.e., to ensure maximum comparability between current and previously obtained data), radiolabelling of H2te1ppH was always carried out in parallel as a control sample; the ligands were radiolabelled with various 64Cu batches and only batches giving the same H2te1ppH radiolabelling efficacy as that previously published (i.e., radiochemical yield, RCY, ∼90 ± 5%)17,25 were considered (Fig. 1). Radiolabelling was carried out at the pH value commonly used for 64Cu radiolabelling (∼6.5) and at room temperature. A low molar excess of the chelators with respect to the amount of radiocopper (only ∼100-times) was used to reach a high specific activity of the radiopharmaceuticals.17,25 | ||
| Fig. 1 Comparison of the radiolabelling efficiency of the title chelators with the established chelators H3nota, H4dota, H2cb-te2a, and H4cb-te2p (Chart 1); H3nota data taken from the literature.17 Data are an average from at least three independent experiments, each done with a freshly prepared batch of 9–11 MBq non-carrier added (NCA) [64Cu]CuCl2. Conditions: 0.5 M MES–NaOH buffer, pH 6.2, 25 °C, ∼100 equiv. of the chelators with respect to the molar amount of 64Cu, labelling time 10 min. | ||
The results show that H3nota, H2cb-te2a, and ligands 1 and 8 with one bis(phosphinic acid) group were radiolabelled poorly, with a maximum radiochemical yield of less than 25%. The presence of one phosphonic acid group in mono-phosphinic–phosphonic acid derivative 3 significantly improved the radiolabelling yield, if compared with chelators with one bis(phosphinic acid) pendant. The efficacy of ligand 3 is comparable with that of H4cb-te2p with two methylphosphonic acid groups, which has already been suggested as a suitable chelator for 64Cu.27
Once two bis(phosphinic acid) pendant arms are present in ligands 2, 9, or 10, radiolabelling is further improved and becomes comparable to that of the simple BPC cyclam chelator with one bis(phosphinate) pendant arm (H2te1ppH, Chart 1).17 This behaviour is in agreement with our previous study of the complexation mechanism, where ligand 2 was shown to complex Cu(II) at a very high rate, approaching the complexation rate of H2te1ppH.30 The comparable radiolabelling of cyclam and cross-bridged cyclam derivatives was observed here for the first time. Similarly to bifunctional BPC,25 efficient radiolabelling is also preserved for bifunctional cb-BPC. Rather surprisingly, derivative 4 with two phosphinic–phosphonic acid pendants binds 64Cu more slowly than other fully substituted derivatives.
The observed radiolabelling behaviour of the chelators could be connected to the mechanism suggested for the complexation of macrocyclic ligands and Cu(II).17,26,30 In the mechanism, a so-called out-of-cage complex (where only pendant arms are bound to the metal ion and the ring amines are protonated) is formed in the first fast step as a kinetic intermediate (the presence of such kinetic intermediates has been proved in complexation mechanisms for a range of metal ion–macrocycle systems).17,26,30,45 The rate-determining step is proton removal from the amine group(s) with simultaneous transfer of the metal ion to the ligand cavity to form the final complex (in-cage complexation). Phosphorus acid pendant arms generally assist proton transfer from the ligand cavity to the bulk solvent due to their ability to form hydrogen bonds and their high hydrophilicity.17,18,26,30 The cb-BPC chelators with two bis(phosphinate) groups are able to “catch” the diluted metal radioisotope to form an out-of-cage complex even better than H4cb-te2p and the groups help with Cu(II) transfer to the inner ligand cavity. The out-of-cage complexes of chelators with only one bis(phosphinate) group or with carboxylate groups are not thermodynamically stable enough and their radiolabelling is not efficient. On the other hand, an out-of-cage complex of ligand 4 with two phosphinate–phosphonate pendants is probably too thermodynamically stable (phosphonates form more stable complexes with metal ions than phosphinates),46 and it might lead to deceleration of the transfer of the metal ion to the inner ligand cavity and, therefore, to a lower radiochemical yield.
The first protonation of the phosphonate group in macrocyclic complexes takes place at around pH 5.5.17,18,30 Phosphonate group protonation should lead to a decrease in the stability of out-of-cage complexes as well as to a lower ability of the phosphorus acid moiety to transfer proton(s) from the ligand cavity to bulk solution. Both effects should lead to a deceleration of the complexation reaction. To check the effect of phosphonate group protonation on radiolabelling efficiency, radiolabelling was carried out at pH 5.6 (Fig. S3†). As this pH is not optimal for fast radiolabelling, the reaction time had to be increased to 1 h. The results agree with the above assumption. Radiolabelling of the phosphonic acid derivatives was worse than that at pH 6.2. The bis(phosphinic acid) groups are fully deprotonated at pH > 3 and their ability to participate in the above complexation mechanism is not altered as much. Therefore, radiolabelling of the bis(phosphinic acid)-containing ligands is not as sensitive to pH. This observation qualitatively agrees with the pH dependences of the complexation rates observed for the ligands at millimolar reactant concentrations.17,18,26,30 It also points to another advantage of bis(phosphinate) pendant arms attached to macrocycles – efficient radiolabelling of such chelators might not require such strict pH control.
To find a limit of specific activity for this chelator family, conditions for quantitative labelling were checked. When radiolabelling was carried out at room temperature and pH 6.2, parent derivative 2 with two bis(phosphinate) pendant arms was quantitatively labelled with only ∼90 equivalents of the chelator with respect to the amount of [64Cu]CuCl2 (9–11 MBq) in 30 min. It led to a very high molar activity of ∼90 GBq per μmol, which was comparable to that of the simple cyclam derivative H2te1ppH.17
The data confirm that the bis(phosphinic acid) group is a suitable moiety to increase the efficiency of metal isotope radiolabelling. They show that even cross-bridged macrocycles can be radiolabelled comparably to common macrocyclic chelators. As only a relatively small excess of the chelator with respect to the amount of radiometal can be used, a very high specific activity is accessible even, with cross-bridged cyclam derivatives under mild radiolabelling conditions.
Stability of 64Cu-radiolabeled complexes
The other aspect relevant for possible in vivo applications is the stability of radiolabelled complexes. One of protocols for how to evaluate the in vitro stability of the 64Cu-labelled complexes is based on the determination of the extent of 64Cu transchelation to human erythrocyte superoxide dismutase (SOD).47 Complexes of several ligands prepared here, and of some other ligands (for comparison), were assessed by the method and the results are shown in Fig. 2. To ensure the quantitative incorporation of 64Cu into the in-cage complex with all ligands, a large molar excess of (∼6000 equiv.) of the chelators with respect to the amount of [64Cu]CuCl2 was used as well as a long labelling time (∼2 h incubation at room temperature; except for H2cb-te2a where heating to 50 °C had to be applied).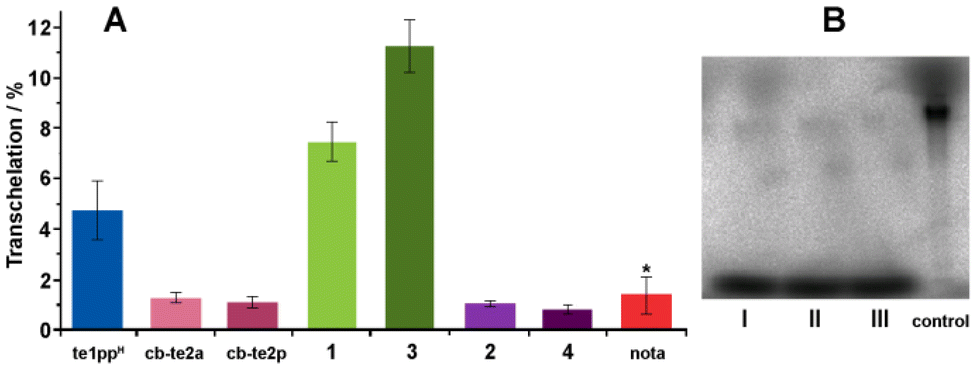 | ||
| Fig. 2 (A) Transchelation (% of control) of 64Cu complexes of 1–4, H2te2ppH, H2cb-te2a, H4cb-te2p and H3nota to human erythrocyte superoxide dismutase (SOD). Data for the 64Cu–H3nota complex were taken from the literature.47 Structures are shown in Chart 1. (B) Example of an audiographic image of the electrophoretic plate (PAGE) obtained for ligand 2 (triplicate, I–III) and the control sample ([64Cu]CuCl2). | ||
It is evident (Fig. 2) that complexes of the cb-cyclam-based ligand with two coordinating pendant arms (i.e., H2cb-te2a, H4cb-te2p, 2, and 4) underwent almost no transchelation (∼1%), suggesting their very high stability, comparable to that of the 64Cu–H3nota complex, which is now generally accepted as stable in vivo.48 In these complexes, an oxygen atom of each pendant arm is bound to the metal ion, leading to octahedral coordination with CN 6.27,30,49 In contrast, the stability of complexes of monosubstituted cb-cyclam-based ligands (i.e., 1 and 3) was significantly lower. This could be explained by the incomplete coordination sphere of their in-cage complexes with CN 5. Coordination of the distant phosphinate/phosphonate group is unlikely due to steric constraints. Thus in complexes of the fully substituted ligands H4cb-te2p, 2, and 4, the Cu(II) ion is completely wrapped by the ligand donor atoms. Another cause of the in vivo instability of copper(II) radiopharmaceuticals is their reduction to monovalent copper. Similarly to the Cu(II)–H2cb-te2a complex, divalent copper should be stabilized in Cu(II)–cb-BPC complexes, compared with complexes of other ligand families. Therefore, only the fully substituted cb-cyclam derivatives are suitable for radiochemical applications.
Conclusions
We recently proved that methylene-bis(phosphinic acid) pendant arms accelerated the formation of copper(II) complexes as well as improving the radiolabelling efficiency of cyclam-based chelators. In this work, we extended these observations to cross-bridged cyclam derivatives. Thus, mono- and disubstituted cb-cyclams with simple bis(phosphinic acid) and/or phosphonic–phosphinic acid moieties were synthesized. The phospha-Mannich reaction of methylene-bis(H-phosphinic acid) is significantly faster than its P-hydroxymethylation and, together with a much higher reactivity of the first amine group of cb-cyclam, it enables monosubstitution using formaldehyde as the reaction-controlling reagent and without any protection of the macrocycle. Yields of double-substituted cb-cyclams depend on the structures of both reagents, on H-phosphinic acid and on the type of first substituent on the cb-cyclam skeleton. The P–H bond on the distant phosphorus atom in the disubstituted derivatives can be fully silylated and used in common reactions of trivalent phosphorus; however, only addition reactions to the double bonds are synthetically useful. A range of bifunctional ligands were obtained. The reactive groups involve amino, carboxylic, isothiocyanate, azide, tetrazine and/or maleimide groups, even in an orthogonally reactive combination. Modification on the distant phosphorus atom(s) not participating in metal ion coordination does not alter the chelating/radiolabelling properties of the bifunctional chelators. Radiochemical experiments with 64Cu showed that only one bis(phosphinate) pendant arm was not sufficient for good radiolabelling. Chelators with two bis(phosphinate)/phosphinate–phosphonate pendant arms are radiolabelled very quickly, even at room temperature and with only a small excess of the chelators. The radiolabelling efficiency is similar to that of the cyclam derivatives and such comparably good radiolabelling of cyclams and cb-cyclams has been observed here for the first time. The stability of the radiolabelled cb-cyclams with two bis(phosphinate) pendant arms is the same as that of 64Cu(II) complexes of other disubstituted cb-cyclams. The results presented in this work confirmed that the bis(phosphinic acid) group was a suitable pendant arm to accelerate in-cage complex formation. The data show that the “cross-bridged bis(phosphinate) cyclam” (cb-BPC) derivatives are a novel class of chelators for the development of radiocopper-based radiopharmaceuticals. Their advantages are very efficient radiolabelling, high stability, high hydrophilicity and the location of the reactive group for conjugations far away from the metal binding site, which does not significantly alter radiolabelling and/or the in vivo stability of the complexes.Experimental section
General
Commercially available (Fluka, Aldrich, CheMatech, Strem) chemicals had synthetic purity and were used as received. Cross-bridged cyclam was purchased as a free base from CheMatech (France) or prepared as the hydrochloride salt by a simplification of the published procedure (details are given in the ESI†).49 Paraformaldehyde was dried and stored over P2O5 in a vacuum desiccator. Methylene-bis(H-phosphinic acid) A50 and (4-nitro-benzyl)(OH)(O)P–CH2–P(O)(OH)(H) C25 (a simplified procedure is described in the ESI†) were obtained according to published procedures. The single-crystal solid-state structure of C was determined (see the ESI† for details). Ligands H2te1ppH, H2cb-te2a, H4cb-te2p, 2·2H2O, H3nota and H4dota·2H2O were available from previous studies.17,30 Syringe filters (PVDF, 0.22 μm pores) were used. The NMR experiments were carried out on Bruker Avance III 600 and 400, Bruker HD850 (1H and 13C{1H}; referenced to external or internal t-BuOH), or Varian S300 (31P and 31P{1H}, referenced to external 85% aq. H3PO4; 19F, referenced to external CF3CH2OH) NMR spectrometers. For the measurements in H2O, pre-saturation of the solvent signal was used. The NMR peaks were assigned through standard 2D 1H–1H/13C correlation experiments. The interaction constants are given in Hz. Analytical HPLC was performed on the C-18 column (Cortecs C-18, 4.6 × 50 mm, 2.7 μm, flow rate 1.2 mL min−1) or C-8 column (ReproSil Gold, 5 μm, 120 Å, 150 × 4.6 mm, flow rate 1.0 mL min−1) using the gradient elution of H2O–MeCN, with or without 0.1% TFA additive (Table S1†). Semi-preparative HPLC was performed on the Waters LC Prep 150 system with C-8 or C-18 (both Phenomenex Luna, 10 μm, 100 Å, 250 × 21.2 mm) columns with flow rates of 15 or 12 mL min−1, respectively, using gradient elution (H2O–MeCN), both with 0.1% TFA additive. Automatic flash chromatography was performed on an Ecom Toy18DAD800 system with C-18 stationary phase (Büchi Sepacore, 25 × 215 mm, 120 g) using gradient elution (0.1% aq. HCl → MeCN, 100:0 → 100:0 → 0:100 → 0:100 over 5, 18, and 3 min, respectively; Table S1†) with a flow rate 50 mL min−1. Standard ESI-MS spectra and analytical HPLC-MS were recorded on a Waters Acquity QDa instrument (ionization with dual orthogonal ESI at atmospheric pressure) with a quadrupolar analyser in the range of m/z 30–1250, with or without a silica gel column (Cortecs C-18, 4.6 × 50 mm, 2.7 μm particle size, dead time of ∼0.4 min) using mobile phases (0.1% aq. TFA and 0.1% TFA in MeCN, various gradients; Table S1†). The conversions were determined by 31P NMR and/or by analytical HPLC. If the compounds were isolated as TFA adducts, the presence of TFA was also confirmed by 13C{1H}/19F NMR. Aluminium foils with silica gel 60 F254 (Merck) were used for TLC. High-resolution MS spectra (Bruker APEX-Q FT-MS; in the positive/negative modes, electro-spray ionization) and elemental analyses are presented as “found (calc.)”. The methodology for the SOD-challenge experiments (human superoxide dismutase and human serum) was adopted from the literature.47
Methylene-[(phosphonic)–(H-phosphinic)]acid (B)
A pre-heated solution (60 °C) of HgCl2 (5.90 g, 21.7 mmol, 1.3 equiv.) in water (60 mL) was added to a pre-heated solution (60 °C) of methylene-bis(phosphinic acid) A (2.50 g, 17.4 mmol) in water (50 mL) and the mixture was stirred at 60 °C for 3 d (conversion to B ∼45% by 31P NMR). The mixture was cooled to room temperature and the aqueous phase was decanted from precipitated Hg2Cl2. The aqueous phase was further filtered via a syringe microfilter (0.22 μm) and the filter was washed with water. The filtrate was saturated with H2S and precipitated HgS was filtered off analogously to that described above and washed with water. The filtrate was evaporated to dryness and the residue was purified by column chromatography (SiO2, 100 g, iPrOH–conc. aq. NH3–H2O 7:3:3; TLC: Rf(B) ∼0.2). Fractions with the pure product were combined, evaporated to dryness and co-evaporated several times with water to remove excess ammonia. The residue was re-dissolved in water and the solution was filtered through a syringe microfilter (0.22 μm; removal of a silica precipitate). The filtrate was further purified on a cation exchange resin (Dowex 50, 50 mL, H+-form, water elution) to remove ammonia quantitatively. The eluate was evaporated to dryness and the resulting oil was further dried to a constant weight (vacuum, 40 °C, 2 d). Product B was obtained as a waxy solid (875 mg, 32%). NMR (H2O + LiOH, pD ≥12): 1H δ 1.93 (CH2, m, 2H), 7.10 (P–H, dm, 1H, 1JHP 526). 13C{1H} δ 34.6 (CH2, dd, 1JCP 114, 1JCP 78). 31P δ 12.3 (PO3H2 td, 2JPH 18, 2JPP 4), 24.9 (HO2P–H, dtd, 1JPH 526, 2JPH 18, 2JPP 4). ESI-MS: (−) 159.0 (159.0, [M − H]−); (+) 161.0 (161.0, [M + H]+). ESI-HR-MS: (−) 158.96154 (158.96177, [CH5O5P2]−). TLC (iPrOH–conc. aq. NH3–H2O 7:3:3): Rf ∼0.2. HPLC (C-18, M2): Rf ∼0.4 min (in dead volume).
Compound 1
:1 HCl (90 mL). Paraformaldehyde (0.28 g, 9.3 mmol, 1 equiv.) was added in one portion and the flask was quickly closed with a stopper. The mixture was vigorously stirred and heated to 60 °C for 2 d. The solution was concentrated under vacuum and the residue was co-evaporated with water (2 × 20 mL). The oily residue was dissolved in water (10 mL) and poured onto a cation exchanger (Dowex 50, 3 × 15 cm, H+-form). The column was washed with water to elute pure acid A in the early fractions. After a delay (>500 mL), pure fully substituted compound 2 was eluted with water. After water evaporation under vacuum, compound 2 was obtained as a viscous oil (∼100 mg, ∼5%; its lyophilisation led to a hydroscopic hexahydrate, 2·6H2O, elemental analysis (calc. for C16H38N4O8P4·6H2O, MR 646.5): C 30.3 (29.7), H 6.6 (7.8), N 8.6 (8.7), P 18.4 (19.2)). Next, the column was washed with 10% aq. pyridine to elute pure product 1. The solvents were removed under vacuum followed by vacuum drying to get a viscous oily residue, which could be directly used in the next reactions. The oil was dissolved in water (250 mL) and the solution was lyophilized to get slightly hydroscopic zwitterionic 1·4H2O (2.6 g, 61%). Elemental analysis (calc. for C14H32N4O4P2·4H2O, MR 454.4): C 37.3 (37.0), H 8.3 (8.9), N 11.8 (12.3), P 12.5 (13.6).
:1 HCl (∼3 mL). To the mixture, paraformaldehyde (6.5 mg, 0.22 mmol, 1 equiv.) was added in one portion and the vial was quickly closed with a stopper. The mixture was vigorously stirred and heated to 80 °C overnight. Then, the solution was concentrated under vacuum and co-evaporated with water (2 × 2 mL). The oily residue was dissolved in water (3 mL) and poured onto an anion exchanger (Amberlite IRA 402, ∼3 × 20 cm, OH−-form). Unreacted cb-cyclam (in a sufficient purity to be re-used) was eluted off with water and the product was eluted off with 10% aq. AcOH. After solvent evaporation under vacuum, the residue was co-evaporated with 6 M aq. HCl (∼5 mL) and then several times with water to remove any remaining AcOH. The oil was dissolved in a minimum amount of aq. HCl (1:1), the solution was evaporated to dryness and the residue was dried under vacuum to obtain 1 as a hydroscopic hydrochloride hydrate, 77 mg (1·2.5HCl·3H2O, 67%). Elemental analysis (calc. for C14H32N4O4P2·2.5HCl·3H2O, MR 527.6): C 31.8 (31.9); H 7.5 (7.7); N 10.6 (10.6). NMR (H2O + CsOH, pD ∼9): 1H δ 1.65–1.75 (CH2–CH2–CH2, m, 1H), 1.71–1.81 (CH2–CH2–CH2, m, 1H), 2.02–2.22 (P–CH2–P, m, 2H), 2.25–2.35 (CH2–CH2–CH2, m, 1H), 2.35–2.45 (CH2–CH2–CH2, m, 1H), 2.50–2.62 (cycle, m, 2H), 2.59–2.67 (cycle, m, 2H), 2.88–2.96 (cycle, m, 3H), 3.01–3.09 (cycle, m, 2H), 3.10–3.18 (cycle, m, 2H), 3.14–3.20 (N–CH2–P, m, 1H), 3.19–3.25 (cycle, m, 1H), 3.26–3.34 (cycle, m, 3H), 3.40–3.48 (cycle, m, 2H), 3.59–3.65 (cycle, m, 1H), 3.71–3.76 (N–CH2–P, m, 1H), 3.75–3.81 (cycle, m, 2H), 7.13 (P–H, d, 1H, 1JHP 533). 13C{1H} δ 18.9 (CH2–CH2–CH2, s), 20.1 (CH2–CH2–CH2, s), 36.0 (P–CH2–P, dd, 1JCP 84, 1JCP 77), 42.2 (cycle, s), 47.7 (cycle, s), 49.5 (cycle, s), 49.9 (cycle, s,), 51.7 (cycle, d, 3JCP 6), 54.0 (N–CH2–P, d, 1JCP 91), 54.4 (cycle, s,), 56.2 (cycle, s), 58.2 (cycle, s), 58.4 (cycle, s), 59.5 (cycle, s). 31P δ 20.6 (P–H, dtd, 1P, 1JPH 533, 2JPH 17, 2JPP 6), 25.7 (P–CH2–N, m, 1P). ESI-MS: (−) 381.2 (381.2, [M − H]−); (+) 383.3 (383.2, [M + H]+), 405.3 (405.2, [M + Na]+). ESI-HR-MS: (−) 381.18246 (381.18260, [C14H31O4N4P2]−). TLC (iPrOH–conc. aq. NH3–H2O 7:3:3): Rf ∼0.7. HPLC (C-18, M2): Rf ∼1.0 min.
Compound 3
In a 500 mL flask, powdered compound 1·4H2O (3.4 g, 7.4 mmol) was dissolved in water (200 mL) and the solution was heated to 75 °C. Then, a hot (75 °C) aq. solution (20 mL) of HgCl2 (3.1 g, 11 mmol, 1.5 equiv.) was added in one portion. The mixture was stirred and heated to 75 °C for 1 d. After completion of the reaction (31P NMR, >95% conversion), the mixture was cooled and the liquid phase was decanted from the precipitate. The aqueous phase was further filtered off through a syringe microfilter (0.22 μm). This clear solution was bubbled with H2S for several minutes. Precipitated HgS was removed by filtration through syringe microfilters (0.22 μm; 2–3 filters were necessary). The solution was then evaporated to dryness under vacuum; the residue was co-evaporated with water (2 × 20 mL) and dried to a constant weight under vacuum. The product 3 hydrochloride was isolated as a viscous oil, which could be directly used in subsequent steps. The oil was dissolved in 6 M aq. HCl, the solution was evaporation to dryness under vacuum and the solid was dried under vacuum to give a hygroscopic yellowish powder of hydrochloride hydrate of 3 (3·7.5HCl·0.5H2O, 4.6 g, 92%). NMR (D2O + CsOD, pD ∼10): 1H δ 1.63–1.69 (CH2–CH2–CH2, m, 1H), 1.72–1.78 (CH2–CH2–CH2, m, 1H), 1.87–1.97 (P–CH2–P, m, 2H), 2.16–2.24 (cycle, m, 1H), 2.29–2.37 (cycle, m, 1H), 2.57–2.63 (cycle, m, 2H), 2.68–2.74 (cycle, m, 2H), 2.88–2.94 (cycle, m, 2H), 2.96–3.05 (cycle, m, 6H), 3.01–3.05 (N–CH2–P, 1H), 3.12–3.18 (cycle, m, 1H), 3.25–3.33 (N–CH2–P and cycle, m, 4H), 3.75–3.83 (cycle, m, 3H), 4.21–4.27 (N–CH2–P, m, 1H). 13C{1H} δ 19.5 (CH2–CH2–CH2, s), 20.6 (CH2–CH2–CH2, s), 33.7 (P–CH2–P, dd, 1JCP 115, 1JCP 88), 42.3 (cycle, s), 48.75 (cycle, s), 50.0 (cycle, s), 50.1 (cycle, s), 52.3 (cycle, d,), 53.2 (cycle, s), 53.4 (N–CH2–P, d, 1JCP 87), 55.0 (cycle, s), 58.7 (cycle, s), 59.1 (cycle, s), 59.4 (cycle, s). 31P{1H} δ 11.3 (H2O3P, d, 2JPP 6.3 Hz, 1P), 27.6 (P–CH2–N, d, 2JPP 6.3 Hz, 1P). ESI-MS: (+) 399.2 (399.2, [M + H]+), 421.2 (421.2; [M + Na]+), 791.4 (791.4 [2M + H]+). ESI-HR-MS: (−) 397.17722 (397.17752, [C14H31O5N4P2]−). TLC (iPrOH–conc. aq. NH3–H2O 7:3:3): Rf ∼0.2. Elemental analysis (calc. for C14H32N4O5P2·7.5HCl·0.5H2O, MR 680.8): C 25.0 (24.7); H 5.8 (6.0); N 8.2 (8.2). HPLC (C-18, M2): Rf ∼0.9 min.
Compound 4
:3:3): Rf ∼0.1. HPLC (C-18, M2): Rf ∼0.6 min.
Compound 5
In 50 mL pear-shaped flask, compound 3·7.5HCl·0.5H2O (1.0 g, 1.5 mmol, 1 equiv.), paraformaldehyde (45 mg, 1.5 mmol, 1 equiv.) and methylene-bis(H-phosphinic acid) A (0.44 g, 3.0 mmol, 2 equiv.) were mixed in aq. HCl (1:1, 30 mL). The mixture was stirred at 60 °C for 4 d in a tightly closed reaction vessel. Then, volatiles were removed under vacuum and the residue was co-evaporated with water several times. The residue was dissolved in water (5 mL) and poured onto a strong cation exchanger (Dowex 50, 10 × 3 cm, H+-form). The column was eluted with water and the starting methylene-bis(H-phosphinic acid) A was obtained in the first fractions in synthetic purity (>90%) to be re-used. In the latter aqueous fraction (after a significant delay), compound 5 was eluted. The combined fractions containing pure 5 were lyophilized to yield waxy compound 5 as the zwitterionic hydrate (5·3.5H2O, 80 mg, ∼10%). The column was finally washed with 10% aq. pyridine and compound 3 (∼0.6 g) was regenerated in synthetic purity (>80%) after removal of volatiles under vacuum; the main impurity (determined by NMR) was compound 5. NMR (H2O, pH 1.2): 1H δ 1.81–2.05 (CH2–CH2–CH2, m, 2H), 2.21–2.35 (CH2–CH2–CH2, m, 2H), 2.37 (P–CH2–P, pseudo-t, 2JHP–2JHP 17, 2H), 2.43 (P–CH2–P, pseudo-t, 2JHP–2JHP 18.5, 2H), 2.83–3.12 (cycle, m, 1H), 3.00–3.12 (cycle, m, 3H), 3.12–3.21 (cycle, m, 2H), 3.21–3.32 (cycle, m, 2H), 3.35–3.61 (cycle and N–CH2–P, m, 10H), 3.61–3.81 (cycle, m, 4H), 7.14 (P–H, d, 1JHP 560, 1H). 13C{1H} δ 20.7 and 20.7 (2× CH2–CH2–CH2, bs), 32.3 (P–CH2–P, dd, 1JCP 125, 1JCP 84), 35.7 (P–CH2–P, pseudo-t, 1JCP–1JCP 80.5), 48.2 (cycle, bs), 49.8–50.8 (cycle, m), 53.9–54.7 (cycle and N–CH2–P, m), 54.7–55.8 (cycle, m), 56.6–57.9 (cycle, m), 57.9–58.8 (cycle and N–CH2–P, m). 31P δ 16.8 (H2O3P, m, 1P), 20.6 (H2O2P, dm, 1P, 1JPH 559), 24.3 (N–C–P, m, 1P), 25.4 (N–C–P, m, 1P). ESI-MS: (−) 553.2 (553.2, [M − H]−); (+) 555.2 (555.2, [M + H]+). ESI-HR-MS: (−) 553.15096 (583.15165, [C16H37O9N4P4]−), 276.07193 (276.07219, [C16H36O9N4P4]2−). Elemental analysis (calc. for C16H38N4O10P4·3.5H2O, MR 617.5): C 31.5 (31.1); H 6.7 (7.4); N 8.9 (9.1), P 19.6 (20.1). HPLC (C-18, M2): Rf ∼0.6 min.
Compound 6
In a 4 mL glass vial, compound 2·6H2O (40 mg, 62 μmol, 1 equiv.) was dissolved in 90% aq. trifluoroacetic acid (TFA, ∼20 mL) and heated to 80 °C. Paraformaldehyde (22 mg, 0.74 mmol, 12 equiv.) was added and the flask was quickly closed with a stopper. After the mixture was stirred at 80 °C for 1 d, another portion of paraformaldehyde (22 mg, 0.74 mmol, 12 equiv.) was added, and the same amount (12 equiv.) of paraformaldehyde was added two more times over 2 d (the mixture was stirred at 80 °C for 4 d in total). Volatiles were removed under vacuum and the residue was co-evaporated several times with water to completely remove TFA. The residue was dissolved in water (2 mL) and poured onto a strong cation exchanger (Dowex 50, 5 × 2 cm, H+-form). The product was eluted with water after a significant delay. Fractions containing pure product were combined and the solvents were removed under vacuum to yield zwitterionic 6·4H2O as a viscous oil (40 mg, yield >90%; purity >95%, the main impurity was compound 7). NMR (D2O, pD 1.8): 1H δ 1.94–2.10 (CH2–CH2–CH2, m, 2H), 2.23–2.35 (CH2–CH2–CH2, m, 2H), 2.47–2.53 (P–CH2–P, m 4H), 2.90–3.07 (cycle, m, 2H), 3.07–3.23 (cycle, m, 6H), 3.23–3.31 (cycle, m, 2H), 3.41–3.62 (cycle and N–CH2–P, m, 10H), 3.67–3.85 (cycle, m, 4H), 3.88 (P–CH2–OH, d, 2JHP 5.2, 4H). 13C{1H} δ 19.7–20.9 (2× CH2–CH2–CH2, bs), 31.6–33.2 (P–CH2–P, m), 46.9–48.7 (cycle, m), 49.7–51.1 (cycle, m), 54.4 (cycle, bs), 54.8–57.5 (cycle and N–CH2–P, m), 57.5–58.9 (cycle, m), 60.8 (P–CH2–OH, d, 2JHP 113). 31P{1H} δ 21.4–26.6 (m, 2P), 36.5–39.0 (m, 2P). ESI-MS: (−) 597.2 (597.2, [M − H]−); (+) 599.2 (599.2, [M + H]+). ESI-HR-MS: (−) 597.17772 (597.17786, [C18H41O10N4P4]−), 268.07454 (268.07473, [C18H40O10N4P4]2−). TLC (iPrOH–conc. aq. NH3–H2O 7:3:3): Rf ∼0.5. Elemental analysis (calc. for C18H42N4O10P4·4H2O, MR 670.5) C 32.3 (32.2); H 7.1 (7.5); N 8.2 (8.4), P 17.8 (18.5). HPLC (C-18, M2): Rf ∼0.6 min.
Compound 7
In a 50 mL pear-shaped flask, compound 3·7.5HCl·0.5H2O (0.40 g, 0.59 mmol, 1 equiv.), paraformaldehyde (35 mg, 1.2 mmol, 2 equiv.) and methylene-bis(H-phosphinic acid) A (0.44 g, 3.0 mmol, 1.5 equiv.) were mixed in conc. aq. HCl (30 mL). The mixture was stirred in a tightly closed reaction vessel at 60 °C for 3 d. Then, volatiles were removed under vacuum and the residue was co-evaporated with water several times. The oily residue was dissolved in ∼90% aq. TFA (30 mL) and the solution was heated to 80 °C. Paraformaldehyde (53 mg, 1.8 mmol, 3 equiv.) was added, the flask was quickly closed with a stopper and the mixture was stirred at 80 °C for 3 h. Then, another portion of paraformaldehyde (159 mg, 5.3 mmol, 9 equiv.) was added and the mixture was stirred at 80 °C for 1 d. Volatiles were removed under vacuum and the residue was co-evaporated several times with water. The residue was dissolved in water (5 mL) and poured onto a strong cation exchanger (Dowex 50, 10 × 3 cm, H+-form). The column was washed with water. In the latter aqueous fraction (after a significant delay), pure compound 7 was obtained. The fractions with pure 7 were combined and lyophilized to yield zwitterionic 7·3H2O as a waxy solid (245 mg, 65% based on compound 3). NMR (D2O, pD 1.6): 1H δ 1.95–2.10 (CH2–CH2–CH2, m, 2H), 2.25–2.37 (CH2–CH2–CH2, m, 2H), 2.47 (P–CH2–P, pseudo-t, 2JHP–2JHP 19, 2H), 2.49–2.55 (P–CH2–P, m, 2H), 2.93–3.08 (cycle, m, 2H), 3.08–3.24 (cycle, m, 6H), 3.24–3.33 (cycle, m, 2H), 3.43–3.64 (cycle and N–CH2–P, m, 10H), 3.68–3.86 (cycle, m, 6H), 3.89 (P–CH2–OH, d, 2JHP 5.2, 2H). 13C{1H} δ 20.5 and 20.8 (2× CH2–CH2–CH2, bs), 31.3–33.1 (2× P–CH2–P, m), 47.9 (cycle, s), 49.6–51.5 (cycle, m), 54.5 (cycle, bs), 54.8–57.8 (cycle and N–CH2–P, m), 58.4 (cycle, bs), 60.7 (P–CH2–OH, d, 1JCP 114). 31P{1H} δ 16.6–18.0 (H2O3P, m, 1P), 20.4–26.7 (m, 2P), 37.5–39.4 (m, 1P). ESI-MS: (−) 583.2 (583.2, [M − H]−); (+) 585.2 (585.2, [M + H]+). ESI-HR-MS: (−) 583.16168 (583.16221, [C17H39O10N4P4]−), 291.07734 (291.07747, [C17H38O10N4P4]2−). TLC (EtOH–conc. aq. NH3 1:1): Rf ∼0.3. Elemental analysis (calc. for C17H40N4O10P4·3H2O, MR 638.5): C 31.8 (32.0); H 6.9 (7.3); N 8.7 (8.8), P 19.0 (19.4). HPLC (C-18, M2): Rf ∼0.6 min.
Compound 8
:1): Rf ∼0.6. HPLC (C-8, M1): Rf ∼5.6 min. HPLC (C-18, M2): Rf ∼4.0 min.
Compound 9
:1): Rf ∼0.1. HPLC (C-8, M1): Rf ∼6.2 min.
Compound 10
:3:3): Rf ∼0.3. HPLC (C-8, M1): Rf ∼4.9 min.
Compound 11
In a 50 mL round-bottomed glass flask, compound 8·3H2O (0.70 g, 1.2 mmol, 1 equiv.), methylene-bis(H-phosphinic acid) A (0.7 g, 7.6 mmol, 4 equiv.) and paraformaldehyde (0.13 g, 4.3 mmol, 3.5 equiv.) were mixed in conc. aq. HCl (40 mL) and the flask was quickly closed with a stopper. The mixture was vigorously stirred at 60 °C for 3 d. Volatiles were removed under vacuum and the residue was co-evaporated several times with water to remove excess HCl. The residue was dissolved in water (5 mL) and poured onto a strong anion exchanger (Dowex 1, 3 × 10 cm bed, OH−-form, 20% aq. AcOH → aq. HCl (1:1) elution). Fractions with the crude product were combined, solvents were removed under vacuum and the residue was co-evaporated several times with water. The residue was subjected to flash chromatography (C-18, M2) leading to a partial purification of 11 and giving pure compound 9 in the later fractions (see also above, 0.11 g, 9%). Fractions containing 11 were evaporated under vacuum and the oily residue was dissolved in water (5 mL), and purified by semi-preparative HPLC (C-18, M4). The fractions containing compound 11 (purity over 90%) were combined and lyophilized to give 11·TFA·H2O as a fluffy solid with sufficient purity (>90%) for the next reactions (0.50 g, 50%); the main impurity was P-hydroxymethylated compound 12, as determined by NMR and MS. NMR (H2O, pH ∼1.8): 1H δ 1.92–2.07 (CH2–CH2–CH2, m, 2H), 2.26–2.37 (CH2–CH2–CH2, m, 2H), 2.46 (P–CH2–P, pseudo-t, 2JHP–2JHP 17, 2H), 2.48 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.89–3.06 (cycle, m, 2H), 3.06–3.24 (cycle, m, 6H), 3.24–3.34 (cycle, m, 2H), 3.34–3.62 (cycle and N–CH2–P, m, 12H), 3.62–3.80 (cycle, m, 4H), 7.23 (P–H, d, 1JHP 568, 1H), 7.54–7.59 (o-phenyl, m, 2H), 8.22–8.27 (m-phenyl, m, 2H). 13C{1H} δ 20.1–21.4 (2× CH2–CH2–CH2, bs), 32.7 (P–CH2–P–CH2–aryl, pseudo-t, 1JCP–1JCP 82.5), 34.1–35.6 (P–CH2–P–H, m), 39.0 (P–CH2–aryl, d, 1JCP 89), 47.5–48.5 (cycle, bs), 49.0–52.0 (cycle, m), 54.1–56.4 (cycle and P–CH2–N, m), 57.5–59.6 (cycle, m), 124.6 (m-phenyl, d, JCP 3.0), 131.6 (o-phenyl, d, JCP 4.7), 141.2 (ipso-phenyl, d, 2JCP 9.2), 147.3 (p-phenyl, d, JCP 4.2). 31P{1H} δ 22.2–22.7 (P–H, m, 1P), 22.0–28.0 (P–CH2–N, m, 2P), 39.5–40.6 (P–CH2–aryl, m, 1P). ESI-MS: (−) 672.2 (672.2, [M − H]−), 335.6 (335.6, [M − 2H]2−); (+) 674.2 (674.2, [M + H]+), 696.2 (696.2, [M + Na]+). ESI-HR-MS: (−) 672.18835 (672.18876, [C23H42N5O10P4]−), 335.59052 (335.59074, [C23H41N5O10P4]2−). Elemental analysis (calc. for C23H43N5O10P4·TFA·H2O, MR 806.6): C 37.1 (37.3); H 6.1 (5.8); N 8.9 (8.7); P 15.8 (15.4). HPLC (C-18, M2): Rf ∼3.8 min.
Compound 12
In a 4 mL glass vial, 11·TFA·H2O (0.12 g, 0.15 mmol, 1 equiv.) was dissolved in ∼90% aq. TFA (∼3 mL) and heated to 80 °C. Paraformaldehyde (26 mg, 0.87 mmol, 6 equiv.) was quickly added and the flask was immediately closed with a stopper. The mixture was stirred at 80 °C for 1 d. Another portion of paraformaldehyde (26 mg, 0.87 mmol, 6 equiv.) was added and the mixture was stirred in the closed vial at 80 °C for another 1 d. The procedure was repeated with another portion of paraformaldehyde (26 mg, 0.87 mmol, 6 equiv.) on the next day. After 3 d, solvents were removed under vacuum and the residue was co-evaporated several times with water to remove the formaldehyde and excess TFA. The oily residue was dissolved in water (∼50 mL) and lyophilized to give fibrous off-white solid 12·1.5TFA·4H2O (0.13 g, >90%, purity >95%, the impurity was assigned to compound 11 by NMR and HPLC). NMR (D2O, pD ∼1.7): 1H δ 1.90–2.08 (CH2–CH2–CH2, m, 2H), 2.20–2.38 (CH2–CH2–CH2, m, 2H), 2.38 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.49 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.88–3.04 (cycle, m, 4H), 3.04–3.35 (cycle, m, 8H), 3.35–3.63 (cycle and N–CH2–P, m, 10H), 3.63–3.81 (cycle, m, 4H), 3.83 (P–CH2–OH, d, 2JHP 5.2, 2H), 7.54–7.60 (o-phenyl, m, 2H), 8.23–8.28 (m-phenyl, m, 2H). 13C{1H} δ 19.9–21.2 (2× CH2–CH2–CH2, bs), 31.6–34.1 (2× P–CH2–P, m), 39.4 (P–CH2–aryl, d, 1JCP 88.6), 47.5–48.9 (cycle, m), 49.5–51.3 (cycle, m), 53.9–59.0 (cycle and N–CH2–P, m), 60.8 (P–CH2–OH, d, 1JCP 113), 124.6 (m-phenyl, s), 131.5 (o-phenyl, d, 3JCP 5.4), 142.0 (p-phenyl), 147.2 (ipso-phenyl, d, 2JCP 3.7). 31P{1H} δ 18.5–29.8 (m, 2P), 38.8–39.8 (m, 2P). ESI-MS: (−) 702.2 (702.2, [M − H]−), 350.6 (350.6, [M − 2H]−); (+) 704.2 (704.2, [M + H]+). ESI-HR-MS: (−) 702.19849 (702.19933, [C24H44O11N5P4]−), 350.59575 (350.59602, [C24H43O11N5P4]2−). Elemental analysis (calc. for C24H45N5O11P4·1.5TFA·4H2O, MR 946.7): C 33.9 (34.3); H 5.8 (5.8); N 7.6 (7.4); P 13.4 (13.1); F 7.7 (9.0). HPLC (C-18, M2): Rf ∼3.8 min.General procedure for compounds 12, 14, and 15a
In an argon-flushed 10 mL glass flask, 11·H2O·TFA (98 mg, 0.12 mmol, 1 equiv.) was mixed with anhydrous CHCl3 (∼5 mL). Then, DIPEA (∼0.2 mL, ∼1.0 mmol, 8 equiv.), BSA (∼0.5 mL, 2.3 mmol, 16 equiv.) and Me3Si-Cl (50 μL, 0.4 mmol, 3 equiv.) were successively added via syringe. The resulting suspension was stirred under Ar at 50 °C for 1 h until the mixture clarified (conversion to silyl esters >95%, by 31P{1H} NMR). Then, paraformaldehyde, acrylonitrile, or phthalimido-N-methylcarbaldehyde, (40 μL, 114 mg or 18 mg, respectively, 0.60 mmol, 5 equiv.) was added and the solution was stirred under an Ar atmosphere at 50 °C overnight. The reaction was quenched with MeOH (0.2 mL), to hydrolyse the silyl esters, and stirred for 5 min. The reaction mixture was extracted with water (3 × 2 mL) to give the crude product in the aqueous phase and the combined aqueous phases were evaporated under vacuum. The oily residue was dissolved in 50% aq. MeOH (3 mL) and the solution was purified by semi-preparative HPLC (C-18; M5). Fractions containing the pure product were combined and lyophilized. The TFA salts of the products (∼95% purity as determined by NMR and HPLC) were obtained as hygroscopic solids (68 mg (∼60%) of 12·1.5TFA·4H2O, 40 mg (35%) of 14·1.5TFA·3H2O and 80 mg (60%) of 15a·1.5TFA·2.5H2O) which were stored in a freezer (−20 °C).Characterization data of 12 were identical to those given above.
Compound 14: NMR (D2O, pD 1.5): 1H δ 1.89–2.05 (CH2–CH2–CH2, m, 2H), 2.20 (P–CH2–CH2–CN, pseudo-ddq, JHP 18, JHP 15, 3JHH 7.5, 2H), 2.25–2.36 (CH2–CH2–CH2, m, 2H), 2.43 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.45–2.51 (P–CH2–P, m, 2H), 2.73 (P–CH2–CH2–CN, dt, 3JHP 15, 3JHH 7.5, 2H), 2.88–3.02 (cycle, m, 2H), 3.02–3.13 (cycle, m, 4H), 3.13–3.20 (cycle, m, 2H), 3.23–3.31 (cycle, m, 2H), 3.41–3.59 (cycle, N–CH2–P and P–CH2–aryl, m, 12H), 3.61–3.73 (cycle, m, 4H), 7.56–7.59 (o-phenyl, m, 2H), 8.24–8.27 (m-phenyl, m, 2H); 13C{1H} δ 11.1 (P–CH2–CH2–CN, d, 2JCP 3.1), 20.2–21.2 (2× CH2–CH2–CH2, bs), 26.7 (P–CH2–CH2–CN, d, 1JCP 97), 33.1 (P–CH2–P, pseudo-t, 1JCP–1JCP 79), 33.7 (P–CH2–P, pseudo-t, 1JCP–1JCP 79), 39.3 (P–CH2–Caryl, d, 1JCP 89), 47.9–48.7 (cycle, m), 50.0–50.6 (cycle, m), 54.2–55.8 (cycle and N–CH2–P, m), 58.0–58.9 (cycle, m), 121.4 (CN, d, 3JCP 13.9), 124.6 (m-phenyl, d, JCP 3.0), 131.6 (o-phenyl, d, JCP 5.5), 141.7 (p-phenyl, bs), 147.3 (ipso-phenyl, d, 2JCP 4.0); 31P{1H} δ 23.2–29.9 (m, 2P), 41.1 (m, 1P), 41.9 (m, 1P). ESI-MS: (−) 725.2 (725.2 [M − H]−); (+) 727.2 (727.2, [M + H]+). ESI-HR-MS: (−) 725.21505 (725.21531, [C26H45O10N6P4]−), 362.10435 (362.10402, [C26H44O10N6P4]2−). Elemental analysis (calc. for C26H46N6O10P4·1.5TFA·3H2O, MR 951.7): C 36.7 (36.6); H 5.4 (5.7); N 8.4 (8.8); P 12.9 (13.0); F 9.7 (9.0). HPLC (C-18, M2): Rf ∼3.9 min.
Compound 15a: NMR (D2O, pD 1.8): 1H δ 1.89–2.10 (CH2–CH2–CH2, m, 2H), 2.21–2.37 (CH2–CH2–CH2, m, 2H), 2.37–2.45 (P–CH2–P, m, 2H), 2.45–2.65 (P–CH2–P, m, 2H), 2.84–3.24 (cycle, m, 8H), 3.24–3.32 (cycle, m, 2H), 3.36–3.62 (cycle + N–CH2–P + P–CH2–aryl, m, 12H), 3.62–3.78 (cycle, m, 4H), 3.84–3.98 (P–CH(OH)–CH2–N, m, 2H), 4.16 and 4.22 (P–CH(OH)–CH2–N, 2 × ddd, 2JHP 11 and 10, 3JHH 7.8 and 3.5, 3JHH 3.5 and 3.8, respectively; 1H), 7.43–7.51 (o-phenyl, m, 2H), 7.77–7.86 (Pht, m, 4H), 8.03–8.15 (m-phenyl, m, 2H); 13C{1H} δ 19.9–21.4 (2× CH2–CH2–CH2, bs), 30.9–32.3 (P–CH2–P, m), 33.1 (P–CH2–P, pseudo-t, 1JCP–1JCP 83), 39.3 (P–CH2–aryl, d, 1JCP 88), 39.5–39.8 (P–CH(OH)–CH2–N, m), 47.3–48.6 (cycle, m), 49.5–50.9 (cycle, m), 54.2–56.1 (cycle and N–CH2–P, m), 57.8–59.1 (cycle, m), 68.7 and 69.0 (P–CH(OH)–CH2–N, d, 1JCP 113 and 111, respectively), 124.1 (Pht, s), 124.3–124.5 (m-phenyl, m), 131.4–131.6 (CH–Pht, m), 131.8 (o-phenyl, d, JCP 8.5), 135.5 (Phth, pseudo-d, JCP 3.4), 141.7–142.0 (p-phenyl, bs), 146.9 (ipso-phenyl, pseudo-dd, J 16, J 3.9), 170.9 (CO, pseudo-d, J 10.7); 31P{1H} δ 18.0–29.5 (bm, 2P), 34.7 and 35.3 (2 × pseudo-s, 1P), 37.3–39.1 (m, 1P). ESI-MS: (−) 861.2 (861.2, [M − H]−), 430.1 (430.1, [M − 2H]2−), 1761.4 (1761.4, [2M − H]−); (+) 863.2 (863.2, [M + H]+), 1725.2 (1725.5, [2M + H]+). ESI-HR-MS: (−) 861.23121 (861.23135, [C33H49O13N6P4]−); (+) 863.24573 (863.24591, [C33H49O13N6P4]+). Elemental analysis (calc. for C33H50N6O13P4·1.5TFA·2.5H2O, MR 1092.8): C 40.0 (40.1); H 4.8 (5.3); N 7.5 (7.8); P 11.8 (11.5); F 7.6 (7.9). HPLC (C-18, M2): Rf ∼4.4 min.
Compound 15
:1 aq. HCl (∼3 mL) and the solution was heated to reflux (120 °C) for 1 d. Solvents were removed under reduced pressure and the oily residue was dissolved in water (∼5 mL). The aqueous phase was washed with AcOEt (3 × 1 mL), evaporated to dryness under vacuum and the residue was purified by semi-preparative HPLC (C-18, M5). Fractions containing the pure product were combined and lyophilized. The product was obtained as a hygroscopic solid as above (6.5 mg, 30%, purity >95% as determined by NMR and HPLC). NMR (D2O, pD 2.3): 1H δ 1.82–2.12 (CH2–CH2–CH2, m, 2H), 2.17–2.41 (CH2–CH2–CH2, m, 2H), 2.31 (P–CH2–P–CH2–aryl, pseudo-t, 2JHP–2JHP 16, 2H), 2.33 (P–CH2–P–CH(OH), m, 2H), 2.83–3.23 (cycle, m, 9H), 3.23–3.31 (cycle, m, 2H), 3.36–3.41 (cycle, m, 1H), 3.44 (P–CH2–aryl, d, 2JHP 17.8, 2H), 3.41–3.65 (cycle and P–CH2–N, m, 10H), 3.65–3.83 (cycle, m, 4H), 4.02–4.11 (P–CH(OH)–CH2, m, 1H), 7.54–7.58 (o-phenyl, m, 2H), 8.22–8.26 (m-phenyl, m, 2H); 13C{1H} δ 19.8–21.1 (2× CH2–CH2–CH2, bs), 33.2 (P–CH2–P–CH2–aryl, pseudo-t, 1JCP–1JCP 79), 33.4–34.2 (P–CH2–P–CH(OH)–CH2, m), 39.7 (P–CH2–aryl, dd, 1JCP 88, 3JCP 5.6), 41.0–41.2 (P–CH(OH)–CH2–N, m), 47.8–48.7 (cycle, m), 49.6–50.9 (cycle, m), 53.8–55.1 (cycle and N–CH2–P, m), 57.6–58.8 (cycle, m), 124.5 (m-phenyl, d, JCP 3.0), 131.5 (o-phenyl, d, JCP 5.2), 142.7 (p-phenyl, bs), 147.1 (ipso-phenyl, d, 2JCP 3.8); 31P{1H} δ 21.4–24.0 (m, 1P), 24.0–26.3 (m, 1P), 29.9–31.5 (m, 1P), 34.2–36.7 (m, 1P). ESI-MS: (−) 731.2 (731.2, [M − H]−), 365.1 (365.1, [M − 2H]2−), 1463.5 (1463.5, [2M − H]−); (+) 733.2 (733.2, [M + H]+). ESI-HR-MS: (−) 731.22600 (731.22588, [C25H47O11N6P4]−), 365.10934 (365.10930, [C25H46O11N6P4]2−); (+) 733.23995 (733.24043, [C25H47O11N6P4]+). HPLC (C-18, M2): Rf ∼3.8 min.
Compound 13
In an argon-flushed 4 mL glass vial, 11·TFA·H2O (80 mg, 0.10 mmol, 1.0 equiv.) was mixed with anhydrous CHCl3 (2 mL). DIPEA (140 μL, 0.80 μmol, 8 equiv.), BSA (0.40 mL, 1.6 mmol, 16 equiv.) and Me3Si-Cl (38 μL, 0.30 mmol, 3 equiv.) were successively added via a syringe. The resulting suspension was stirred at 50 °C for 1 h until the mixture clarified (conversion >95% by 31P{1H} NMR). Then, t-butyl acrylate (79 μL, 0.50 mmol, 5 equiv.) was added and the solution was stirred at 50 °C overnight. The reaction was quenched with MeOH (0.2 mL) to hydrolyse the silyl esters. After stirring for 5 min, water (∼2 mL) was added and the crude product was extracted (3 × 2 mL) into the aqueous phase. Combined aqueous phases were evaporated under vacuum. The oily residue was dissolved in 50% aq. MeOH (2 mL), and the solution was purified by semi-preparative HPLC (C-18, M4). Fractions containing the pure product were combined and the solvents were removed under vacuum. Product 13a was obtained as a colourless sticky oil (∼60 mg, purity ≥95% as determined by NMR; the impurity was assigned to compound 13), which was directly used in the next step.Compound 13a: NMR (D2O, pD ∼1.6): 1H δ 1.44 (H3C–C, s, 9H), 1.86–2.03 (CH2–CH2–CH2, m, 2H), 2.06–2.22 (P–CH2–CH2–C(O), m, 2H), 2.23–2.39 (CH2–CH2–CH2, m, 2H), 2.39–2.64 (P–CH2–CH2–C(O) and 2× P–CH2–P, m, 6H), 2.83–3.00 (cycle, m, 2H), 3.00–3.19 (cycle and N–CH2–P, m, 6H), 3.19–3.34 (cycle, m, 2H), 3.34–3.57 (cycle and P–CH2–aryl, m, 12H), 3.57–3.77 (cycle, m, 4H), 7.51–7.62 (o-phenyl, d, 3JHH 8.1, 2H), 8.20–8.30 (m-phenyl, d, 3JHH 8.2, 2H); 31P{1H} δ 24.0–27.0 (bm, 2P), 39.2 (d, 2JPP 9.7, 1P), 45.7 (d, 2JPP 10.0, 1P). ESI-MS: (−) 800.3 (800.3, [M − H]−); (+) 802.3 (802.3, [M + H]+), 824.3 (824.3, [M + Na]+).
The oil from the previous step (TFA salt of 13a, 60 mg, ∼50 μmol) was dissolved in TFA (5 mL) and the solution was stirred at room temperature overnight. The solvents were removed under vacuum and the residue was co-evaporated several times with water to get quantitatively a TFA salt of 13 as a colourless sticky oil (∼51 mg, ∼50% if calculated as mono-TFA salt; two steps, based on 11·TFA·H2O).
Compound 13: NMR (D2O, pD ∼1.1): 1H δ 1.89–2.00 (CH2–CH2–CH2, m, 2H), 2.13–2.22 (P–CH2–CH2–CO, dm, 2JHP 15, 2H), 2.27–2.37 (CH2–CH2–CH2, m, 2H), 2.46 (P–CH2–P, pseudo-t, 2JHP 16, 2H), 2.51–2.59 (P–CH2–P, pseudo-t, 2JHP 16, 2H), 2.62 (P–CH2–CH2–C(O), dt, 3JHP 12.6, 3JHH 7.9, 2H), 2.86–3.00 (cycle, m, 2H), 3.04–3.20 (cycle and N–CH2–P, m, 6H), 3.23–3.31 (cycle, m, 2H), 3.34–3.57 (cycle and P–CH2–aryl, m, 12H), 3.57–3.73 (cycle, m, 4H), 7.54–7.58 (o-phenyl, m, 2H), 8.22–8.26 (m-phenyl, dm, 3JHH 8.4, 2H); 13C{1H} δ 20.9 and 21.0 (2× CH2–CH2–CH2, bs), 26.2 (P–CH2–CH2–C(O), d, 1JCP 98), 27.2 (P–CH2–CH2–C(O), s), 32.2–33.5 (2× P–CH2–P, m), 39.2 (P–CH2–aryl, d, 1JCP 89), 47.7–48.6 (cycle, m), 49.9–50.6 (cycle, m), 54.3–55.9 (cycle, m), 57.4–59.4 (cycle, m), 128.6 (m-phenyl, d, JCP 3), 131.6 (o-phenyl, d, JCP 5), 141.4 (p-phenyl, bs), 147.3 (ipso-phenyl, d, JCP 4), 177.1 (CO, d, 3JCP 16); 31P{1H} δ 23.6–28.3 (m, 2P), 39.5 (d, 2JPP 9.8, 1P), 45.5 (d, 2JPP 8.9, 1P). ESI-MS: (−) 744.2 (744.2, [M − H]−); (+) 746.2 (746.2, [M + H]+). ESI-HR-MS: (−) 744.20940 (744.20898, [C26H46O12N5P4]−), 371.60101 (371.60131, [C26H45O12N5P4]2−). HPLC (C-18, M2): Rf ∼3.9 min.
Compound 16
In a 100 mL glass flask, 9·1.5HCl·3.5H2O (110 mg, 120 μmol, 1 equiv.) and 10% Pd/C (22 mg, 20% w/w) were mixed with ∼90% aq. AcOH (50 mL). The flask was briefly degassed by using a pump and then gently flushed with hydrogen from a balloon. The reaction mixture was vigorously stirred at 50 °C for 1 d. The solids were filtered off using a syringe microfilter (0.22 μm) and the filtrate was concentrated under reduced pressure. The oily residue was dissolved in 1:1 aq. HCl (∼5 mL), and the solvents were removed under vacuum. The oil of 16 obtained in this way was immediately used in the next reactions (see below). To obtain a sample for characterization studies, the residue was dissolved in MeOH (∼5 mL) and acetone (∼50 mL) was added to the solution. The formed suspension was briefly sonicated and the precipitate was filtered off (S4) and washed with acetone (∼5 mL) and Et2O (2 × 5 mL). The product was briefly dried in air and under high vacuum to yield the hydrochloride derivative of light-sensitive compound 16·4HCl·7H2O (98 mg, 80%), which slowly decomposed upon standing, even in the dark, and the compound could not be stored. NMR (D2O, pD ∼2): 1H δ 1.79–1.99 (cycle, m, 2H), 1.99–2.19 (P–CH2–P, m, 4H), 2.21–2.42 (cycle, m, 2H), 2.54–3.16 (cycle, m, 8H), 3.16–3.30 (cycle and P–CH2–aryl, m, 6H), 3.30–3.86 (cycle and N–CH2–P, m, 14H), 7.32–7.36 (m-phenyl, d, 2JHH 8, 2H), 7.44–7.48 (o-phenyl, d, 2JHH 8, 2H). 13C{1H} δ 20.6 (cycle, bs), 34.2 (P–CH2–P, pseudo-t, 1JCP 82), 39.3 (P–CH2–Caryl, d, 1JCP 90), 49.1 (cycle, s), 49.9 (cycle, bs), 53.9 (cycle, bs), 52.5–57.7 (P–CH2–N and cycle, m), 57.7–59.2 (cycle, bs), 123.6 (m-phenyl, s), 128.8 (p-phenyl, s), 131.9 (o-phenyl, d, 3JCP 5), 136.3 (ipso-phenyl, d, 2JCP 8). 31P{1H} δ 22.8 (N–CH2–P–CH2, m, 2P), 32.6 (CH2–P–Bn, m, 2P). ESI-MS: (−) 747.3 (747.2, [M − H]−); 373.1 (373.1, [M − 2H]2−). ESI-HR-MS: (−) 747.27179 (747.27243, [C30H50O8N6P4]−), 373.13228 (373.13258, [C30H50O8N6P4]2−). Elemental analysis (calc. for C30H52N6O8P4·4HCl·7H2O, MR 1020.6): C 35.1 (35.3); H 6.3 (6.9); N 8.4 (8.2); P 12.0 (12.1); Cl 12.6 (13.9). HPLC (C-18, M2): Rf ∼3.4 min.
Compound 17
Compound 16 (obtained from 9·1.5HCl·3.5H2O, 76 mg, 70 μmol) was dissolved in 0.25 M aq. HCl (1.12 mL, 280 μmol, 4.0 equiv.), the solution was cooled in an ice bath (5 °C) and freshly prepared 0.1 M aq. NaNO2 (1.65 mL, 165 μmol, 2.4 equiv.) was added in one portion. The mixture was stirred for 5 min and freshly prepared 0.1 M aq. NaN3 (2.45 mL, 245 μmol, 3.5 equiv.) was added. The vial was removed from the ice bath and the mixture was stirred at room temperature for 3 h (conversion ∼95%). The reaction mixture was purified by semi-preparative HPLC (C-8, M1). Fractions with the pure product were pooled, water was added (final volume ∼50 mL) and the solution was lyophilized to give 17·TFA·2H2O as a white foam (38 mg, 57%; over two steps). NMR (D2O, pD ∼2): 1H δ 1.89–2.03 (cycle, m, 2H), 2.27–2.39 (cycle and P–CH2–P, m, 6H), 2.60–4.07 (cycle, P–CH2–aryl and N–CH2–P, m, 28H), 7.02–7.06 (o-phenyl, d, 2JHH 8, 4H), 7.28–7.32 (m-phenyl, d, 2JHH 8, 4H). 13C{1H} δ 20.4 (cycle, bs), 32.0–34.0 (P–CH2–P, pseudo-t, 1JCP 82), 38.1 (P–CH2–aryl, d, 1JCP 91), 48.2 (cycle, s), 50.2 (cycle, s), 54.4 (cycle, s), 55.2 (cycle, bs), 56.0–58.2 (P–CH2–N and cycle, m), 58.2 (cycle, s), 119.8 (m-phenyl, s), 129.8 (ipso-phenyl, d, 2JCP 8 Hz), 131.9 (o-phenyl, d, 3JCP 6), 139.0 (p-phenyl, s). 31P{1H} δ 22.4 (N–CH2–P–CH2, m, 2P); 40.4 (CH2–P–Bn, m, 2P). ESI-MS: (−) 799.0 (799.3, [M − H]−); (+) 801.2 (801.3, [M + H]+), 823.2 (823.3, [M + Na]+); (−) 799.3 (799.3, [M − H]−), 399.1 (399.1, [M − 2H]2−). ESI-HR-MS: (−) 799.25267 (799.25343, [C30H47N6O8P4]−), 399.12250 (399.12308, [C30H46N6O8P4]2−). Elemental analysis (calc. for C30H48N10O8P4·TFA·2H2O, MR 950.7): C 40.3 (40.4); H 5.5 (5.6); N 15.0 (14.7). HPLC (C-8, M1): Rf ∼6.5 min. HPLC (C-18, M2): Rf ∼4.9 min.Compound 18
Compound 16 (obtained from 9·1.5HCl·3.5H2O, 250 mg, 261 μmol) was dissolved in water (30 mL) and CSCl2 (120 μL, 1.57 mmol, ∼6 equiv.) dissolved in CCl4 (30 mL) was added. The two-phase mixture was vigorously vortexed at room temperature for 16 h. The mixture was diluted with water (50 mL) and extracted with CH2Cl2 (50 mL). The aqueous layer was separated and evaporated to dryness under vacuum. The residue was purified by semi-preparative HPLC (C-8, M1). Fractions with the pure product were pooled, diluted to 300 mL with water and the solution was lyophilized to give 18·2.5TFA·1.5H2O as an off-white product (164 mg, 55%, over two steps). NMR (D2O, pD ∼2): 1H δ 1.63–2.04 (cycle, m, 2H); 2.04–2.54 (cycle and P–CH2–P, m, 6H); 2.54–4.08 (cycle, P–CH2–aryl and P–CH2–N, m, 28H); 7.06–7.10 (phenyl, m, 4H); 7.19–7.23 (phenyl, m, 4H). 13C{1H} δ 20.4 (cycle, s); 32.1 (P–CH2–P, pseudo-t, 1JCP 85); 38.0 (P–CH2–aryl, d, 1JCP 91); 47.6 (cycle, s); 49.5 (cycle, s); 52.8–55.6 (P–CH2–N and cycle, m); 56.4–59.5 (cycle, m); 125.9 (phenyl, s); 129.3 (phenyl, s); 131.2 (phenyl, s); 132.0 (phenyl, s); 134.8 (NCS, s). 31P{1H} δ 24.9 (N–CH2–P, bs), 39.3 (P–CH2–aryl, s). ESI-MS: (−) 831.2 (831.2, [M − H]−); (+) 833.2 (833.2, [M + H]+). ESI-HR-MS: (+) 833.19947 (833.19983, [C32H49N6O8P4S2]+); (−) 831.18577 (831.18527, [C32H47N6O8P4S2]−). Elemental analysis (calc. for C32H48N6O8P4S2·2.5TFA·1.5H2O, MR 1144.9): C 38.7 (38.8); H 4.3 (4.7); N 7.1 (7.3). HPLC (C-18, M2): Rf ∼5.4 min.Compound 19
In a 100 mL flask, the pyridinium salt of 10 (10·py·3.5H2O, 1.4 g, 1.8 mmol) was dissolved in ∼75% aq. AcOH (50 mL), Pd/C (0.14 g, 10% w/w) was added and the flask was well-flushed with hydrogen gas. A hydrogen balloon was attached and the suspension was vigorously stirred at 50 °C for 2 d. The reaction mixture was cooled to room temperature, the solids were filtered off through a syringe microfilter (0.22 μm) and the solvents were removed under vacuum. The oily residue was successively co-evaporated with toluene (10 mL), conc. aq. HCl (10 mL), water (10 mL) and MeOH (10 mL). The oily residue was dissolved in MeOH (5 mL) and acetone (50 mL) was slowly added leading to the precipitation of a solid. The solid was filtered off, washed with Et2O (3 × 10 mL) and immediately dissolved in water (5 mL). The solution containing the crude product was purified by flash chromatography (C-18, M2). Fractions containing the pure product were combined and the solvents were evaporated under vacuum. The oily residue was dissolved in water (200 mL) and the solution was lyophilized to give zwitterionic 19·4H2O as a fluffy solid (0.95 g, 72%). NMR (D2O, pD ∼2): 1H δ 1.90–2.08 (cycle, m, 2H), 2.15–2.29 (P–CH2–P, pseudo-t, 2JHP–2JHP 16.2, 2H), 2.19–2.35 (cycle, m, 2H), 2.25–2.37 (P–CH2–P, pseudo-t, 2JHP–2JHP 18, 2H), 2.70–4.19 (cycle, N–CH2–P and P–CH2–aryl, m, 26H); 7.37–7.41 (o-phenyl, d, 2H, 3JHH 8), 7.48–7.52 (m-phenyl, d, 2H, 3JHH 8); 13C{1H} δ 20.2 (cycle, s), 20.5 (cycle, s), 32.9 (P–CH2–P, dd, 1JCP 118, 1JCP 82), 33.0–34.2 (P–CH2–P, pseudo-t, 1JCP 83), 38.8 (P–CH2–aryl, d, 1JCP 91), 47.3–58.7 (cycle, m), 123.7 (m-phenyl, d, 4JCP 3), 128.8 (p-phenyl, s), 131.9 (o-phenyl, d, 3JCP 5), 135.6 (ipso-phenyl, d, 2JCP 7); 31P{1H} δ 15.0–15.6 (PO3H2, m, 1P), 22.2–22.8 (P–CH2–N, m, 1P), 23.1–23.7 (P–CH2–N, m, 1P), 35.1–35.7 (P–CH2–aryl, m, 1P). ESI-MS: (−) 658.2 (658.2, [M − H]−), (+) 660.2 (660.2, [M + H]+), 682.2 (682.2 [M + Na]+). ESI-HR-MS: (−) 658.20912 (658.20950, [C23H44O9N5P4]−); (+) 680.19127 (680.19144, [C23H44O9N5P4Na]+). TLC (i-PrOH–conc. aq. NH3–H2O 7:3:3): Rf ∼0.1. Elemental analysis (calc. for C23H45N5O9P4·4H2O, MR 731.6): C 37.6 (37.8), H 7.1 (7.3), N 9.6 (9.6), P 16.1 (16.9). HPLC (C-18, M2): Rf ∼1.8 min.
Compound 20
Compound 19·4H2O (250 mg, 0.34 mmol, 1 equiv.) was dissolved in 1% aq. HCl (20 mL) and the solution was cooled in an ice bath. An aqueous solution (2 mL) of NaNO2 (35 mg, 0.51 mmol, 1.5 equiv.) was added and, after a few minutes, an aqueous solution (3 mL) of NaN3 (45 mg, 0.69 mmol, 2 equiv.) was added. The reaction mixture was left to warm up to room temperature over 3 h. The volatiles were evaporated under vacuum. The oily residue was dissolved in water (2 mL) and the crude product was purified by flash chromatography (C-18, M2). Fractions containing the pure product were combined and the solvents were evaporated to dryness under vacuum. The oily residue was dissolved in water (50 mL) and lyophilized. The product was obtained as 20·2HCl·4H2O (fluffy solid, 180 mg, 63%). NMR (D2O, pD ∼3): 1H δ 1.92–2.08 (cycle, m, 2H), 2.23–2.37 (P–CH2–P, pseudo-t, 2JHP–2JHP 15.9, 2H), 2.27–2.39 (cycle, m, 2H), 2.30–2.42 (P–CH2–P, pseudo-t, 2JHP–2JHP 19, 2H), 2.71–4.12 (cycle, N–CH2–P and P–CH2–aryl, m, 26H), 7.10–7.14 (o-phenyl, d, 2H, 3JHH 8), 7.36–7.40 (m-phenyl, d, 2H, 3JHH 8); 13C{1H} δ 20.4 (cycle, s), 20.6 (cycle, s), 31.9–34.1 (P–CH2–P, m), 38.4 (P–CH2–aryl, d, 1JCP 92), 46.9–60.1 (cycle, m), 119.8 (m-phenyl, s), 130.3 (p-phenyl, s), 132.0 (o-phenyl, d, 3JCP 5), 139.0 (ipso-phenyl, d, 2JCP 4); 31P{1H} δ 15.7–16.3 (PO3H2, m, 1P), 21.4–23.4 (P–CH2–N, m, 1P), 22.1–24.7 (P–CH2–N, m, 1P), 38.2–39.8 (P–CH2–C, m, 1P). ESI-MS: (−) 684.2 (684.2, [M − H]−), 341.6 (341.6, [M − 2H]2−); (+) 686.2 (686.2, [M + H]+), 708.2 (708.2, [M + Na]+). ESI-HR-MS: (+) 684.19969 (684.20000, [C23H42O9N7P4] +), 341.59624 (341.59636, [C23H41O9N7P4]2+). TLC (iPrOH–conc. aq. NH4OH–H2O 7:3:3): Rf ∼0.2. Elemental analysis (calc. for C23H43N7O9P4·2HCl·4H2O, MR 830.5): C 33.2 (33.3); H 6.0 (6.4); N 11.4 (11.8), P 14.8 (14.9), Cl 9.1 (8.5). HPLC (C-8, M1): Rf ∼5.7 min.
Compound 21
Compound 19·4H2O (150 mg, 0.205 mmol, 1 equiv.) was dissolved in 1% aq. HCl (4 mL) and a solution of thiophosgene (47 μL, 0.61 mmol, 3 equiv.) in CCl4 (4 mL) was added. The mixture was vigorously stirred at room temperature for 1 d. The aqueous phase was separated and evaporated under vacuum. The oily residue was dissolved in water (2 mL) and the crude product was purified by flash chromatography (C-18, M2). Fractions containing the pure product were combined and the solvents were evaporated to dryness under vacuum. The oily residue was dissolved in water (50 mL) and lyophilized. Product 21·2HCl·4H2O was isolated as a fluffy solid (105 mg, 60%). NMR (D2O, pD ∼2): 1H δ 1.94–2.10 (CH2–CH2–CH2, m, 2H), 2.22–2.36 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.26–2.38 (CH2–CH2–CH2, m, 2H), 2.31–2.43 (P–CH2–P, pseudo-t, 2JHP–2JHP 19, 2H), 2.86–3.92 (cycle, N–CH2–P and P–CH2–C, m, 26H), 7.35–7.39 (o-phenyl, d, 2H, 3JHH 8 Hz), 7.37–7.41 (m-phenyl, m, 2H); 13C{1H} δ 20.8 (CH2–CH2–CH2, s), 21.3 (CH2–CH2–CH2, s), 31.7 (P–CH2–P, dd, 1JCP 120, 1JCP 83), 31.7–33.0 (P–CH2–P, m), 38.4 (P–CH2–aryl, d, 1JCP 91), 46.6–59.2 (cycle, m), 126.7 (m-phenyl, s), 130.1 (p-phenyl, s), 131.8 (o-phenyl, s), 132.2 (ipso-phenyl, d, 2JCP 8 Hz), 134.8 (CS, s); 31P{1H} δ 16.0–16.6 (PO3H2, m, 1P), 22.4–24.4 (P–CH2–N, m, 1P), 22.4–24.8 (P–CH2–N, m, 1P), 37.7–39.1 (P–CH2–aryl, m, 1P). ESI-MS: (−) 700.2 (700.2, [M − H]−), 349.6 (349.6, [M − 2H]2−); (+) 702.2 (702.2, [M + H]+). ESI-HR-MS: (−) 700.16563 (700.16592, [C24H42O9N5P4S]−), 349.57921 (349.57932, [C24H41O9N5P4S]2−). Elemental analysis (calc. for C24H43N5O9P4S·2HCl·4H2O, Mw 846.6): C 34.7 (34.1), H 6.1 (6.3), N 8.1 (8.3), P 14.6 (14.6), S 3.3 (3.8), Cl 8.8 (8.4). HPLC (C-8, M3): Rf ∼16 min.
Compound 22
The trifluoroacetate salt of 13 obtained above (∼51 mg) was dissolved in ∼75% aq. AcOH (10 mL) and Pd/C (9 mg, 20% w/w) was added. The flask was briefly degassed and then flushed with hydrogen gas. A hydrogen balloon was attached and the suspension was vigorously stirred at 50 °C for 4 h. The reaction mixture was cooled to room temperature, the solids were filtered off through a syringe microfilter (0.22 μm) and the solvents were removed under vacuum. The oily residue was successively co-evaporated with toluene (5 mL), conc. aq. HCl (5 mL) and water (5 mL). Compound 22 was isolated as an oil (∼30 mg), which was used directly in the next step. NMR (D2O, pD <1): 1H δ 1.88–2.04 (CH2–CH2–CH2, m, 2H), 2.15–2.40 (CH2–CH2–CH2 and P–CH2–CH2, m, 4H), 2.44–2.53 (P–CH2–P, m, 2H), 2.53–2.72 (CH2–CH2–CO and P–CH2–P, m, 4H), 2.87–3.04 (cycle, m, 2H), 3.06–3.23 (cycle and N–CH2–P, m, 6H), 3.24–3.33 (cycle, m, 2H), 3.45 (P–CH2–aryl, d, 2JHP 17.6, 2H), 3.33–3.74 (cycle, m, 14H), 7.40–7.45 (o-phenyl, d, 2H, 3JHH 7.8 Hz), 7.46–7.52 (m-phenyl, m, 2H); 13C{1H} δ 21.0 (2× CH2–CH2–CH2, bs), 26.2 (P–CH2–CH2–CO, d, 1JCP 101), 27.2 (P–CH2–CH2–CO, bs), 2× 31.4–33.7 (2× P–CH2–P, m), 38.3 (P–CH2–aryl, d, 1JCP 91), 47.6–48.4 (cycle, m), 49.9–50.8 (cycle, m), 54.3–56.0 (cycle, m), 57.2–59.2 (cycle, m), 124.1 (m-phenyl, bs), 129.4 (o-phenyl, bs), 132.2 (o-phenyl, bs), 134.0 (i-phenyl, bs), 177.1 (CO, d, 3JCP 15); 31P{1H} δ 23.8–28.2 (m, 2P), 41.3–42.1 (m, 1P), 45.4–46.2 (m, 1P). ESI-MS: (−) 714.2 (714.2, [M − H]−), 356.6 (356.6, [M − 2H]2−); (+) 716.2 (716.2, [M + H]+). ESI-HR-MS: (−) 714.23547 (714.23571, [C26H48O10N5P4]−), 356.61398 (356.61422, [C26H47O10N5P4]2−). HPLC (C-18, M2): Rf ∼3.5 min.
Compound 23
The oily hydrochloride salt of 22 obtained above (∼30 mg) was dissolved in 1% aq. HCl (5 mL) and the solution was cooled in an ice bath. An aqueous solution (1 mL) of NaNO2 (4.0 mg, 58 μmol, 1.5 equiv.) was added and, after a few minutes, an aqueous solution (1 mL) of NaN3 (5.0 mg, 77 μmol, 2 equiv.) was added. The solution was left to warm up to room temperature over 3 h. The solution was concentrated under vacuum to ∼2 mL and the crude product was purified by semi-preparative HPLC (C-18, M5). Fractions containing the pure product were combined and directly lyophilized. The product was isolated as 23·1.5TFA·H2O (fluffy solid, 13 mg, yield ∼18%, over four steps, based on 11·TFA·H2O, 80 mg). NMR (D2O, pD ∼1.6): 1H δ 1.86–2.01 (CH2–CH2–CH2, m, 2H), 2.15 (P–CH2–CH2, dt, 2JHP 14.9, 3JHH 7.5, 2H), 2.26–2.38 (CH2–CH2–CH2, m, 2H), 2.41 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.47 (P–CH2–P, dd, 2JPH 16.1, 2JPH 12.9, 2H), 2.62 (CH2–CH2–CO, dt, 3JHP 12.2, 3JHH 8.0, 2H), 2.81–2.99 (cycle, m, 2H), 2.97–3.20 (cycle and N–CH2–P, m, 6H), 3.22–3.32 (cycle, m, 2H), 3.35 (P–CH2–aryl, d, 2JHP 17.1, 2H), 3.40–3.59 (cycle, m, 10H), 3.60–3.72 (cycle, m, 4H), 7.08–7.14 (o-phenyl, dm, 2H, 3JHH 8.2 Hz), 7.33–7.40 (m-phenyl, m, 2H); 13C{1H} δ 20.7 (2× CH2–CH2–CH2, bs), 26.3 (P–CH2–CH2–CO, d, 1JCP 98), 27.4 (P–CH2–CH2–CO, bs), 31.9–33.9 (2× P–CH2–P, m), 38.2 (P–CH2–aryl, d, 1JCP 91), 47.8–48.7 (cycle, m), 49.8–50.7 (cycle, m), 54.2–55.7 (cycle, m), 57.3–59.0 (cycle, m), 120.0 (m-phenyl, d, 4JCP 3), 129.5 (p-phenyl, s), 132.1 (o-phenyl, d, 3JCP 5), 139.3 (ipso-phenyl, d, 2JCP 4), 177.3 (CO, d, 3JCP 16); 31P{1H} δ 21.9–25.8 (bm, 2P), 41.0–42.0 (m, 1P), 43.5–44.7 (m, 1P). ESI-MS: (−) 740.2 (740.2, [M − H]−), 369.6 (369.2, [M − 2H]2−); (+) 742.2 (742.2, [M + H]+), 764.2 (764.2, [M + Na]+). ESI-HR-MS: (−) 740.22574 (740.22621, [C26H46O10N7P4]−), 369.60920 (369.60947, [C26H45O10N7P4]2−). Elemental analysis (calc. for C26H47N7O10P4·1.5TFA·H2O, Mw 930.6): C 37.8 (37.4), H 5.4 (5.5), N 10.1 (10.5). HPLC (C-18, M2): Rf ∼4.1 min.
Compound 25
In a 4 mL glass vial, 19·4H2O (33 mg, 45.5 μmol) and the NHS-ester of 3-[4-(carboxymethyl)-phenyl]-6-methyl-1,2,4,5-tetrazine (16.4 mg, 50.1 μmol, 1.1 equiv.) were dissolved in a mixture of MES–NaOH aq. buffer (1.0 M, pH 6.2, 1.35 mL, 1.35 mmol, 25 equiv.) and MeCN (2.0 mL). Water (870 μL) was added, and the reaction mixture was stirred at room temperature for 2 d. The volatiles were evaporated, and the reaction mixture was purified by semi-preparative HPLC (C-8, M1). Fractions containing the pure product were transferred to a 100 mL round-bottomed flask with water (∼20 mL) and the solution (∼60 mL) was lyophilized. The product (≥99% purity as determined by NMR) was obtained as a fine pink powder of 25·0.5TFA·1.5H2O (19 mg, 42%). NMR (D2O, pD ∼2): 1H δ 1.92–2.02 (CH2–CH2–CH2, m, 2H), 2.21–2.31 (CH2–CH2–CH2, m, 2H), 2.25–2.37 (P–CH2–P, pseudo-t, 2JHP–2JHP 16, 2H), 2.30–2.42 (P–CH2–P, pseudo-t, 2JHP–2JHP 19, 2H), 2.71–3.86 (cycle, N–CH2–P, P–CH2–aryl and tetrazine-CH3, m, 29H), 3.93 (CH2–CO, s, 2H), 7.36–7.40 (phenyl, dm, 2H, 3JHH 8), 7.44–7.48 (phenyl, dm, 2H, 3JHH 8), 7.65–7.69 (phenyl, dm, 2H, 3JHH 8), 8.41–8.45 (phenyl, dm, 2H, 3JHH 8); 13C{1H} δ 20.4 (CH2–CH2–CH2, s), 20.6 (CH2–CH2–CH2, s), 20.7 (CH3, s), 31.6–34.0 (2× P–CH2–P, m), 38.6 (P–CH2–aryl, d, 1JCP 91), 43.6 (CH2–CO, s), 47.4–58.7 (cycle, m), 126.7 (m-phenyl, s), 130.1 (p-phenyl, s), 131.8 (o-phenyl, s), 132.2 (ipso-phenyl, d, 2JCP 8), 164.7 (N2–C–N2, s), 167.9 (N2–C–N2, s), 173.2 (CO, s). 31P{1H} δ 15.7–16.3 (PO3H2, m, 1P), 21.0–23.0 (P–CH2–N, m, 1P), 22.0–25.0 (P–CH2–N, m, 1P), 38.0–39.6 (P–CH2–aryl, m, 1P). ESI-MS: (−) 870.3 (870.3, [M − H]−); (+) 872.5 (872.3, [M + H]+), 894.5 (894.3, [M + Na]+). Elemental analysis (calc. for C34H53N9O10P4·0.5TFA·1.5H2O, MR 955.8): C 43.9 (44.0), H 5.5 (6.0), N 12.8 (13.2). HPLC (C-8, M1): Rf ∼6.2 min.Compound 26
In a 4 mL glass vial, 18·2.5TFA·1.5H2O (45 mg, 39 μmol, 1 equiv.) and N-(2-aminoethyl)maleimide hydrochloride (7.0 mg, 39 μmol, 1 equiv.) were dissolved anhydrous DMF (∼2 mL) and anhydrous iPr2NEt (35 μL, 200 μmol, 5 equiv.) was added. The mixture was vigorously stirred at room temperature for 5 h, during which time the solution turned into a cloudy suspension. Then, 0.1% aq. TFA (∼2 mL) was added and the mixture was stirred until the solution became clear. The aqueous solution was washed with AcOEt (2 × 2 mL) and then directly purified by semi-preparative HPLC (C-18, M7). Fractions containing the pure products were combined and directly lyophilized. The products 26a·2TFA·3H2O (13 mg, 24%; obtained as a mixture of two conformers) and 26·2TFA·2H2O (21 mg, 43%) were isolated as fluffy solids. Some unreacted 18·2.5TFA·1.5H2O (9 mg, 20%) was regenerated after purification.Compound 26: NMR (D2O, pD ∼1.8): 1H δ 1.65–2.05 (CH2–CH2–CH2, m, 2H), 2.11–2.56 (CH2–CH2–CH2 and P–CH2–P, m, 6H), 2.64–3.15 (cycle, m, 8H), 3.15–3.33 (cycle, m, 8H), 3.33–3.91 (cycle, N–CH2–P and C(S)NH–CH2–CH2–NC(O), m, 16H), 6.65–6.91 (CH–CO, bs, 4H), 7.09–7.22 (phenyl, m, 4H), 7.22–7.38 (phenyl, m, 4H). 13C{1H} δ 20.0–21.9 (2× CH2–CH2–CH2, bs), 31.7–33.9 (2× P–CH2–P, m), 37.9 (CH2–NHCS, s), 38.0–39.1 (2× P–CH2–aryl, m), 43.6 (CH2–NCO, s), 47.8–48.8 (cycle, m), 49.6–51.0 (cycle, m), 54.2–55.8 (cycle and P–CH2–N, m), 58.2–59.5 (cycle, m), 126.4–127.0 (phenyl, m), 129.4–130.0 (phenyl, m), 131.5–132.3 (phenyl, m), 132.7–133.2 (phenyl, m), 135.0–135.5 (CH–CO, s), 173.4 (NCO, s), 180.7 (NHCS, s). 31P{1H} δ 21.2–27.6 (P–CH2–N, m, 2P), 37.6–40.1 (P–CH2–aryl, m, 1P), 40.1–41.9 (P–CH2–aryl, m, 1P). ESI-MS: (−) 971.2 (971.2, [M − H]−), 485.1 (485.1, [M − 2H]2−). ESI-HR-MS: (−) 971.24303 (971.24385, [C38H55N8O10P4S2]−). Elemental analysis (calc. for C38H56N8O10P4S2·2TFA·2H2O, MR 1237.0): C 40.6 (40.8); H 4.7 (5.0); N 8.9 (9.1); P 9.9 (10.0); S 5.2 (5.2). HPLC (C-18, M2): Rf ∼4.9 min.
Compound 26a (a major conformer): NMR (D2O, pD ∼1.8): 1H δ 1.79–2.06 (CH2–CH2–CH2, m, 2H), 2.18–2.36 (CH2–CH2–CH2, m, 2H), 2.36–2.50 (P–CH2–P, m, 4H), 2.76–3.19 (cycle, m, 8H), 3.19–3.36 (cycle, m, 8H), 3.36–3.60 (cycle and N–CH2–P, m, 12H), 3.60–3.86 (C(S)NH–CH2–CH2–NC(O), m, 8H), 6.85 (CH–C(O), s, 4H), 7.10–7.24 (phenyl, m, 4H), 7.29–7.40 (phenyl, m, 4H). 13C{1H} δ 20.0–21.4 (CH2–CH2–CH2, bs), 32.1–33.5 (P–CH2–P, m), 37.9 (CH2–NHC(S), s), 38.4 (P–CH2–aryl, d, 1JCP 90), 43.5 (CH2–NC(O), s), 44.2–45.3 (cycle, m), 47.7–48.7 (cycle, m), 49.8–50.8 (cycle, m), 54.2–55.8 (cycle and P–CH2–N, m), 58.0–59.6 (cycle, m), 126.5–126.8 (phenyl, m), 131.6–132.1 (phenyl, m), 132.0–132.4 (phenyl, m), 135.3 (CH–C(O), s), 135.5–136.0 (phenyl, m), 173.7 (NC(O), s), 180.5 (NHC(S)NH s). 31P{1H} δ 17.5–27.5 (P–CH2–N, m, 2P), 36.8–43.5 (P–CH2–aryl, m, 2P). ESI-MS: (−) 1111.3 (1111.3, [M − H]−), 555.1 (555.1, [M − 2H]2−). ESI-HR-MS: (−) 1111.30173 (1111.30243, [C44H61N10O12P4S2]−), 555.14685 (555.14758, [C44H60N10O12P4S2]2−). Elemental analysis (calc. for C44H64N10O12P4S2·2TFA·3H2O, MR 1395.2): C 41.1 (41.3); H 5.0 (5.2); N 10.1 (10.0). HPLC (C-18, M2): Rf ∼4.5 min.
Radiolabelling experiments
For ligand labelling, stock solutions (1 mM) of the ligands were prepared from the solid samples by dissolving them in water. These solutions were carefully diluted to obtain stock solutions (10–600 μM) for the labelling experiments. For ligand comparative studies, aq. solutions (1 mM) of the ligands were prepared from the solids and they were diluted to obtain stock solutions (100 μM) for the labelling experiments. A solution of a ligand in MES buffer (prepared by mixing 1 μL of the ligand stock solution with cL = 100 μM and 10 μL of the MES stock solution with pH 6.2/5.6 and c = 0.5 M) was pre-heated to 25 °C. To this solution, freshly prepared (within less than 1 h) [64Cu]CuCl2 in aq. HCl (6 μL, 9–11 MBq) was added to get the final samples (17 μL, Cu:L molar ratio approx. 1:85–1:95). The samples were incubated for 10 min (pH 6.2) or 1 h (pH 5.6) at 25 °C. For all ligands, the experiments were always done with three different batches of [64Cu]CuCl2. The labelling progress was checked by TLC analysis (0.5 μL of the mixture, together with 0.5 μL [64Cu]CuCl2 stock solution) performed in aq. EDTA (0.1 M, pH ∼5): Rf 0.8–0.9 (free Cu), start (complexes of the phosphorus ligands), 0.1–0.2 (carboxylate complexes).
SOD stability test
To ensure quantitative incorporation of 64Cu into the in-cage complex with all ligands in this experiment, ∼6000 equiv. of the chelators with respect to the molar amount of [64Cu]CuCl2 was used as well as a long radiolabelling time (∼2 h incubation at room temperature was used for all complexes, except that of H2cb-te2a where heating to 50 °C had to be applied). The SOD stability test was carried according to the literature procedure.47 Briefly, human erythrocyte superoxide dismutase (SOD) was reconstituted in water to a protein concentration of 1 μg μL−1 (∼4 units per μL) and stored in aliquots (10 μL, 0.3 nmol) at −20 °C. For the SOD experiments, aliquots were thawed on ice and [64Cu]Cu-labelled ligands 1–4 and the “standard” ligands (0.1 nmol, 1.5 μL) or [64Cu]CuCl2 as a reference were added to SOD (0.3 nmol, 10 μg). The mixtures were incubated at 37 °C for 1 h followed by adding one volume of native sample buffer (Bio-Rad Laboratories, cat. #161-0738). The samples were separated using non-reducing and non-denaturing polyacrylamide gel electrophoresis (PAGE) with acrylamide concentrations of 15% in the resolving gel and 5% in the stacking gel; 20 μL of each sample were loaded into each well of the gel. The native PAGE was run at room temperature and 80 V until the dye front reached the resolving gel and then increased up to 140–160 V. After electrophoresis, the gel was washed with water for 1 min and exposed to a reusable imaging plate (Fujifilm) for 10 min. Following electronic autoradiography using a radioluminography laser scanner, the gel was stained with PageBlue protein staining solution (Thermo Fisher Scientific) according to the manufacturer's instructions. Quantitative analysis of average band intensities was performed with the Advanced Image Data Analysis (AIDA) program (Raytest).Author contributions
TD, VK and PH conceptualized the project and VK, HJP and PH supervised different parts of the realization. PU, TD and VH conducted syntheses and characterization. TD and HJP conducted the radiochemical experiments. PU, TD and VK wrote the original draft, and all co-authors contributed to the reviewing and editing process.Data availability
Crystallographic data for compound C (pno2ppinb) has been deposited at the CCDC (https://www.ccdc.cam.ac.uk/) under no. 2341020.†The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare the following competing financial interest(s): T. David, V. Kubíček and P. Hermann have filed a patent application containing some data presented in the manuscript.Acknowledgements
We thank Dr I. Císařová and Prof. J. Kotek for X-ray data collection and data fitting. We also thank Dr M. Drexler for collection of the 850 MHz NMR data. The work was supported by TAČR (TA78020003) and the I2PAD project (Horizon 2020).References
- T. W. Price, J. Greenman and G. J. Stasiuk, Dalton Trans., 2016, 45, 15702–15724 RSC.
- L. E. McInnes, S. E. Rudd and P. S. Donnelly, Coord. Chem. Rev., 2017, 352, 499–516 CrossRef CAS.
- A. J. Amoroso, I. A. Fallis and S. J. A. Pope, Coord. Chem. Rev., 2017, 340, 198–219 CrossRef CAS.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- J. A. Jackson, I. N. Hungnes, M. T. Ma and C. Rivas, Bioconjugate Chem., 2020, 31, 483–491 CrossRef CAS PubMed.
- B. M. Paterson and P. S. Donnelly, Adv. Inorg. Chem., 2016, 68, 223–251 CrossRef CAS.
- A. Ahmedova, B. Todorov, N. Burdzhiev and C. Goze, Eur. J. Med. Chem., 2018, 157, 1406–1425 CrossRef CAS PubMed.
- A. Boschi, P. Martini, E. Janevik-Ivanovska and A. Duatti, Drug Discovery Today, 2018, 23, 1489–1501 CrossRef CAS PubMed.
- S. Howard and V. N. Starovoitova, Appl. Radiat. Isotop., 2015, 96, 162–167 CrossRef CAS PubMed.
- O. Keinänen, K. Fung, J. M. Brennan, N. Zia, M. Harris, E. van Dam, C. Biggin, A. Hedt, J. Stoner, P. S. Donnelly, J. S. Lewis and B. M. Zeglis, Proc. Natl. Acad. Sci. U. S. A, 2020, 117, 28316–28327 CrossRef PubMed.
- R. J. Hicks, P. Jackson, G. Kong, R. E. Ware, M. S. Hofman, D. A. Pattison, T. A. Akhurst, E. Drummond, P. Roselt, J. Callahan, R. Price, C. M. Jeffery, E. Hong, W. Noonan, A. Herschtal, L. J. Hicks, A. Hedt, M. Harris, B. M. Paterson and P. S. Donnelly, J. Nucl. Med., 2019, 60, 777–785 CrossRef CAS PubMed.
- M. Loft, E. A. Carlsen, C. B. Johnbeck, H. H. Johannesen, T. Binderup, A. Pfeifer, J. Mortensen, P. Oturai, A. Loft, A. K. Berthelsen, S. W. Langer, U. Knigge and A. Kjaer, J. Nucl. Med., 2021, 62, 73–80 CrossRef PubMed.
- D. L. Bailey, K. P. Willowson, M. Harris, C. Biggin, A. Aslani, N. A. Lengkeek, J. Stoner, M. E. Eslick, H. Marquis, M. Parker, P. J. Roach and G. P. Schembri, J. Nucl. Med., 2023, 64, 704–710 CrossRef CAS PubMed.
- V. Kubíček, Z. Böhmová, R. Ševčíková, J. Vaněk, P. Lubal, Z. Poláková, R. Michalicová, J. Kotek and P. Hermann, Inorg. Chem., 2018, 57, 3061–3072 CrossRef PubMed.
- J. Kotek, P. Lubal, P. Hermann, I. Císařová, I. Lukeš, T. Godula, I. Svobodová, P. Táborský and J. Havel, Chem. – Eur. J., 2003, 9, 233–248 CrossRef CAS PubMed.
- T. David, V. Kubíček, O. Gutten, P. Lubal, J. Kotek, H.-J. Pietzsch, L. Rulíšek and P. Hermann, Inorg. Chem., 2015, 54, 11751–11766 CrossRef CAS PubMed.
- M. Paúrová, T. David, I. Císařová, P. Lubal, P. Hermann and J. Kotek, New J. Chem., 2018, 42, 11908–11929 RSC.
- D. N. Pandya, J. Y. Kim, J. C. Park, H. Lee, P. B. Phapale, W. Kwak, T. H. Choi, G. J. Cheon, Y.-R. Yoon and J. Yoo, Chem. Commun., 2010, 46, 3517–3519 RSC.
- D. N. Pandya, N. Bhatt, A. V. Dale, J. Y. Kim, H. Lee, Y. S. Ha, J.-E. Lee, G. I. An and J. Yoo, Bioconjugate Chem., 2013, 24, 1356–1366 CrossRef CAS PubMed.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 4829–4833 CrossRef CAS.
- A. V. Dale, G. J. An, D. N. Pandya, Y. S. Ha, N. Bhatt, N. Soni, H. Lee, H. Ahn, S. Sarkar, W. Lee, P. T. Huynh, J. Y. Kim, M.-R. Gwon, S. H. Kim, J. G. Park, Y.-R. Yoon and J. Yoo, Inorg. Chem., 2015, 54, 8177–8186 CrossRef CAS PubMed.
- L. M. P. Lima, Z. Halime, R. Marion, N. Camus, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2014, 53, 5269–5279 CrossRef CAS PubMed.
- X. Sun, M. Wuest, G. R. Weisman, E. H. Wong, D. P. Reed, C. A. Boswell, R. Motekaitis, A. E. Martell, M. J. Welch and C. J. Anderson, J. Med. Chem., 2002, 45, 469–477 CrossRef CAS PubMed.
- T. David, V. Hlinová, V. Kubíček, R. Bergmann, F. Striese, N. Berndt, D. Szöllősi, T. Kovács, D. Máthé, M. Bachmann, H.-J. Pietzsch and P. Hermann, J. Med. Chem., 2018, 61, 8774–8796 CrossRef CAS PubMed.
- L. Pazderová, M. Benešová, J. Havlíčková, M. Vojtíčková, J. Kotek, P. Lubal, M. Ullrich, M. Walther, S. Schulze, C. Neuber, S. Rammelt, H.-J. Pietzsch, J. Pietzsch, V. Kubíček and P. Hermann, Dalton Trans., 2022, 51, 9541–9555 RSC.
- D. J. Stigers, R. Ferdani, G. R. Weisman, E. H. Wong, C. J. Anderson, J. A. Golen, C. Moore and A. L. Rheingold, Dalton Trans., 2010, 39, 1699–1701 RSC.
- R. Ferdani, D. J. Stigers, A. L. Fiamengo, L. Wei, B. T. Y. Li, J. A. Golen, A. L. Rheingold, G. R. Weisman, E. H. Wong and C. J. Anderson, Dalton Trans., 2012, 41, 1938–1950 RSC.
- N. Bhatt, N. Soni, Y. S. Ha, W. Lee, D. N. Pandya, S. Sarkar, J. Y. Kim, H. Lee, S. H. Kim, G. I. An and J. Yoo, ACS Med. Chem. Lett., 2015, 6, 1162–1166 CrossRef CAS PubMed.
- L. Pazderová, T. David, V. Hlinová, J. Plutnar, J. Kotek, P. Lubal, V. Kubíček and P. Hermann, Inorg. Chem., 2020, 59, 8432–8443 CrossRef PubMed.
- Y. Guo, R. Ferdani and C. J. Anderson, Bioconjugate Chem., 2012, 23, 1470–1477 CrossRef CAS PubMed.
- N. Camus, Z. Halime, N. Le Bris, H. Bernard, C. Platas-Iglesias and R. Tripier, J. Org. Chem., 2014, 79, 1885–1899 CrossRef CAS PubMed.
- Z. Halime, M. Frindel, N. Camus, P.-Y. Orain, M. Lacombe, M. Chérel, J.-F. Gestin, A. Faivre-Chauvet and R. Tripier, Org. Biomol. Chem., 2015, 13, 11302–11314 RSC.
- R. J. Motekaitis, I. Murase and A. E. Martell, Inorg. Nucl. Chem. Lett., 1971, 7, 1103–1107 CrossRef CAS.
- G. Sravya, A. Balakrishna, G. V. Zyryanov, G. Mohan, C. S. Reddy and N. B. Reddy, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196, 353–381 CrossRef CAS.
- L. Wozniak and J. Chojnowski, Tetrahedron, 1989, 45, 2465–2524 CrossRef CAS.
- J. Dussart, J. Deschamp, E. Migianu-Griffoni and M. Lecouvey, Org. Process Res. Dev., 2020, 24, 637–651 CrossRef CAS.
- J. Dussart, N. Guedeney, J. Deschamp, M. Monteil, O. Gager, T. Legigan, E. Migianu-Griffoni and M. Lecouvey, Org. Biomol. Chem., 2018, 16, 6969–6979 RSC.
- J. Dussart, J. Deschamp, M. Monteil, O. Gager, E. Migianu-Griffoni and M. Lecouvey, Synthesis, 2019, 421–432 CAS.
- J. Dussart-Gautheret, J. Deschamp, M. Monteil, O. Gager, T. Legigan, E. Migianu-Griffoni and M. Lecouvey, J. Org. Chem., 2020, 85, 14559–14569 CrossRef CAS PubMed.
- J. K. Thottathil, D. E. Ryono, C. A. Przybyla, J. L. Moniot and R. Neubeck, Tetrahedron Lett., 1984, 25, 4741–4744 CrossRef CAS.
- M. Kalyva, A. L. Zografos, E. Kapourani, E. Giambazolias, L. Devel, A. Papakyriakou, V. Dive, Y. G. Lazarou and D. Georgiadis, Chem. – Eur. J., 2015, 21, 3278–3289 CrossRef CAS PubMed.
- J. K. Thottathil, C. A. Przybyla and J. L. Moniot, Tetrahedron Lett., 1984, 25, 4137–4740 CrossRef.
- S. Nishimura, Handbook of Hetergogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001, p. 254 Search PubMed.
- E. Brücher, G. Tircsó, Z. Baranyai, Z. Kovács and A. D. Sherry, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, 2nd ed, ed. A. Merbach, L. Helm and É. Tóth, Wiley, Chichester, U.K., 2013, pp. 157–208 Search PubMed.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216–217, 287–312 CrossRef.
- K. Zarschler, M. Kubeil and H. Stephan, RSC Adv., 2014, 4, 10157–10164 RSC.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS PubMed.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K.-C. Lam, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 10561–10572 CrossRef CAS.
- C. King, D. M. Roundhill and F. R. Fronczek, Inorg. Chem., 1986, 25, 1290–1292 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis of cb-cyclam, compound C and phthaloyl-glycine aldehyde, gradients used in chromatographic separations, structure of bis-1, 31P NMR spectrum of fully silylated 11, reduction of nitrile-containing compound 14 to compounds 24 and 24a, additional radiolabelling data, description of the solid-state structure of compound C with X-ray experimental details, and characterization data of the synthesized compounds. CCDC 2341020. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ob01473a |
| ‡ Current address: Institute of Organic Chemistry and Biochemistry, Academy of Science of the Czech Republic, Flemingovo náměstí 2, 160 00 Prague 6, Czech Republic. |
| This journal is © The Royal Society of Chemistry 2025 |